

Abstract
Background
Cancer is a highly complex disease, which involves the cooperation of tumor cells with multiple types of host cells and the extracellular matrix. Cancer studies that rely solely on static measurements of individual cell types are insufficient to dissect this complexity. In the last two decades, intravital microscopy has established itself as a powerful technique that can significantly improve our understanding of cancer by revealing the dynamic interactions governing cancer initiation, progression, and treatment effects in living animals. This review focuses on intravital multiphoton microscopy (IV‐MPM) applications in mouse models of cancer.
Recent findings
IV‐MPM studies have already enabled a deeper understanding of the complex events occurring in cancer at the molecular, cellular, and tissue levels. Multiple cell types present in different tissues influence cancer cell behavior via activation of distinct signaling pathways. As a result, the boundaries in the field of IV‐MPM are continuously being pushed to provide an integrated comprehension of cancer. We propose that optics, informatics, and cancer (cell) biology are coevolving as a new field. We have identified four emerging themes in this new field. First, new microscopy systems and image processing algorithms are enabling the simultaneous identification of multiple interactions between the tumor cells and the components of the tumor microenvironment. Second, techniques from molecular biology are being exploited to visualize subcellular structures and protein activities within individual cells of interest and relate those to phenotypic decisions, opening the door for “in vivo cell biology”. Third, combining IV‐MPM with additional imaging modalities or omics studies holds promise for linking the cell phenotype to its genotype, metabolic state, or tissue location. Finally, the clinical use of IV‐MPM for analyzing efficacy of anticancer treatments is steadily growing, suggesting a future role of IV‐MPM for personalized medicine.
Conclusion
IV‐MPM has revolutionized visualization of tumor‐microenvironment interactions in real time. Moving forward, incorporation of novel optics, automated image processing, and omics technologies in the study of cancer biology, will not only advance our understanding of the underlying complexities but will also leverage the unique aspects of IV‐MPM for clinical use.
Keywords: cancer microscopy, fate mapping, intravital microscopy, label‐free, tumor microenvironment, two‐photon
1. INTRODUCTION
Understanding the mechanisms essential for cancer initiation and progression holds great potential for developing new therapies. Among these, the mechanisms controlling motility and invasion of cancer cells are of particular importance, as they are necessary for metastasis.1 However, motility and invasion are exhibited by only a small portion of tumor cells. As a result, most static measurements done by averaging across broad areas of a tumor or collected at the tissue level are not able to detect or accurately quantify the behaviors of the small number of motile and invading cells. To overcome this problem and thus permit the visualization and measurement in real time of single cells in a living animal, intravital microscopy techniques have been increasingly used in cancer research. Our review focuses on intravital multiphoton microscopy (IV‐MPM) of mouse models of cancer, which has evolved to be the gold standard imaging technique for preclinical cancer research.
Over the last decade, IV‐MPM studies have led to significant discoveries in cancer cell biology. However, continuing this pace of discovery is becoming a challenge for the following reasons: (a) multiple and large areas need to be imaged as the primary tumor and metastatic sites are located in different organs; (b) the acquisition time must range from milliseconds (eg, visualization of cell signaling) to weeks (eg, cancer cell fate mapping), while maintaining the high spatial resolution needed to resolve subcellular structures; (c) IV‐MPM generates large 5D data sets that are not supported by currently available image processing tools; (d) IV‐MPM is limited by the availability of fluorescent reporters and dyes. Therefore, to embrace the complexity of cancer and continue advancing our understanding of the disease, innovations in optics and informatics are being rapidly implemented into cancer (cell) biology, and the coevolution of these disciplines is pushing the frontiers of IV‐MPM of cancer.
This review discusses four areas of the coevolution of IV‐MPM and other disciplines. We propose first, that recent advancements in microscopy systems and image processing algorithms are allowing simultaneous identification of multiple interactions between the tumor cells and the components of the tumor microenvironment. Second, protein chimeras and molecular biosensors are being developed to visualize subcellular structures and protein activities within individual cells and to relate those to metastatic behavior, opening the door for “in vivo cell biology”. Third, IV‐MPM can be combined with additional imaging modalities or omics studies to link cell behavior to its genotype, metabolic state, or tissue location. Finally, the preclinical and clinical applications of IV‐MPM are steadily growing, suggesting that IV‐MPM will have a growing role in personalized medicine. Each section also includes a few speculations about how IV‐MPM will evolve in the near future.
2. TOWARDS MULTIPARAMETRIC IV‐MPM OF CANCER: NEW IMAGING AND IMAGE PROCESSING TOOLS
Most IV‐MPM studies to date have neglected the multiplicity of cell‐cell and cell‐ECM interactions in cancer, reducing the complexity of tumors to two components. Such studies have relied on simultaneous detection of one or two channels, commonly cancer cells and one host cell type or the extracellular matrix (ECM). Collectively, this work has revealed important information about cancer cell communication with stromal cells, such as fibroblasts, immune cells, including neutrophils, macrophages, and T‐cells, as well as cancer cell interactions with collagen fibers in the ECM (reviewed in Calvo and Sahai, Charras and Sahai, Fein and Egeblad, Conklin and Keely, Alexander et al, te Boekhorst et al, Bragado et al, and Ramamonjisoa and Ackerstaff2, 3, 4, 5, 6, 7, 8, 9). However, to elucidate key mechanisms governing cancer progression and therapeutic efficacy, ideally all these interactions should be monitored simultaneously through three‐dimensional (3D) space and over time in their native environment/or in living animals. In this section, we report on technical advances in microscope setups that allow multicolor IV‐MPM. Then, we turn to analysis of multicolor images and review benefits and limitations of different spectral unmixing techniques. Finally, we describe how semiautomated image processing and machine learning classifiers are making new inroads into the problem of cell segmentation in densely packed tissues, allowing an integrated view of the multiple components affecting cancer cell behavior in vivo.
2.1. Next generation of multiphoton microscope systems
IV‐MPM is increasingly used to gain insights into the dynamic behavior of cancer cells in living animals. In the two‐photon (2P) fluorescence microscopy (most commonly used version of multiphoton microscopy), fluorophore excitation occurs when two photons are simultaneously absorbed by the fluorophore molecule, each of the photons carrying half of the energy necessary to excite the fluorophore. The probability of this concurrent interaction is highest within the focal volume, where the photon flux is at its maximum. This unique property prevents out‐of‐focus fluorescence and increases 3D resolution without the use of pinholes, necessary in single photon confocal fluorescence microscopy.10 To visualize fluorescent proteins and dyes in the visible spectrum, single photon excitation requires lasers with wavelengths ranging from approximately 350 to 650 nm, while 2P‐excited fluorescence requires infrared (IR) or near‐IR light (750‐1800 nm; note that the energy of a photon is inversely proportional to its wavelength). 2P fluorescence microscopy has become the gold standard for intravital imaging because of its multiple benefits over confocal fluorescence microscopy. First, in 2P fluorescence microscopy, more excitation photons reach a deep focus level inside thick samples than possible with confocal fluorescence microscopy.11 This is due to the fact that the near‐IR and IR light is not absorbed as strongly by water, hemoglobin, melanin, fat and is less scattered than the shorter‐wavelength light used in confocal fluorescence imaging. Second, the low energy IR light produces less photo damage compared with the higher‐energy light used in confocal fluorescence microscopy. This allows for time‐lapse imaging in which the region of interest is monitored over time without fluorophore bleaching or tissue damage. Third, the IR light additionally induces second harmonic generation (SHG) in collagen fibers and third harmonic generation (THG) at water‐lipid and water‐protein interfaces,12 enabling label‐free detection of additional cell types and ECM components in tissues (see section 5.2).
Commercially available Titanium:Sapphire (Ti:S) lasers are the most commonly used light sources for multiphoton microscopy because they provide a robust, flexible, platform for generation of mode‐locked femtosecond pulses tunable from approximately 700 to 1000 nm in wavelength with pulse durations configurable over a range from approximately 10 to 300 fs.13 However, two shortcomings limit the utility of these lasers. First, this wavelength range restricts imaging to blue and green fluorescent protein derivatives, as most yellow and red fluorescent protein derivatives would require 2P excitation at wavelengths longer than 1000 nm. Second, although a single wavelength can be used to image multiple fluorophores in 2P fluorescence microscopy, this requires a compromise in the efficiency of excitation among the fluorophores. Hence, the simultaneous imaging of multiple fluorophores in vivo requires an optimized excitation scheme.
To broaden the spectral coverage of the excitation source, custom microscope systems were developed either by splitting the light from one laser source into two beams14, 15 or by using two lasers.16, 17 Each of these options is now commercially available from Olympus, Nikon, Leica, and LaVision. In the latter arrangement, the two‐source system consists of a tunable Ti:S (750‐950 nm) and a second Ti:S used as the second 2P excitation beam in the 960‐1040 nm range or, alternatively, as the pump for an optical parametric oscillator (OPO). The OPO “signal” output can be used as the second 2P excitation beam in the 1100‐1600 nm range. Spatio‐temporal alignment of the near‐IR and IR pulse trains allows for simultaneous excitation of multiple fluorophores, ranging from blue to far red. For example, simultaneous imaging of collagen fibers, Dendra2‐expressing tumor cells, and two injectable probes (Texas Red Dextran and MMPSense 680) was achieved in mouse xenograft tumors by tuning the first Ti:S to 880 nm and the selected OPO output to 1250 nm.18 However, as the dual beam is limited to two excitation wavelengths, yellow fluorophores will not be efficiently excited by neither the beam optimized for blue/green fluorophores nor the one optimized for far‐red fluorophores.
To simultaneously excite blue/green, yellow, and far‐red fluorophores, two‐color 2P‐excited fluorescence (2c‐2PEF) can be used. In this setup, the pulse train from a single Ti:S laser is split into two with one portion used directly to excite the blue/green fluorophores in the sample, while the other is used to pump an OPO. The OPO signal wave is then used to excite far‐red fluorophores. In addition, the spatio‐temporal alignment of the pulse trains coming directly from the Ti:S or from the OPO can lead to the production of an additional “virtual” excitation wavelength for 2P‐excited fluorescence (Figure 1A).19 In virtual excitation, 2P excitation occurs as a result of absorption of one photon from each of the two beams (ie, near IR from the Ti:S laser and IR from the OPO). Such wavelength mixing allows triple excitation of the sample. Using the mixing approach, one‐shot multicolor imaging of brain tissues from transgenic Brainbow mice, which stochastically express mCerulean, mEYFP, tdTomato, and mCherry, was achieved.19 Triple excitation of a broad range of fluorophores was also demonstrated by IV‐MPM in the lymph node germinal centers, where distinct immune cell types were labeled with Hoechst, eGFP, Kusabira Orange, CMTPX Red, QD655, and Atto 680 (Figure 1B).15 One limitation of wavelength mixing is that it restricts imaging to the superficial layers of tissue (~400 μm in Mahou et al19), as the efficient operation of the OPO requires the Ti:S laser wavelength to be set below 880 nm.
Figure 1.
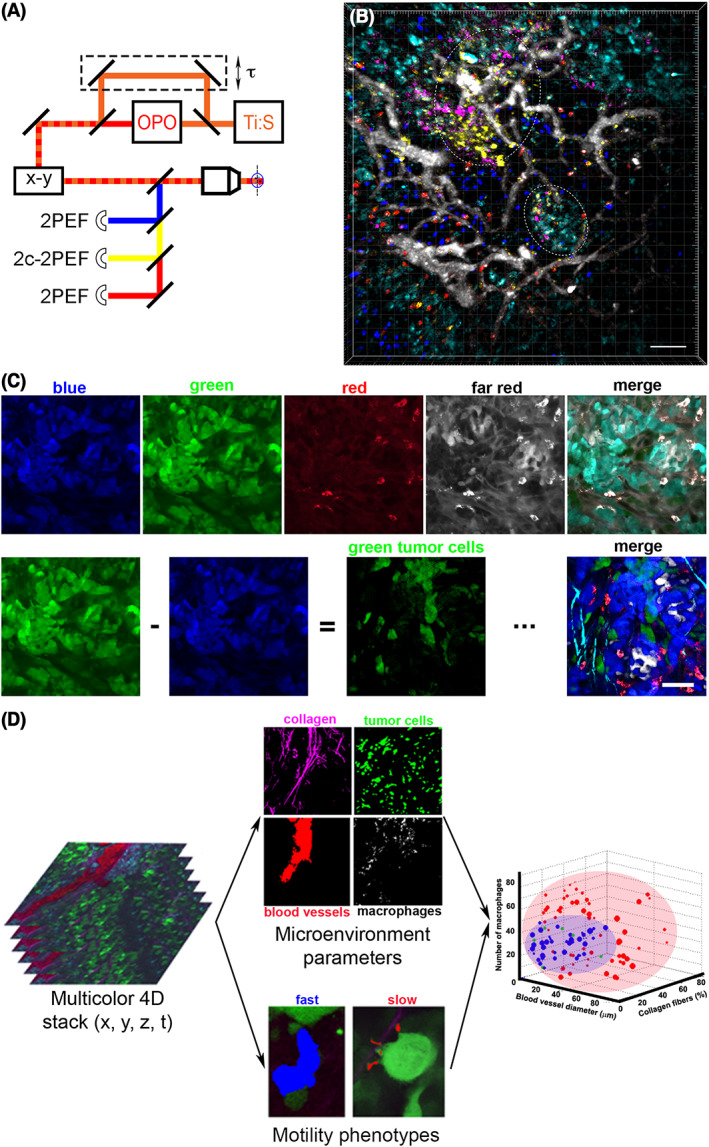
New multiphoton microscope systems, spectral unmixing techniques, and machine learning classifiers allow for multiparametric intravital multiphoton microscopy (IV‐MPM) of cancer. (A) Microscope setup for two‐color two‐photon imaging by wavelength mixing. The pulse trains from a Ti:S laser (orange) and an optical parametric oscillator (OPO, red) are synchronized (τ) and coaligned (dashed line). In addition to the blue and red two‐photon‐excited fluorescence (2PEF), a third yellow fluorophore can be accessed by two‐color two‐photon‐excited fluorescence (2c‐2PEF). Adapted from Mahou et al.19 (B) Visualization of seven distinct cellular and tissue compartments in vivo. IV‐MPM micrograph shows the germinal centers (dashed areas) in the popliteal lymph node of a mouse. Blue—naïve B cells, green—plasma blasts, orange—B cells, red—T cells, white—blood vessels, violet—follicular dendritic cells, cyan—macrophages. Scale bar, 50 μm. Reprinted with permission from Rakhymzhan et al.15 (C) Spectral unmixing by color subtraction. The emission signal from a mouse mammary tumor is simultaneously acquired by four detectors: blue, green, red, and far red (top left panels). The unprocessed, merged image of the four channels does not allow separation of the multiple cell types and stromal features of the tissue (top far right panel). Spectral bleed through of CFP into the GFP channel can be eliminated by subtracting the blue from the green (bottom left panels). Repetition of this operation leads to a merge image with five separated channels: cyan—SHG signal from collagen fibers, blue—CFP‐labeled tumor cells, green—GFP‐labeled tumor cells, red—macrophages, white—TagRFP657‐labeled tumor cells (bottom right panel). Scale bar, 60 μm. Adapted and reprinted with permission from Entenberg et al.16 (D) Workflow for machine learning classification of an IV‐MPM data set. Multicolor three‐dimensional (3D) stacks (at a single time point) are separated into four channels, and microenvironment parameters were extracted (top middle panels). Three‐dimensional (3D) time lapses of motile tumor cells (green channel) are separated into fast or slow locomotion on the basis of cell segmentation and tracking (bottom middle panels). Finally, support vector machine (SVM) classification is done, linking motility phenotypes with microenvironment parameters (right panel). Adapted and reprinted with permission from Gligorijevic et al.18
To image deep tissue structures, a novel wavelength mixing approach was developed, circumventing the use of OPOs. This novel system uses an ytterbium (Yb) fiber amplifier emitting at 1055 nm to pump a diamond Raman laser, in place of the OPO, resulting in an emission at 1240 nm as well as 1140 nm (from wavelength mixing).20 The authors demonstrated imaging of the cortical vasculature of living mice up to 1 mm deep. Due to its IR excitation light, this triple‐color excitation scheme can be ideal for applications where multiple red and/or far‐red probes need to be imaged simultaneously. In addition, while dual/triple output Ti:S sources can be costly, the Yb fiber laser approach is more affordable, opening wavelength mixing to smaller laboratories.
