

Abstract
Protein phosphorylation is a critical post-translational modification (PTM). Despite recent technological advances in reversed-phase liquid chromatography (RPLC)-mass spectrometry (MS)-based proteomics, comprehensive phosphoproteomic coverage in complex biological systems remains challenging, especially for hydrophilic phosphopeptides with enriched regions of serines, threonines, and tyrosines that often orchestrate critical biological functions. To address this issue, we developed a simple, easily implemented method to introduce a commonly used tandem mass tag (TMT) to increase peptide hydrophobicity, effectively enhancing RPLC-MS analysis of hydrophilic peptides. Different from conventional TMT labeling, this method capitalizes on using a nonprimary amine buffer and TMT labeling occurring before C18-based solid phase extraction. Through phosphoproteomic analyses of MCF7 cells, we have demonstrated that this method can greatly increase the number of identified hydrophilic phosphopeptides and improve MS detection signals. We applied this method to study the peptide QPSSSR, a very hydrophilic tryptic peptide located on the C-terminus of the G protein-coupled receptors (GPCR) CXCR3. Identification of QPSSSR has never been reported, and we were unable to detect it by traditional methods. We validated our TMT labeling strategy by comparative RPLC-MS analyses of both a hydrophilic QPSSSR peptide library as well as common phosphopeptides. We further confirmed the utility of this method by quantifying QPSSSR phosphorylation abundances in HEK 293 cells under different treatment conditions predicted to alter QPSSSR phosphorylation. We anticipate that this simple TMT labeling method can be broadly used not only for decoding GPCR phosphoproteome but also for effective RPLC-MS analysis of other highly hydrophilic analytes.
Graphical Abstract
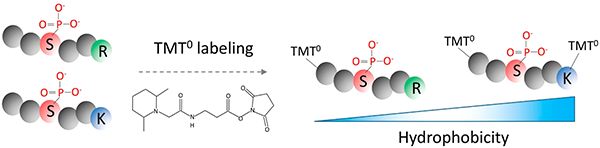
The ability to detect phosphorylation patterns is critical to understanding biological function. Improved phosphoproteome profiling workflows which integrate optimized immobilized metal affinity chromatography (IMAC) or metal oxide chromatography (MOC) with LC-MS enable identification of more than 10,000 phosphorylation sites in a single LC-MS/MS analysis (or 30,000 sites with multidimensional separation approaches).1–6 Despite these advances, significant gaps in phosphoproteome coverage still exist, especially for small, hydrophilic phosphopeptides with low abundance. One reason is that trypsin, the protease of choice for most shotgun proteomics studies,7 generates peptides well suited for ESI-MS.8 However, certain peptides containing hydrophilic phosphate groups can be too hydrophilic to be retained by the separation of peptide mixtures prior to MS. The RP retention of hydrophilic peptides can be enhanced by using a low column temperature (0°C) during sample loading,9 but this method often requires additional equipment for cooling the LC column which is not always feasible.
We sought to address these challenges in phosphoproteome profiling of hydrophilic peptides by studying a previously unidentified short and hydrophilic tryptic peptide, QPSSSR, and its phosphorylation status. QPSSSR is located on the C-terminus of the G protein-coupled receptor (GPCR) CXCR3 and plays a central role in T cell immune responses.10–14 GPCRs are the largest family of mammalian receptors15 and often undergo post-translational modifications (PTMs) to regulate their signaling. GPCRs are commonly expressed at densities as low as a few hundred femtomoles per milligram of protein,16 complicating analyses of this important receptor class that is the target of >30% of FDA-approved medications. All GPCRs share a common architecture that consists of an extracellular N-terminal sequence, seven transmembrane domains, and an intracellular C-terminal domain that is often enriched with serine and threonine residues. The active receptor is phosphorylated by G protein-coupled receptor kinases (GRKs) and other kinases,17 and it is thought that different phosphorylation patterns direct distinct intracellular signaling responses. An increasing number of ligands have been described that selectively activate some GPCR signaling pathways while not activating others downstream of a receptor. These “biased agonists” often display unique signaling and therapeutic profiles that correlate with distinct receptor conformations.18,19 Determining if such biased agonists lead to distinct GPCR phosphorylation patterns that direct these unique GPCR conformational patterns is an area of active research. We chose to study the GPCR CXCR3 because of its biased signaling properties and high therapeutic potential, as well as evidence that the C-terminus of CXCR3 is differentially phosphorylated following treatment with different agonists.10 However, of the two tryptic peptides with putative phosphorylation sites located on the C-terminus of CXCR3, only one phosphorylation site (S355) in one of two tryptic peptides (DSSWSETTEASYLGL) has been reported in the public phosphorylation site database.20 No potential phosphorylation sites on the second short and hydrophilic tryptic peptide (QPSSSR) have been reported.
To address the current gap in the ability to detect short, hydrophilic tryptic peptides/phosphopeptides with current LC-MS analytical workflows, we have developed a simple TMT-assisted method for significantly improving peptide hydrophobicity and hence LC-MS detectability. TMT reagents are isobaric labeling chemical compounds that use N-hydroxysuccinimide (NHS) chemistry to label primary amines (N-terminus and ε-amine group of lysine) with chemical tags and allow for sample multiplexing. Using the highly hydrophilic tryptic peptide QPSSSR as a case study, we tested the applicability of this method to expand our ability to detect hydrophilic peptides that often undergo post-translational modifications. By using a combinatorial QPSSSR peptide library with heavy isotope labeled reference standards combined with parallel reaction monitoring (PRM) analyses, our results demonstrate that TMT labeling can effectively increase the hydrophobicity of peptides. This method enabled the rapid and quantitative measurement of phosphorylation of the QPSSSR peptide that could not be detected to date with current techniques. We anticipate this method can be applied to detect other previously undetected hydrophilic phosphor peptides.
EXPERIMENTAL SECTION
Reagents.
3M Empore C18 membrane disks were obtained from 3M (St. Paul, MN, USA). Ni-NTA silica resins were purchased from Qiagen (Hilden, Germany). Sequencing grade-modified trypsin was from Promega (Madison, WI, USA). Urea, dithiothreitol (DTT), iodoacetamide, iron chloride, ammonium bicarbonate, trifluoroacetic acid (TFA), ethyl enediaminetetraacetic acid (EDTA), ammonium phosphate monobasic (NH4H2PO4), iron-(III) chloride (FeCl3), and formic acid (FA) were obtained from Sigma (St. Louis, MO). Fmoc-protected amino acids, 1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexa-fluorophosphate (HATU), were obtained from AAPPTec (Louisville, KY). A heavy isotope-labeled Fmoc-Arg(Pbf)-OH (13C6, 99%; 15N4, 99%) was obtained from Cambridge Isotope Labolatories (Tewksbury, MA, USA). 2-Chlorotrityl chloride resin (Cl-CP(Cl) ProTide) was obtained from CEM Corporation (Matthews, NC, USA). TMTzero (TMT0) labeling reagent was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Water was obtained from a Millipore Milli-Q system (Bedford, MA, USA).
