

Abstract
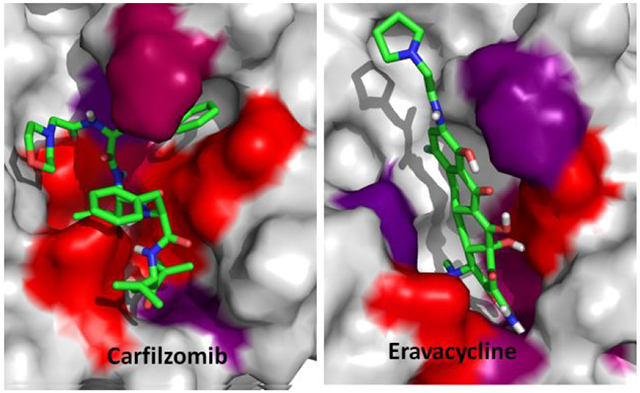
The recent outbreak of novel coronavirus disease −19 (COVID-19) calls for and welcomes possible treatment strategies using drugs on the market. It is very efficient to apply computer-aided drug design techniques to quickly identify promising drug repurposing candidates, especially after the detailed 3D-structures of key virous proteins are resolved. The virus causing COVID-19 is SARS-Cov-2. Taking the advantage of a recently released crystal structure of SARS-Cov-2 main protease in complex with a covalently-bonded inhibitor, N3,1 I conducted virtual docking screening of approved drugs and drug candidates in clinical trials. For the top docking hits, I then performed molecular dynamics simulations followed by binding free energy calculations using an endpoint method called MM-PBSA-WSAS (Molecular Mechanics-Poisson Boltzmann Surface Area-Weighted Solvent-Accessible Surface Area).2-4 Several promising known drugs stand out as potential inhibitors of SARS-Cov-2 main protease, including Carfilzomib, Eravacycline, Valrubicin, Lopinavir and Elbasvir. Carfilzomib, an approved anti-cancer drug acting as a proteasome inhibitor, has the best MM-PBSA-WSAS binding free energy, −13.8 kcal/mol. The second-best repurposing drug candidate, eravacycline, is synthetic halogenated tetracycline class antibiotic. Streptomycin, another antibiotic and a charged molecule, also demonstrates some inhibitory effect, even though the predicted binding free energy of the charged form (−3.8 kcal/mol) is not nearly as low as that of the neutral form (−7.9 kcal/mol). One bioactive, PubChem 23727975, has a binding free energy of −12.9 kcal/mol. Detailed receptor-ligand interactions were analyzed and hot spots for the receptor-ligand binding were identified. I found that one hotspot residue HIS41, is a conserved residue across many viruses including SARS-Cov, SARS-Cov-2, MERS-Cov, and HCV. The findings of this study can facilitate rational drug design targeting the SARS-Cov-2 main protease.
Graphical Abstract
1. Introduction
A great application of drug repurposing is to identify drugs which were developed for treating other diseases to treat a new disease. Drug repurposing can be achieved by conducting systematic drug-drug target interaction (DTI) and drug-drug interaction (DDI) analyses. We have conducted a survey on DTIs collected by the DrugBank database5 and found that on average each drug has 3 drug targets and each drug target has 4.7 drugs.6 The analysis demonstrates that polypharmacology is a common phenomenon. It is important to identify potential DTIs for both approved drugs and drug candidates, which serves as the basis of repurposing drugs and selection of drug targets without DTIs that may cause side-effects. Polypharmacology opens novel avenues to rationally design next generation of more effective but less toxic therapeutic agents. Computer-aided drug design (CADD) has been playing essential roles in modern drug discovery and development. To balance the computational efficiency and accuracy, a hieratical strategy employing different types of scoring functions are applied in both the drug lead identification and optimization phases. A docking scoring function, such as the one employed by the Glide docking program,7 is very efficient and thus can be utilized to screen a large library, but it is not very accurate. On the other hand, the molecular mechanical force field (MMFF)-based scoring functions, are physical and more accurate, but much less efficient. With the ever increasing computer power, MMFF-based free energy calculation methods, such as the endpoint MM-PBSA and MM-GBSA (Molecular Mechanics Generalized Born Surface Area) methods2, 3, 8-21 and the alchemical thermodynamic integration (TI) and free energy perturbation (FEP) methods,22, 23 have been extensively applied in structure-based drug discovery projects. We have developed a hierarchical virtual screening (HVS)to balance the efficiency and accuracy and improve the success rate of rational drug design.8, 24 The newly released crystal structure of SARS-Cov-2 main protease1 provides a solid structural basis for identification of drugs that might interact with this protein target. In this work, I applied multiscale modeling techniques to identify drugs that may be repurposed to target SARS-Cov-2 main protease. Flexible docking and MM-PBSA-WSAS were applied as the 1st and 2nd filters, respectively, to improve the efficiency and accuracy of HVS in inhibitor identification for SARS-Cov-2 main protease. Compared to the experimental means, CADD-based approaches are more efficient in providing possible treatment solutions for epidemic disease outbreaks like COVID-19. The detailed ligand-residue interaction profile as well as the decomposition of binding free energy into different components provide insight into rationally designing potent and selective inhibitors of ARS-Cov-2 main protease.
2. Methodologies.
I conducted a hierarchical virtual screening (HVS) using the newly resolved crystal structure of SARS-Cov-2 main protease (Resolution 2.16Å).1 Later on more crystal structures of SARS-Cov-2 man protease were resolved.25 Two types of HVS filters were employed: Glide7 flexible docking followed by MM-PBSA-WSAS.2, 4 Detailed computational methods are described below.
