

ABSTRACT
Background
The protein kinase target of rapamycin (mTOR) in complex 1 (mTORC1) is activated by amino acids and in turn upregulates anabolic processes. Under nutrient-deficient conditions, e.g., amino acid insufficiency, mTORC1 activity is suppressed and autophagy is activated. Intralysosomal amino acids generated by autophagy reactivate mTORC1. However, sustained mTORC1 activation during periods of nutrient insufficiency would likely be detrimental to cellular homeostasis. Thus, mechanisms must exist to prevent amino acids released by autophagy from reactivating the kinase.
Objective
The objective of the present study was to test whether mTORC1 activity is inhibited during prolonged leucine deprivation through ATF4-dependent upregulation of the mTORC1 suppressors regulated in development and DNA damage response 1 (REDD1) and Sestrin2.
Methods
Mice (8 wk old; C57Bl/6 × 129SvEV) were food deprived (FD) overnight and one-half were refed the next morning. Mouse embryo fibroblasts (MEFs) deficient in ATF4, REDD1, and/or Sestrin2 were deprived of leucine for 0–16 h. mTORC1 activity and ATF4, REDD1, and Sestrin2 expression were assessed in liver and cell lysates.
Results
Refeeding FD mice resulted in activation of mTORC1 in association with suppressed expression of both REDD1 and Sestrin2 in the liver. In cells in culture, mTORC1 exhibited a triphasic response to leucine deprivation, with an initial suppression followed by a transient reactivation from 2 to 4 h and a subsequent resuppression after 8 h. Resuppression occurred concomitantly with upregulated expression of ATF4, REDD1, and Sestrin2. However, in cells lacking ATF4, neither REDD1 nor Sestrin2 expression was upregulated by leucine deprivation, and resuppression of mTORC1 was absent. Moreover, in cells lacking either REDD1 or Sestrin2, mTORC1 resuppression was attenuated, and in cells lacking both proteins resuppression was further blunted.
Conclusions
The results suggest that leucine deprivation upregulates expression of both REDD1 and Sestrin2 in an ATF4-dependent manner, and that upregulated expression of both proteins is involved in resuppression of mTORC1 during prolonged leucine deprivation.
Keywords: leucine, mTOR, REDD1, Sestrin2, ATF4
Introduction
The mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase that regulates cell growth and differentiation by controlling a variety of cellular processes, including protein synthesis and carbohydrate and nucleotide metabolism (1). The kinase forms 2 complexes in mammalian cells, referred to as mTOR complex 1 and 2 (mTORC1 and mTORC2, respectively), and the complexes have distinct substrate preferences. For example, when mTOR is present in complex 1, but not complex 2, it phosphorylates proteins such as the 70-kDa ribosomal protein S6 kinase 1 (p70S6K1), eukaryotic initiation factor 4E binding protein 1 (4E-BP1), and Unc-51-like autophagy activating kinase 1 (ULK1). Thus, activation of mTORC1 promotes cell growth both by upregulating anabolic processes, e.g., protein synthesis, and by suppressing catabolic processes, e.g., autophagy.
The activity of mTORC1 is regulated in part by hormones and nutrients, e.g., insulin and the branched-chain amino acid leucine, respectively. For example, refeeding (RF) a feed-deprived (FD) rat or mouse activates mTORC1 and upregulates mRNA translation in the liver (2, 3). Oral administration of leucine to FD rats has a similar effect (4). Likewise, leucine readdition to cells in culture deprived of the amino acid activates mTORC1 and upregulates protein synthesis (5). Both in vivo and in cells in culture, rapamycin treatment blocks both the leucine-induced activation of mTORC1 and upregulation of protein synthesis, showing that activation of mTORC1 is required for the response.
In contrast to the upregulation of anabolic processes associated with mTORC1 activation, suppressed mTORC1 activity, e.g., in response to nutrient deprivation, leads to activation of catabolic processes, e.g., autophagy, to provide metabolic substrates for energy production. Indeed, one of the best-characterized mechanisms through which autophagy is induced is deprivation of amino acids (6). Activation of autophagy leads to degradation of macromolecules such as proteins, resulting in the release of amino acids and other substrates that can be further metabolized. However, if the amino acids released during autophagy were to reactivate mTORC1, autophagy would be suppressed even though extracellular amino acids remain limiting in amino acid–deprived cells. Thus, mechanisms must exist to prevent the amino acids released during autophagic protein degradation from reactivating mTORC1 while nutrients are limiting.
One of the best-characterized suppressors of mTORC1 activation is a protein referred to as regulated in development and DNA damage response 1 (REDD1) (7). REDD1 expression is upregulated in response to a variety of stresses, including food deprivation in vivo (8) and serum deprivation of cells in culture (9). REDD1 acts to repress mTORC1 in part by binding to protein phosphatase 2A (PP2A) and targeting it to Akt (also known as protein kinase B) leading to dephosphorylation and inactivation of the kinase (10). In addition to REDD1, the Sestrin proteins have also been shown to act as mTORC1 suppressors (11). Similar to REDD1, expression of the Sestrins is upregulated in response to a variety of stresses (12). However, in contrast to REDD1, the Sestrins do not act through the Akt pathway to repress mTORC1, but instead bind to and activate AMPK (11). AMPK subsequently phosphorylates a subunit of the mTORC1 complex referred to as regulatory associated protein of mTOR (Raptor), leading to suppression of mTORC1 activity (13).