Once fluorophores are excited, they start emitting fluorescent light. Most typically, the emission light is guided through the optical path by filters and mirrors to the photon detectors called photomultiplier tubes (PMTs), one for each of the fluorophores/channels. Alternatively, a 16‐channel PMT array can be used to acquire hyperspectral images that are then reconstructed using complex algorithms.21 As the number of fluorophores increases, an overlap in excitation/emission wavelengths becomes unavoidable, causing bleed through between the channels. Hence, unambiguous separation of emission signals requires further processing of the data, as described in the following section.
2.2. Spectral unmixing techniques
Multispectral (ie, multicolor) imaging requires image processing to isolate the emission signal from each of the fluorophores. As emission spectra of most fluorescent proteins are 50 to 150 nm wide, an increasing level of cross‐over is to be expected in samples labeled with three or more fluorophores. To overcome this limitation and resolve emission signals, spectral unmixing methods have been developed.
The most straightforward unmixing technique is color subtraction. Instead of requiring algorithms, color subtraction exploits the “Image Calculator” tool on Fiji/ImageJ to remove bleed through from one channel into another.16, 22 This step creates a separate channel and can be repeated for all overlapping channels. Color subtraction was recently used to visualize five components in breast xenografts, including three clonal subpopulations of tumor cells, macrophages, and collagen fibers (Figure 1C).16
A more advanced approach includes spectral (λ) imaging (ie, lambda scan) of each pixel followed by linear unmixing. This method assumes that each spectrum is distinguishable from all others and contributes to the multispectral image in a linear fashion.23, 24 Reference images acquired during spectral imaging are used to measure the relative contribution of each spectrum to all channels. The obtained spectral signature is then used to correct the experimental images. Both commercial (Zeiss and Nikon) and open‐source softwares (J. Walter Plugin on Fiji/ImageJ) take advantage of this algorithm. One limitation of linear spectral unmixing is that it does not account for changes in the fluorophore emission spectra that might arise from interactions between colocalized fluorophores (eg, Föster energy resonance transfer [FRET]).
Tunable emission filters required for lambda scans are not used in 2P excitation, making spectral imaging inapplicable to IV‐MPM. As an alternative to linear unmixing, spectral deconvolution can be used for IV‐MPM, as it does not require acquisition of a reference spectral image.25 In the work of Ricard et al, using sequential excitation at 800 nm and then at 940 nm, six cellular and ECM components of the cortex of a living mouse bearing brain tumor were imaged. At both excitation wavelengths, the fluorescence intensity of a fluorophore was measured in each of the five detectors, providing a reference channel (excitation wavelength/detector combination) in which the intensity of the fluorescent reporter is the highest. Spectral unmixing of a fluorophore was achieved by subtracting the contributions of all other fluorophores in the reference channel of the fluorophore of interest.
To achieve color separation using information collected on all available channels, a new algorithm called similarity unmixing (SIMI) was developed.15 From each image and for each unknown fluorophore, the relative signal distribution on each photodetector is extracted pixel by pixel and matched to known fluorophore spectral fingerprints based on similarities. The fluorophore is then identified based on the closest match with one of the fingerprints. The key requirement for separating fluorophores is that their fingerprints must be distinct. Unlike the color subtraction and the linear spectral unmixing approaches, the SIMI method is independent from the cross talk between the fluorophores present in the sample. An added benefit of SIMI is that the solving strategy of the SIMI algorithm enables identification of a larger number of fluorophores than detectors present, increasing the number of colors that can be identified in the multicolor images.
After the individual emission signals from cell types and ECM components present in an image are successfully discriminated, a thorough understanding of the cell‐cell and cell‐ECM interactions can be gained using quantitative analysis of the parameters extracted from the 5D image (x, y, z, t, λ), as discussed in the next section.
2.3. (Semi)automated image processing and machine learning classifiers
The most commonly used approach for image segmentation is thresholding of the fluorescence intensity, which performs optimally in tissues where individual cells can be resolved.26, 27 However, the tight packing of cells within tumors, combined with cytoplasmic labeling of cells with fluorescent proteins, can mask the borders of neighboring cells when cells have similar expression levels of the fluorescent proteins. This complicates cell segmentation and tracking analysis of IV‐MPM data. For this reason, most IV‐MPM studies of cancer have relied on manual methods for segmentation and tracking. While this approach has produced many valuable measurements of cancer and immune cell motility,14, 15, 28, 29, 30, 31 cell‐cell interaction frequencies,32, 33 and vascular permeability,33, 34, 35 it is time consuming, and throughput is limited to a small number of measurements.
A few approaches have been developed to address the challenges caused by dense cell packing. For example, a semiautomated Fiji/ImageJ plug‐in was created in which the user manually segments the cell or region of interest at each time‐point, facilitating the 3D tracking and analysis steps.16 While this program simplifies the quantification of motility parameters for objects that may cross different imaging planes over time, it remains semiautomated and requires significant user input.
To fully automate cell segmentation, cells can be labeled with multiple fluorophores, which improves contrast between neighboring cells and eliminates packing issues. For example, Coffey et al generated multicolored cell populations, to ease the segmentation process.36 Once segmented, cells can be automatically tracked, analyzed, and classified using machine learning algorithms.36, 37
Considerable variability in nuclear size can complicate automated segmentation and of cell nuclei in tumors because of the high cell density and the large variability in nuclear size. Marker‐controlled watersheds followed by a machine learning method was used to efficiently identify cell nuclei in xenograft tumors imaged by IV‐MPM.38
LEVER‐3D is the first user‐friendly software developed to visualize and validate the automated analysis of large 5D IV‐MPM data sets.39 Its unique workflow includes a validation step, during which the processed image is displayed and the user can edit the results to enhance the accuracy of the analysis. The revisions provided by the user are then utilized to automatically correct similar errors in the analysis.
While most of these approaches have yet to be directly implemented into cancer biology studies, a semiautomated image processing and a machine learning classification for IV‐MPM were recently applied to migrating tumor cells. By segmenting and tracking motile cancer cells, two distinct motility phenotypes were identified: a slow phenotype that exhibited invadopodia and degraded the ECM and a fast phenotype that exhibited no invadopodia and was contact guided along the collagen fibers.18 In addition, Gligorijevic et al characterized the locations where each of the two motility phenotypes occurred, by extracting microenvironmental parameters from the images18, 40 and classifying them by machine learning‐based support vector machine (SVM) classification. Once trained, SVM classification was able to accurately predict motility phenotype from the microenvironmental parameters (Figure 1D). Use of this approach, titled intravital systems microscopy was instrumental in demonstrating a direct link between the local microenvironment parameters in tumors, the assembly of invadopodia in tumor cells, and the ability of those tumor cells to intravasate and metastasize in vivo.41, 42 This work also indicated that tumor cell behavior is a result of complex interactions among cues present in the tumor microenvironment and that machine learning algorithms have the ability to grasp this complexity.18, 43
In neuroscience and developmental biology, the field of computer science is contributing new image processing and analysis workflows, advancing large‐scale brain mapping44, 45, 46 and analysis of tissue morphodynamics.47, 48, 49 Implementing similar unbiased strategies for IV‐MPM studies of cancer will allow us to accelerate the pace of discoveries.
2.4. Future perspectives of multiparametric IV‐MPM of cancer
While not yet applied to IV‐MPM of cancer in mice, several recent approaches offer new opportunities for future multicolor IV‐MPM studies. For example, as an alternative to dual beam output using OPOs, fibers can be used as a simultaneous up‐and‐down wavelength convertor.50 In addition, the generation of three excitation wavelengths using fiber lasers and solitons are gaining popularity.51, 52
To correct for diffraction‐induced aberrations during deep in vivo imaging, adaptive optics have been combined with IV‐MPM to study neuronal structures in mice53 and image cancer cells inside the zebrafish vasculature in combination with lattice light‐sheet microscopy.54
Finally, spectral phasor analysis has been used for robust unmixing of seven overlapping fluorophores acquired by single photon excitation within a living zebrafish.55 While spectral phasor analysis is promising, the noisy emission signals inherent in 2P microscopy may make it challenging to apply Fourier transformation essential for this approach to IV‐MPM data.
3. CELL FATE MAPPING, IMAGING MOLECULAR ACTIVITIES, AND CELL‐CELL COMMUNICATION BY HIGH‐RESOLUTION IV‐MPM OF CANCER
IV‐MPM studies have revealed that only a small portion of cancer cells exhibit motility and that only an even smaller group metastasizes to secondary sites. In this section, we describe how fluorescent proteins are being used to trace cells of interest from the primary tumor to secondary tumor sites and demonstrate that this body of work has uncovered heretofore unknown (sub)phenotypes of cancer cells. To better understand the principles underlying such heterogeneities in tumor cell behaviors, we then cover the use of biosensors to image cell signaling in vivo. Finally, we show how IV‐MPM studies have led to unique discoveries in cancer cell cooperation, by revealing how extracellular vesicles and microtubes enable communication between cancer cells.
3.1. Fate mapping of cancer cells to reveal their metastatic potential
In mouse models, metastasis occurs over a time scale of days, requiring the use of longitudinal imaging to follow cells from primary to secondary tumor sites. However, tissue remodeling caused by tumor cell proliferation, angiogenesis, and ECM restructuring makes it impossible to follow the same cells over repeated imaging sessions without specifically marking them. Photoconvertible fluorescent proteins, such as Dendra2, have been useful in breaking this constraint as they allow monitoring of chosen tumor cell populations on a scale of days (Figure 2A),56, 61 and thus allow the mapping of cell fate from primary to secondary tumor sites (Figure 2A').18, 40 Using longitudinal IV‐MPM of photoconverted Dendra2 in Dendra2‐expressing cells in organoid‐derived tumors, a recent study compared the cell dispersion in tumors expressing different combinations of the driver mutations. Results showed that alteration of four signaling pathways (Wnt, EGFR, P53, and TGFβ) is required for migration of colon cancer cells in vivo.62
Figure 2.
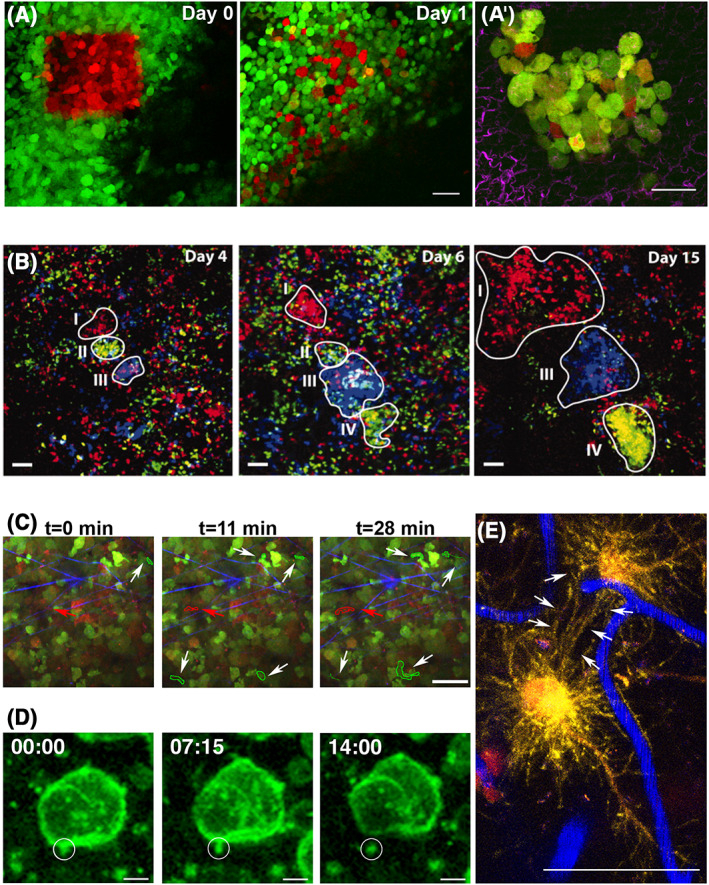
High‐resolution intravital multiphoton microscopy (IV‐MPM) and new fluorescent proteins allow cell fate mapping and studying cell‐cell communication mechanisms in cancer. Photoswitchable fluorescent proteins allow tracking of individual cells in vivo. (A) Longitudinal IV‐MPM imaging of photoswitched Dendra2 (green to red) expressed by mammary tumor cells shows cell dispersion adjacent to a blood vessel (dark region) after 1 day. Scale bar, 30 μm. Adapted and reprinted with permission from.56 (A') A lung metastatic colony is observed ex vivo, 5 days post‐photoconversion of Dendra2 expressed by mammary tumor cells in the primary tumor. Red cells arrived in the lung at 0 to 5 days, orange or yellow cells divided and synthesized additional green Dendra2. Green cells cannot be traced. SHG signal from collagen fibers is shown in magenta. Scale bar, 50 μm. Adapted and reprinted with permission from Gligorijevic et al.18 (B) In vivo lineage tracing. Series of IV‐MPM images of a growing carcinoma at the indicated time points after induction of the confetti constructs (blue: CFP+ cells, green: YFP+ cells, red: RFP+ cells). Different growth patterns of clones were observed (outlined and indicated by I‐IV). Region I displays continuous growth, regions II and III show growth before disappearance or regression, respectively, and region IV shows a delayed onset of clonal outgrowth. Scale bars, 50 μm. Adapted and reprinted with permission from Zomer et al.57 (C) Motility analysis of hypoxic cells in vivo. IV‐MPM images of a xenograft mammary tumor at 0, 11, and 28 min. Tumor cells were engineered to switch from expressing a green fluorophore to a red fluorophore in hypoxic conditions. White arrows point to motile normoxic cells (green outline). Red arrow points to a motile hypoxic cell (red outline). SHG signal from collagen fibers is shown in blue. Scale bar, 50 μm. Adapted and reprinted with permission from Wang et al.58 (D) Following extracellular vesicles (EVs) in vivo. IV‐MPM time‐lapse recordings show a tethered vesicle (circle) released in the extracellular compartment. Tumor cells were labeled with membrane GFP (green). Time is shown in min:sec. Scale bars, 5 μm. Adapted and reprinted with permission from Lai et al.59 (E) Brain cancer cells communicate via tumor microtubes. IV‐MPM of glioblastoma cells (red) in the mouse brain reveals tumor microtubes, rich in actin (yellow) that interconnect single tumor cells (arrows). Blood vessels are shown in blue. Scale bar, 50 μm. Adapted and reprinted with permission from Osswald et al.60 [Correction added on 12 July 2019, after first online publication: the Figure 2 caption has been updated.]