QPSSSR Peptide Library Synthesis.
The peptide library was synthesized on 2-chlorotrityl chloride resin (loading: 0.5 mmol/g) using Fmoc solid phase peptide synthesis and the “split and combine” approach. Each Fmoc group removal was performed with 20% piperidine in DMF, each coupling with 10 eq Fmoc-amino acid, 10 eq HATU, and 30 eq DIEA in DMF. The C-terminal residue was introduced by 2 h incubation of a heavy isotope-labeled Fmoc-Arg(Pbf)-OH (10 μmol) and DIEA (30 μmol) in DCM with 15 μmol of resin; the remaining resin active sites were capped by incubation with 20% methanol and 10% DIEA in DCM. For each S/pS residue, the resin was split into two separate reaction vessels, and 1 h coupling with Fmoc-Ser(tBu)-OH or Fmoc-Ser(PO(OBzl)-OH)-OH was performed, confirmed with a ninhydrin test. After four washes with DMF, both portions of resin were combined for each Fmoc group removal. The peptide library was cleaved from resin and fully deprotected by 2 h incubation with a cleavage mixture of TIS/water/TFA (2.5:2.5:95). The cleavage solution was evaporated under reduced pressure on a rotary evaporator, and the remaining solid was washed five times with diethyl ether. The peptide library was dissolved in 50% acetonitrile and lyophilized to obtain 8.3 mg of final product.
Cell Culture and Protein Digestion.
The preparation of MCF-7 cells has been described in a previous report.21 Cells were harvested and washed with ice-cold phosphate-buffered saline (PBS), lysed in a lysis buffer containing 100 mMNH4HCO3, pH 8.0, 8 M urea, and a 1% phosphatase inhibitor, and sonicated in an ice bath for 3 min. The protein concentrations were determined via a Pierce BCA protein assay (Thermo Fisher Scientific). The lysates were digested based on a previous report.21 After digestion, samples were acidified by TFA, desalted by C18 SPE extraction, and concentrated for BCA assay analysis. Then, 300 μg of peptide was aliquoted and dried for TMT labeling.
CXCR3 Immunoprecipitation from HEK 293 Cells.
FLAG-CXCR3 was generated and transiently overexpressed in HEK 293 cells as previously described.11 To induce C-terminal phosphorylation, cells were treated with either vehicle or CXCL10 (100 nM) for 5 min. Cells were lysed using a buffer consisting of 50 mM Tris HCl, 150 mM NaCl, and 1% NP-40 at pH 7.5 containing phosphatase inhibitors: 100× inhibitor cocktail (Sigma), 100 mM Na3VO4 (Sigma), 500 mM NaF, 200 mM dithiothreitol (DTT), 300 mM fresh iodoacetamide (IAA), and 1 M trimethylammonium bicarbonate (TMAB). FLAG-CXCR3 was eluted with 0.1 M glycine HCl buffer (pH 3) and then neutralized with 0.5 M Tris HCl, pH 7.4, with 1.5 M NaCl (volume of elution:volume of Tris HCl = 7:1). The proteins were precipitated with acetone, and the precipitates were air-dried. Lysates were incubated for at least 4 h at 4°C with anti-FLAG magnetic beads (Thermo Fisher) and washed according to manufacturer protocol.
TMT0 Labeling.
Prior to phosphopeptide enrichment, the TMT labeling was performed as previously described. The tryptic peptides were dissolved with 200 mM HEPES (pH 8.5) and then mixed with a TMT0 reagent in 100% ACN. The recently optimized ratio of TMT to peptide amount, 1:1 (w/w), was used (i.e., 300 μg of peptides labeled by 300 μg of TMT0 reagent) and is significantly lower than standard 8:1 for reducing potential TMT modifications.22 While for tryptic peptides from CXCR3 immunoprecipitation, ~60 μg of TMT0 reagent was used for the TMT labeling because their concentration is too low to be determined via a Pierce BCA assay (Thermo Fisher Scientific). After incubation for 1 h at room temperature, the reaction was terminated by adding 5% hydroxylamine for 15 min. The TMT-labeled peptides were then acidified and diluted with the resultant ACN concentration of 4% before desalting. Then, the labeling peptides were desalted by C18 SPE (tryptic peptides from MCF-7 cell lysates) or C18 StageTip23 (synthesized heavy peptide library and tryptic peptides from CXCR3 immunoprecipitation).
IMAC Enrichment.
The procedure for phosphopeptide enrichment by an IMAC tip was similar to previous reports24–26 with minor modifications. Briefly, Ni2+ ions were removed by adding 50 mM EDTA in 1 M NaCl. The tip was then activated with 100 mM FeCl3. Tryptic peptides were reconstituted in 1% acetic acid and loaded onto the IMAC tip. After successive washes with 80%ACN/1%TFA and 1% (v/v) acetic acid, bound phosphopeptides were eluted with 200 mM phosphate salt and transferred to a C18 StageTip.23 Next, the C18 StageTip was washed with 1% FA, and peptides were eluted by 80%ACN/0.1%TFA. Eluted phosphopeptides were dried with SpeedVac and stored at −80°C until LC-MS/MS analysis.
LC-MS/MS Analysis.
Lyophilized peptides were recon stituted in 0.1%FA with 2% ACN and analyzed by LC-MS/217 MS. Details of LC-MS/MS analysis are described in the SI Methods.
Data Analysis.
Raw MS/MS data was processed with MaxQuant.27,28 The searching types were set to “Standard” for label free and “Reporter ion MS2” with “TMT0” for isobaric label measurements. A peptide search was performed with full tryptic digestion and allowed a maximum of two missed cleavages against a human UniProt database (version May 20, 2015) or CXCR3 only databases (including contaminates) for targeting search. The acetylation (protein N-term), oxidation (M), and phospho (STY) were set as variable modifications, and the carbamidomethyl (C) was set as a fixed modification. The heavy labeling of Lys8 and Arg10 were set as variable modifications for synthetic heavy peptide library. The false discovery rate (FDR) was set to 1% at the level of proteins, peptides, and modifications. The minimal peptide length for identification was set as five amino acids for targeted searching of peptides fromCXCR3.