2.1. Docking Screening
The crystal structure was first treated using the protein structure preparation wizard provided by the Schrodinger software, followed by docking grid generation. Glide flexible docking was performed using the default settings except that the formation of intramolecular hydrogen bonds was rewarded and the enhancement of planarity of conjugated pi groups was turned on. The co-crystal ligand, N3, was covalently bonded to CYS145. I generated a new version of N3, N3’ by breaking the covalent bond and filling in open valence. I then evaluated whether Glide flexible docking can reproduce the native binding pose. In addition, a dataset of approved drugs was prepared using DrugBank,5 and a set of PubChem compounds which are structurally similar to Lopinavir were enriched for docking screenings. Lopinavir, a potent inhibitor of HIV-1 protease,26 was found effective in treating COVID-19 patients. Top hits from the docking screenings were advanced to the next HVS filter – MM-PBSA-WSAS. The 3D-structures of the screening compounds were generated using the OpenBabel software.27
2.2. System setup for molecular dynamics (MD) simulation and free energy calculation
MD simulations were first performed for a docking hit for two purposes: (1) studying the relative stability of the ligand residing in the binding pocket; (2) sampling a set of conformations for MM-PBSA-WSAS binding free energy calculations and MM-GBSA residue-ligand binding free energy decomposition analysis. A MD system consisted of one copy of SARS-Cov-2 main protease, one copy of docked ligand, 17597 TIP3P28 water molecules, about 50 Na+ and Cl− ions depending on the charge state of the ligand. The whole system was neutralized. For the force field parameters, the partial atomic charges of ligands were derived using the RESP29 program to fit the HF/6-31G* electrostatic potentials generated using the GAUSSIAN 16 software package30. The other force field parameters of ligands come from GAFF31 and the AMBER FF14SB32 force field was employed to model the viral protein. The residue topologies for ligands were prepared using the Antechamber module33 implemented in the AMBER software package.34 For the covalently-bonded N3 ligand, I applied the residuegen program to generate non-standard amino acid residue topology.
2.3. MD Simulation Protocols.
For a protein-ligand complex, the MD system was first relaxed through a series of minimization procedures. The mainchain atoms of the receptor and the bound ligand were restrained using a harmonic potential and its force constant decreased from 20 to 10, 5, 1 and 0 kcal/mol/Å2, progressively in five 10,000-step minimizations. Note that the last step applied no restraint at all as the force constant is 0. The system was further relaxed by a set of 100-picosecond atomistic MD simulations with the same restrain setting of minimizations.
There were three phases for a MD simulation: the relaxation phase, the equilibrium phase, and the sampling phase. In the relaxation phase, the simulation system was heated up progressively from 50 K to 250 K at steps of 50 K. At each temperature, a 1-nanosecond MD simulation was performed without any restraints or constraints. In the next equilibrium phase, the system was equilibrated at 298 K, 1 bar for 10 ns. Finally, a 100-nanosecond MD simulation was performed at 298 K, 1 bar to produce NTP (constant temperature and pressure) ensembles. In total, 10,000 snapshots were recorded from the last simulation. 200 snapshots were evenly selected for the MM-PBSA-WSAS binding free energy calculation and 5000 were selected for the MM-GBSA ligand-protein binding free energy decomposition analysis. Additional settings for constant pressure MD simulations performed in this work are listed as follows: temperature was regulated using Langevin dynamics35 with a collision frequency of 5 ps−1; pressure was regulated using the isotropic position scaling algorithm with the pressure relaxation time being set to 1.0 ps; integration of the equations of motion was conducted at a time step of 1 fs for the relaxation phase and 2 fs for the equilibrium and sampling phases. The Particle Mesh Ewald (PME) method36 was used to calculate the full electrostatic energy of a unit cell in a macroscopic lattice of repeating images. All bonds were constrained using the SHAKE algorithm37 in both the minimization and MD simulation stages. All MD simulations were performed using the pmemd program in AMBER 18.34
2.4. MM-PBSA-WSAS Binding Free Energy Calculation
200 MD snapshots were evenly selected for the binding free energy calculations. For each selected MD snapshot, the molecular mechanical (MM) energy (EMM) and the MM-PBSA solvation free energy were calculated without further minimization.8, 10, 11, 38-40 Key parameters controlling the MM-PBSA-WSAS analyses are listed as follows: external dielectric constant: 80; internal dielectric constant: 4; and the surface tension for estimating the nonpolar solvation energy by using solvent assessible surface area: 0.054. The Parse radii41 were used in the MM-PBSA solvation calculation using the Delphi package (http://compbio.clemson.edu/delphi). The entropic term was estimated using a method coined WSAS (weighted solvent accessible surface area) described elsewhere.4 It is noted that the entropic contribution cannot be neglected for this protein target as most ligands are large and have many rotatable bonds.
2.5. MM-GBSA Ligand-Residue Free Energy Decomposition Analysis
I conducted ligand-residue free energy decomposition analysis for 5000 snapshots evenly selected from the sampled snapshots. Besides the electrostatic and van der Waals interactions, the solvation effect was taken into account using a Generalized GB model developed by Hawkins et al.42 The ligand-residue MM-GBSA interaction energies were calculated using the Sander program in AMBER18.34 Data analysis was performed using an internal program developed by us. A hotspot residue is recognized when its ligand-residue MM-GBSA interaction is stronger than −1.0 kcal/mol.
3. Results
In this work, I have performed two-step hierarchical virtual screenings to identify repurposing drugs targeting SARS-Cov-2 main protease from a set of 2201 approved drugs downloaded from DrugBank.5 In Step 1, Glide docking,7 an efficient but less accurate method was applied to enrich repurposing drug candidates; in Step 2, the docking hits were further evaluated using a more accurate but less efficient method, MM-PBSA-WSAS. The final repurposing drug candidates were selected based on the MM-PBSA-WSAS binding free energies.