To track single circulating tumor cells (CTCs) in vivo, Nedoskin et al developed a methodology where Dendra2 was photoconverted in the Dendra2‐expressing CTCs present in the bloodstream.63 This approach enables monitoring the distribution and destinations of CTCs.
While most IV‐MPM studies to date used a single photoconvertible fluorescent protein, it is now possible to track two cell populations simultaneously, by using multiple photoconvertible proteins (eg, blue‐to‐green photoswitchable cyan fluorescent protein 2 (PS‐CFP2)64 combined with orange‐to‐far‐red PS‐mOrange65).
Alternatively, to expand the color scheme available for cell tracking within tumors, a genetic mouse model of breast cancer was developed so that one of four confetti constructs (cyanfluorescent protein (CFP), green fluorescent protein (GFP), yellow fluorescent protein (YFP), and red fluorescentprotein (RFP)) is randomly expressed by cancer cells.57 As the expression of a confetti construct is maintained in the progeny of each cell, this allowed tracing the lineage of individual cancer cells over time. Interestingly, at the adenoma stage, color distribution was stochastic, while at the carcinoma stage, cells were clustering into single‐color patches. Longitudinal IV‐MPM of the carcinoma tumors showed that different regions of the tumor have distinct growth patterns, ie, either continuous, delayed onset, or stasis (Figure 2B). These results have revealed that only a small fraction of cancer cells, possibly cancer stem cells, expands clonally and that proliferation capabilities of individual cells are highly diverse.
In some cases, the use of chimeric proteins, in place of ubiquitous cytoplasmic markers, is more appropriate to visualize cell behaviors in the primary tumor. For example, the epithelial‐mesenchymal transition (EMT) of cancer cells is a highly studied transition between the epithelial (high E‐cadherin expression level) and mesenchymal (low E‐cadherin expression level) phenotypes. Currently, there is a controversy regarding the respective metastatic potential of epithelial and mesenchymal cells in vivo.66 By creating transgenic mice that develop metastatic breast tumors in which endogenous E‐cadherin is fluorescently tagged, Beerling et al were able to distinguish two populations of cancer cells based on their E‐cadherin expression level: high vs low E‐cadherin.67 The authors found that motile cells in the primary tumor carried low E‐cadherin expression and mesenchymal features, while epithelial cells were nonmigratory. Moreover, while cells migrating in the primary tumor were all in mesenchymal state, cells in secondary tumor sites completely reverted to the epithelial state. This finding, made possible by imaging E‐cadherin, implies that the reversibility of the EMT may eliminate the initial differences in metastatic potential between the epithelial and mesenchymal states.
A different study reported a similar phenotypic reversibility between the primary and the secondary tumor sites in a melanoma model.68 Using an engineered fluorescent reporter for melanocyte differentiation, Brn2, Pinner et al demonstrated that only the undifferentiated cells were motile within the primary tumor and that these cells redifferentiated once arrived at the secondary site.
In contrast to breast and melanoma models, in invasive pancreatic cancers, loss of E‐cadherin expression is not required for tumor cell dissemination.69 A recent IV‐MPM study using fluorescence recovery after photo‐bleaching (FRAP) found that the genetic mutations driving invasive pancreatic cancer triggered E‐cadherin mobility at cell‐cell junctions.70 Interestingly, the existence of heterotypic interactions between cancer cells expressing E‐cadherin and cancer‐associated fibroblasts expressing N‐cadherin has been suggested in vivo.71 Collectively, this may indicate that a subpopulation of tumor cells with high E‐cadherin mobility has fluid cell‐cell junctions, allowing heterotypic interactions with surrounding fibroblasts, which increases the metastatic potential of cells with high E‐cadherin mobility compared with cells with low E‐cadherin mobility.
In summary, longitudinal tracking of cancer cells at a single cell resolution has allowed for cancer cell behaviors and metastatic fate to be described with more precision, leading to the observations of previously unknown (sub)phenotypes. IV‐MPM of cancer was also instrumental to further understand the mechanisms underlying different phenotypes, by monitoring molecular activities within the cancer cells.
3.2. Imaging molecular activities of cancer cells
To image intracellular protein/protein interactions and protein activity, numerous molecular biosensors were developed based on the principle of FRET. In FRET, energy transfer occurs between two fluorescent proteins from a donor to an acceptor, and its efficiency is dependent on their local proximity, disappearing at distances greater than 10 nm.72 Most commonly, FRET is detected by collecting emission signals of donor and acceptor proteins, following donor excitation. However, intensity‐based methods are susceptible to variations in protein expression levels and depend on molecular diffusion, requiring multiple controls for each experiment. To avoid the shortcomings of intensity‐based FRET, FRET detection can be done via fluorescence lifetime imaging microscopy (FLIM)‐FRET. The lifetime of the donor, which is shortened if FRET occurs, is measured. Advantage of the FLIM‐FRET approach, compared with intensity‐based methods, is that it is internally calibrated and less affected by external factors. However, physical motion of the imaged tissue, due for example to peristalsis, respiration, or the heartbeat, is a challenge for FLIM, as it requires long acquisition times for quantitative analysis. To correct for physiological motion in FRET‐FLIM IV‐MPM data, Warren et al developed a new open source software tool, Galene.73
Previously, the elevated activity of the small G‐protein Rac1 was shown to facilitate the hyperproliferation of cells at the base of intestinal crypts, leading to the initiation of colorectal cancer.74, 75 To test this hypothesis in vivo, a Rac‐FRET mouse exhibiting pancreatic ductal adenocarcinoma was developed.76 FLIM‐FRET IV‐MPM showed that Rac activity was significantly higher in the pancreas of animals with cancer compared with control animals, confirming the link between the Rac1 activity and cell hyperproliferation.
IV‐MPM studies revealed that motile tumor cells are unevenly distributed within the primary tumor, suggesting that signaling pathways that regulate cell motility are activated locally.18, 77, 78 One major controller of the motility pathways is RhoA, which promotes contractility of the cell rear. Use of FLIM‐FRET IV‐MPM of a RhoA biosensor on pancreatic tumors identified RhoA activity not only in the cell rear but also at the leading edge of migrating cells, which was not seen in vitro.79 This polar regulation of RhoA activity might be causing cell contraction and/or protrusion formation enhancing cell motility. Using the similar RhoA biosensor, it was later demonstrated that the blood vasculature contributes to the local RhoA activation, which could explain why RhoA activity was not observed in in vitro measurements.80 By generating a RhoA‐FRET biosensor mouse, Nobis et al confirmed the deregulation of RhoA signaling in both breast and pancreatic cancers.81
In addition to RhoA, two other members of the RhoGTPase family, namely, Rac1 and Cdc42, are critical for cell motility.82 Recently, IV‐MPM FRET showed that the activity balance among these GTPases determines two distinct motility phenotypes of brain cancer cells.83 Namely, cells that invade along blood vessels move rapidly and in a straight fashion, whereas invasive cells in the brain parenchyma are slow. Interestingly, cells infiltrating the brain parenchyma had high Rac1 and Cdc42, but low RhoA activity, while the cells along blood vessels had low Rac1 and Cdc42, but high RhoA activity. Consistently, knockdown of a Rac1 and Cdc42 activator led to cell clustering in the perivascular regions, suggesting that the slow cell phenotype is responsible for shuffling cells to the perivascular niche. This contrasts with findings in breast carcinoma, where the fast phenotype migrates along collagen fibers towards the perivascular niche in which the slow invasive cells are present.18 At this point, the link between the RhoGTPase activity, the two invasion modes, and progression of glioblastoma remains unclear.
Tumor cell motility can also be promoted by EMT regulators, such as the transforming growth factor β (TGFβ).84 To link cell motility with the activity of the TGFβ signaling pathway in vivo, breast carcinoma cells were engineered to express fluorescent chimeras of TGFβ signaling regulators (Smad2, CAGA12) and then visualized by IV‐MPM.78 Results demonstrated that only single but not cohesively migrating cells show active TGFβ signaling. Blocking of TGFβ signaling was able to induce the switch from single cell to cohesive migration. Importantly, active TGFβ signaling and single cell motility, while necessary for intravasation, did not favor the growth of cells, which reached the lung, consistent with previous findings that activation of TGFβ signaling promotes growth arrest.85 These combined data demonstrated that transient activation of TGFβ signaling is required for lung colonization.
In melanoma tumors, less than 10% of cells are motile, and cell motility occurs as single cell migration or cell streaming.86 Using IV‐MPM of fluorescent reporters for Notch and SRF signaling pathways, both pathways were shown to be active in motile cells, and this activity was regulated by levels of the histone methyltransferase EZH2, which initiates the motility in melanoma. At metastatic sites, SRF but not Notch activity was maintained.
Numerous microenvironmental cues can trigger transcriptional changes in cancer cells, which modulate their behavior (reviewed in Calvo and Sahai, Charras and Sahai, Fein and Egeblad, Conklin and Keely, Alexander et al, te Boekhorst et al, Bragado et al, and Ramamonjisoa and Ackerstaff2, 3, 4, 5, 6, 7, 8, 9). A recent IV‐MPM study used a fluorescent reporter for hypoxia in breast carcinoma cells to demonstrate that cancer cell invasiveness increases in hypoxia compared with normoxia.58 Surprisingly, hypoxic cells were mostly found near blood vessels moving slowly with abundant invadopodia that degraded the surrounding collagen fibers, while normoxic cells were among the fast subpopulation, according to the categories defined in Gligorijevic et al18 (Figure 2C). As the presence of invadopodia was associated with the metastatic population,18 this work suggested that hypoxic microenvironments might increase metastasis in vivo.
Imaging molecular activities of cancer cells has allowed a better understanding of the heterogeneity in cell signaling within tumor microenvironments. Until recently, most studies considered cells as independent entities, disregarding cell‐cell communication. A new level of tumor complexity is now being recognized, revealing that cell invasion also involves extensive cooperation between cancer cells via specialized structures.
3.3. Direct and indirect interactions between cancer cells: extracellular vesicles and microtubes
Multiple in vitro studies have demonstrated that extracellular vesicles (EVs) and their cargoes secreted by both cancer and stromal cells, facilitate long‐range intercellular communication in tumors, and can contribute to heterogeneity of cancer cell behaviors.87 EVs originate from the plasma membrane and have diameters ranging from 30 to 150 nm.87 To image these small structures in vivo, Lai et al engineered thymoma cells with fluorescently labeled plasma membranes.59 Cells were then implanted under the dorsal skinfold chamber of mice, and IV‐MPM revealed the existence of different categories of EVs with distinct trafficking behaviors (Figure 2D). Interestingly, most EVs accumulated at the tumor margin, suggesting that the stromal tissue may influence EV biogenesis and uptake. Compared with commonly used membrane dyes that decorate only the periphery of the lipid bilayer, this labeling approach allowed better visualization of EVs in vivo, most likely because only the inner EV membrane is labeled, eliminating interference with EV surface molecules that are critical for EV binding and uptake by recipient cells. Unfortunately, as cells that took up EVs could not be distinguished from cells that did not, studying EV transfer and its effects was not possible in this experiment. Moreover, simply labeling EVs does not differentiate functional from nonfunctional transfer, ie, if the EV content is degraded by lysosomes inside the recipient cell. Determining the functional consequences of EV uptake required further innovation.
A strategy based on the Cre‐LoxP was developed to distinguish cells that have taken up EVs from those that have not using IV‐MPM.88, 89 When the EV‐mediated Cre transfer occurs and the cargo is released into the cytoplasm, cells switch from DsRed to eGFP labeling (see Zomer et al89 for details). One major advantage of this protocol is that the Cre+ and the reporter+ cell populations can be defined a priori, allowing the study of EVs originating from specific cell types. For example, the release of EVs from melanoma cells in mice ubiquitously expressing the Cre‐LoxP reporter tdTomato demonstrated that both immune cells and nonimmune cells take up EVs released by the tumor cells. Coinjection of highly and less‐malignant mammary tumor cells demonstrated local and systemic transfers of EVs from malignant to less malignant cells. Strikingly, motility analysis revealed that the uptake of EVs rendered the less malignant tumor cells more migratory, implying the EV uptake increased their metastatic potential. While this study suggests that cancer cells can phenocopy behavior through EV transfer, how such small cargos can mediate these changes is debated, especially in tumors where all cells belong to the same subtype with little molecular heterogeneity.
Recently, Steenbeek et al revealed that EVs exchanged between the syngeneic melanoma cell lines B16F1 (nonmetastatic) and B16F10 (metastatic) in vivo contain thousands of RNAs and proteins involved in migration.90 This suggests that upon uptake of EVs released by B16F10 cells, the phenotypic switch of B16F1 cells to a migratory behavior is mediated by upregulation of entire pathways rather than specific genes.
Circulating tumor EVs were also shown to be internalized by endothelial cells and patrolling macrophages. So far, these observations were made in zebrafish embryos and remains to be tested in mice.91
A novel type of distant cell‐cell communication was identified in brain tumor cells that were shown to interconnect using long membrane protrusions (LMPs) or microtubes forming a multicellular network (Figure 2E).60, 92 IV‐MPM in a patient‐derived xenograft revealed that LMPs are highly dynamic and that their density increases to 50% of the tumor volume as the tumor progresses. After mitosis, LMPs can function as tracks for travelling nuclei, suggesting that they are used for efficient colonization of brain tissue. To test the function of microtubes in vivo, calcium waves were visualized, as they propagated along the tumor microtubes of interconnected cells. The synchronicity of the Ca2+ waves was significantly perturbed after knock down of the Cx43 gap junction protein. The newly identified LMP network appeared to be self‐repairing, as laser ablation within the network led to the extension of new microtubes towards the site of injury.
Altogether, new findings made possible by IV‐MPM reveal that tumor cells communicate and cooperate with each other using a multiplicity of routes. These discoveries need to be taken into consideration in the development of future anticancer treatments.