A recent study (14) showed that depriving mouse embryo fibroblasts (MEFs) of leucine led to a rapid, i.e., within 30 min, suppression of mTORC1 activity followed by a gradual recovery over the next several hours. However, with more prolonged deprivation, e.g., 16–24 h, mTORC1 activity was again suppressed, concomitant with increased expression of activating transcription factor 4 (ATF4) and Sestrin2. In MEFs deficient in ATF4, Sestrin2 was not induced and mTORC1 activity remained elevated, even after 24 h of leucine deprivation. This finding shows that the suppression of mTORC1 in response to prolonged leucine deprivation requires ATF4 and suggests that ATF4-induced upregulation of Sestrin2 expression is involved in the effect. However, we (15) and others (16) showed that ATF4 also upregulates REDD1 expression. Moreover, we showed that REDD1 expression is induced by prolonged leucine deprivation in HeLa cells (17). Whether or not upregulated expression of both proteins is necessary for maintenance of mTORC1 suppression during prolonged leucine deprivation, and if so the relative contribution of each to maintaining the suppression, is unknown. Consequently, the hypothesis tested in the present study was that mTORC1 activity is suppressed during prolonged leucine deprivation by combined upregulation of REDD1 and Sestrin2, and that the response occurs through an ATF4-dependent mechanism. Herein, we show that prolonged leucine deprivation leads to upregulated expression of ATF4, REDD1, and Sestrin2, concomitant with resuppression of mTORC1 activity. We also show that the activity of mTORC1 is resistant to resuppression in cells deficient in either REDD1 or Sestrin2. Overall, the results suggest that resuppression of mTORC1 during prolonged leucine deprivation is mediated through ATF4-induced upregulation of both REDD1 and Sestrin2.
Methods
Animals
The animal facilities and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of The Penn State College of Medicine. Wild-type C57Bl/6 × 129SvEv mice were housed in a temperature (25°C) and light (12:12 h light:dark cycle) controlled environment. Mice were provided standard rodent chow (Harlan-Teklad, catalog number: 8604; 24.3% crude protein, 4.7% fat, and 40.2% carbohydrate) and water ad libitum. Mice (8 wk of age; 8 male and 8 female) were food deprived for 24 h, and one-half of the mice were refed for 1 h. Mice were anesthetized by isoflurane inhalation (EZ-Anesthesia), and their livers were rapidly removed and flash frozen between aluminum blocks cooled to the temperature of liquid nitrogen and stored at −80°C until use. Tissues were processed as described previously (18). A Bradford protein assay (BioRad, catalog number: 5000006) was performed on an aliquot of the supernatant according to the manufacturer's protocol, and samples were adjusted to 1.0 µg protein/µl SDS sample buffer prior to electrophoresis performed as described below.
Cell culture
Wild-type and REDD1−/− mouse embryo fibroblasts (MEFs; kindly provided by Dr. Leif Ellison, Harvard Medical School) were maintained in high-glucose DMEM (Gibco, catalog number: 11965-092) supplemented with 10% FBS (Atlas Biologicals, catalog number: EF-0500-A) and 1% Pen/Strep (Gibco, catalog number: 15070). ATF4−/− MEFs (kindly provided by Drs. David Ron and Heather Harding, Cambridge Institute for Medical Research) were maintained similarly, except the medium additionally contained 55 μM β-mercaptoethanol (Gibco, catalog number: 21985-023) and 1 × MEM nonessential amino acids (Sigma Aldrich, catalog number: M7145). All cells were maintained at 37°C and 5% CO2. Two days prior to experimentation, cells were seeded onto 6-well plates and grown to 50–70% confluency. On the day of the experiment, cells were rinsed once with media lacking leucine (DMEM 4.5, Atlanta Biologicals, catalog number: D22215L) supplemented with 10% FBS and then incubated in this media for the time indicated in the figures. Cells were rinsed once with ice-cold PBS and harvested in 200 µl lysis buffer consisting of 20 mM HEPES (pH 7.4), 2 mM EGTA, 50 mM NaF, 100 mM KCl, 0.2 mM EDTA·Na2, 50 mM β-glycerophosphate, 2.5% Triton X-100, 0.25% deoxycholate, 1 mM DTT, 1 mM benzamidine, 0.5 mM sodium vanadate, and 10 μl/ml protease inhibitor cocktail. The suspension was rocked for 30 min at 4°C, and then centrifuged at 11,570 × g in an Eppendorf 5415R centrifuge for 10 min at 4°C. The protein content of the supernatant fraction was quantified by the Bradford method and equal quantities of protein were used for Western blot analysis.
Lentiviral packaging and infection
A short hairpin RNA (shRNA) targeting the sequence of the mouse Sestrin2 gene (TRCN0000087791, sense sequence: CCGGCGTAGGATGTACAACCTCTTTCTCGAGAAAGAGGTTGTACATCCTACGTTTTTG) was obtained from the Human and Mouse Whole Genome shRNA Library Core at the Penn State College of Medicine (TRC Lentiviral Genome shRNA Library, catalog number: RMM4013, Dharmacon), and TRC Lentiviral Non-targeting shRNA Control was purchased from Dharmacon (catalog number: RHS6848). Lentiviral constructs were transfected into 293FT cells using Lipofectamine 2000 (Invitrogen, catalog number: 11668-019) and lentivirus was packaged using a ViraPower Kit (Invitrogen, catalog number: K497500) following the manufacturer's instructions. Medium was collected, filtered through a 0.45-μm Millex-hv filter, and stored at −80°C until use. Wild-type and REDD1−/− MEFs were infected with lentivirus and selected with 3 μg/ml and 5 μg/ml puromycin, respectively, for 5 d. Cells were diluted to a concentration of 5 cells/ml, and 100 µl/well of the suspension was plated in 96-well plates to generate individual colonies. The extent of Sestrin2 knockdown was assessed by Western blot, and a positive Sestrin2 knockdown clone was selected for each cell line. These clones are referred to as MEF shSestrin2 and MEF REDD1−/− shSestrin2, respectively.