3.1. Docking Screenings.
I first evaluated the docking power of Glide program for the co-crystal ligand of SARS-Cov-2 main protease, N3. The ligand RMSD of the best docking pose based on docking score (−9.4), 3.3 Å, was acceptable for a big ligand of about 100 atoms in flexible docking. I then applied the docking setting to conduct docking screenings. All the drug molecules that had docking scores better than −8.5 kcal/mol, roughly corresponding to 1μM, were selected as hits and advanced to the next filter – MM-PBSA-WSAS. By utilizing this cutoff, the number of hits accounted for about 1% of total screening compounds
3.2. MD Simulations
For the promising docking hits, I conducted molecular dynamics simulations using the AMBER software package.34 In total 39 ligands including the co-crystal N3 ligand, and 5 bio-actives which are structurally similar to Lopinavir, were studied in the second phase of HVS. The Top 5 approved neutral drugs that have excellent MM-PBSA-WSAS binding free energies (ΔGbind <=−5.0 kcal/mol) are shown in Figure 1. The 2D structures of charged drugs with at least one form achieved ΔGbind <=−5.0 kcal/mol are shown in Figure 2. I also found two bio-actives (Figure 3), have excellent binding free energies (Section 3.3). It is noted that Lopinavir was observed to be effective in treating COVID-19 patients.
Figure 1.

2D-Structures of promising repurpose drugs. All five approved drugs are in neural form under physiological conditions.
Figure 2.

2D-Structures of promising repurposing drugs. All three approved drugs are in charged form under physiological conditions.
Figure 3.

2D-Structures of promising bio-actives which are structurally similar to Lopinavir. PubChem 88143175, although studied in neutral form, bears −3 charges under physiological conditions.
I explored the MD stability of each MD system. Figure 4 showed the RMSD fluctuations along the MD simulation time. It is shown that the mainchain atoms of the receptor (black curves) and the secondary structures (red curves) reached equilibrium after 20 nanoseconds. The least-square (LS) fitted RMSDs of the ligands (green curves) are around 2 Å, which is reasonable for large ligands like SARS-Cov-2 main protease inhibitors. A ligand’s No-Fit RMSD was calculated by first performing LS-fitting for the main chain atoms of the receptor and the resulting translation-rotation matrix was applied to the ligand, and then the RMSD was calculated directly. Evidently, the ligand No-Fit RMSDs measures not only the conformational changes, but also its translational and rotational movements inside the binding pocket. The ligand No-Fit RMSDs (blue curves) are larger than the LS-Fit values, however, those values around 3-4 Å are still acceptable for large ligands. It is pointed out that sometimes, the LS-Fit and No-Fit RMSD values are overestimated due to rotation of symmetrical functional groups of a ligand. In summary, the RMSD fluctuation analysis suggests that the MD trajectories are overall stable during the sampling phase for all the studied MD systems.
Figure 4.

Plots of Root-mean-square deviations of receptor main chain atoms and ligand heavy atoms along the MD simulation time for (A) co-crystal ligand N3, (B) DB08889, (C) DB12329, (D) DB00385, (E) DB01601 and (F) DB11574.
I first generated the average structure of the collected snapshots, and then the MD snapshot that had the smallest main chain atom RMSD against the average structure was chosen as the representative conformation. The comparisons between the crystal structure and the representative MD conformations are shown in Figure 5 for the native ligand N3, and Figures 6-8 for the other ligands. As shown in Figure 5, the benzene motif located in the dashed red cycle was inserted between two hotspot residues (HIS41 and MET49) for the MD structure, which is quite distinct from the crystal structure, which shows that the benzene motif has no direct interactions with HIS41 and MET49.1 believe that under physiological conditions, the benzene motif becomes less solvent exposed and has more favorable interactions with HIS41 and MET49 by inserting itself between the side chains of the two residues. It is known that histidine has versatile roles in protein interactions.43 After the sulfide bond is formed as a result of nucleophilic attack on catalytic CYS145 by the N3 ligand, HIS41 can stabilize the ligand-protein interaction by forming π-π stacking interactions between the HIS41 and the benzene motif of N3. As shown below, both HIS41 and MET49 were hotspot residues in our MM-GBSA free energy decomposition analysis.
Figure 5.

Structural comparison between the crystal structure and a representative MD structure of SARS-Cov-2 main protease bound to the known ligand, N3. The crystal structure is shown as blue cartoon with the co-crystal ligand shown as brown sticks, while the representative MD structure is shown in grey cartoon and the ligand as green sticks (Panel A). The hotspot residues (ΔGLig-Res < −3.0 kcal/mol) revealed by MM-GBSA analysis are shown in Panel B; the more bluish a residue is colored, the stronger the interaction between the residue and the ligand.
Figure 6.

Structural comparison between the crystal structure and a representative MD structure of SARS-Cov-2 main protease bound to three neutral ligands DB08889, DB12329, and DB00385. The crystal structure is shown as blue cartoon with the docked ligand shown as brown sticks, while the representative MD structure is shown in grey cartoon and the ligand as green sticks. A: DB08889, B: DB12329, and C: DB00385. The detailed ligand-receptor interactions are shown in the bottom panel (D-F). All the hotspot residues (ΔGLig-Res < −3.0) revealed by MM-GBSA analyses are labeled and colored by a blue to red spectrum, the more bluish a residue is colored, the stronger the interaction between the residue and the ligand. D: DB08889, E: DB12329, and F: DB00385.
Figure 8.

Structural comparison between the crystal structure and a representative MD structure of SARS-Cov-2 main protease bound to three charged ligands DB01082, DB03147, and DB11184. The crystal structure is shown as blue cartoon with the docked ligand shown as brown sticks, while the representative MD structure is shown in grey cartoon and the ligand as green sticks. A: DB01082, B: DB03147, and C: DB11184. The detailed ligand-receptor interactions are shown in the bottom panel (D-F). All the hotspot residues (ΔGLig-Res < −3.0) revealed by MM-GBSA analyses are labeled and colored by a blue to red spectrum, the more bluish a residue is colored, the stronger the interaction between the residue and the ligand. D: DB01082, E: DB03147, and F: DB11184.