3.4. Future perspectives on single cell and subcellular IV‐MPM of cancer
Light manipulation for controlling protein activity via optogenetics has been heavily employed in neuroscience and is making its way to cancer biology, so far primarily in in vitro studies. For example, Zhou et al developed an optogenetic tool that induces EMT and subsequent cell migration of lung cancer cells.93 Next, the development of light‐sensitive receptor tyrosine kinases promises a better understanding of the mitogenic and morphogenic behaviors induced by growth factors in the future.94 A few in vivo studies were also recently published. In one of them, using a mouse model of subcutaneous melanoma, Kim et al showed that optical stimulation of calcium signaling in adoptive transferred T cells activated T cell immune response at the tumor site.95 In glioblastomas, an optogenetic strategy was used to inhibit tumor growth and increase the survival of animals bearing glioma.96 While optogenetics remains extremely challenging, it could theoretically be implemented into IV‐MPM of cancer.
As described in section 3.2, the study of protein‐protein interactions and protein activities in cells using FRET biosensors has revolutionized the field of imaging. However, accurate FRET measurement is challenging, in part because of low signal and the necessity for multiple control experiments.97 As an alternative, FRET biosensors can be designed based on reversibly photoswitchable proteins, a method called photochromic FRET. For example, Subach et al developed a photoswitchable red FRET acceptor protein that can be reversibly switched on and off, offering an intrinsic and internal signal control for FRET.98
An additional way of controlling molecular activity using light is to use photostatins: photoswitchable microtubule inhibitors.99 Using photostatins, Borowiak et al showed optical control of microtubule dynamics in vivo, modulating mitosis at a single cell level. Interestingly, the cytotoxic effect of photostatins is significantly increased upon blue light illumination that makes them a promising tool for precision chemotherapeutics.
4. EXTENDING AND COMPLEMENTING THE CAPABILITIES OF IV‐MPM
IV‐MPM provides us with a unique ability to measure real‐time cell behaviors, protein activities, and cell‐cell interactions. However, IV‐MPM used alone has its limitations: it cannot inform about gene expression within imaged tissues; it is limited to a single level of magnification; longitudinal imaging is challenging; it relies on available fluorescent molecules; and it provides relatively low‐throughput data. In the following section, we discuss how IV‐MPM can be complemented with omics platforms and/or single cell isolation techniques towards molecular profiling of imaged tumor cells. We also show how correlative imaging approaches can be used to expand the spatial scales of tumor imaging and by reviewing some emerging tools that facilitate imaging over time, provide new fluorescent molecules, and improve the throughput of IV‐MPM data (Figure 3A).
Figure 3.
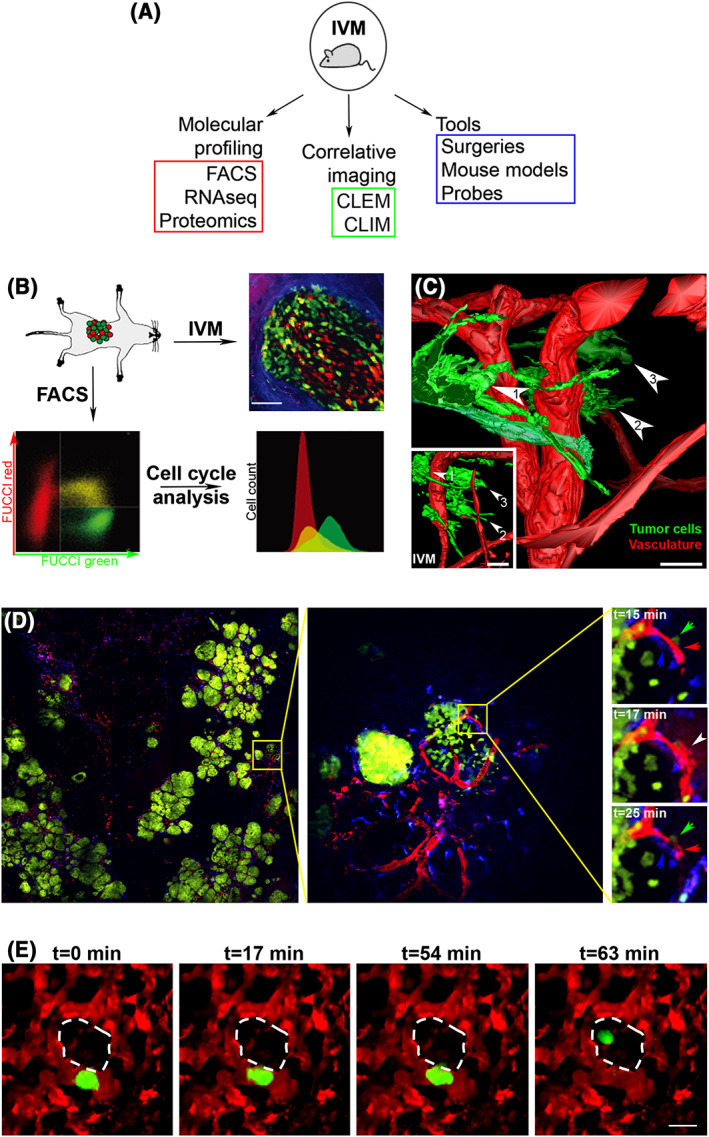
Extending the capabilities of intravital multiphoton microscopy (IV‐MPM) with correlative imaging, molecular profiling, and other emerging tools. (A) Methods that extend the potential of IV‐MPM. IV‐MPM: intravital multiphoton microscopy; FACS: fluorescence‐activated cell sorting; RNAseq: RNA sequencing; CLEM: correlative light and electron microscopy; CLIM: cryosection labeling and intravital microscopy. (B) Molecular profiling of cells imaged in vivo using fluorescence‐activated cell sorting (FACS). Fluorescent ubiquitination‐based cell cycle indicator (FUCCI)‐expressing invasive colon cancer cells were inoculated into the cecum of mice (top left panel) and observed by IV‐MPM (top right panel). Tumor dissociation and fluorescence‐activated cell sorting (FACS) enables analysis of the cell cycle profiles (bottom panels). Green: FUCCI‐green, S/G2; red: FUCCI‐red, G1; blue: SHG signal from collagen fibers. Scale bar, 75 μm. Adapted and reprinted with permission from Kagawa et al.100 (C) Intravital correlative light and electron microscopy (CLEM). Three‐dimensional (3D) model obtained from serial transmission electron microscopy imaging of a subcutaneous xenograft in the mouse ear. Three tumor cells (green) from the invasive front imaged with IV‐MPM have been retrieved and highlighted (numbered arrowheads). The inset shows a 3D model from the IV‐MPM dataset. Blood vessels are shown in red. Scale bars, 20 μm. Adapted and reprinted with permission from Karreman et al.101 (D) Large‐volume high‐resolution intravital imaging (LVHR‐IVI). Left panel: a single z plane showing large area imaging of a mammary tumor within a transgenic mouse expressing fluorescent proteins. Green: Dendra2‐expressing tumor cells, red: fluorescent dextran labeled vasculature, blue: CFP expressing macrophages. Scale, 4 × 4 mm. Middle panel: one individual tile (yellow square) from which the mosaic is composed. Image shows two groups of cancer cells with distinct growth patterns. Scale, 512 × 512 μm. Right panels: time lapse imaging of the subregion (yellow square). A macrophage (blue arrow), a tumor cell (green arrow), and an endothelial cell (red arrow) induce transient vascular leakage (white arrow). Scale, 133 × 133 μm. Adapted and reprinted with permission from Entenberg et al.102 (E) Lung IV‐MPM. Stills from a time‐lapse IV‐MPM imaging video of a spontaneously metastasizing tumor cell (green) in the lung vasculature (red), 1 day after window implantation. The cell crosses the endothelium into the alveolar space (dashed white outline) by t = 63 min. Scale bar, 15 μm. Adapted and reprinted with permission from Entenberg et al.103
4.1. Molecular profiling of tumors
Microarray analysis of tumor tissues following IV‐MPM can enable a deeper understanding of the observed tumor cell behavior. One of the first such studies observed that cells within metastatic breast tumors polarize and migrate towards blood vessels, while cells within nonmetastatic breast tumors migrate in a random fashion. This suggested that growth factors diffusing from the vasculature produced a chemotaxis response in metastatic cells only.104 By following IV‐MPM with gene expression analysis of metastatic vs nonmetastatic tumors, it was demonstrated that the nonmetastatic tumor cells expressed higher level of FGFR and IGFBP, but very low levels of EGFR compared with metastatic tumor cells. These results revealed that among all growth factors released by blood vessels, EGF is the main inducer of tumor cell chemotaxis.
Fluorescence‐activated cell sorting (FACS) combined with IV‐MPM can also complement dynamic data by providing phenotypic information. In a recent study, IV‐MPM of breast tumors in mice showed that while some myeloid cells are motile, others are sessile.29 By imaging the endocytic activity of myeloid cells, following dextran injection into the circulation, the authors found that only the stationary myeloid cells performed endocytosis. To further characterize the phenotype of the stationary myeloid cells, IV‐MPM was followed by FACS analysis of the dextran+ cells, which demonstrated that this population consisted of both tumor‐associated macrophages and dendritic cells. While a number of previous studies had referred to either tumor‐associated macrophages105, 106 or dendritic cells107 as dextran+ cells, this work proved that dextran uptake alone is not sufficient to discriminate between the two populations.
Combined IV‐MPM and FACS has also been used to analyze the fate of myeloid cells in lung metastases.108 Direct observation of the arrival of circulating tumor cells to the lung via IV‐MPM showed that tumor cells fragment and shed microparticles that are then ingested by myeloid cells. Further FACS analysis revealed that the identity of the myeloid cells that were loaded with tumor cell fragments changed over time. Many of the tumor cell‐ingesting myeloid cells extravasated and accumulated in the lung, together with the tumor cells, promoting metastases. Conversely, a population of resident dendritic cells among the last to interact with the tumor microparticles was found to restrict metastasis. Following IV‐MPM, cell fate analysis using FACS demonstrated the existence of the competition between phagocytic myeloid cells during lung metastasis.
FACS can also follow IV‐MPM imaging to isolate cell subpopulations and analyze the protein expression in the isolated cells. For example, Kagawa et al showed that the migration and invasion of colon cancer cells are cell cycle dependent via the action of the RhoGTPase‐activating protein ARHGAP11A.100 Using IV‐MPM to monitor colon cancer cells expressing the fluorescent ubiquitination‐based cell cycle indicator (FUCCI) reporters, cells in the S/G2 phases were found to migrate faster compared with cells in G1. FACS of the FUCCI‐red and FUCCI‐green cells (Figure 3B), followed by western blotting, revealed that cells in the S/G2 phases have higher level of the RhoGTPase activating protein ARHGAP11A compared with cells in the G1 phase of the cell cycle. Subcutaneous injection of ARHGAP11A‐knockdown cells followed by IV‐MPM showed that these cells were less motile, had a lower extravasation rate, and formed smaller tumors than control cells. This work highlighted the potential role of RhoGTPases in coordinating cell cycle with cell motility.
Although FACS is a high‐throughput isolation technique that allows for the collection and robust analysis of specific cell types, it requires tissue dissociation that separates cells from their microenvironment, leading to the loss of valuable contextual information. To isolate specific subpopulations of cells from tissue sections, laser capture microdissection (LCM) can be used.109, 110 As an alternative to LCM, spatial transcriptomics was recently developed in which tissue section is placed on a chip containing arrays of capture probes, which can bind cellular RNA from permeabilized cells. When combined with single‐cell RNA sequencing, spatial transcriptomics was able to provide a spatial map of all the cell types present in pancreatic ductal adenocarcinomas.111 In theory, either LCM or spatial transcriptomics could potentially improve upon FACS as a follow‐up to IV‐MPM, to analyze the gene expression profile of cells of interest while maintaining the spatial organization of the tumor tissue. The development of new methodologies for IV‐MPM followed by LCM or spatial transcriptomics will advance our understanding of cancer.
4.2. Correlative imaging: IV‐MPM followed by immunofluorescence (CLIM) or electron microscopy (CLEM)
The availability of a large collection of antibodies makes immunofluorescence a powerful technique for studying the composition of tissues, albeit in static conditions. To take advantage of both IV‐MPM and immunofluorescence imaging technologies, a correlative microscopy method called cryosection labeling and IV‐MPM (CLIM) was developed.112 After performing IV‐MPM, the area is marked by photo tattooing: a near‐IR 2P laser is used to place a label inside or next to the field of view. After cutting the tissue, all the cryosections containing the photo tattoo (visualized by autofluorescence) are used to reconstruct the area of interest and match the region to the corresponding IV‐MPM image. Alternatively, in vivo photoconversion can be used to label the region of interest.40 Using CLIM, the migratory behavior of mammary tumor cells was linked to the presence of CD4+ T and CD8+ T cell subpopulations.112
To gather information on structures at the nanoscale level, IV‐MPM can be followed by electron microscopy (EM), using a method called intravital correlative light and EM (intravital CLEM).113 To locate the region of interest imaged by IV‐MPM, serial tissue sections are imaged individually using light microscopy, searching for regions morphologically similar to IV‐MPM images.114 Unfortunately, the sample preparation for EM alters the tissue structure, complicating the identification of the exact same region of interest. To address this challenge, a novel protocol was developed to rapidly and precisely move from in vivo imaging to three‐dimensional EM (3DEM).101 After performing IV‐MPM, the surface of the specimen is marked using near‐IR light, registering the xy coordinates of the area imagined by IV‐MPM. To accelerate the process of retrieving the z‐position of the region of interest, microscopic X‐ray computed tomography (microCT) of the entire resin‐embedded sample is done.101 Using this workflow, single tumor cells imaged by IV‐MPM in the mouse ear skin were located in EM sections (Figure 3C). Among invasive cells, one cell was found to extend membrane protrusions enriched in cytoskeletal filaments (Figure 3C, cell 1), while another cell shows bleb‐like structures that are filament free but rich in intracellular vesicles (Figure 3C, cell 3) that allowed the authors to hypothesize that these two cells interact with distinct environment. This methodology also revealed that metastasizing tumor cells trapped inside brain capillaries have extensions that either intercalate between or invaginate into endothelial cells.101
4.3. Emerging tools for IV‐MPM
4.3.1. Method for large‐volume high‐resolution IV‐MPM
In the clinic, pathologists routinely switch from low‐ to high‐magnification objectives to observe tissue sections at different magnifications and establish a diagnosis. While low magnification identifies abnormal regions within a healthy organ, high magnification can detect morphological changes at single cell levels. In contrast, typical uses of IV‐MPM commonly capture images only at high magnification, making it difficult to analyze the tissue context. Recently, a method called large‐volume high‐resolution intravital imaging (LVHR‐IVI) was developed to address the need for IV‐MPM where both the tissue‐wide context and individual cells are visualized simultaneously.102 This method requires serial acquisition of high‐magnification images followed by mosaic stitching that allows for reconstruction at the tissue level. When complete, LVHR‐IVI provides a coherent picture of the tissue architecture, while maintaining high‐resolution of the individual fields of view. Using LVHR‐IVI in the MMTV‐PyMT mouse model, Entenberg et al were able to visualize large tissue volumes and revealed that at the early carcinoma stage, mammary ducts can have different morphologies, highlighting the tumor heterogeneities (Figure 3D, left and middle panels). These large mosaic images were acquired at subcellular resolution, allowing authors to monitor dynamics of cell morphologies and cell‐cell interactions (Figure 3D, right panel).