RESULTS AND DISCUSSION
QPSSSR Peptide Library without TMT0 Labeling.
Using a standard proteomic sample processing protocol,4 in-depth global phosphoproteomic analysis of HEK 293 cells overexpressing the GPCR CXCR3 could not identify any hydrophilic QPSSSR phosphopeptides within the intracellular C-terminus of CXCR3 (data not shown). To facilitate identification of potential phosphorylation sites of QPSSSR, we synthesized a corresponding heavy isotope-labeled peptide library that contained all eight possible combinations of nonphosphorylated and phosphorylated forms of the three serine (23) residues (i.e., the combinatorial QPSSSR peptide247 library). Phosphorylation patterns were confirmed by direct infusion analysis (Figure S1a). However, initial analysis of the QPSSSR peptide library by the commonly used RPLC-MS/MS proteomics platforms yielded poor results due to the low hydrophilicity of the peptide library (Figure S1b). To reduce potential peptide losses during RPLC-MS/MS analysis, the front-end C18 trap column was removed, and the QPSSSR peptide library was directly loaded onto the capillary C18 analytical column. The extracted ion chromatograms (XICs) at the MS1 level demonstrated that nonphosphorylated and singly and doubly phosphorylated QPSSSR peptides could be detected with RPLC-MS (Figure 1a). However, the peptides eluted at the very early LC gradient (≤5 min) displayed broad diffusion peaks (Figure 1b), consistent with being in the unretained “void volume”, precluding specific peptide identification and quantification.
Figure 1.
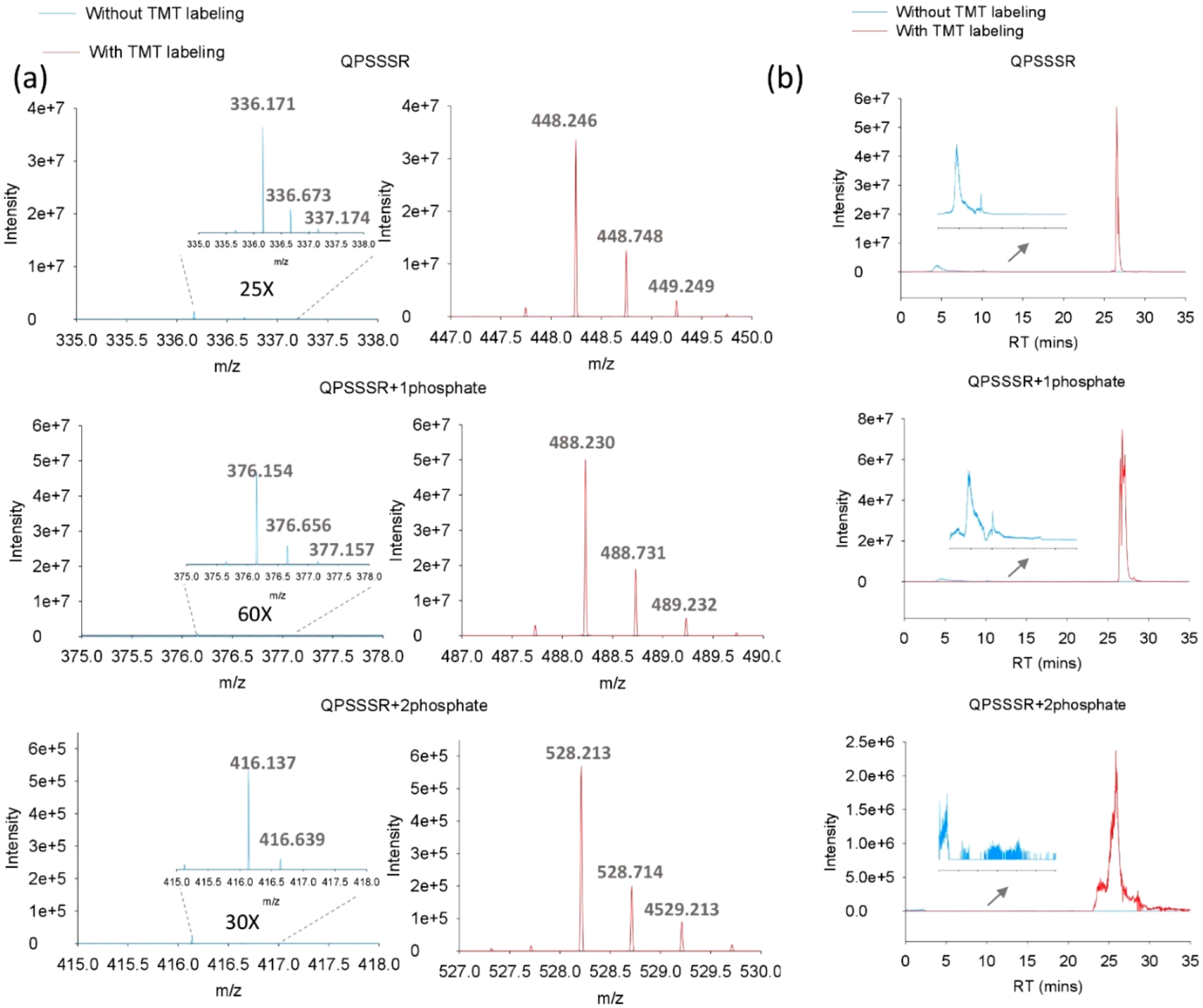
TMT labeling increases MS1 single intensity and alters retention time. The synthetic QPSSSR library phosphopeptides with (red color) and without (blue color) TMT labeling were directly analyzed by LC-MS/MS without using a trapping column. (a) Representative MS1 signals.(b) Extracted ion chromatograms (XICs) for the QPSSSR phosphopeptide library with and without TMT0 labeling. RT, retention time.
Increasing Peptide Hydrophobicity with TMT Label ing.