Western blot analysis
Samples were subjected to electrophoresis on Criterion TGX precast Gels (Bio-Rad, catalog numbers: 5671034, 5671084, and 5671083) and proteins in the gel were transferred to a polyvinylidene difluoride membrane (Merck Millipore Ltd, catalog number: IPVH00010). The membrane was blocked and then incubated overnight at 4°C with primary antibody as described previously (19). Antibody against p70S6K1 was purchased from Bethyl Laboratories, Inc. (catalog number: A300-510A); antibody to phospho-T389 p70S6K1 was obtained from Cell Signaling Technology (catalog number: 9205); antibodies to REDD1 (catalog number: 10638-1-AP), Sestrin1 (catalog number: 21668-1-AP), and Sestrin2 (catalog number: 10795-1-AP) were from Proteintech Group; antibody to Sestrin3 was from Abgent (catalog number: AP12471C); antibody to GAPDH was from Santa Cruz Biotechnology (catalog number: sc-32233); and ATF4 antibody was generously provided by Dr. Michael Kilberg, University of Florida College of Medicine. Following incubation with appropriate secondary antibodies, the antigen–antibody interaction was visualized with Clarity Western ECL blotting substrate (Bio-Rad, catalog number: 1705060) using a Fluorchem M imaging system (ProteinSimple). Blots were quantified using Image J software (NIH).
qRT-PCR
Wild-type or knockout MEFs were harvested before 8 or 16 h after the medium was changed to one lacking leucine. RNA was extracted from cells using Trizol reagent (Invitrogen, catalog number: 15596-018) according to the manufacturer's protocol. RNA quality and concentration were determined using a NanoDrop Spectrophotometer (ND-1000, Thermo Fisher Scientific). RNA (1 μg) was reverse transcribed and subjected to qRT-PCR using the following primers: Atf4 forward, 5′-GGAATGGCCGGCTATGG-3′, reverse, 5′-TCCCGGAAAAGGCATCCT-3′; Sesn2 forward, 5′-TAGCCTGCAGCCTCACCTAT-3′, reverse, 5′-TATCTGATGCCAAAGACGCA-3′; and Ddit4 (the gene encoding REDD1) forward, 5′-TGGTGCCCACCTTTCAGTTG-3′, reverse, 5′-GTCAGGGACTGGCTGTAACC-3′. β-actin (Actb) primers were purchased from Qiagen (catalog number: QT00095242). mRNA expression levels were normalized to β-actin mRNA using the 2–∆∆Ct method (20).
Statistical methods
Figures were generated and statistical analysis was performed using GraphPad Prism 7.0c (Graphpad Software Inc.). Data are presented as means ± SEM. Student's t test was used for analysis when 2 groups were compared. Otherwise, 1- or 2-way ANOVA was used with Tukey or Sidak correction for multiple comparisons, respectively. Statistical significance was set at P < 0.05.
Results
In agreement with previous studies (2, 3), mTORC1 was activated in the liver in response to RF mice that were FD overnight (Figure 1A and B). The refeeding–induced activation of mTORC1 was accompanied by a rapid, i.e., within 1 h, suppression of both REDD1 (Figure 1A and C) and Sestrin2 (Figure 1A and D) expression, consistent with their contributing to suppression of mTORC1 activity in response to overnight food deprivation.
FIGURE 1.

Activation of mTORC1 in response to refeeding FD mice is associated with downregulation of REDD1 and Sestrin2 expression. Mice were fasted overnight and the next morning one-half of the animals were refed for 1 h. Blots shown are mean ± SEM of 7–8 animals/condition. ***P < 0.0005 compared with FD; **P < 0.005 compared with FD. FD, food deprived; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; p70S6K1, 70 kDa ribosomal protein S6 kinase 1; p-p70S6K1, p70S6K1 phosphorylated on Thr389; REDD1, regulated in development and DNA damage responses 1.
To further assess a possible role for REDD1 and/or Sestrin2 in suppressing mTORC1 activity in response to nutrient deprivation, MEFs were deprived of leucine for various periods of time and mTORC1 activity was assessed. mTORC1 activity exhibited a triphasic response to leucine deprivation in MEFs and was rapidly (within 15 min) suppressed, followed by a transient reactivation from 2 to 4 h and a subsequent resuppression after 8 h of deprivation (Figure 2A and B). Notably, upregulation of both REDD1 and Sestrin2 (Figure 2C and D), respectively, but not Sestrin1 or Sestrin3, expression occurred concomitantly with resuppression of mTORC1. In addition, ATF4 expression (Figure 2E) was also upregulated during prolonged (e.g., ≥8 h) leucine deprivation, suggesting that it might mediate the increase in REDD1 and Sestrin2 expression that occurs in response to ≥8 h of leucine deprivation.
FIGURE 2.