4.3.2. Prolonged visualization of internal organs using imaging windows
Surgical exposure of tissues is routinely used for IV‐MPM of internal organs; however, such “nonsurvival” surgeries permit only a single, 6 to 24‐hour imaging session, even when animal vitals are carefully controlled. To allow repeated IV‐MPM in the brain, implantable imaging windows suitable for longitudinal imaging sessions (several weeks) were developed. These have allowed long‐term imaging of the mammary gland and the abdomen.61, 115 Recently, a longitudinal imaging lung window was added to this list.103 This lung window made it possible for the first time to visualize tumor cell extravasation in real time (Figure 3E).
4.3.3. Microenvironment manipulation during IV‐MPM
Simultaneous perturbation of the tumor microenvironment and in vivo imaging has been limited because tools such as needles filled with matrigel and growth factors were not compatible with high‐resolution IV‐MPM.116 An improved version of this approach for collecting tumor cells uses the Induction Nano Intravital Device (iNANIVID) in place of needles.117, 118 iNANIVID is fully implanted in the tumor mass and has a fluorescently labeled opening that allows the user to locate and image the region where the device is creating a chemotactic gradient within the tumor microenvironment. Williams et al demonstrated that the iNANIVID filled with EGF establishes a stable gradient of the chemoattractant and induces tumor cell chemotaxis in vivo. iNANIVID is an attractive tool to manipulate the tumor microenvironment and record the effect of various agents (chemokines, growth factors, chemotherapeutics) in real time using IV‐MPM.
4.3.4. New far‐red fluorescent probes suitable for IV‐MPM
The development of new laser sources has broadened the available range of wavelengths from the near‐IR to the IR region of the light spectrum (see section 2.1), creating the need for new fluorescent proteins and dyes suitable for 2P excitation at such longer wavelengths. Bacterial phytochrome photoreceptors are attractive templates for designing such fluorescent proteins excited by near‐IR and IR light, as their absorption spectrum is the most red shifted. One such example is iRFP670119 that was used in an IV‐MPM study15 (Figure 1B). Recently, a red‐shifted‐mCherry variant was developed, opening the door to further improvement of red fluorescent proteins for IV‐MPM.120 In addition to fluorescent proteins, fluorescent dyes and far‐red quantum dots can be used in IV‐MPM studies. For example, the brain vasculature of mice was imaged with quantum‐dots 655 (molecular probes).121 However, some concerns were raised regarding the toxicity that quantum dots accumulation may have on organs and tissues.122 As an alternative, a novel class of dyes, semiconducting polymer dot (pdot), appeared as promising candidates: they are bright, stable, and have low biotoxicity.123 Using pdots, Hassan et al recently visualized the cortical microvasculature of mice at depths greater than 1 mm.124
4.3.5. New mouse models for IV‐MPM of cancer
Development of transgenic mouse models engineered to express fluorescent proteins in cancer and/or host cell types is rapidly evolving. Until recently, new genetic mouse models were mostly tailored to research interests by crossing mice with different genotypes. This approach is extremely time consuming (1‐2 y), directing most studies to employ xenograft models and develop cancer cells lines expressing fluorescent proteins of interest. With the recent emergence of genome editing techniques (eg, CRISPR/Cas9, TALEN125), transgenic mice with multiple knock‐out and knock‐in proteins, including point mutations and fluorescent proteins can easily be introduced into mouse embryos. Consequently, in the near future, we can expect a significant increase in the number of mouse models suitable for IV‐MPM of cancer.
4.4. Future technologies that will extend and complement the capabilities of IV‐MPM
Several new strategies for tissue profiling hold great promise for future integration with IV‐MPM:
4.4.1. Mass spectrometry imaging and in situ decellularization of tissues
The new technology of mass spectrometry imaging (MSI) is able to analyze the molecular signatures of cancerous tissues in an unbiased and label‐free manner.126 Using MSI, proteins, peptides, lipids, glycans, and metabolites can be detected and correlated with histological staining. Recently, Dilillo et al proved the potential use of MSI based on matrix assisted laser desorption/ionization (MALDI) to detect tumor‐specific proteoforms and proteins in a mouse model of high‐grade glioma.127 However, most of MSI studies employ fixatives that alter tissue integrity, followed by tissue sectioning or tissue clearing systems that are not optimized for imaging the extracellular matrix (ECM) architecture at high resolution.128 Hence, while MSI is extremely useful for measuring cellular protein levels, it cannot offer information on the ECM structure. Towards complementing MSI, in situ decellularization of tissues (ISDoT) was developed to decellularize organs while preserving the ECM topography and allowing downstream proteomic analysis.129 This unique methodology provides information on the ECM structure at submicron precision, as well as information on molecular composition. Mass spectrometry imaging of decellularized tissues offers the opportunity to correlate intracellular molecules and the ECM architecture with the behavior of cells imaged by IV‐MPM.
4.4.2. Microfluidic sorting and RNAseq of EVs
EVs carry diverse biomolecules like RNA, DNA, proteins, and lipids, which modulate the behavior of a recipient cell locally or at distant sites.87 They also can be used to get information on the genetic status of the tumor. Recent IV‐MPM studies have provided preliminary data on EV dynamics and EV effects (see Section 3.3), but the full composition of EVs is unknown. A recent microfluidic platform designed to isolate tumor‐derived EVs from blood was used to provide patient‐derived EVs for next‐generation RNA sequencing, demonstrating the heterogeneity of glioblastomas.130 A similar approach may be used in the future to provide detailed functional analysis of tumor EVs imaged by IV‐MPM.
4.4.3. Quantification of cell‐cell interactions in vivo
Whereas the dynamics of cell‐cell interactions can be visualized directly using IV‐MPM, the engagement of receptors and ligands in this contact must be assayed indirectly. A novel system called labeling immune partnerships by SorTagging intercellular contacts (LIPSTIC) was introduced to monitor and precisely quantify cell‐cell interactions in vivo.131 In this procedure, the ligand and receptor pair of interest are designed so that a label (fluorophore or biotin) is transferred from the ligand onto the receptor when they are in close proximity. The signal is then quantified by flow cytometry ex vivo. This methodology enables isolation of cells based on their intercellular interaction in vivo, making it a potentially valuable complement to IV‐MPM.
5. TRANSLATIONAL POTENTIAL OF IV‐MPM FOR CANCER
A large portion of chemotherapeutic agents fail to deliver the promised results in clinical trials, despite their potency in in vitro assays and animal models.132, 133 This lack of success is due, in part, to the fact that drug efficacy is based on endpoint measures, such as reduction in tumor size and/or decrease in the number of metastases. However, the dynamics and spatial heterogeneity in cell behaviors significantly influence treatment response but remain unknown. In this section, we demonstrate how IV‐MPM is uniquely positioned to provide real‐time insights into the pharmacokinetics (drug delivery), pharmacodynamics (drug effect), and resistance to chemotherapeutic agents. Additionally, we cover the development of new label‐free imaging methods, which may boost the clinical utility of IV‐MPM by guiding researchers and clinicians in designing more efficient screenings and cancer therapies for patients.
5.1. Monitoring chemotherapy distribution, effects, and resistance in preclinical models
IV‐MPM can be used to monitor the distribution of fluorescent drugs. Doxorubicin, a cytotoxic drug used to treat human cancers,134 is naturally fluorescent, providing a direct way to image its pharmacokinetics in vivo. Using the transgenic breast carcinoma model MMTV‐PyMT and IV‐MPM, Nakasone et al measured changes in sensitivity to and distribution of doxorubicin during tumor progression.33 The autofluorescence of this drug combined with IV‐MPM allowed them to demonstrate that distribution and retention of drugs in a tumor can be heavily influenced by the tumor stage, a key finding that is valuable to optimize drug delivery.
However, naturally fluorescent drugs are rare. Therefore, fluorescent analogs of chemotherapeutic agents with preserved functions need to be created to image drug effects in vivo.135, 136 Laughney et al generated a fluorescently tagged analog of the microtubule inhibitor eribulin.35 At the time, eribulin appeared to be a promising chemotherapeutic agent for advanced, taxane‐resistant breast cancers,137 but only particular patient subgroups were benefiting from this drug, raising the question of its mechanism of action.138 IV‐MPM showed that eribulin is progressively delivered from the blood vessels into the tumor tissue over 2 hours and that eribulin uptake is blocked in cells with high expression of the multidrug resistance protein 1 (MDR1) known to confer resistance to taxanes by increasing drug efflux (Figure 4A). On the basis of these results, the authors redesigned the therapeutic strategy such that an MDR1 inhibitor, HM30181, which sensitizes the MDR1‐high resistant cells to eribulin, was coadministered with eribulin. In this example, IV‐MPM at a single cell resolution was able to demonstrate heterogeneity in MDR1 expression within the tumor cell population, pointing at a strategy for potentiating eribulin efficacy.
Figure 4.
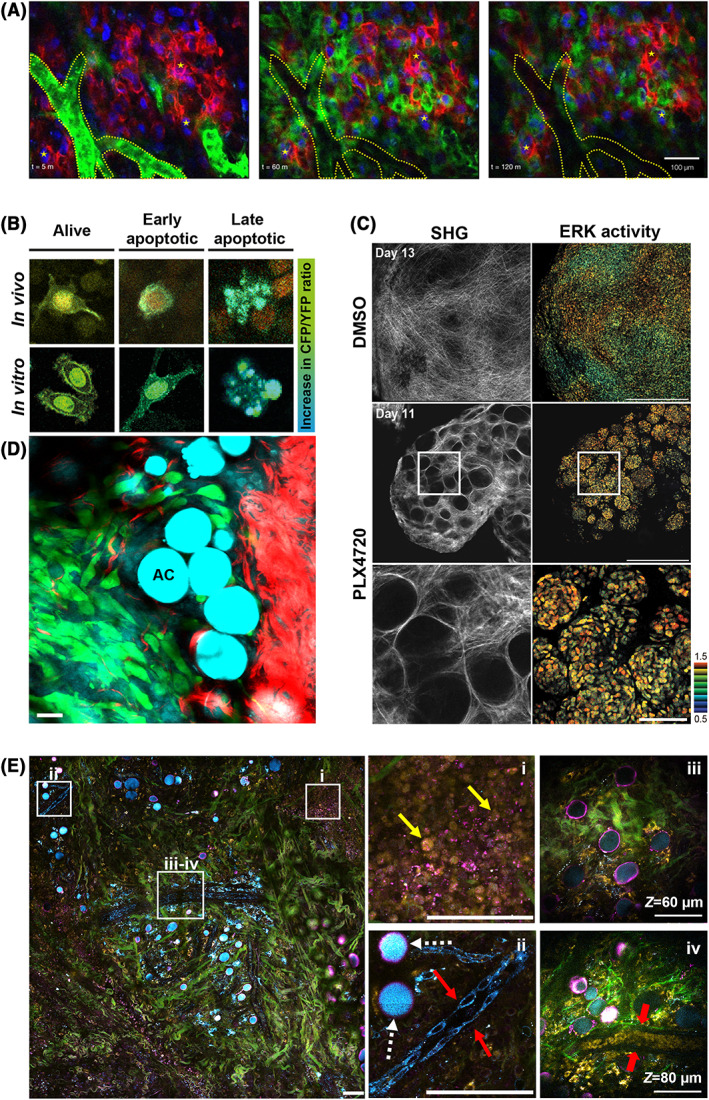
Translational potential of intravital multiphoton microscopy (IV‐MPM) of cancer. (A) Imaging pharmacokinetics using fluorescently labeled chemotherapeutics. IV‐MPM images of a taxane‐resistant HT1080 xenografts engineered to heterogeneously express MDR1‐mApple (red) and ubiquitously express H2B‐iRFP (blue). Intravenously injected BODIPY‐mesylate (green) is dynamically monitored over the course of 2 h. Blood vessels are segmented in dotted lines. Fiducial markers (stars) are placed for visual registration between frames. Scale bar, 100 μm. Reprinted with permission from Dubach et al.139 (B) Imaging pharmacodynamics. Representative images of cells in vitro and in vivo showing an increase in the CFP/YFP ratio of the Caspase‐3 Föster energy resonance transfer (FRET) biosensor as apoptosis progresses. Reprinted with permission from Janssen et al.140 (C) Imaging the role of the microenvironment in drug resistance. Representative IV‐MPM images of ERK biosensor‐expressing WM266.4 cells in nude mice treated with DMSO control (top panels) and PLX4720 (middle panels) for the indicated days. The high magnification images of the areas in white boxes are shown in bottom panels. In PLX4720‐treated mice, there are “safe havens” of thick collagen fibers (SHG signal, left panels) containing tumor cells with high ERK activity (FRET biosensor, right panels). Scale bars, 500 μm (low magnification), 100 μm (high magnification). Reprinted with permission from Janssen et al.140 (D) Label‐free imaging with combined 2‐photon/coherent anti‐Stokes Raman spectroscopy (CARS) microscopy. Direct interaction between adipocytes (AC, CARS, cyan) and tumor cells (exogenous labeling, green) during local invasion. SHG signal from collagen fibers is shown in red. Scale bar, 25 μm. Adapted and reprinted with permission from Lee et al.141 (E) Label‐free imaging of the tumor microenvironment using simultaneous label‐free autofluorescence‐multiharmonic (SLAM) microscopy. Pseudo‐colored image of a 1.5 mm × 1.5 mm field of view of a rat mammary tumor showing collagen fibers (SHG signal, green), interfaces (THG signal, magenta), flavin adenine dinucleotide (FAD) (2P autofluorescence, yellow) and nicotinamide adenine dinucleotide (NADH) (three‐photon autofluorescence, cyan). The zoom ins of boxed areas are displayed on the right. (i) NADH signal allows visualization of tumor cells (yellow arrows). (ii) Due to their shape, the FAD signal is stronger in macrophages (white dashed arrows) than in vascular endothelial cells (red arrows). (iii‐iv) the deeper z‐sections of the area show a mature blood vessel containing red blood cells (red arrows). Adapted and reprinted with permission from You et al.142
IV‐MPM of fluorescent drugs provides invaluable information about drug distribution; however, the data do not provide information about the specificity of action of the drug or whether the drug is engaged with its target. Recently, Dubach et al developed a method to determine the unbound vs bound state of a drug based on fluorescence polarization/fluorescence anisotropy, a technique called multiphoton fluorescence anisotropy microscopy (MALM).139 Measurements of anisotropy indicate the rotational diffusion rate of molecules, the value of which increases as a function of the drug binding its target. The authors used BODIPY‐AZD2281, a fluorescently labeled version of olaparib, a PARP1 inhibitor that is currently in phase III clinical trials (ClinicalTrials.gov Identifier: NCT03286842). As PARP1 is a large protein, binding of olaparib to PARP1 (ie, “target‐bound” state) yields a significant increase in anisotropy compared with unbound drug (ie, “free” state, if the drug is in the extracellular space or “intracellular” state). Following drug administration, Dubach et al saw an immediate engagement of the drug with PARP1 in the nucleus. This nuclear “target‐bound” drug pool remained stable while the free and intracellular drug pools were cleared over time, highlighting the longevity and specificity of the drug‐target interaction. By being able to assess drug‐target interaction, MALM is a promising tool to accelerate testing of chemotherapies in preclinical mouse models.143
After monitoring the delivery of chemotherapeutic agents and the interaction between these agents and their targets, IV‐MPM can be used to ensure that the tumor mass is eliminated. To be able to monitor the fate of tumor cells post‐therapy, some of the single cell and subcellular strategies presented in section 3 can be used as a real‐time readout of the host response to therapies. One such example is assessing the efficacy of a widely used microtubule modulator, paclitaxel, which causes mitotic arrest and leads to cell death. To understand whether the single cell, in vitro response to paclitaxel was similar to the in vivo response, Orth et al used a cell line expressing H2B‐mRFP (with fluorescent chromatin) and mEGFP‐α‐tubulin (with fluorescent mitotic spindle).144 IV‐MPM imaging through the mouse dorsal skinfold chamber revealed that paclitaxel is significantly less apoptotic in vivo than in vitro: in vivo, tumor cells remained arrested and survived for longer after mitotic arrest than did paclitaxel‐treated cells in vitro. However, these observations relied on the morphological signs of apoptosis via monitoring chromatin morphology, which typically appear in the late stages of apoptosis. To assess early stages of apoptosis specifically, Janssen et al used a FRET biosensor for caspase‐3 activity, along with the photoswitchable protein H2B‐Dendra2, to monitor mitotic progression and the onset of apoptosis within the same cell, following docetaxel treatment (Figure 4B).140 Strikingly, IV‐MPM performed upon docetaxel treatment revealed that the vast majority of apoptosis originated from mitosis‐independent effects of docetaxel, indicating that the drug had different mechanisms of action in vitro and in vivo. In separate IV‐MPM studies, the antimitotic drugs eribulin, paclitaxel, and ispinesib were also shown to exhibit nonmitotic effects and reduced efficacy compared with effects measured in vitro.38 Overall, measuring responses to chemotherapeutic agents by IV‐MPM may help explain varied and limited responses in clinical trials and improve current cancer treatment strategies.