3.3. MM-PBSA-WSAS Binding Free Energy Calculations
I measured the ligand binding affinity using the endpoint MM-PBSA method. Considering the ligands of SARS-Cov-2 main protease are flexible molecules with large sizes, the contribution of the binding free energy from the conformational entropy change due to protein-ligand binding cannot be neglected. Instead of applying normal mode analysis to estimate the entropic effect, I applied an efficient method called WSAS4 to calculate this energy term. This scoring function is therefore called MM-PBSA-WSAS. The calculated binding free energies and the Glide docking scores are summarized in Table 1. The calculated entropic term, TΔS, is quite different for different ligands as shown in Table 1, suggesting the necessity of including this term in binding free energy calculations. The structures of the promising drug repurposing candidates, which have both excellent docking scores and MM-PBSA-WSAS binding affinities are shown in Figures 1-3. All the known drugs shown in Figure 1 are neutral and have a better MM-PBSA-WSAS affinity than −5.0 kcal/mol. It should be noted that the cocrystal ligand, N3 is covalently bonded to the receptor, therefore its binding free energy is not directly comparable to those non-covalent ligands. The individual terms of MM-PBSA-WSAS binding free energies of other less potent ligands were summarized in Table S1.
Table 1. List of Glide docking scores and MM-PBSA-WSAS binding free energies for potential inhibitors binding to SARS-Cov-2 main protease (in kcal/mol).
PubChem IDs are listed for the two bio-actives. The entropic contribution was estimated using T of 298.15 K.
| Compound Name | Docking Score | ΔEEEL | ΔEVDW | ΔGPB | ΔGSA | TΔS | ΔGbind |
|---|---|---|---|---|---|---|---|
| Co-crystal ligand covalently bonds to sulfur of CYS145 | - | −75.1 ± 0.3 | −82.0 ± 0.4 | 84.0 ± 0.2 | −6.5 ± 0.0 | −29.8 ± 0.1 | −38.8 ± 0.6 |
| Co-crystal ligand (no covalent bond formed) | −9.4 | −52.3 ± 0.3 | −26.9 ± 0.3 | 56.3 ± 0.2 | −4.8 ± 0.0 | −24.1 ± 0.1 | −3.6 ± 0.4 |
| Neutral Approved Drugs | |||||||
| DB08889 | −8.6 | −75.7 ± 0.4 | −40.9 ± 0.1 | 78.3 ± 0.4 | −6.0 ± 0.0 | −30.4 ± 0.1 | −13.8 ± 0.2 |
| DB12329 | −8.8 | −45.7 ± 0.4 | −25.5 ± 0.6 | 45.0 ± 0.3 | −3.4 ± 0.0 | −21.8 ± 0.0 | −7.7 ± 0.5 |
| DB00385 | −9.2 | −59.8 ± 0.2 | −21.5 ± 0.2 | 52.8 ± 0.4 | −4.6 ± 0.0 | −25.9 ± 0.1 | −7.2 ± 0.1 |
| DB01601 (Lopinavir) | −9.8 | −52.5 ± 0.3 | −20.1 ± 0.6 | 46.6 ± 0.6 | −4.6 ± 0.0 | −23.9 ± 0.0 | −6.6 ± 0.3 |
| DB11574 | −9.9 | −70.6 ± 0.4 | −21.8 ± 0.4 | 65.6 ± 0.6 | −6.4 ± 0.0 | −26.6 ± 0.2 | −6.5 ± 0.3 |
| Charged Approved Drugs | |||||||
| DB01082 (NC=0) | −8.6 | −46.0 ± 0.4 | −71.7 ± 1.0 | 89.4 ± 0.9 | −3.8 ± 0.0 | −24.2 ± 0.0 | −7.9 ± 0.4 |
| DB01082 (NC=2) | −6.9 | −33.4 ± 0.6 | −280.0 ± 1.6 | 291.7± 1.3 | −3.4 ± 0.0 | −21.2 ± 0.1 | −3.8 ± 0.5 |
| DB03147 (NC=0) | −10.2 | −67.8 ± 0.3 | −67.6 ± 0.9 | 106.6 ± 0.6 | −5.0 ± 0.0 | −26.3 ± 0.1 | −7.5 ± 0.5 |
| DB03147 (NC=−2) | −8.3 | −52.9 ± 0.1 | 123.0 ± 0.3 | −74.3 ± 0.5 | −4.5 ± 0.0 | −23.2 ± 0.1 | 14.6 ± 0.3 |
| DB11184 (NC=0) | −8.6 | −57.4 ± 0.8 | −52.2 ± 0.4 | 82.2 ± 0.3 | −4.9 ± 0.0 | −25.4 ± 0.1 | −6.8 ± 0.3 |
| DB11184 (NC=−4) | −7.4 | −50.1 ± 0.4 | 175.4 ± 1.6 | −148.7 ± 0.8 | −4.4 ± 0.0 | −22.9 ± 0.1 | −5.0 ± 0.5 |
| Bio-actives Structurally Similar to Lopinavir | |||||||
| 23727975 | −8.8 | −63.8 ± 0.1 | −50.1 ± 0.7 | 77.91± 0.1 | −5.2 ± 0.0 | −28.3 ± 0.1 | −12.9 ± 0.5 |
| 88143175 (NC = 0) | −10.0 | −73.9 ± 0.2 | −104.5 ± 1.3 | 148.9 ± 0.9 | −6.6 ± 0.0 | −30.5 ± 0.0 | −5.6 ± 0.6 |
The values of each energy term, van der Waals (ΔEVDW), electrostatics (ΔEVDW + ΔGPB), nonpolar solvation term (ΔGSA), and entropy (TΔS), vary significantly from one system to another (Table 1 and Table S1), suggesting there is no single energy term that dominates the protein-ligand interaction.