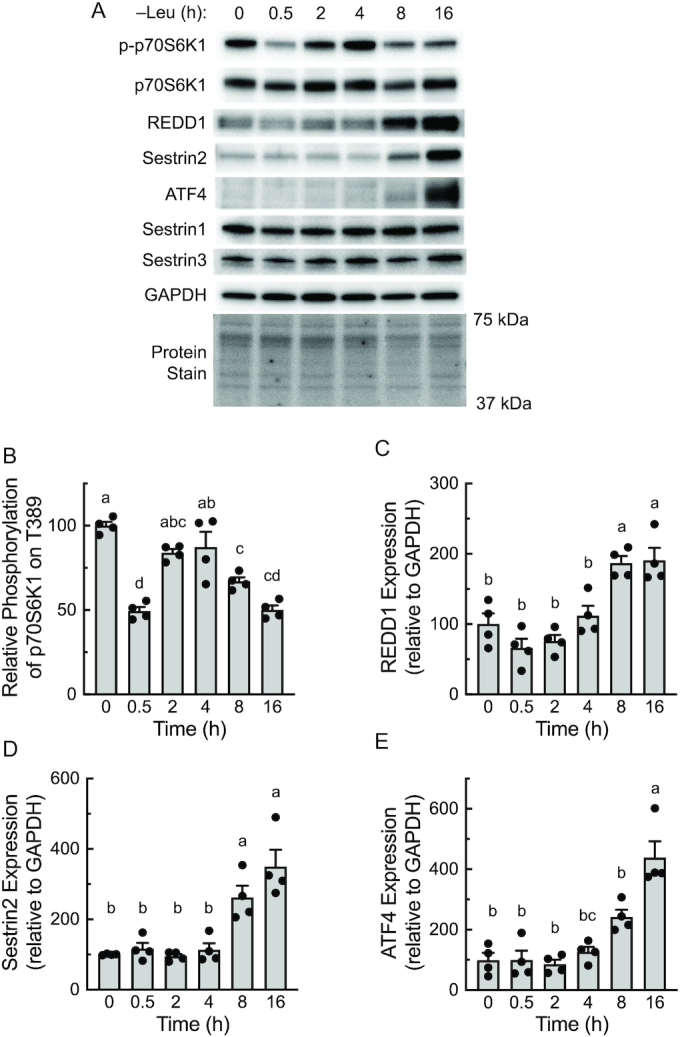
Resuppression of mTORC1 following prolonged leucine deprivation coincides with increased ATF4 expression. (A) Representative blots. p-p70S6K1, p70S6K1 phosphorylated on Thr389. (B–E) Quantitation of Western blots. Values are means ± SEM, n = 4. Labeled means without a common letter differ, P < 0.05. ATF4, activating transcription factor 4; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; p70S6K1, 70 kDa ribosomal protein S6 kinase 1; REDD1, regulated in development and DNA damage responses 1.
To evaluate the role ATF4 plays in mediating leucine deprivation–induced upregulation of REDD1 and Sestrin2 expression, wild-type and ATF4–/– MEFs were deprived of leucine for 8 or 16 h, and expression of the Atf4, Redd1, and Sesn2 mRNAs was assessed. As shown in Figure 3, in wild-type cells, expression of all 3 mRNAs was increased after 8 h of leucine deprivation and further increased at the 16-h time point. However, in cells lacking ATF4, Sesn2 mRNA (Figure 3B) expression was not altered by leucine deprivation and Redd1 mRNA expression (Figure 3C) was downregulated rather than upregulated after 16 h of leucine deprivation, showing that the upregulated expression of both mRNAs occurs through an ATF4-dependent mechanism. As expected based on the change in mRNA expression, the leucine deprivation-induced suppression of mTORC1 activity (Figure 4A and B) was associated with a progressive upregulation of ATF4 (Figure 4A and C), REDD1 (Figure 4A and D), and Sestrin2 (Figure 4A and E). In contrast, leucine deprivation had no effect on mTORC1 activity or expression of REDD1 or Sestrin2 proteins in ATF4–/– MEFs. Moreover, compared to wild-type cells, mTORC1 activity was elevated in ATF4–/– MEFs at each time point.
FIGURE 3.
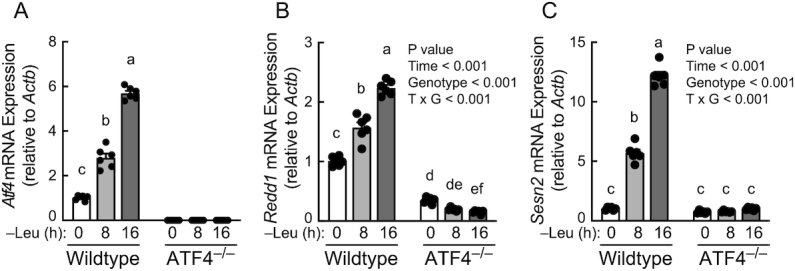
Leucine deprivation upregulates Redd1 and Sesn2 mRNA expression in an ATF4-dependent manner. RNA was isolated and subjected to qRT-PCR analysis for (A) Atf4, (B) Redd1, and (C) Sesn2 mRNA expression. Values are means ± SEM, n = 6. Panel A: 1-way ANOVA. Panels B and C: 2-way ANOVA. Labeled means without a common letter differ, P < 0.001. ATF4, activating transcription factor 4; REDD1, regulated in development and DNA damage responses 1.
FIGURE 4.