To increase peptide library hydrophobicity, we sought to introduce a hydrophobic tag to the peptides through well-established conjugation chemistry with a NHS ester reacting with primary amines at the N-terminus and ε-amine group of lysine residues. This conjugation reaction has been broadly used in modern MS-based proteomics.4,6,29 For example, a tandem mass tag (TMT) is a chemical label that utilizes NHS chemistry for high-throughput labeling and multiplexed quantitative proteomic analyses.4,6 Before introducing the TMT tag into the QPSSSR peptide library, we systematically evaluated the effect of TMT labeling on peptide RPLC retention time using the phosphoproteome from MCF-7 cell lysates. Equal amounts of tryptic peptides from MCF-7 cell lysates were labeled with and without TMT tags (i.e., TMT0), respectively, followed by purification with IMAC (Figure S2a).Global phosphoproteome profiling demonstrated that the number of identified phosphopeptides with TMT0 labeling (N = 10,192) was comparable to that without TMT0 labeling (N = 11,122) (Figure S2b). A total of 15,906 unique phosphopeptides were identified from the two sets of samples with 5408 (34%) phosphopeptides in common (Figure S2c), an overlap that is significantly lower than the typical overlap for replicate analysis of phosphopeptides (~50%), suggesting that TMT labeling allowed for the identification of phosphopetides that otherwise would have been overlooked with traditional analytical strategies. Indeed, evaluation of the changes in peptide retention time before and after TMT0 labeling showed that TMT0 labeling led to greatly increased peptide retention time (Figure 2a) and thus identify phosphopeptides with lower hydrophobicity (Figure 2b). The Kyte–Doolittle scale30 was used to determine the hydrophobicity of peptides. After TMT labeling, more hydrophilic peptides (median value = −0.97) can be detected when compared to peptides without TMT labeling (median value = −0.87) (Figure 2b). We also noted that the median retention time values of phosphopeptides containing lysine residues (~14.7 min) were significantly higher than those without lysine residues (~6.4 min) (Figure 2c) due to the presence of an additional TMT0 tag on a lysine.The increased phosphopeptide hydrophobicity is primarily determined by the introduced TMT tag. This was further confirmed by this observation that the number of phosphategroup has no obvious effect on the retention time shift (Figure S3).
Figure 2.
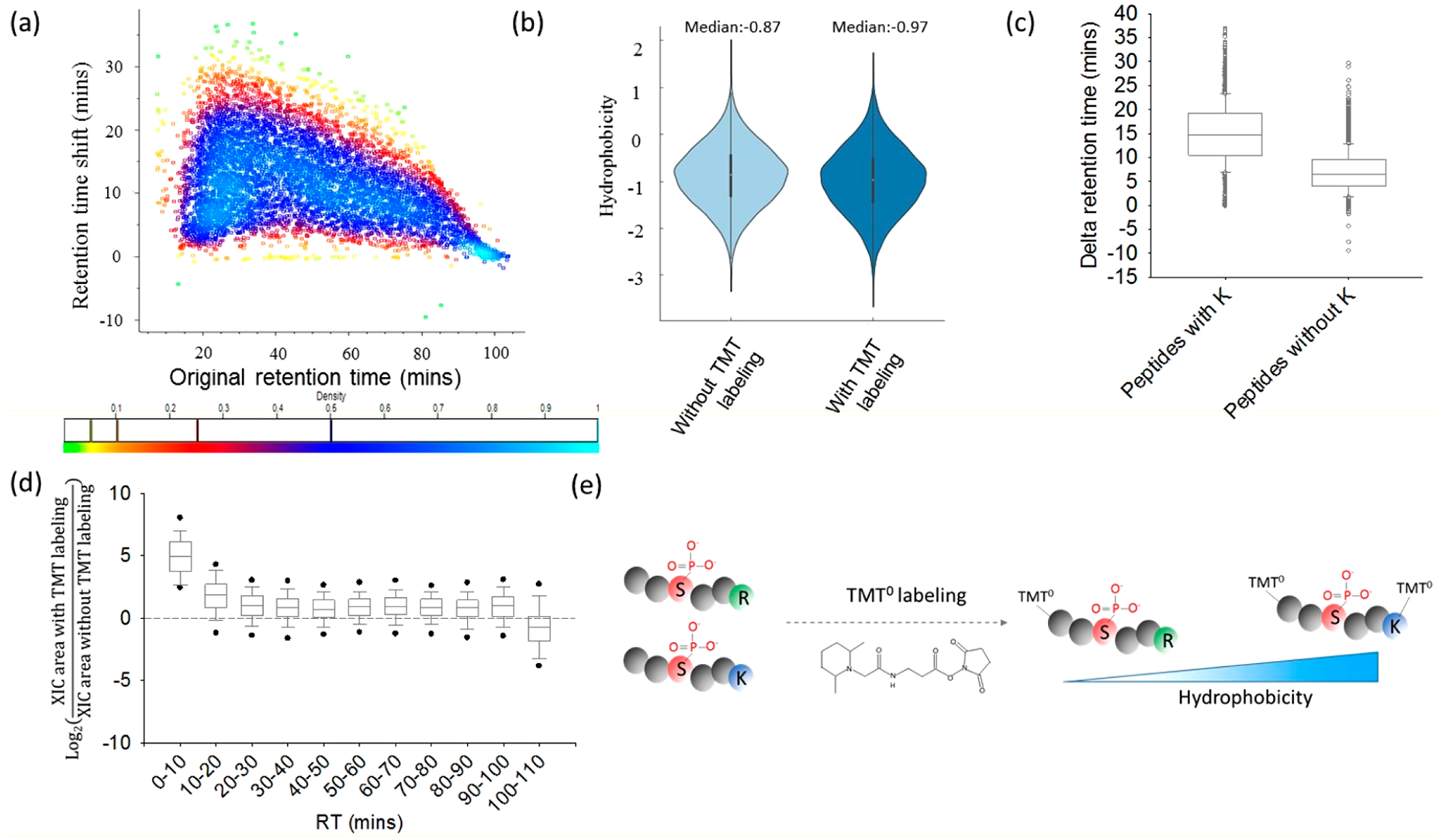
TMT labeling provides a greater shift in retention time for peptides containing lysine. (a) Retention time shift of phosphopeptides after TMT labeling (light blue, data points with the highest density; bright green, data points with the lowest density). (b) Hydrophobicity distribution of identified phosphopeptides. (c) Median retention time shift of phosphopeptides with or without lysine (K) residues. (d) Peak area ratio of phosphopeptides before and after TMT labeling. (e) Schematic diagram of increasing peptide hydrophobicity with TMT labeling.
Besides that, after systematic analysis of the shared phosphopeptides between with and without TMT labeling, the TMT labeling was also found to improve the MS signal (the median value of 0.93 at log2 scale), especially for early eluted hydrophilic phosphopeptides (Figure 2d). In addition to a TMT tag, other isobaric tags such as iTRAQ 4-plex were observed to lead to differences in LC elution times.31 These findings demonstrate that the addition of TMT tags on the peptides significantly increased peptide hydrophobicity (Figure 2e).