For the charged drug molecules, caution should be taken in result interpretation. For example, the neural form of Streptomycin (DB01082) has a MM-PBSA-WSAS binding free energy of −7.92 kcal/mol, much better than the charged form (−3.82 kcal/mol). The difference is caused by the distinct electrostatic properties between the neutral and charged molecules. However, the charged form is dominant under physiological conditions,44 we therefore should use the result of the charged form or take the penalty of protonation into consideration when using the result of the neutral form.
3.4. MM-GBSA Free Energy Decomposition.
I performed MM-GBSA binding free energy decomposition to identify the hotspot residues which make substantial contributions to the protein-ligand binding. The identified hotspots could enable us to rationally design potent and selective inhibitors of this drug target. To obtain statistically meaningful results, I studied 5000 MD snapshots for each system, and both the average ligand-residue interaction energies (ΔGlig-res) and their RMSD values were calculated.
A hotspot residue is defined as a residue with ΔGlig-res equal to or smaller than −1.0 kcal/mol. The identified hotspots of each ligand are summarized in Table 2. The most significant hotspot residues (ΔGlig-res<−3.0) are illustrated in Figures 5-8. The common significant hotspot residues for most ligands (in bold in Table 2) are as follows: HIS41, MET49, ASN142, HIS164, MET165, GLU166, and GLN189.
Table 2: Ligand-residue MM-GBSA interaction energies (kcal/mol).
PubChem IDs are listed for the two bio-actives. The common hotspots for all the ligands are: HIS41, MET49, ASN142, HIS164, MET165, GLU166, and GLN189.
| Residue ID |
Residue Type |
Co- crystal Ligand |
Neutral Approved Drug | Charged Approved Drug | Bioactive | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DB088 89 |
DB123 29 |
DB003 85 |
DB016 01 |
DB115 74 |
DB010 82 |
DB031 47 |
DB111 84 |
237279 75 |
881431 75 |
|||
| 24 | THR | −0.1 | −0.2 | −0.0 | −0.0 | −0.1 | −0.9 | −0.0 | −0.1 | −0.3 | −0.1 | −1.4 |
| 25 | THR | −1.5 | −1.5 | −0.2 | −0.6 | −1.6 | −4.0 | −0.1 | −1.6 | −3.2 | −0.6 | −3.7 |
| 26 | THR | −0.6 | −0.9 | −0.2 | −0.2 | −0.3 | −6.2 | −0.1 | −3.1 | −4.5 | −0.2 | −4.3 |
| 27 | LEU | −3.7 | −1.2 | −0.5 | −1.0 | −1.3 | −2.2 | −0.7 | −2.6 | −2.5 | −0.2 | −1.6 |
| 28 | ASN | −4.8 | −0.1 | −0.0 | −0.1 | −0.1 | −0.2 | −0.1 | −0.2 | −0.1 | −0.1 | −0.1 |
| 39 | PRO | −0.8 | −0.2 | −0.1 | −0.1 | −0.2 | −0.2 | −0.3 | −0.3 | −0.1 | −0.1 | −0.2 |
| 41 | HIS | −6.7 | −6.6 | −2.9 | −5.4 | −3.6 | −3.8 | −5.4 | −7.8 | −4.2 | −3.0 | −3.0 |
| 44 | CYS | −1.0 | −0.1 | −0.1 | −0.1 | −1.4 | −0.7 | −0.0 | −2.5 | −0.4 | −0.1 | −0.4 |
| 45 | THR | −0.6 | −0.3 | −0.2 | −0.1 | −1.1 | −0.6 | −0.0 | −1.8 | −0.3 | −0.2 | −0.5 |
| 46 | SER | −0.9 | −1.8 | −1.5 | −0.6 | −4.3 | −1.4 | −0.1 | −3.5 | −1.3 | −1.8 | −2.7 |
| 49 | MET | −4.4 | −2.4 | −3.1 | −2.1 | −5.6 | −4.2 | −1.1 | −5.5 | −2.2 | −2.2 | −3.9 |
| 52 | PRO | −0.8 | −0.1 | −0.1 | −0.1 | −0.0 | −0.2 | −0.0 | −0.1 | −0.0 | −0.1 | −0.0 |
| 54 | TYR | −0.6 | −0.1 | −0.4 | −0.2 | −0.1 | −0.5 | −0.2 | −0.2 | −0.1 | −0.1 | −0.0 |
| 119 | ASN | −0.2 | −0.0 | −0.1 | −0.1 | −0.0 | −1.4 | −0.0 | −0.4 | −0.0 | −0.0 | −0.0 |
| 140 | PHE | −0.2 | −1.0 | −0.0 | −0.1 | −0.0 | −0.1 | −0.8 | −0.1 | −0.2 | −0.4 | −1.8 |
| 141 | LEU | −0.3 | −1.2 | −0.1 | −0.2 | −0.0 | −0.1 | −1.0 | −0.0 | −0.3 | 0.5 | −2.6 |
| 142 | ASN | −3.5 | −4.4 | −3.5 | −3.7 | −0.6 | −2.9 | −6.7 | −4.6 | −4.6 | −5.5 | −6.8 |
| 143 | GLY | −2.2 | −0.8 | −1.0 | −0.4 | −0.4 | −2.3 | −1.9 | −3.8 | −1.5 | −2.5 | −2.1 |
| 144 | SER | −13.0 | −1.4 | −0.1 | −0.2 | −0.1 | −0.3 | −2.1 | −1.2 | −0.6 | −5.3 | −2.3 |
| 145 | CYS | −66.3 | −3.2 | −0.7 | −1.5 | −1.6 | −2.1 | −4.0 | −4.3 | −1.5 | −3.0 | −3.8 |
| 146 | GLY | −10.2 | −0.0 | −0.0 | −0.0 | −0.0 | −0.0 | −0.1 | −0.0 | −0.0 | −0.1 | −0.1 |
| 147 | SER | −0.6 | −0.1 | −0.0 | −0.0 | −0.0 | −0.0 | −0.1 | −0.0 | −0.0 | −0.0 | −0.1 |
| 163 | HIS | −3.9 | −1.6 | −0.1 | −0.4 | −0.1 | −0.2 | −2.8 | −0.4 | −0.6 | −1.2 | −1.2 |
| 164 | HIS | −3.9 | −4.9 | −1.4 | −3.6 | −1.8 | −1.6 | −5.3 | −5.4 | −1.3 | −2.5 | −2.5 |