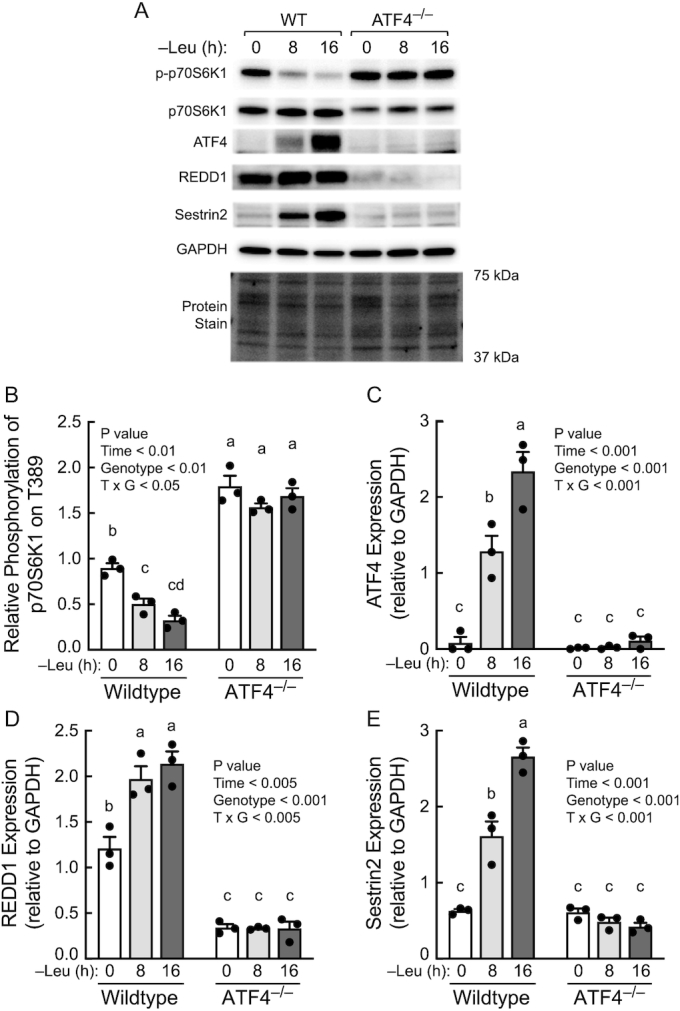
Resuppression of mTORC1 activity during prolonged leucine deprivation occurs through an ATF4-dependent process. (A) Representative blots. (B–E) Quantitation of Western blots. Values are means ± SEM, n = 3. Labeled means without a common letter differ, P < 0.05. ATF4, activating transcription factor 4; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; REDD1, regulated in development and DNA damage responses 1; WT, wildtype.
The role REDD1 plays in mediating resuppression of mTORC1 during prolonged leucine deprivation was further assessed using REDD1−/− MEFs. Consistent with the results shown in Figure 4, leucine deprivation–induced suppression of mTORC1 activity (Figure 5A and B) was associated with upregulation of ATF4 (Figure 5A and C), REDD1 (Figure 5A and D), and Sestrin2 (Figure 5A and E) expression in wild-type (i.e., REDD1+/+) MEFs. ATF4 and Sestrin2 abundances were similar in both wild-type and REDD1−/− MEFs after 16 h of leucine deprivation. However, mTORC1 activity was higher in REDD1−/− than in wild-type cells maintained in leucine-containing (hereafter referred to as complete) medium and remained higher at each time point after leucine deprivation, suggesting that REDD1 plays an important role in limiting basal mTORC1 activity in cells maintained in complete medium. Moreover, the results suggest that upregulated expression of Sestrin2 alone does not account entirely for the resuppression of mTORC1 activity induced by prolonged leucine deprivation.
FIGURE 5.
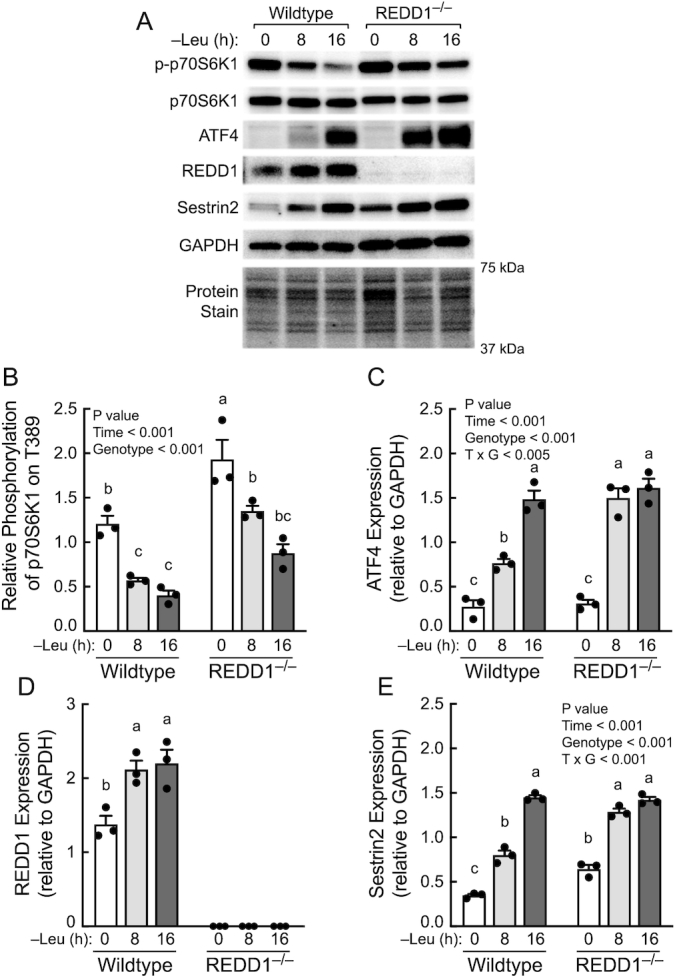
Contribution of REDD1 to the resuppression of mTORC1 activity during prolonged leucine deprivation. (A) Representative blots. (B–E) Quantitation of Western blots. Values are means ± SEM, n = 3. Panels B, C, and E: 2-way ANOVA. Panel D: 1-way ANOVA. Labeled means without a common letter differ, P < 0.05. ATF4, activating transcription factor 4; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; REDD1, regulated in development and DNA damage responses 1.