QPSSSR Peptide Library with TMT0 Labeling.
Given the increased phosphopeptide hydrophobicity following TMT labeling, we proceeded to apply the TMT labeling strategy to the QPSSSR peptide library. TMT labeling shifted the peptide library retention time from ≤5 min to ~25 min with the same LC gradient (Figure 1. Furthermore, the increased hydrophobicity led to stronger retention of the QPSSSR phosphopeptides on the C18 column, resulting in greater than 25-fold higher peptide signal intensity at the MS1 level than that without TMT0 labeling (Figure 1b). Interestingly, with TMT labeling, the high-resolution RPLC separation even allowed for partially resolving isomeric phosphopeptides with one peak for nonphosphopeptide, three peaks for singlyphosphopeptide, and three peaks for doubly phosphopeptide (Figure 1b), corresponding to every theoretical phosphorylation state of QPSSSR aside from the QPpSpSpSR, which given steric hindrance is unlikely to form under biological conditions. This observation was further confirmed with LC-MS/MS sequencing of the peptide library (Table 1). In addition, due to its increased hydrophobicity, the TMT0-labeled peptide library can still be detected even with the C18 trap column in place (data not shown). These results demonstrate that the highly hydrophilic QPSSSR and its corresponding phosphopeptides can be detected and resolved utilizing TMT labeling combined with RPLC-MS/MS analyses that would otherwise be impossible under standard analytical conditions.
Table 1.
Summary of Identified QPSSSR Peptide Library after TMT0 Labeling by Maxquanta
| m/z | z | Modified sequence | Site probability | Score | RT (min) |
|---|---|---|---|---|---|
| 448.25 | 2 | QPSSR | 52 | 25.95 | |
| 488.23 | 2 | QPpSSSR | QPS(0.764)S(0.118)S(0.118)R | 46 | 38.36 |
| 488.23 | 2 | QPSpSSR | QPS(0.049)S(0.759)S(0.192)R | 70 | 26.77 |
| 488.23 | 2 | QPSSpSR | QPS(0.001)S(0.28)S(0.719)R | 64 | 29.37 |
| 528.21 | 2 | QPpSSpSR | QPS(0.84)S(0.302)S(0.857)R | 52 | 29.27 |
| 528.21 | 2 | QPpSpSSR | QPS(0.741)S(0.959)S(0.3)R | 42 | 31.09 |
| 528.21 | 2 | QPSpSpSR | QPS(0.183)S(0.913)S(0.904)R | 97 | 31.6 |
The number included in parentheses indicates the probability of phosphorylation at the indicated serine residue. The score is the Andromeda score for the best associated MS/MS spectrum. No triply phosphorylated peptides were identified.
Detection and Quantification of Endogenous Non-phosphorylated and Singly Phosphorylated QPSSSR Peptides.
Given the success of TMT0 labeling for detecting and (partially) separating distinct phosphopetides generated in a library, we next applied this method to more complex biological conditions. We again focused on the tryptic peptide QPSSSR located on the C-terminus of the GPCR CXCR3, which is known to be phosphorylated by agonist treatment, although the specific serine residues on the C-terminus that are phosphorylated are currently unknown. To promote phosphorylation of CXCR3, we stimulated CXCR3 with CXCL10, a member of the CXC chemokine family which acts as anagonist of CXCR3.32 FLAG-CXCR3 was overexpressed inHEK 293 cells, treated with either vehicle or CXCL10, and immunoprecipitated (IP) from whole cell lysates. We applied our recently developed single-tube digestion method33 for sample processing to minimize losses, and a nonprimary amine buffer (i.e., HEPES buffer34,35) was used for effective sample labeling with the TMT0 reagent. TMT0 labeling was performed prior to C18-based SPE to increase peptide hydrophobicity and prevent phosphopeptide losses (Figure 3a). Tryptic peptides generated from digestion of CXCR3 IP samples treated with either vehicle or CXCL10 were analyzed by LC-MS/MS in the parallel reaction monitoring (PRM) mode. The MS1 signal of the endogenous peptide was detected based on the same elution time, Δm/z of 5.004 different from the synthesized heavy isotopic peptide (Figure 3b) and similar to the MS/MS patterns between the endogenous peptides and their corresponding heavy standard peptides (Figure 3c). Besides that, a neutral loss of a phosphate group with a reduction of 98 Da in mass is very common for MS/MS sequencing of phosphopeptides. Thus, the loss of 98 Da from the precursor was used as a signature for phosphopeptide identification (Figure 3c). The high similarity in the MS/MS spectra (R2 > 0.99) between heavy isotope library-generated QPSSSR peptides and endogenous QPSSSR peptides generated through tryptic digestion of CXCR3 further confirmed endogenous detection by correlation of relative transition intensities (Figure S4). With the use of Maxquant for quantification (i.e., iBAQ, Intensity Based Absolute Quantification), the expression levels of CXCR3 were similar between the two sets of samples (Figure S5), suggesting that the IP enrichment efficiency was consistent across agonist treatment conditions. Based on these results, we are able to confidently identify a singly phosphorylated QPSSSR peptide in both vehicle and CXCL10-treated HEK 293 cells. To our knowledge, this is the first report of a QPSSSR phosphopeptide identified from CXCR3.
Figure 3.