The development of therapy resistance is a key contributor to therapeutic failures, caused in large part by interactions between tumor cells and their microenvironment. Therefore, to understand how resistance develops over time, it is essential to longitudinally image drug response in the native tumor microenvironment. Hirata et al established the chronology of biochemical events in response to the BRAF inhibitor PLX4720, in the BRAF‐mutant melanoma mouse model.145 In this model, a rapid initial response to the drug was observed, followed by an emergence of resistance. Interestingly, no genetically resistant subclones were detected prior to the treatment, hinting at extrinsic factors being involved in the observed response. To understand the emergence of resistance in this model, the activity of the ERK/MAPK pathway, which mediates BRAF function, was monitored by a FRET‐based biosensor.146 IV‐MPM revealed that upon PLX4720 treatment, fibroblast deposition and ECM remodeling increased, creating niches of resistant tumor cells with high ERK/MAPK signaling activity. Via ECM‐derived signals activating the integrin β1/FAK signaling axis, ERK/MAPK signaling was upregulated independently of BRAF. This allowed for large numbers of cells to survive BRAF inhibition (Figure 4C). These results pointed to a combination therapy in which for BRAF inhibition and FAK inhibition synergized to eliminate melanoma tumors and prevent the emergence of resistance. Interestingly, the tumor‐stroma dynamics proved to be integral in the development of resistance to other inhibitors as well. For example, melanoma cells adjacent to bundled collagen fibers survived after treatment with the MEK inhibitor trametinib.147 Vascular leakage and infiltration of myeloid cells were important for the response to doxorubicin or cisplatin in breast carcinomas.33 These findings have underscored the importance of considering the role of the tumor microenvironment when designing chemotherapeutic strategies.148
IV‐MPM can be used to monitor therapy response and improve already effective therapies, such as immunotherapy. Adoptive cell therapy and cyclophosphamide (ACT‐CTX) is one of the most effective treatments against melanoma.149, 150 ACT is the isolation, expansion, and reinfusion of tumor antigen‐specific lymphocytes from patients, which triggers a powerful antitumor response by tumor‐specific cytotoxic T lymphocytes (CTLs). CTX is a potent immunosuppressant that selectively targets and depletes regulatory T cells in the tumor microenvironment, which can weaken the activity of CTLs.150, 151, 152 However, it is unclear how these two therapies synergize and no spatio‐temporal dynamics have been described in vivo. By imaging implanted melanoma tumors, Qi et al showed that regulatory T cells form an “immunosuppressive ring” around the solid tumor that prevents infiltration by the adoptive CTLs and other anticancer immune cells.153 Pretreatment with CTX before ATC not only led to the removal of the immunosuppressive barrier but also promoted the accumulation of adoptive CTLs and endogenous immune cells in the tumor area. Guided by their IV‐MPM data, the authors proposed a novel regimen of three rounds of metronomic CTX‐ACT treatments that yields the most efficient results.
In summary, IV‐MPM can be employed to explain the successes or failures of cancer therapies and help design more effective strategies. However, applying discoveries from preclinical models to humans still requires caution. For example, measurement of tumor vasculature using IV‐MPM in mice underestimated the size of tumor vessels in humans,154 leading to potential differences in pharmacokinetics.
5.2. Label‐free IV‐MPM
Label‐free, nonlinear optical methodologies, such as multiharmonic signals (SHG and THG) and detection of autofluorescence are increasingly used in IV‐MPM of cancer to visualize tissue features. These eliminate the need for injectable fluorescent markers (dyes, quantum dots, nanoparticles154, 155) or cells and animals expressing fluorescent proteins (see section 4.3), which can introduce unexpected perturbations to cellular behavior and/or the microenvironment.
In biomedicine, SHG is used to visualize noncentrosymmetric structures such as collagen fibers and striated myofibers.156 Using SHG to image collagen architecture at the tumor‐stroma interface in preclinical models, the Keely lab demonstrated how ECM organization and density affect tumor progression. They outlined three tumor‐associated collagen signatures, termed TACS‐1, ‐2, ‐3, which accompany breast cancer progression in a predictable manner.157, 158 Importantly, their work further showed that the invasion‐associated signature, TACS‐3, can predict disease recurrence and survival in human patients.159
THG primarily occurs at local transitions of optical refractive index in the tissue, which can include water‐lipid or water‐protein interfaces providing additional information beyond SHG about tissue structure and organization.17 The THG signal is primarily induced by wavelengths greater than 1200 nm, which typically requires the use of OPOs to extend the laser wavelength (see section 2.1) or custom build fiber lasers.160 Due to limited applications of such high‐price light sources outside of THG in microscopy, THG is not as extensively used as SHG to date (this is likely to change with the recent development of fiber lasers50, 51, 52). In IV‐MPM of cancer, THG so far has aided in the imaging of the blood vessel walls and blood cells161 and provided outlines of adipocytes162 and extracellular vesicles.142
Autofluorescence allows imaging of metabolites and structural tissue components. Metabolic cofactors nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) are of particular interest, as they emit the strongest autofluorescence signals. Given that changes in the metabolic state contribute to tumorigenesis, imaging NADH and FAD levels can serve as cancer biomarkers.163 Furthermore, the abundance and redox state of these metabolites can be used to distinguish between different cell types present in the tumor microenvironment. Notably, the identification of tumor‐associated macrophages, which are high in FAD has been enabled by quantitative FLIM‐based imaging.164 The infiltration and recruitment of macrophages to the tumor tissue are associated with poor prognosis.165, 166, 167, 168 The ability to noninvasively image and quantify macrophages without the use for exogenous labels meets the critical need in the clinic for assessment of fresh biopsies. In addition to the intracellular metabolites, structural tissue components, such as elastin, can be visualized. While elastin autofluorescence has been useful for ex vivo studies in the lung,169 so far, no IV‐MPM study of lung elastin has been reported. With the development of the lung imaging window103 (see section 4.3.2), we can expect that more longitudinal studies will harness the autofluorescence properties of elastin, abundant in the lung.
Another label‐free technique allows imaging of chemical bonds present in biological specimens. The nonlinear optical technique called coherent anti‐Stokes Raman scattering (CARS) microscopy is based on molecular vibrational spectroscopy and provides detailed information on the chemical bonds present in biological specimens.170 In tumor biology, it has been used in combination with other imaging modalities to detect changes in adipose tissue of breast cancer,171, 172 brain tumor delineation,173 and blood flow in the tumor microenvironment.141
Combined use of SHG/THG, 2P excited fluorescence, and CARS provides an excellent platform to image tumor cell interactions with their immediate surroundings, such as blood vessels and adipocytes. For example, fluorescently labeled tumor cells were shown to directly interact with adipocytes during local invasion141 (Figure 4D). Furthermore, imaging of a carcinogen‐induced breast carcinoma in rats, using contrasts from molecular vibration (CARS microscopy), harmonic generation, and autofluorescence, revealed that a concurrent enrichment of EVs as well as a shift in the metabolic signature accompanied tumorigenesis.172 Further development of CARS‐based imaging, either alone or in combination with other imaging modalities will greatly benefit research and clinical applications.
Widespread use of entirely label‐free imaging has been hindered by the challenge of combining different technologies to provide good contrast and minimal phototoxicity. For example, the combination of 2P and 3P excitation requires sequential imaging because of the different wavelengths required, resulting in longer acquisition times and increased photodamage. To overcome such challenges, You et al designed a novel imaging platform called simultaneous label‐free autofluorescence multiharmonic (SLAM) microscopy.142 This methodology allows simultaneous imaging of NADH and FAD via 2P and 3P excited autofluorescence, respectively, as well as imaging of noncentrosymmetric structures via SHG signal and interfacial features with THG signal using a single excitation source. Using this platform, the authors were able to obtain a comprehensive and detailed view of the tumor tissue composition and organization, including collagen fibers, tumor cells (high FAD because of increased glycolytic activity), endothelial cells, and adipocytes (Figure 4E). Imaging deeper into the tissue captured mature blood vessels and red blood cells, identified by their high FAD signal and absence of nuclei (Figure 4Eiv). Thus, the SLAM microscopy platform provides fast multimodal visualization of unlabeled tissues at the molecular level with spatio‐temporal dynamics. The use of a single excitation source and the label‐free imaging capabilities make this system adaptable for clinical use. Further development of entirely label‐free imaging modes will not only allow for the visualization of additional endogenous features but has potential for the use in the clinic.
5.3. Clinical uses of IV‐MPM of cancer
The recent advances in near‐IR intravital microscopes (see section 2.1), together with the development of novel label‐free imaging methods (see section 5.2), have opened the door to IV‐MPM in cancer patients.174, 175, 176, 177, 178, 179 For example, the DermInspect and MPTflex devices (JenLab) were used for melanoma detection in patients.177, 178, 179 Confocal laser endomicroscopy is used intravitally, in place of classical histology, for gastrointestinal and bladder urothelial tumors.180, 181, 182, 183, 184 And in two current pilot clinical trials, intraoperative microscopy is being used to directly assess tumor vasculature in melanoma patients (ClinicalTrials.gov identifiers NCT01886235, NCT02857374). While the use of IV‐MPM in humans is still in its infancy, the translational potentials of this technology in intraoperative diagnosis and detection of tumor edges and/or vasculature are undeniable. With the continuous development of multiphoton microscopy systems and image analysis software, we anticipate that in the future, IV‐MPM will become an integral tool not only for diagnosis and tumor resection but also for personalized medicine.
CONFLICT OF INTEREST
The authors declare no competing or financial interests.
AUTHORS CONTRIBUTIONS
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, L.P., B.B., B.G.; Investigation, L.P., B.B.; Writing ‐ Original Draft, L.P. (sections 1, 2, 3, 4), B.B. (section 5); Writing ‐ Review & Editing, L.P., B.G.; Visualization, L.P., B.B.; Supervision, B.G.; Funding Acquisition, B.G.
ACKNOWLEDGMENTS
This study was funded by R00 CA172360/R01 CA230777, National Institutes of Health and Concern Foundation Conquer Cancer Now/Young Investigator Award to B.G. We apologize to colleagues whose work we could not cite because of manuscript space constraints. We are grateful to the authors and the journals that contributed images to this review. We thank Dr Karin Wang and Dr Chetan Patil from the Temple Bioengineering Department and Dr Rachel E. Locke for their revisions and valuable comments.
Perrin L, Bayarmagnai B, Gligorijevic B. Frontiers in intravital multiphoton microscopy of cancer. Cancer Reports. 2020;3:e1192. 10.1002/cnr2.1192
Contributor Information
Louisiane Perrin, Email: louisiane.perrin@temple.edu.
Bojana Gligorijevic, Email: bojana.gligorijevic@temple.edu.