To further assess the role Sestrin2 plays in mediating resuppression of mTORC1 activity, MEFs with stable knockdown of Sestrin2 were generated. In wild-type MEFs, the Sestrin2 shRNA prevented the upregulation of Sestrin2 expression induced by prolonged leucine deprivation and in REDD1−/− MEFs with stable Sestrin2 suppression the upregulation was markedly attenuated (Figure 6A and D). Similar to the results shown in Figure 5, the time course for mTORC1 resuppression was similar in REDD1−/− compared to wild-type MEFs stably expressing the control shRNA, although mTORC1 activity was numerically higher at each time point in REDD1−/− compared to wild-type MEFs (Figure 6A and B). In wild-type MEFs deficient in Sestrin2 resuppression of mTORC1 was not observed until 16 h of leucine deprivation (Figure 6A and C). Interestingly, mTORC1 activity was upregulated in REDD1−/− MEFs deficient in Sestrin2 after 8 h of leucine deprivation compared to cells in complete medium. However, mTORC1 was partially, but not completely suppressed in cells deficient in both proteins after 16 h of leucine deprivation, perhaps due to an incomplete suppression of Sestrin2 expression in those cells. Combined, the results strongly suggest that both REDD1 and Sestrin2 play critical roles in maintaining mTORC1 in a suppressed state during prolonged leucine deprivation.
FIGURE 6.
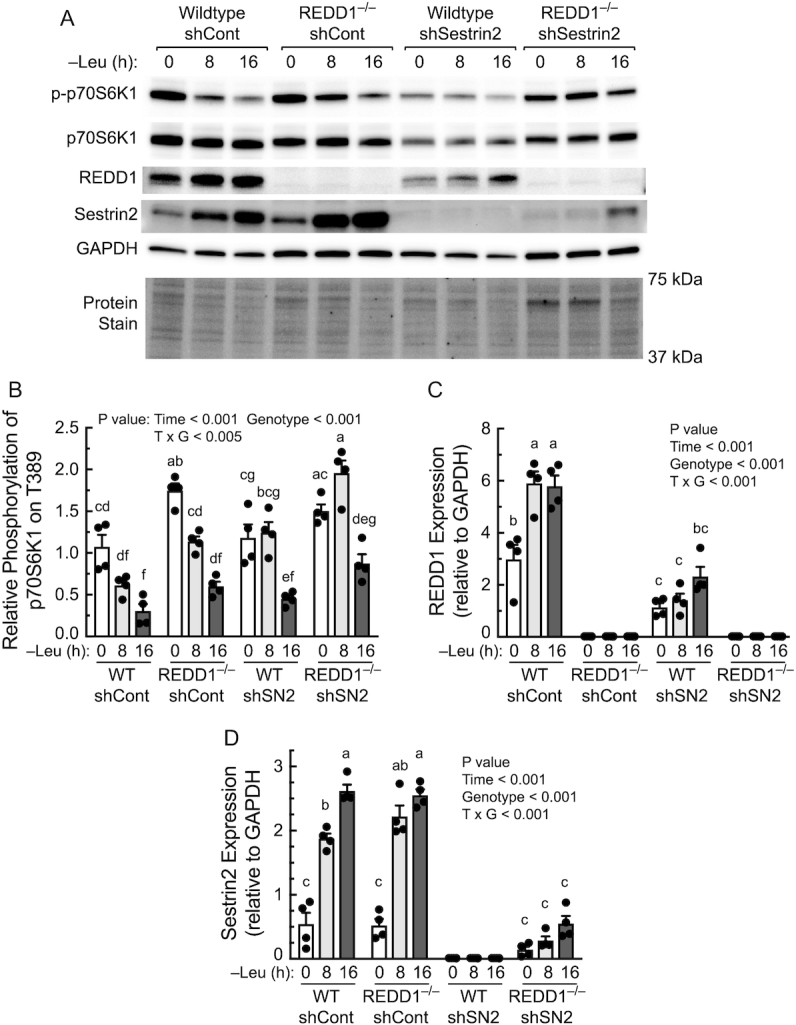
Upregulated expression of both REDD1 and Sestrin2 contribute to resuppression of mTORC1 activity during prolonged Leu deprivation. REDD1+/+ (WT) and REDD1−/− MEFs stably expressing an shCont or shSN2 were generated as described in the Methods section. (A) Representative blots. (B–E) Quantitation of Western blots. p-p70S6K1, p70S6K1 phosphorylated on Thr389. Values are means ± SEM, n = 4. Labeled means without a common letter differ, P < 0.05. Leu, leucine; MEF, mouse embryo fibroblast; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; p70S6K1, 70 kDa ribosomal protein S6 kinase 1; REDD1, regulated in development and DNA damage responses 1; shCont, scrambled control shRNA; shRNA, short hairpin RNA; shSN2, shRNAs targeting Sestrin2; WT, wildtype.