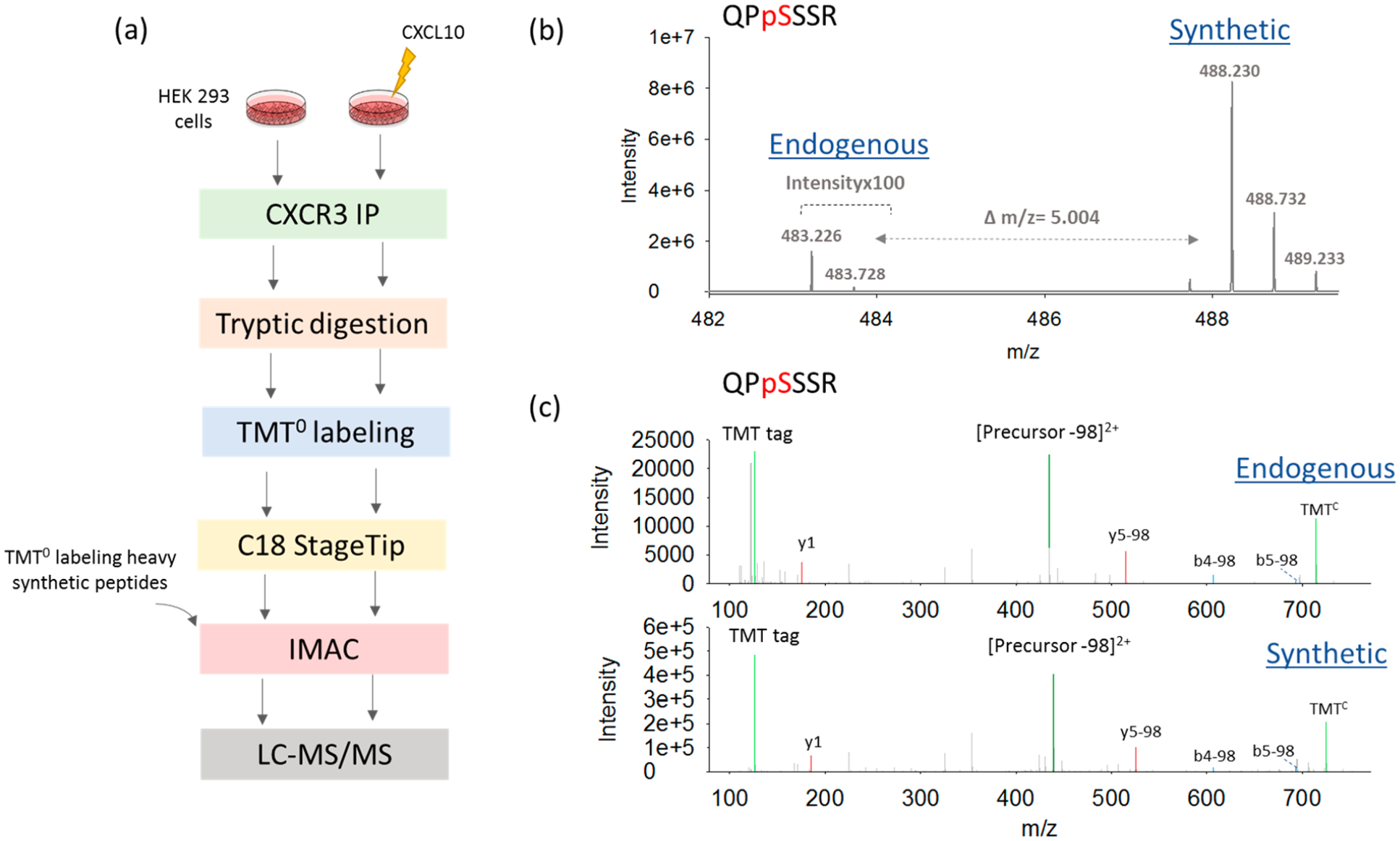
Endogenous detection of singly phosphorylated QPSSSR peptides. (a) Workflow for proteomic analysis of immunoprecipitated CXCR3 samples. Evidence for detection of endogenous singly phosphorylated QPSSSR peptides at (b) the MS1 level and (c) the MS2 level. The Δm/z of5.004 is the m/z difference between endogenous and synthesized heavy isotopic peptides. Neutral loss of phosphate group with a reduction of 98 Da in mass is very common for MS/MS sequencing of phosphopeptides, and thus, it was used as a signature for phosphopeptide identification.
In addition to defining specific sites that undergo PTMs, it is important to know the relative abundance of such PTMs. To estimate the stoichiometric changes of QPSSSR phosphorylation, we proceeded to spike the heavy isotope-labeled peptide library into CXCR3 IP samples prior to IMAC enrichment. This strategy makes use of PRM-based targeted quantification to define the absolute degree of QPSSSR phosphorylation using the heavy-labeled QPSSSR phosphopeptides from the library as a reference. The bound and the flow-through fractions were analyzed by PRM to quantify endogenous levels of both the singly phosphorylated peptides and their nonphosphorylated versions (Figure 4. For accurate determination of phosphorylation stoichiometries, the apex intensities of four abundant product ions (b3, b5, y5, and TMTc ions, complement reporter ions36) without interferences were used for quantification, as the peptide library408 contains singly phosphorylated QPSSSR peptide isomers (i.e., QPpSSSR, QPSpSSR, or QPSSpSR) with similar elution times. These measurements showed the endogenous peptides to have detectable levels of phosphorylation in these samples at a single site. In contrast to the endogenous samples, the peptide library showed broader and unresolved peaks for the three phosphopeptides (Figure 4a), posing a challenge for PRM quantification (i.e., the inability to reliably distinguish the three phosphosites). However, such confounding issues do not exist for the nonphosphorylated QPSSSR which displayed a similar peak width in the peptide library and endogenous QPSSSR peptide analyses (Figure 4a). The stoichiometries of singly phosphorylated peptides are shown in Figure 4b. As shown in Figure 4c, the precision of the PRM analyses enabled the detection of a significant increase in phosphorylation stoichiometry for S349 of CXCR3 (QPpSSSR) after CXCL10 treatment (from 26% to 33%, p = 0.03 based on t test analysis). This result is consistent with prior findings demonstrating that phosphorylation of the CXCR3 C-terminus increases with agonist stimulation.10,11 To the best of our knowledge, the C-terminus of CXCR3, and likely many other GPCR proteins, have been overlooked with traditional LC-MS-based analytical techniques. Our results demonstrate the broad utility of this TMT0 labeling method for rescuing highly hydrophilic phosphopeptides that can likely aid in expanding our knowledge of biological regulation through PTMs.
Figure 4.