| 165 | MET | −5.5 | −7.8 | −4.2 | −4.6 | −3.5 | −3.6 | −5.3 | −4.9 | −4.7 | −6.5 | −4.4 |
| 166 | GLU | −7.3 | −6.7 | −6.0 | −6.8 | −0.8 | −1.8 | −17.1 | −6.4 | −4.9 | −14.4 | −23.8 |
| 167 | LEU | −1.7 | −1.4 | −1.3 | −2.1 | −0.6 | −1.3 | −1.9 | −0.8 | −1.3 | −2.1 | −1.2 |
| 168 | PRO | −2.7 | −1.2 | −1.1 | −2.5 | −1.3 | −2.7 | −2.6 | −0.4 | −1.2 | −2.2 | −0.9 |
| 170 | GLY | −0.0 | −0.1 | −0.0 | −0.0 | −0.0 | −0.1 | −0.5 | −0.0 | −0.0 | −0.1 | −0.2 |
| 172 | HIS | −0.2 | −1.2 | −0.1 | −0.2 | −0.1 | −0.1 | −0.7 | −0.1 | −0.3 | −1.1 | −1.1 |
| 186 | VAL | −0.3 | −0.6 | −0.3 | −0.2 | −0.3 | −0.4 | −0.1 | −0.6 | −0.3 | −0.3 | −0.3 |
| 187 | ASP | −1.5 | −1.3 | −2.2 | −1.4 | −3.7 | −1.9 | −2.8 | −2.0 | −1.6 | −1.3 | −1.2 |
| 188 | ARG | −1.6 | −2.0 | −1.9 | −1.7 | −1.7 | −2.3 | −0.6 | −1.8 | −1.8 | −1.8 | −1.2 |
| 189 | GLN | −5.2 | −13.8 | −5.9 | −11.5 | −8.1 | −7.7 | −2.3 | −6.1 | −8.8 | −10.1 | −6.9 |
| 190 | THR | −1.4 | −1.6 | −3.9 | −2.4 | −1.6 | −3.8 | −0.0 | −1.1 | −3.2 | −1.0 | −0.6 |
| 191 | ALA | −1.6 | −2.5 | −0.2 | −0.7 | −1.5 | −1.6 | −0.0 | −0.2 | −0.8 | −0.5 | −0.1 |
| 192 | GLN | −2.2 | −3.5 | −4.0 | −2.3 | −1.1 | −1.8 | −0.2 | −1.2 | −6.3 | −1.7 | −0.8 |
4. Discussion
The outbreak of highly infectious diseases such as COVID-19 demands to work out multiple treatment plans as soon as possible. Computational drug repurposing study can provide treatment options in a short period of time. For this study, amounts of computational time used for individual tasks are as follows. Docking screenings of all the 2201 approved drugs with a single CPU core (Intel Xeon CPU E5-2683) took 11 hours. For each docking hit, we need to perform ab initio calculations to derive point charges. The ab initio calculation using wB97XD/6-31G*//HF/6-31G* consumed about 1 day using four CPU cores; then it took us about 1.2 days to sample 120 nanoseconds using one GTX-1080 ti GPU; the following MM-PBSA-WSAS calculation consumed one day. Therefore, equipped with sufficient numbers of CPUs and GPUs and the current hardware, we can finish the drug repurposing screenings within four to five days using a reliable HVS strategy. Given that the inhibitors of SARS-Cov-2 main protease have relatively large sizes, the screening time can be even shorter for other drug targets with smaller ligands.
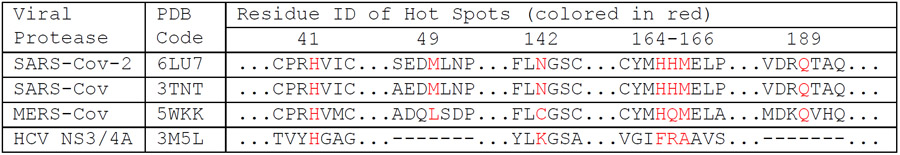
Another consideration is the availability of high-quality drug target structures. Luckily, a high-resolution crystal structure of SARS-Cov-2 protease in complex with a ligand was resolved timely, allowing us to conduct this drug repurpose screening. If no high-quality structure is available, one can rely on homology modeling technique, probably with a reduced success rate of identifying repurposing drugs. Take SARS-Cov-2 main protease as an example, I performed structural alignments using an internal program which takes a multiple-sequence-alignment (MSA) as an input. The MSA was generated by using the Promals3D web server.45 The structure of SARS-Cov-2 main protease is found to be most similar to those of SARS-Cov protease (PDB Code 3TNT46) and less similar to MERS-Cov protease (PDB Code 5WKK47) (Figure 9A). In comparison, the structure of HCV NS3/4A (PDB Code 3M5L48) is quite different from the main proteases of coronaviruses: the RMSD of 2.26 Å between HCV and SARS-Cov-2 is much larger and with only 108 residues participating the least-square fitting (Figure 9B). I also compared the sequences of the four proteases around the seven hotspot residues, which are colored in red in Table 3. It is shown that SARS-Cov-2 and SARS-Cov share all the seven hotspot residues. MERS-Cov and SARS-Cov-2 have four of the seven common hotspot residues, while HCV NS3/4A and SARS-Cov-2 have only one common hotspot residue (H41). Even though the sequence identity is low between SARS-Cov-2 main protease and HCV NS3/4A, as shown in Figure 9B, the co-crystal ligands, N3 (green sticks) for SARS-Cov-2 and ITMN-191 (brown sticks) for HCV NS3/4A, largely overlap. This suggests that homology models can be constructed using Modeller49 with SARS-Cov, MERS-Cov and even HCV NS3/4A as templates.