Discussion
mTORC1 is often referred to as a master regulator of cell growth due to its pleiotropic effects on cell metabolism. For example, activation of mTORC1 upregulates a variety of anabolic reactions leading to enhanced synthesis of protein, nucleic acids, and lipids while simultaneously suppressing catabolic processes such as autophagy and proteasome assembly (21). Perhaps the most intensely studied of the upstream activators of mTORC1 are amino acids. Recent studies have identified proteins that act as intracellular “sensors” for amino acids such as leucine, arginine, and the methionine metabolite, S-adenosylmethionine, i.e., the Sestrin (22), Castor (23), and SAMTOR (24) proteins, respectively, that mediate amino acid–induced mTORC1 activation. Solute carrier 38A9 (SLC38A9) also acts as an arginine sensor and transports several essential amino acids, including leucine and methionine, from the lumen of the lysosome to the cytoplasm (25). Indeed, SLC38A9 is essential for the leucine produced during autophagic protein degradation to activate mTORC1. Thus, mTORC1 activity is initially suppressed in response to amino acid deprivation, an effect likely mediated in part by the aforementioned amino acid sensors. Continued amino acid deprivation leads to activation of autophagy and the amino acids produced by autophagic protein degradation in the lysosome lead to subsequent reactivation of mTORC1 (26). However, reactivation of mTORC1 and the consequent stimulation of energy-consuming processes such as protein synthesis would likely be detrimental to the cell if extracellular nutrients remained limiting. Consequently, it is unsurprising that mechanisms for resuppressing mTORC1 during prolonged amino acid deprivation exist.
A recent study linked upregulated Sestrin2 expression to resuppression of mTORC1 during prolonged leucine deprivation (14). The Sestrin family is comprised of 3 proteins termed Sestrin 1, 2, and 3, and all 3 bind leucine (27) and are induced under a variety of conditions associated with downregulation of mTORC1 signaling, e.g., DNA damage, oxidative stress, and glucocorticoid treatment (28, 29). Indeed, overexpression of either Sestrin1 or 2 leads not only to inhibition of mTORC1 activity, but to a severe blunting of responsiveness to amino acid–induced mTORC1 activation (27, 30, 31). In contrast, cells deficient in the 3 Sestrins exhibit elevated mTORC1 activity independent of amino acid availability (22, 27). In the present study, mTORC1 activity was the same in Sestrin2-deficient compared to wild-type cells maintained in complete medium. Moreover, compared to wild-type MEFs, mTORC1 activity was higher in both ATF4–/– and REDD1−/− MEFs maintained in complete medium with no change in Sestrin2 expression. Combined, these findings suggest that in the absence of an external stress, e.g., leucine deprivation, Sestrin2 abundance is not sufficient to significantly suppress mTORC1 activity, and instead it acts to modulate mTORC1 activity in a leucine-dependent manner.
In contrast to Sestrin2-deficient MEFs, mTORC1 activity was higher in REDD1−/− MEFs than in wild-type cells maintained in complete medium. REDD1 acts to suppress Akt activity, leading to activation of TSC2 and suppression of hormone, e.g., insulin and growth factor, signaling to mTORC1 (7). TSC2 subsequently inhibits mTORC1 by activating Rheb GTPase activity, leading to reduced abundance of the active, GTP-loaded form of the protein (32, 33). Thus, a likely explanation for the upregulated mTORC1 activity observed in REDD1−/− compared to wild-type cells maintained in complete medium is that the abundance of REDD1 in wild-type cells is sufficient to maintain TSC2 in a partially active state, leading to a reduction in Rheb GTP loading in wild-type compared to REDD1−/− MEFs.
In the present study, resuppression of mTORC1 was observed after 8 h of leucine deprivation in both REDD1−/− MEFs and in MEFs deficient in Sestrin2, but not in cells deficient for both proteins. One interpretation of these data is that both REDD1 and Sestrin2 contribute to mTORC1 resuppression after 8 h of leucine deprivation. The finding that mTORC1 activity is upregulated after 8 h of leucine deprivation in cells deficient in both proteins, but not in cells deficient for either one alone, is also consistent with this interpretation. Interestingly, during more prolonged, i.e., 16 h, of leucine deprivation, mTORC1 was re-suppressed in REDD1−/− MEFs expressing Sestrin2 shRNA in association with partial restoration of Sestrin2 expression. However, the magnitude of the restoration was small. Indeed, Sestrin2 abundance after 16 h of leucine deprivation in REDD1−/− MEFs expressing the Sestrin2 shRNA was similar to that observed in control cells maintained in complete medium, and markedly less than in control cells deprived of leucine. Curiously, in cells with undetectably low levels of Sestrin2 expression, the resuppression of mTORC1 activity after 16 h of leucine deprivation was likewise associated with restoration of REDD1 expression to levels similar to those in control cells maintained in complete medium. It is unclear how, or if, such low levels of REDD1 and Sestrin2 might cause suppression of mTORC1 activity, or if another mTORC1 suppressor is involved in the effect. This will be an important topic for future investigations.
The results of the present study suggest that REDD1 expression in cells maintained in complete medium is sufficient to partially suppress mTORC1 activity and that ATF4 contributes significantly to both basal and leucine deprivation–induced REDD1 expression. Overall, the results are consistent with a model in which upregulated expressions of both REDD1 and Sestrin2 are involved in mediating resuppression of mTORC1 during prolonged leucine deprivation. The finding that expression of both REDD1 and Sestrin2 is suppressed in the liver after refeeding overnight FD mice suggests that both proteins may also contribute to maintaining hepatic mTORC1 suppression in food-deprived animals, although studies using mice deficient in both proteins are required to provide more definitive evidence supporting such a conclusion.