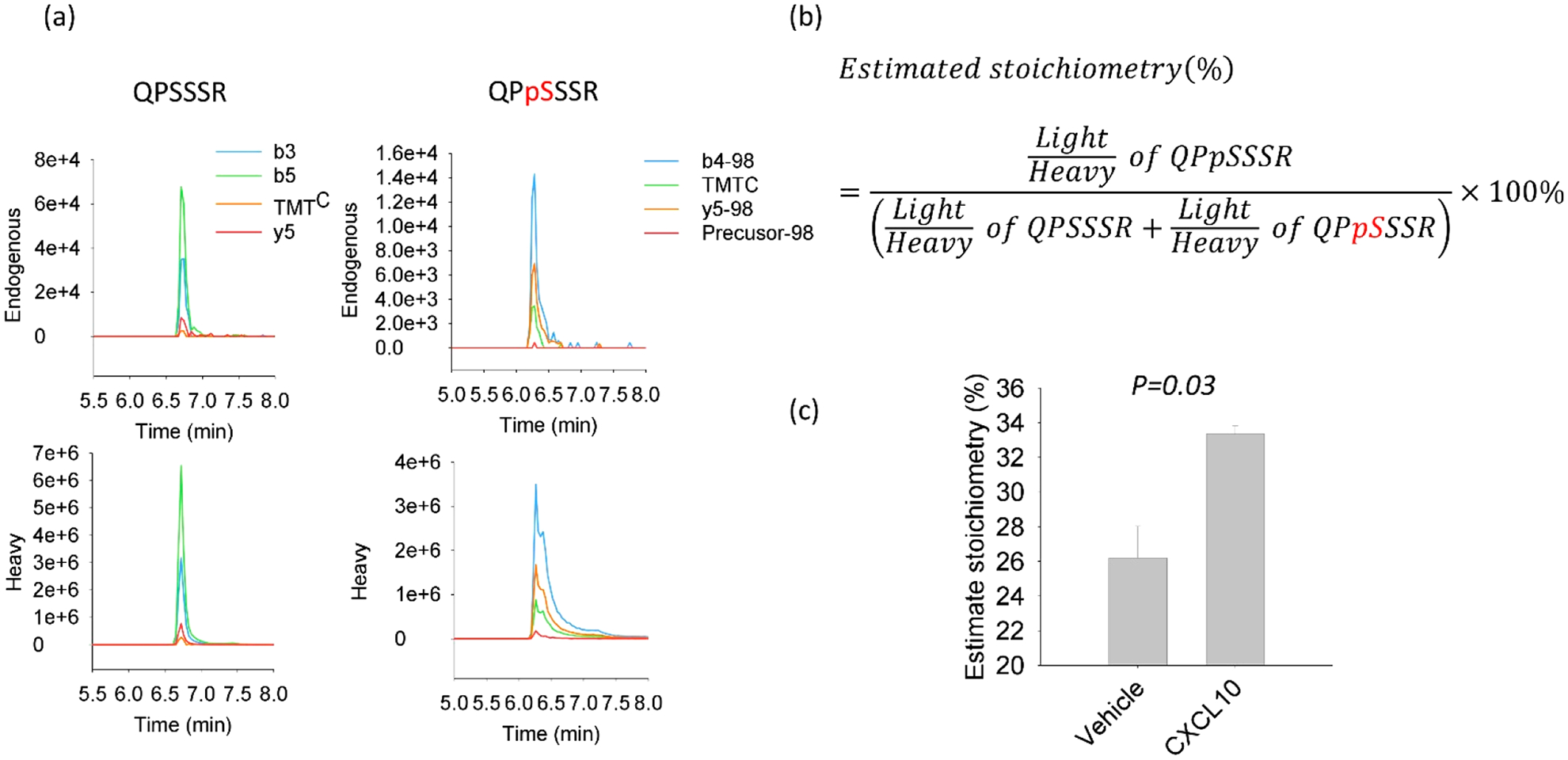
Quantification of endogenous QPSSSR peptides. QPSSSR phosphopeptides obtained through immunoprecipitation and tryptic digestion of CXCR3 were referenced to spiked-in as heavy QPSSSR library standards. (a) XICs of fragment ions. (b) Equation utilized for estimating phosphorylation stoichiometry of endogenous QPSSSR. (c) Estimated phosphorylation stoichiometry for singly phosphorylated QPSSSR peptides for samples treated either with vehicle or CXCL10. Error bars indicate mean ± SD. The p-value calculated with t test was 0.03.
Alternatives To Address Hydrophilic and/or Isomeric Phosphopeptide Measurements.
TMT reagents are commercially available and routinely used for multiplexed quantitative proteomics with improved MS detection sensitivity. Thus, the TMT labeling method should have broad utility for addressing challenges of effective detection and quantification of highly hydrophilic peptides and phosphopeptides and, particularly, multiply phosphorylated peptides. Besides the TMT reagents, other hydrophobic reagents (e.g., succinimidyl 3-(perfluorooctyl)propionate and 2,4,6-trinitro-benzenesulfonic acid) may also have utility for increasing peptide hydrophobicity. An additional challenge for isomeric phosphopeptides with current LC-MS based proteomics platforms is the need for site-specific product ions for their differentiation especially for multisite phosphopeptides. Different fragmentation modes such as electron transfer dissociation (ETD) or ultraviolet photodissociation (UVPD), or their combination, may be useful to facilitate the differentiation of the TMT-labeled isomeric phosphopeptides. Another promising technology is ion mobility separations, particularly when using ultrahigh-resolution SLIM (Structures for Lossless Ion Manipulations)-based IMS (ion-mobility spectrometry). SLIM-based IMS separations are highly orthogonal to RPLC as well as more highly reproducible,37 improving their discrimination of isomeric hydrophilic phosphopeptides and alleviating the potential drawback in phosphopeptide identification of using MS/MS after TMT labeling.
CONCLUSION
We have developed a simple, easily implemented TMT labeling method to rescue and increase identification of hydrophilic phosphopeptides that would otherwise be overlooked by current LC-MS analytical strategies. Different from standard TMT labeling sample preparation, this method capitalizes on using a nonprimary amine buffer and TMT0 labeling occurring before (rather than after) C18-based SPE to avoid hydrophilic peptide losses. By increasing the hydrophobicity of very hydrophilic peptides for RPLC-MS analysis through TMT0 labeling, we were able to identify a previously unreported peptide from the GPCR CXCR3, QPSSSR, that regulates important biological functions. This method allowed for the detection of the unphosphorylated and phosphorylated forms of QPSSSR from both a synthesized library as well as complex biological samples. With QPSSSR analyses as a proof-of-concept, we anticipate that our TMT0 labeling method will have broad utility for improving RPLC-MS analysis of very hydrophilic peptides (e.g., phosphopeptides with multiple phosphorylation sites) that are undersampled and under-reported to date.
Supplementary Material
ACKNOWLEDGMENTS
Portions of the research were supported by P41GM103493 (R.D.S), R21CA223715 (T.S.), T32GM7171 (J.S.S.), the Duke Medical Scientist Training Program (J.S.S.), R01GM122798 (S.R.), and Burroughs Wellcome Career Award for Medical Scientists (S.R.). The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, a national scientific user facility sponsored by the United States of America Department of Energy under Contract DE-AC05-76RL0 1830.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b01814.