Figure 9.

Structural comparison of proteases among three coronavirus viruses (SARS-Cov-2, SARS-Cov and MERS-Cov) (A), and between SARS-Cov-2 and hepatitis C NS3/4A proteases (B). The SARS-Cov-2 main protease is colored in grey and its ligands are shown as green sticks. The following are the color codes for the other proteases: SARS-Cov protease and its co-crystal ligand – brown, MERS-Cov protease and its co-crystal ligand – blue, HCV NS3/4A – blue, co-crystal ligand of HCV NS3/4A – brown. Backbone RMSD between SARS-Cov and SARS-Cov-2 is 0.4711 Å, with 284 residues participating in the least-square fitting and 22 omitted, and the backbone RMSD between MERS-Cov and SARS-Cov-2 is 0.41 Å, but with 195 residues participating in the least-square fitting and 104 omitted. In contrast, the backbone RMSD between SARS-Cov-2 main protease and HCV NS3/4A is 2.2632 Å, with 108 residues participating in the least-square fitting and 43 omitted.
Table 3. Sequence comparison around hotspot residues for proteases of four types of viruses.
|
In this work, we have conducted computational drug repurposing study for the SARS-COV-2 main protease. To find robust treatments of COVID-19 particularly after the virus has developed different variations, it is necessary to screen repurposing drugs targeting other proteins which is essential in the life cycle of the virus.
5. Conclusion
In this study, I took advantage of the recently released crystal structure of SARS-Cov-2 main protease and conducted multiscale drug repurposing screenings. Five neutral drugs, namely, Carfilzomib, Eravacycline, Valrubicin, Lopinavir and Elbasvir, are identified to have inhibitory activities against SARS-Cov-2 main protease. Streptomycin, a charged molecule may also be an inhibitor of this SARS-Cov-2 main protease. Our study suggests that computational drug repurposing screening is very efficient and it can provide potential repurposing drug candidates in less than five days. A set of hotspot residues which make substantial contributions to the protein-ligand binding are also identified, which can facilitate us to rationally design novel selective inhibitors targeting SARS-Cov-2 main protease.
Supplementary Material
Figure 7.

Structural comparison between the crystal and a representative MD structure of SARS-Cov-2 main protease bound to two neutral ligands DB01601 and DB11574. The crystal structure is shown as blue cartoon with the docked ligand shown as brown sticks, while the representative MD structure is shown in grey cartoon and the ligand as green sticks. A: DB01601 and B: DB11574. The detailed ligand-receptor interactions are shown in the bottom panel (C-D). All the hotspot residues (ΔGLig-Res < −3.0) revealed by MM-GBSA analyses are labeled and colored by a blue to red spectrum, the more bluish a residue is colored, the stronger interaction between the residue and the ligand. C: DB01601 and D: DB11574.
ACKNOWLEDGMENT
The author gratefully acknowledges the funding support from the National Institutes of Health (R01GM079383, R21GM097617 and P30DA035778). The author also thanks for the computing resources provided by the Center for Research Computing (CRC) at University of Pittsburgh. The author is grateful to Dr. Xiang Liu at Nankai University who kindly provided the PDB file of 6LU7 to me prior its release at www.rcsb.org.
Footnotes
Supporting Information.
Table S1 lists the MM-PBSA-WSAS binding free energies for other less promising docking hits with ΔGlig-res are worse than −5.0 kcal/mol.
The author declares no competing financial interest.
REFERENCES
- 1.Liu X; Zhang B; Jin Z; Yang H; Rao Z, The crystal structure of COVID-19 main protease in complex with an inhibitor N3. 2020. [Google Scholar]
- 2.Wang E; Sun H; Wang J; Wang Z; Liu H; Zhang JZH; Hou T, End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev 2019, 119, 9478–9508. [DOI] [PubMed] [Google Scholar]
- 3.Wang JM; Hou TJ; Xu XJ, Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput.-Aided Drug Des 2006, 2, 287–306. [Google Scholar]
- 4.Wang J; Hou T, Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Info. Model 2012, 52, 1199–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law V; Knox C; Djoumbou Y; Jewison T; Guo AC; Liu Y; Maciejewski A; Arndt D; Wilson M; Neveu V; Tang A; Gabriel G; Ly C; Adamjee S; Dame ZT; Han B; Zhou Y; Wishart DS, DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42 (Database issue), D1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J; Ge Y; Xie XQ, Development and Testing of Druglike Screening Libraries. J. Chem. Info. Model 2019, 59, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 8.Wang J; Morin P; Wang W; Kollman PA, Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc 2001, 123, 5221–5230. [DOI] [PubMed] [Google Scholar]
- 9.Swanson JM; Henchman RH; McCammon JA, Revisiting free energy calculations: a theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J 2004, 86 (1 Pt 1), 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn B; Gerber P; Schulz-Gasch T; Stahl M, Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem 2005, 48, 4040–4048. [DOI] [PubMed] [Google Scholar]
- 11.Hou T; Wang J; Li Y; Wang W, Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Info. Model 2011, 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L; Sun H; Li Y; Wang J; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [DOI] [PubMed] [Google Scholar]
- 13.Sun H; Li Y; Shen M; Tian S; Xu L; Pan P; Guan Y; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys 2014, 16, 22035–22045. [DOI] [PubMed] [Google Scholar]
- 14.Sun H; Li Y; Tian S; Xu L; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys 2014, 16, 16719–16729. [DOI] [PubMed] [Google Scholar]
- 15.Chen F; Liu H; Sun H; Pan P; Li Y; Li D; Hou T, Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys 2016, 18, 22129–22139. [DOI] [PubMed] [Google Scholar]