REFERENCES
- 1. Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3(12):921‐930. [DOI] [PubMed] [Google Scholar]
- 2. Calvo F, Sahai E. Cell communication networks in cancer invasion. Curr Opin Cell Biol. 2011;23(5):621‐629. [DOI] [PubMed] [Google Scholar]
- 3. Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol. 2014;15(12):813‐824. [DOI] [PubMed] [Google Scholar]
- 4. Fein MR, Egeblad M. Caught in the act: revealing the metastatic process by live imaging. Dis Model Mech. 2013;6(3):580‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conklin MW, Keely PJ. Why the stroma matters in breast cancer: insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adh Migr. 2012;6(3):249‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alexander S, Weigelin B, Winkler F, Friedl P. Preclinical intravital microscopy of the tumour‐stroma interface: invasion, metastasis, and therapy response. Curr Opin Cell Biol. 2013;25(5):659‐671. [DOI] [PubMed] [Google Scholar]
- 7. te Boekhorst V, Preziosi L, Friedl P. Plasticity of cell migration in vivo and in silico. Annu Rev Cell Dev Biol. Oct. 2016;32(1):491‐526. [DOI] [PubMed] [Google Scholar]
- 8. Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre‐Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012;195:69‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramamonjisoa N, Ackerstaff E. Characterization of the tumor microenvironment and tumor–stroma interaction by non‐invasive preclinical imaging. Front Oncol. Jan. 2017;7:28‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson JP. Chapter 4 principles of confocal microscopy. Methods Cell Biol. 2001;63:89‐106. [DOI] [PubMed] [Google Scholar]
- 11. Tromberg BJ, Shah N, Lanning R, et al. Non‐invasive in vivo characterization of breast tumors using photon migration spectroscopy. Neoplasia. 2000;2(1–2):26‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedl P, Wolf K, Harms G, von Andrian UH. Biological Second and Third Harmonic Generation Microscopy. In: Current Protocols in Cell Biology. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2007:1‐21. [DOI] [PubMed] [Google Scholar]
- 13. Klein J, Kafka JD. The Ti:Sapphire laser: the flexible research tool. Nat Photonics. 2010;4(5):289. [Google Scholar]
- 14. Herz J, Siffrin V, Hauser AE, et al. Expanding two‐photon intravital microscopy to the infrared by means of optical parametric oscillator. Biophys J. 2010;98(4):715‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rakhymzhan A, Leben R, Zimmermann H, et al. Synergistic strategy for multicolor two‐photon microscopy: application to the analysis of germinal center reactions in vivo. Sci Rep. 2017;7(1):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Entenberg D, Wyckoff J, Gligorijevic B, et al. Setup and use of a two‐laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat Protoc. 2011;6(10):1500‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weigelin B, Bakker G‐J, Friedl P. Intravital third harmonic generation microscopy of collective melanoma cell invasion. IntraVital. 2012;1(1):32‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gligorijevic B, Bergman A, Condeelis J. Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 2014;12(11):e1001995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahou P, Zimmerley M, Loulier K, et al. Multicolor two‐photon tissue imaging by wavelength mixing. Nat Methods. 2012;9(8):815‐818. [DOI] [PubMed] [Google Scholar]
- 20. Perillo EP, Jarrett JW, Liu Y‐L, et al. Two‐color multiphoton in vivo imaging with a femtosecond diamond Raman laser. Light Sci Appl. 2017;6(11):e17095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng F, Ding C, Martin JC, et al. Spatial‐spectral multiplexing for hyperspectral multiphoton fluorescence imaging. Opt Express. 2017;25(26):32243. [Google Scholar]
- 22. Sahai E, Wyckoff J, Philippar U, Segall JE, Gertler F, Condeelis J. Simultaneous imaging of GFP, CFP and collagen in tumors in vivo using multiphoton microscopy. BMC Biotechnol. 2005;5:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lansford R, Bearman G, Fraser SE. Resolution of multiple green fluorescent protein color variants and dyes using two‐photon microscopy and imaging spectroscopy. J Biomed Opt. 2001;6(3):311. [DOI] [PubMed] [Google Scholar]
- 24. Zimmermann T, Rietdorf J, Girod A, Georget V, Pepperkok R. Spectral imaging and linear un‐mixing enables improved FRET efficiency with a novel GFP2‐YFP FRET pair. FEBS Lett. 2002;531(2):245‐249. [DOI] [PubMed] [Google Scholar]
- 25. Ricard C, Debarbieux FC. Six‐color intravital two‐photon imaging of brain tumors and their dynamic microenvironment. Front Cell Neurosci. 2014;8:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klauschen F, Ishii M, Qi H, et al. Quantifying cellular interaction dynamics in 3D fluorescence microscopy data. Nat Protoc. 2009;4(9):1305‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klauschen F, Qi H, Egen JG, Germain RN, Meier‐Schellersheim M. Computational reconstruction of cell and tissue surfaces for modeling and data analysis. Nat Protoc. 2009;4(7):1006‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60(9):2504‐2511, May. [PubMed] [Google Scholar]
- 29. Lohela M, Casbon A‐J, Olow A, et al. Intravital imaging reveals distinct responses of depleting dynamic tumor‐associated macrophage and dendritic cell subpopulations. Proc Natl Acad Sci U S A. 2014;111(47):E5086‐E5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Looney MR, Thornton EE, Sen D, Lamm WJ, Robb W, Krummel MF. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8(1):91‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Germain RN, Robey EA, Cahalan MD. Immune system a decade of imaging cellular motility and interaction dynamics in the a decade of imaging cellular motility and interaction dynamics in the immune system. Sci (New York, NY). 2012;336(6089):1676‐1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arlauckas SP, Garris CS, Kohler RH, et al. In vivo imaging reveals a tumor‐associated macrophage–mediated resistance pathway in anti–PD‐1 therapy. Sci Transl Med. 2017;9(389):eaal3604, May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakasone ES, Askautrud HA, Kees T, et al. Imaging tumor‐stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21(4):488‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harney AS, Arwert EN, Entenberg D, et al. Real‐time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage–derived VEGFA. Cancer Discov. 2015;5(9):932‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laughney AM, Kim E, Sprachman MM, et al. Single‐cell pharmacokinetic imaging reveals a therapeutic strategy to overcome drug resistance to the microtubule inhibitor eribulin. Sci Transl Med. 2014;6(261):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coffey SE, Giedt RJ, Weissleder R. Automated analysis of clonal cancer cells by intravital imaging. IntraVital. 2013;2(3):e26138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khorshed RA, Hawkins ED, Duarte D, et al. Automated identification and localization of hematopoietic stem cells in 3D intravital microscopy data. Stem Cell Reports. 2015;5(1):139‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chittajallu DR, Florian S, Kohler RH, et al. In vivo cell‐cycle profiling in xenograft tumors by quantitative intravital microscopy. Nat Methods. 2015;12(6):577‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wait E, Winter M, Bjornsson C, et al. Visualization and correction of automated segmentation, tracking and lineaging from 5‐D stem cell image sequences. BMC Bioinformatics. 2014;15(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bayarmagnai B, Perrin L, Esmaeili Pourfarhangi K, Gligorijevic B. Intravital Imaging of Tumor Cell Motility in the Tumor Microenvironment Context. In: Wells CM, Parsons M, eds. Cell Migration: Methods and Protocols. Vol.769 Totowa, NJ: Humana Press; 2018:175‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N‐WASP‐mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125(3):724‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roh‐Johnson M, Bravo‐Cordero JJ, Patsialou A, et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene. 2014;33(33):4203‐4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pourfarhangi KE, Bergman A, Gligorijevic B. ECM cross‐linking regulates invadopodia dynamics. Biophys J. 2018;114(6):1455‐1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soda P. BioImage informatics: the challenge of knowledge extraction from biological images. DT 2014‐10th Int. Conf. Digit. Technol. 2014; 2014:311–320.
- 45. Peng H. Bioimage informatics: a new area of engineering biology. Bioinformatics. 2008;24(17):1827‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meijering E, Carpenter AE, Peng H, Hamprecht FA, Olivo‐Marin JC. Imagining the future of bioimage analysis. Nat Biotechnol. 2016;34(12):1250‐1255. [DOI] [PubMed] [Google Scholar]
- 47. Dufour AC, Jonker AH, Olivo‐Marin JC. Deciphering tissue morphodynamics using bioimage informatics. Philos Trans R Soc B Biol Sci. 2017;372(1720):20150512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blanchard GB. Taking the strain: quantifying the contributions of all cell behaviours to changes in epithelial shape. Philos Trans R Soc B Biol Sci. 2017;372(1720):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mao Y, Green JBA. Systems morphodynamics: understanding the development of tissue hardware. Philos Trans R Soc B Biol Sci. 2017;372(1720):2‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li K‐C, Huang LLH, Liang J‐H, Chan M‐C. Simple approach to three‐color two‐photon microscopy by a fiber‐optic wavelength convertor. Biomed Opt Express. 2016;7(11):4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang K, Liu T‐M, Wu J, Horton NG, Lin CP, Xu C. Three‐color femtosecond source for simultaneous excitation of three fluorescent proteins in two‐photon fluorescence microscopy. Biomed Opt Express. 2012;3(9):1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He J, Du Y, Zhuang Z, et al. Wavelength separation tunable 2‐color soliton generation and its application to 2‐color fluorescence multiphoton microscopy. J Light Technol. 2018;36(16):3249‐3253. [Google Scholar]
- 53. Rodríguez C, Ji N. Adaptive optical microscopy for neurobiology. Curr Opin Neurobiol. 2018;50:83‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu T‐L, Upadhyayula S, Milkie DE, et al. Observing the cell in its native state: imaging subcellular dynamics in multicellular organisms. Science (80‐). April 2018;360(6386):eaaq1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cutrale F, Trivedi V, Trinh LA, et al. Hyperspectral phasor analysis enables multiplexed 5D in vivo imaging. Nat Methods. 2017;14(2):149‐152. [DOI] [PubMed] [Google Scholar]
- 56. Gligorijevic B, Entenberg D, Kedrin D, Segall J, van Rheenen J, Condeelis J. Intravital imaging and photoswitching in tumor invasion and intravasation microenvironments. Micros Today. Jan. 2010;18(01):34‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zomer A, Ellenbroek SIJ, Ritsma L, Beerling E, Vrisekoop N, Van Rheenen J. Brief report: intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells. 2013;31(3):602‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y, Wang H, Li J, et al. Direct visualization of the phenotype of hypoxic tumor cells at single cell resolution in vivo using a new hypoxia probe. IntraVital. 2016;5(2):e1187803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lai CP, Kim EY, Badr CE, et al. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun. 2015;6(May):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528(7580):93‐98. [DOI] [PubMed] [Google Scholar]
- 61. Kedrin D, Gligorijevic B, Wyckoff J, et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods. 2008;5(12):1019‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fumagalli A, Drost J, Suijkerbuijk SJE, et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci. 2017;114(12):E2357‐E2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nedosekin DA, Verkhusha VV, Melerzanov AV, Zharov VP, Galanzha EI. In vivo photoswitchable flow cytometry for direct tracking of single circulating tumor cells. Chem Biol. 2014;21(6):792‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol. 2004;22(11):1435‐1439. [DOI] [PubMed] [Google Scholar]
- 65. Subach OM, Patterson GH, Ting LM, Wang Y, Condeelis JS, Verkhusha VV. A photoswitchable orange‐to‐far‐red fluorescent protein, PSmOrange. Nat Methods. 2011;8(9):771‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nieto MA, Huang RYYJ, Jackson RAA, Thiery JPP. Emt: 2016. Cell. 2016;166(1):21‐45. [DOI] [PubMed] [Google Scholar]
- 67. Beerling E, Seinstra D, de Wit E, et al. Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis‐enhancing stem cell capacity. Cell Rep. 2016;14(10):2281‐2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pinner S, Jordan P, Sharrock K, et al. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69(20):7969‐7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. David JM, Rajasekaran AK. Dishonorable discharge: the oncogenic roles of cleaved e‐cadherin fragments. Cancer Res. 2012;72(12):2917‐2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Erami Z, Herrmann D, Warren SC, et al. Intravital FRAP imaging using an E‐cadherin‐GFP mouse reveals disease‐ and drug‐dependent dynamic regulation of cell‐cell junctions in live tissue. Cell Rep. 2016;14(1):152‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Labernadie A, Kato T, Brugués A, et al. A mechanically active heterotypic E‐cadherin/N‐cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19(3):224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hirata E, Kiyokawa E. Future perspective of single‐molecule FRET biosensors and intravital FRET microscopy. Biophys J. 2016;111(6):1103‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Warren SC, Nobis M, Magenau A, et al. Removing physiological motion from intravital and clinical functional imaging data. Elife. 2018;7:1‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer‐based single‐molecule probes in the membrane of living cells. Mol Cell Biol. 2002;22(18):6582‐6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Myant KB, Cammareri P, McGhee EJ, et al. ROS production and NF‐κB activation triggered by RAC1 facilitate WNT‐driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12(6):761‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Johnsson AKE, Dai Y, Nobis M, et al. The Rac‐FRET mouse reveals tight spatiotemporal control of rac activity in primary cells and tissues. Cell Rep. 2014;6(6):1153‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992‐1009. [DOI] [PubMed] [Google Scholar]
- 78. Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11(11):1287‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Timpson P, McGhee EJ, Morton JP, et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71(3):747‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McGhee EJ, Morton JP, von Kriegsheim A, et al. FLIM‐FRET imaging in vivo reveals 3D‐environment spatially regulates RhoGTPase activity during cancer cell invasion. Small GTPases. 2011;2(4):239‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nobis M, Herrmann D, Warren SC, et al. A RhoA‐FRET biosensor mouse for intravital imaging in normal tissue homeostasis and disease contexts. Cell Rep. 2017;21(1):274‐288. [DOI] [PubMed] [Google Scholar]
- 82. Tozluoğlu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol. 2013;15(7):751‐762. [DOI] [PubMed] [Google Scholar]
- 83. Hirata E, Yukinaga H, Kamioka Y, et al. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho‐family GTPases in glioblastoma cell invasion. J Cell Sci. 2012;125(4):858‐868. [DOI] [PubMed] [Google Scholar]
- 84. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119(6):1420‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Donovan J, Slingerland J. Transforming growth factor‐β and breast cancer: cell cycle arrest by transforming growth factor‐β and its disruption in cancer. Breast Cancer Res. 2000;2(2):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Manning CS, Hooper S, Sahai EA. Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene. 2015;34(33):4320‐4332, Aug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell. 2016;30(6):836‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zomer A, Maynard C, Verweij FJ, et al. In vivo imaging reveals extracellular vesicle‐mediated phenocopying of metastatic behavior. Cell. 2015;161(5):1046‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zomer A, Steenbeek SC, Maynard C, Van Rheenen J. Studying extracellular vesicle transfer by a Cre‐loxP method. Nat Protoc. 2016;11(1):87‐101. [DOI] [PubMed] [Google Scholar]
- 90. Steenbeek SC, Pham TV, de Ligt J, et al. Cancer cells copy migratory behavior and exchange signaling networks via extracellular vesicles. EMBO J. 2018;37(15):e98357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hyenne V, Ghoroghi S, Collot M, et al. Studying the fate of tumor extracellular vesicles at high spatio‐temporal resolution using the zebrafish embryo. bioRxiv. 2018;37:380238. [DOI] [PubMed] [Google Scholar]
- 92. Winkler F, Wick W. Harmful networks in the brain and beyond. Science (80‐). March 2018;359(6380):1100‐1101. [DOI] [PubMed] [Google Scholar]