Acknowledgements
The authors’ responsibilities were as follows—DX, LSJ, MDD, and SRK: designed research; DX, LK, and HAL: conducted research; DX and SRK: analyzed data; DX, LSJ, MDD, and SRK: wrote manuscript; SRK: had primary responsibility for final content; and all authors: read and approved the final manuscript.
Notes
Supported by NIH grants R01-DK13499 and R01-DK15658 (to SRK) and R01-EY029702 (to MDD).
Author disclosures: SRK is a member of the Journal's Editorial Board. All other authors report no conflicts of interest.
Abbreviations used: AMPK, AMP-activated protein kinase; ATF4, activating transcription factor 4; MEF, mouse embryo fibroblast; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; p70S6K1, 70 kDa ribosomal protein S6 kinase 1; PP2A, protein phosphatase 2A; Raptor, regulatory associated protein of mTOR; REDD1, regulated in development and DNA damage responses 1; Rheb, ras homolog enriched in brain; shRNA, short hairpin RNA; SLC38A9, solute carrier 38A9; TSC, tuberous sclerosis complex; ULK1, Unc-51-like autophagy activating kinase 1; 4E-BP1, eukaryotic initiation factor 4E binding protein 1.
References
- 1. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. [DOI] [PubMed] [Google Scholar]
- 2. Naito T, Kuma A, Mizushima N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J Biol Chem. 2013;288(29):21074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoshizawa F, Kimball SR, Jefferson LS. Modulation of translation initiation in rat skeletal muscle and liver in response to food intake. Biochem Biophys Res Commun. 1997;240:825–31. [DOI] [PubMed] [Google Scholar]
- 4. Anthony TG, Anthony JC, Yoshizawa F, Kimball SR, Jefferson LS. Oral administration of leucine stimulates ribosomal protein mRNA translation but not global rates of protein synthesis in the liver of rats. J Nutr. 2001;131:1171–6. [DOI] [PubMed] [Google Scholar]
- 5. Kimball SR, Shantz LM, Horetsky RL, Jefferson LS. Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J Biol Chem. 1999;274(17):11647–52. [DOI] [PubMed] [Google Scholar]
- 6. Dikic I, Elazar Z.. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–64. [DOI] [PubMed] [Google Scholar]
- 7. Lipina C, Hundal HS.. Is REDD1 a metabolic eminence grise? Trends Endocrinol Metab. 2016;27(12):868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGhee NK, Jefferson LS, Kimball SR. Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor REDD1. J Nutr. 2009;139:828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dennis MD, McGhee NK, Jefferson LS, Kimball SR. Regulated in DNA damage and development 1 (REDD1) promotes cell survival during serum deprivation by sustaining repression of signaling through the mechanistic target of rapamycin in complex 1 (mTORC1). Cell Signal. 2013;25(12):2709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal. 2014;7(335):ra68 doi: 10.1126/scisignal.2005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Budanov AV, Karin M. p53 Target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang M, Xu Y, Liu J, Ye J, Yuan W, Jiang H, Wang Z, Jiang H, Wan J. Recent insights into the biological functions of Sestrins in health and disease. Cell Physiol Biochem. 2017;43(5):1731–41. [DOI] [PubMed] [Google Scholar]
- 13. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molec Cell. 2008;30(2):214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, Koumenis C, Thompson CB. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Develop. 2015;29(22):2331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem Biophys Res Commun. 2009;379(2):451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin H-O, Seo S-K, Woo S-H, Kim E-S, Lee H-C, Yoo D-H, An S, Choe T-B, Lee S-J, Hong S-I et al.. Activating transcription factor 4 and CCAAT/enhancer-binding protein-[beta] negatively regulate the mammalian target of rapamycin via REDD1 expression in response to oxidative and endoplasmic reticulum stress. Free Radical Biol Med. 2009;46(8):1158–67. [DOI] [PubMed] [Google Scholar]
- 17. Black AJ, Gordon BS, Dennis MD, Jefferson LS, Kimball SR. Regulation of protein and mRNA expression of the mTORC1 repressor REDD1 in response to leucine and serum. Biochem Biophys Rep. 2016;8:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu D, Shimkus KL, Lacko HA, Kutzler L, Jefferson LS, Kimball SR. Evidence for a role for Sestrin1 in mediating leucine-induced activation of mTORC1 in skeletal muscle. Am J Physiol Endocrinol Metab. 2019;316:E817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grainger DL, Kutzler L, Rannels SL, Kimball SR. Validation of a commercially available anti-REDD1 antibody using RNA interference and REDD1-/- mouse embryonic fibroblasts. F1000Research. 2016;5:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–8. [DOI] [PubMed] [Google Scholar]
- 21. Saxton RA, Sabatini DM. mTOR Signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, Sabatini DM. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2015;351:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165(1):153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, Sabatini DM. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358(6364):813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, Sabatini DM. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171(3):642–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F et al.. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell. 2014;159(1):122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Almon RR, DuBois DC, Yao Z, Hoffman EP, Ghimbovschi S, Jusko WJ. Microarray analysis of the temporal response of skeletal muscle to methylprednisolone: comparative analysis of two dosing regimens. Physiol Genomics. 2007;30(3):282–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ho A, Cho CS, Namkoong S, Cho US, Lee JH. Biochemical basis of Sestrin physiological activities. Trends Biochem Sci. 2016;41(7):621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9(4):1281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garami A, Zwartkruis FJT, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP1 signaling, is inhibited by TSC1 and 2. Molec Cell. 2003;11:1457–66. [DOI] [PubMed] [Google Scholar]
- 33. Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Develop. 2003;17(15):1829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]