- 16.Karami M; Jalali C; Mirzaie S, Combined virtual screening, MMPBSA, molecular docking and dynamics studies against deadly anthrax: An in silico effort to inhibit Bacillus anthracis nucleoside hydrolase. J. Theor. Biol 2017, 420, 180–189. [DOI] [PubMed] [Google Scholar]
- 17.Chen F; Sun H; Wang J; Zhu F; Liu H; Wang Z; Lei T; Li Y; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 8. Predicting binding free energies and poses of protein-RNA complexes. RNA 2018, 24, 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra SK; Koca J, Assessing the Performance of MM/PBSA, MM/GBSA, and QM-MM/GBSA Approaches on Protein/Carbohydrate Complexes: Effect of Implicit Solvent Models, QM Methods, and Entropic Contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [DOI] [PubMed] [Google Scholar]
- 19.Sun H; Duan L; Chen F; Liu H; Wang Z; Pan P; Zhu F; Zhang JZH; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys 2018, 20, 14450–14460. [DOI] [PubMed] [Google Scholar]
- 20.Wang E; Weng G; Sun H; Du H; Zhu F; Chen F; Wang Z; Hou T, Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein-protein Interactions. Phys. Chem. Chem. Phys 2019, 21, 18958–18969. [DOI] [PubMed] [Google Scholar]
- 21.Weng G; Wang E; Chen F; Sun H; Wang Z; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 9. Prediction reliability of binding affinities and binding poses for protein-peptide complexes. Phys. Chem. Chem. Phys 2019, 21, 10135–10145. [DOI] [PubMed] [Google Scholar]
- 22.Lee TS; Hu Y; Sherborne B; Guo Z; York DM, Toward Fast and Accurate Binding Affinity Prediction with pmemdGTI: An Efficient Implementation of GPU-Accelerated Thermodynamic Integration. J. Chem. Theory Comput 2017, 13, 3077–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L; Wu Y; Deng Y; Kim B; Pierce L; Krilov G; Lupyan D; Robinson S; Dahlgren MK; Greenwood J; Romero DL; Masse C; Knight JL; Steinbrecher T; Beuming T; Damm W; Harder E; Sherman W; Brewer M; Wester R; Murcko M; Frye L; Farid R; Lin T; Mobley DL; Jorgensen WL; Berne BJ; Friesner RA; Abel R, Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc 2015, 137, 2695–2703. [DOI] [PubMed] [Google Scholar]
- 24.Wang J; Kang X; Kuntz ID; Kollman PA, Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MM-PB/SA. J. Med. Chem 2005, 48, 2432–2444. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Capparelli EV; Holland D; Okamoto C; Gragg B; Durelle J; Marquie-Beck J; van den Brande G; Ellis R; Letendre S; Group H, Lopinavir concentrations in cerebrospinal fluid exceed the 50% inhibitory concentration for HIV. AIDS 2005, 19, 949–952. [DOI] [PubMed] [Google Scholar]
- 27.O'Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR, Open Babel: An open chemical toolbox. J. Cheminf 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926. [Google Scholar]
- 29.Bayly CI; Cieplak P; Cornell WD; Kollman PA, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges- the RESP model. J. Phys. Chem 1993, 97, 10269–10280. [Google Scholar]
- 30.Frisch MJ T. GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ, Gaussian 16, Revision B.01 Gaussian, Inc., Wallingford CT: 2016. [Google Scholar]
- 31.Wang JM; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 32.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J; Wang W; Kollman PA; Case DA, Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph Model 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- 34.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham I,TE; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; D.M. Y; Kollman PA, AMBER 2018. University of California, San Francisco: 2018. [Google Scholar]
- 35.Larini L; Mannella R; Leporini D, Langevin stabilization of molecular-dynamics simulations of polymers by means of quasisymplectic algorithms. J. Chem. Phys 2007, 126, 104101. [DOI] [PubMed] [Google Scholar]
- 36.Darden T; Perera L; Li L; Pedersen L, New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, R55–60. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto S; Kollman PA, Settle - an analytical version of the Shake and Rattle algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- 38.Page CS; Bates PA, Can MM-PBSA calculations predict the specificities of protein kinase inhibitors? J. Comput. Chem 2006, 27, 1990–2007. [DOI] [PubMed] [Google Scholar]
- 39.Kongsted J; Ryde U, An improved method to predict the entropy term with the MM/PBSA approach. J. Comput.-Aided Mol. Des 2009, 23, 63–71. [DOI] [PubMed] [Google Scholar]
- 40.Hou T; Wang J; Li Y; Wang W, Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem 2011, 32, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sitkoff D; Sharp KA; Honig B, Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem 1994, 98, 1978–1988. [Google Scholar]
- 42.Hawkins GD; Cramer CJ; Truhlar DG, Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem 1996, 100, 19824–19839. [Google Scholar]
- 43.Liao SM; Du QS; Meng JZ; Pang ZW; Huang RB, The multiple roles of histidine in protein interactions. Chem. Cent. J 2013, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wray R; Iscla I; Gao Y; Li H; Wang J; Blount P, Dihydrostreptomycin Directly Binds to, Modulates, and Passes through the MscL Channel Pore. PLoS biol. 2016, 14, e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei J; Grishin NV, PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinfo. 2007, 23, 802–808. [DOI] [PubMed] [Google Scholar]
- 46.Hilgenfeld R, From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galasiti Kankanamalage AC; Kim Y; Damalanka VC; Rathnayake AD; Fehr AR; Mehzabeen N; Battaile KP; Lovell S; Lushington GH; Perlman S; Chang KO; Groutas WC, Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem 2018, 150, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romano KP; Ali A; Royer WE; Schiffer CA, Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U S A 2010, 107, 20986–20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webb B; Sali A, Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci 2016, 86, 5.6.1–5.6.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.