Figures and tables. (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Humphrey SJ; Azimifar SB; Mann M Nat. Biotechnol 2015, 33, 990–995. [DOI] [PubMed] [Google Scholar]
- (2).Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC;Haldrup C; Bramsen JB; Sorensen KD; Hoyer S; Orntoft TF; Andersen CL; Nielsen ML; Olsen JV Cell Syst 2017, 4, 587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wakabayashi M; Kyono Y; Sugiyama N; Ishihama Y Anal. Chem 2015, 87, 10213–10221. [DOI] [PubMed] [Google Scholar]
- (4).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA; Petyuk VA; Thomas SN; Mani DR; Mundt F; Moore RJ; Hu Y; Zhao R; Schnaubelt M; Keshishian H; Monroe ME; Zhang Z; Udeshi ND; Mani D; Davies SR; Townsend RR; Chan DW; Smith RD; Zhang H; Liu T; Carr SA Nat. Protoc 2018, 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hogrebe A; von Stechow L; Bekker-Jensen DB; Weinert BT; Kelstrup CD; Olsen JV Nat. Commun 2018, 9, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yi L; Tsai CF; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian WJ Anal. Chem 2019, 91 (9), 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Giansanti P; Aye TT; van den Toorn H; Peng M; van Breukelen B; Heck AJ Cell Rep. 2015, 11, 1834–1843. [DOI] [PubMed] [Google Scholar]
- (8).Olsen JV; Ong SE; Mann M Mol. Cell. Proteomics 2004, 3, 608–614. [DOI] [PubMed] [Google Scholar]
- (9).Young C; Podtelejnikov AV; Nielsen ML J. Proteome Res 2017, 16, 2307–2317. [DOI] [PubMed] [Google Scholar]
- (10).Colvin RA; Campanella GS; Sun J; Luster AD J. Biol. Chem 2004, 279, 30219–30227. [DOI] [PubMed] [Google Scholar]
- (11).Smith JS; Alagesan P; Desai NK; Pack TF; Wu JH; Inoue A; Freedman NJ; Rajagopal S Mol. Pharmacol 2017, 92, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smith JS; Nicholson LT; Suwanpradid J; Glenn RA; Knape NM; Alagesan P; Gundry JN; Wehrman TS; Atwater AR; Gunn MD; MacLeod AS; Rajagopal S Sci. Signaling 2018,11 (pii), eaaq1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rashighi M; Agarwal P; Richmond JM; Harris TH; Dresser K; Su MW; Zhou Y; Deng A; Hunter CA; Luster AD; Harris JE Sci. Transl. Med 2014, 6, 223ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chow MT; Ozga AJ; Servis RL; Frederick DT; Lo JA; Fisher DE; Freeman GJ; Boland GM; Luster AD Immunity 2019, 50, 1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Santos R; Ursu O; Gaulton A; Bento AP; Donadi RS; Bologa CG; Karlsson A; Al-Lazikani B; Hersey A; Oprea TI; Overington JP Nat. Rev. Drug Discovery 2017, 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Andressen KW; Norum JH; Levy FO; Krobert KA Mol. Pharmacol 2006, 69, 207–215. [DOI] [PubMed] [Google Scholar]
- (17).Shenoy SK; Lefkowitz RJ Trends Pharmacol. Sci 2011, 32, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Viscusi ER; Webster L; Kuss M; Daniels S; Bolognese JA; Zuckerman S; Soergel DG; Subach RA; Cook E; Skobieranda F Pain 2016, 157, 264–272. [DOI] [PubMed] [Google Scholar]
- (19).Manglik A; Lin H; Aryal DK; McCorvy JD; Dengler D; Corder G; Levit A; Kling RC; Bernat V; Hubner H; Huang X-P; Sassano MF; Giguere PM; Lober S; Da Duan; Scherrer G;Kobilka BK; Gmeiner P; Roth BL; Shoichet BK Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hornbeck PV; Zhang B; Murray B; Kornhauser JM; Latham V; Skrzypek E Nucleic Acids Res. 2015, 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yi L; Shi T; Gritsenko MA; X’Avia Chan CY; Fillmore TL; Hess BM; Swensen AC; Liu T; Smith RD; Wiley HS; Qian WJ Anal. Chem 2018, 90, 5256–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zecha J; Satpathy S; Kanashova T; Avanessian SC; Kane MH; Clauser KR; Mertins P; Carr SA; Kuster B Mol. Cell. Proteomics 2019, 18, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rappsilber J; Mann M; Ishihama Y Nat. Protoc 2007, 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- (24).Tsai CF; Wang YT; Yen HY; Tsou CC; Ku WC; Lin PY; Chen HY; Nesvizhskii AI; Ishihama Y; Chen YJ Nat. Commun 2015, 6, 6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsai CF; Hsu CC; Hung JN; Wang YT; Choong WK; Zeng MY; Lin PY; Hong RW; Sung TY; Chen YJ Anal. Chem 2014, 86, 685–693. [DOI] [PubMed] [Google Scholar]
- (26).Tsai CF; Wang YT; Chen YR; Lai CY; Lin PY; Pan KT; Chen JY; Khoo KH; Chen YJ J. Proteome Res 2008, 7, 4058–4069. [DOI] [PubMed] [Google Scholar]
- (27).Cox J; Mann M Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (28).Tyanova S; Temu T; Cox J Nat. Protoc 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- (29).Dean RA; Overall CM Mol. Cell. Proteomics 2007, 6, 611–623. [DOI] [PubMed] [Google Scholar]
- (30).Kyte J; Doolittle RF J. Mol. Biol 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- (31).Pichler P; Kocher T; Holzmann J; Mazanek M; Taus T; Ammerer G; Mechtler K Anal. Chem 2010, 82, 6549–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Taub DD; Lloyd AR; Conlon K; Wang JM; Ortaldo JR; Harada A; Matsushima K; Kelvin DJ; Oppenheim JJ J. Exp. Med 1993, 177, 1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang P; Gaffrey MJ; Zhu Y; Chrisler WB; Fillmore TL; Yi L; Nicora CD; Zhang T; Wu H; Jacobs J; Tang K; Kagan J; Srivastava S; Rodland KD; Qian WJ; Smith RD; Liu T; Wiley HS; Shi T Anal. Chem 2019, 91, 1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Isasa M; Rose CM; Elsasser S; Navarrete-Perea J; Paulo JA; Finley DJ; Gygi SP J. Proteome Res 2015, 14, 5306–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA J. Proteome Res 2018, 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wuhr M; Haas W; McAlister GC; Peshkin L; Rad R; Kirschner MW; Gygi SP Anal. Chem 2012, 84, 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chouinard CD; Nagy G; Webb IK; Shi T; Baker ES; Prost SA; Liu T; Ibrahim YM; Smith RD Anal. Chem 2018, 90, 10889–10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.