- 93. Zhou X, Wang J, Chen J, et al. Optogenetic control of epithelial‐mesenchymal transition in cancer cells. Sci Rep. 2018;8(1):14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Grusch M, Schelch K, Riedler R, et al. Spatio‐temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014;33(15):1713‐1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kim K‐D, Bae S, Capece T, et al. Targeted calcium influx boosts cytotoxic T lymphocyte function in the tumour microenvironment. Nat Commun. 2017;8:15365, May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang F, Tu J, Pan JQ, et al. Light‐controlled inhibition of malignant glioma by opsin gene transfer. Cell Death Dis. 2013;4(10):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32(9):407‐414. [DOI] [PubMed] [Google Scholar]
- 98. Subach FV, Zhang L, Gadella TWJ, Gurskaya NG, Lukyanov KA, Verkhusha VV. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem Biol. 2010;17(7):745‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Borowiak M, Nahaboo W, Reynders M, et al. Photoswitchable inhibitors of microtubule dynamics optically control mitosis and cell death. Cell. 2015;162(2):402‐411. [DOI] [PubMed] [Google Scholar]
- 100. Kagawa Y, Matsumoto S, Kamioka Y, et al. Cell cycle‐dependent Rho GTPase activity dynamically regulates cancer cell motility and invasion in vivo. PLoS One. 2013;8(12):e83629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Karreman MA, Mercier L, Schieber NL, et al. Fast and precise targeting of single tumor cells in vivo by multimodal correlative microscopy. J Cell Sci. 2016;129(2):444‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Entenberg D, Pastoriza JM, Oktay MH, et al. Time‐lapsed, large‐volume, high‐resolution intravital imaging for tissue‐wide analysis of single cell dynamics. Methods. 2017;128:65‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Entenberg D, Voiculescu S, Guo P, et al. A permanent window for the murine lung enables high‐resolution imaging of cancer metastasis. Nat Methods. 2018;15(1):73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wang W, Wyckoff JB, Frohlich VC, et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62(21):6278‐6288, Nov. [PubMed] [Google Scholar]
- 105. Egeblad M, Ewald AJ, Askautrud HA, et al. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Dis Model Mech. 2008;1(2–3):155‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ojalvo LS, King W, Cox D, Pollard JW. High‐density gene expression analysis of tumor‐associated macrophages from mouse mammary tumors. Am J Pathol. 2009;174(3):1048‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Engelhardt JJ, Boldajipour B, Beemiller P, et al. Marginating dendritic cells of the tumor microenvironment cross‐present tumor antigens and stably engage tumor‐specific t cells. Cancer Cell. 2012;21(3):402‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Headley MB, Bins A, Nip A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531(7595):513‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Brasko C, Smith K, Molnar C, et al. Intelligent image‐based in situ single‐cell isolation. Nat Commun. 2018;9(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ezzoukhry Z, Henriet E, Cordelières FP, et al. Combining laser capture microdissection and proteomics reveals an active translation machinery controlling invadosome formation. Nat Commun. 2018;9(1):2031, Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Moncada R, Wagner F, Chiodin M, et al. Building a tumor atlas: integrating single‐cell RNA‐Seq data with spatial transcriptomics in pancreatic ductal adenocarcinoma. bioRxiv, May 2018;531:254375. [Google Scholar]
- 112. Ritsma L, Vrisekoop N, Van Rheenen J. In vivo imaging and histochemistry are combined in the cryosection labelling and intravital microscopy technique. Nat Commun. 2013;4:1‐8. [DOI] [PubMed] [Google Scholar]
- 113. Karreman MA, Hyenne V, Schwab Y, Goetz JG. Intravital correlative microscopy: imaging life at the nanoscale. Trends Cell Biol. 2016;26(11):848‐863. [DOI] [PubMed] [Google Scholar]
- 114. Karreman MA, Mercier L, Schieber NL, Shibue T, Schwab Y, Goetz JG. Correlating intravital multi‐photon microscopy to 3D electron microscopy of invading tumor cells using anatomical reference points. PLoS One. 2014;9(12):1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Alieva M, Ritsma L, Giedt RJ, Weissleder R, van Rheenen J. Imaging windows for long‐term intravital imaging. IntraVital. 2014;3(2):e29917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wyckoff JB, Segall JE, Condeelis JS. The collection of the motile population of cells from a living tumor advances in brief the collection of the motile population of cells from a living tumor. Cancer Res. 2000;60(19):5401‐5404. [PubMed] [Google Scholar]
- 117. Raja WK, Gligorijevic B, Wyckoff J, Condeelis JS, Castracane J. A new chemotaxis device for cell migration studies. Integr Biol. 2010;2(11–12):696‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Williams JK, Entenberg D, Wang Y, et al. Validation of a device for the active manipulation of the tumor microenvironment during intravital imaging. IntraVital. 2016;5(2):e1182271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shcherbakova DM, Verkhusha VV. Near‐infrared fluorescent proteins for multicolor in vivo imaging. Nat Methods. 2013;10(8):751‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Shen Y, Chen Y, Wu J, Shaner NC, Campbell RE. Engineering of mCherry variants with long stokes shift, red‐shifted fluorescence, and low cytotoxicity. PLoS One. 2017;12(2):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ricard C, Lamasse L, Jaouen A, Rougon G, Debarbieux F. Combination of an optical parametric oscillator and quantum‐dots 655 to improve imaging depth of vasculature by intravital multicolor two‐ photon microscopy. Biomed Opt Express. 2016;7(6):2362‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hardman R. A toxicologic review of quantum dots: toxicity depends on physicochemical and environmental factors. Environ Health Perspect. 2006;114(2):165‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yu J, Rong Y, Kuo C‐T, Zhou X‐H, Chiu DT. Recent advances in the development of highly luminescent semiconducting polymer dots and nanoparticles for biological imaging and medicine. Anal Chem. 2017;89(1):42‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hassan AM, Wu X, Jarrett JW, et al. Polymer dots enable deep in vivo multiphoton fluorescence imagingof microvasculature. Biomed Opt Express. 2019;10(2):584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Carroll D. Genome editing: past, present, and future. Yale J Biol Med. 2017;90(4):653‐659. [PMC free article] [PubMed] [Google Scholar]
- 126. Arentz G, Mittal P, Zhang C, et al. Applications of mass spectrometry imaging to cancer. Adv Cancer Res. 2017;134:27‐66. [DOI] [PubMed] [Google Scholar]
- 127. Dilillo M, Ait‐Belkacem R, Esteve C, et al. Ultra‐high mass resolution maldi imaging mass spectrometry of proteins and metabolites in a mouse model of glioblastoma. Sci Rep. 2017;7(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Byron A, Humphries JD, Humphries MJ. Defining the extracellular matrix using proteomics. Int J Exp Pathol. 2013;94(2):75‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mayorca‐Guiliani AE, Madsen CD, Cox TR, Horton ER, Venning FA, Erler JT. ISDoT: in situ decellularization of tissues for high‐resolution imaging and proteomic analysis of native extracellular matrix. Nat Med. 2017;23(7):890‐898. [DOI] [PubMed] [Google Scholar]
- 130. Reátegui E, van der Vos KE, Lai CP, et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor‐specific extracellular vesicles. Nat Commun. 2018;9(1):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pasqual G, Chudnovskiy A, Tas JMJ, et al. Monitoring T cell–dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature. 2018;553(7689):496‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Johnson JI, Decker S, Zaharevitz D, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424‐1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583‐592. [DOI] [PubMed] [Google Scholar]
- 134. Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678‐5685. [DOI] [PubMed] [Google Scholar]
- 135. Thurber GM, Reiner T, Yang KS, Kohler RH, Weissleder R. Effect of small‐molecule modification on single‐cell pharmacokinetics of PARP inhibitors. Mol Cancer Ther. 2014;13(4):986‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Thurber GM, Yang KS, Reiner T, et al. Single‐cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nat Commun. 2013;4:1504‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cortazar P, Justice R, Johnson J, Sridhara R, Keegan P, Pazdur R. US Food and Drug Administration approval overview in metastatic breast cancer. J Clin Oncol. 2012;30(14):1705‐1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Saji S. Evolving approaches to metastatic breast cancer patients pre‐treated with anthracycline and taxane. BioDrugs. 2013;27(5):469‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dubach JM, Vinegoni C, Mazitschek R, Fumene Feruglio P, Cameron LA, Weissleder R. In vivo imaging of specific drug‐target binding at subcellular resolution. Nat Commun. 2014;5(May):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Janssen A, Beerling E, Medema R, van Rheenen J. Intravital FRET imaging of tumor cell viability and mitosis during chemotherapy. PLoS One. 2013;8(5):e64029, May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lee M, Downes A, Chau YY, et al. In vivo imaging of the tumor and its associated microenvironment using combined CARS/2‐photon microscopy. IntraVital. 2015;4(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. You S, Tu H, Chaney EJ, et al. Intravital imaging by simultaneous label‐free autofluorescence‐multiharmonic microscopy. Nat Commun. 2018;9(1):2125, Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vinegoni C, Dubach JM, Feruglio PF, Weissleder R. Two‐photon fluorescence anisotropy microscopy for imaging and direct measurement of intracellular drug target engagement. IEEE J Sel Top Quantum Electron. 2016;22(3):179‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single‐cell pharmacodynamics. Cancer Res. 2011;71(13):4608‐4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hirata E, Girotti MR, Viros A, et al. Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27(4):574‐588, Apr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Harvey CD, Ehrhardt AG, Cellurale C, et al. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci. 2008;105(49):19264‐19269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Brighton HE, Angus SP, Bo T, et al. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res. 2018;78(2):542‐557, Jan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Celià‐Terrassa T, Kang Y. Metastatic niche functions and therapeutic opportunities. Nat Cell Biol. 2018;20(8):868‐877. [DOI] [PubMed] [Google Scholar]
- 149. Pittet MJ, Grimm J, Berger CR, et al. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc Natl Acad Sci U S A. 2007;104(30):12457‐12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4 + CD25 + Foxp3 + regulatory T cells by low‐dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010;70(12):4850‐4858, Jun. [DOI] [PubMed] [Google Scholar]
- 152. Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol. 2013;171(1):36‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Qi S, Li H, Lu L, et al. Long‐term intravital imaging of the multicolor‐coded tumor microenvironment during combination immunotherapy. Elife. 2016;5(e14756):1‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Stroh M, Zimmer JP, Duda DG, et al. Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo. Nat Med. 2005;11(6):678‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Mousa S, Bharali D, Khalil M, Gurbuz M, Simone TM, Mousa SA. Nanoparticles and cancer therapy: a concise review with emphasis on dendrimers. Int J Nanomedicine. January 2009;4:1. [PMC free article] [PubMed] [Google Scholar]
- 156. Campagnola PJ, Millard AC, Terasaki M, Hoppe PE, Malone CJ, Mohler WA. Three‐dimensional high‐resolution second‐harmonic generation imaging of endogenous structural proteins in biological tissues. Biophys J. 2002;82(1):493‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor‐stromal interface facilitates local invasion. BMC Med. 2006;4:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Provenzano PP, Inman DR, Eliceiri KW, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Conklin MW, Eickhoff JC, Riching KM, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178(3):1221‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Chen S‐Y, Chen S‐U, Wu H‐Y, Lee W‐J, Liao Y‐H, Sun C‐K. In vivo virtual biopsy of human skin by using noninvasive higher harmonic generation microscopy. IEEE J Sel Top Quantum Electron. 2010;16(3):478‐492. [Google Scholar]
- 161. Dietzel S, Pircher J, Nekolla AK, et al. Label‐free determination of hemodynamic parameters in the microcirculaton with third harmonic generation microscopy. PLoS One. June 2014;9(6):e99615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Rehberg M, Krombach F, Pohl U, Dietzel S. Label‐free 3D visualization of cellular and tissue structures in intact muscle with second and third harmonic generation microscopy. PLoS One. November 2011;6(11):e28237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Skala MC, Riching KM, Gendron‐Fitzpatrick A, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci. 2007;104(49):19494‐19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Szulczewski JM, Inman DR, Entenberg D, et al. In vivo visualization of stromal macrophages via label‐free FLIM‐based metabolite imaging. Sci Rep. 2016;6:25086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. October 1996;56(20):4625‐4629. [PubMed] [Google Scholar]
- 166. Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12(1):306, Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tang X. Tumor‐associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. May 2013;332(1):3‐10. [DOI] [PubMed] [Google Scholar]
- 168. Qiu S, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schröder CP. Tumor‐associated macrophages in breast cancer: innocent bystander or important player? Cancer Treat Rev. November 2018;70(August):178‐189. [DOI] [PubMed] [Google Scholar]
- 169. Tilbury K, Hocker J, Wen BL, Sandbo N, Singh V, Campagnola PJ. Second harmonic generation microscopy analysis of extracellular matrix changes in human idiopathic pulmonary fibrosis. J Biomed Opt. 2014;19(8):086014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Evans CL, Xie XS. Coherent anti‐stokes raman scattering microscopy: chemical imaging for biology and medicine. Annu Rev Anal Chem. 2008;1(1):883‐909. [DOI] [PubMed] [Google Scholar]
- 171. Le TT, Rehrer CW, Huff TB, Nichols MB, Camarillo IG, Cheng J‐X. Nonlinear optical imaging to evaluate the impact of obesity on mammary gland and tumor stroma. Mol Imaging. 2007;6(3):205‐211. [PMC free article] [PubMed] [Google Scholar]
- 172. Tu H, Liu Y, Marjanovic M, et al. Concurrence of extracellular vesicle enrichment and metabolic switch visualized label‐free in the tumor microenvironment. Sci Adv. 2017;3(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Le V‐H, Yoo SW, Yoon Y, et al. Brain tumor delineation enhanced by moxifloxacin‐based two‐photon/CARS combined microscopy. Biomed Opt Express. 2017;8(4):2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Perry SW, Burke RM, Brown EB. Two‐photon and second harmonic microscopy in clinical and translational cancer research. Ann Biomed Eng. 2012;40(2):277‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. König K. Clinical multiphoton tomography. J Biophotonics. 2008;1(1):13‐23. [DOI] [PubMed] [Google Scholar]
- 176. Gabriel EM, Fisher DT, Evans S, Takabe K, Skitzki JJ. Intravital microscopy in the study of the tumor microenvironment: from bench to human application. Oncotarget. 2018;9(28):20165‐20178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Dimitrow E, Ziemer M, Koehler MJ, et al. Sensitivity and specificity of multiphoton laser tomography for in vivo and ex vivo diagnosis of malignant melanoma. J Invest Dermatol. July 2009;129(7):1752‐1758. [DOI] [PubMed] [Google Scholar]
- 178. Balu M, Kelly KM, Zachary CB, et al. Distinguishing between benign and malignant melanocytic nevi by in vivo multiphoton microscopy. Cancer Res. May 2014;74(10):2688‐2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Balu M, Zachary CB, Harris RM, et al. In vivo multiphoton microscopy of basal cell carcinoma. JAMA Dermatology. October 2015;151(10):1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wallace MB, Meining A, Canto MI, et al. The safety of intravenous fluorescein for confocal laser endomicroscopy in the gastrointestinal tract. Aliment Pharmacol Ther. 2010;31(5):548‐552. [DOI] [PubMed] [Google Scholar]
- 181. Wu K, Liu JJ, Adams W, et al. Dynamic real‐time microscopy of the urinary tract using confocal laser endomicroscopy. Urology. 2011;78(1):225‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kiesslich R, Gossner L, Goetz M, et al. In vivo histology of barrett's esophagus and associated neoplasia by confocal laser endomicroscopy. Clin Gastroenterol Hepatol. 2006;4(8):979‐987. [DOI] [PubMed] [Google Scholar]
- 183. Xie X, Li C, Zuo X, et al. Differentiation of colonic polyps by confocal laser endomicroscopy. Endoscopy. February 2011;43(02):87‐93. [DOI] [PubMed] [Google Scholar]
- 184. Nakao M, Yoshida S, Tanaka S, et al. Optical biopsy of early gastroesophageal cancer by catheter‐based reflectance‐type laser‐scanning confocal microscopy. J Biomed Opt. 2008;13(5):054043. [DOI] [PubMed] [Google Scholar]