

Abstract
Nearly half (45%) of the human genome is composed of transposable elements, or “jumping genes.” Since Barbara McClintock’s original discovery of transposable elements in 1950, we have come to appreciate that transposable element mobilization is a major driver of evolution, that transposons are active in the germline and the soma, and that transposable element dysregulation is causally associated with many human disorders. In the present review, we highlight recent studies investigating transposable element activation in the adult brain and in the context of neurodegeneration. Collectively, these studies contribute to a greater understanding of the frequency of complete retrotransposition in the adult brain as well as the presence of transposable element-derived RNA and protein in brain and fluids of patients with neurodegenerative disorders. We discuss therapeutic opportunities and speculate on the larger implications of transposable element activation in regard to current hot topics in the field of neurodegeneration.
Introduction
Transposable elements are a diverse superfamily of genomic DNA species that have the ability to either copy themselves and insert the DNA copy into a new genomic location (retrotransposons) or excise themselves from the genome and insert in a new genomic location (transposons). Over the course of human evolution, most retrotransposons and all DNA transposons have become inert, or non-mobile, due to truncation and mutation. Some human retrotransposons, however, retain mobilization potential, including specific long and short interspersed nuclear element (LINE and SINE, respectively) subfamilies [1]. Since select retrotransposons retain mobilization ability and retrotransposons outnumber DNA transposons 13 to 1 in the human genome [2], we focus on retrotransposons in the current review.
Together with a longstanding literature focused on retrotransposition in the human brain and retrotransposon dysregulation in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), new studies implicate transposable element activation in the context of tau-mediated neurodegeneration, including Alzheimer’s disease. This review highlights our current understanding of retrotransposon activation in the human brain, new insights into the involvement of retrotransposition and retrotransposon-derived products in TDP-43, C9orf72 and tau-associated neurodegenerative disorders, including breakdown of cellular mechanisms that regulate transposable element activation in neurodegenerative disorders, as well as current and future therapeutic directions. A recent comprehensive review by Tam and colleagues extends the discussion of retrotransposon activation in neurodegenerative disorders to Multiple Sclerosis and Acardi-Goutiéres Syndrome [3].
Retrotransposon biology
Retrotransposons are structurally akin to retroviruses (Fig. 1). Retrotransposon mobilization occurs through a copy-and-paste mechanism that involves transcription of endogenous retrotransposon DNA into RNA, reverse transcription of the nascent retrotransposon RNA into a new DNA copy, and reinsertion of the new DNA copy into the genome [1]. Retrotransposition is completed using proteins encoded by retrotransposon RNA. “Endogenous retroviruses” (ERVs) are a family of transposable elements that are similar to some exogenous retroviruses in that they are flanked by Long Terminal Repeats (LTRs) and harbor protein-coding gag and pol domains. The gag protein assembles into a structural matrix in which reverse transcription by a pol-encoded reverse transcriptase occurs. Pol also encodes an endonuclease and integrase that facilitate reinsertion of the newly-formed DNA into the genome. Some intact human ERVs (HERVs) such as HERV-K(HML) also harbor an env domain that encodes a surface glycoprotein similar to the retroviral envelope. Intact members of the LINE family of retrotransposons encode an endonuclease and reverse transcriptase that facilitate mobilization. Current estimates suggest that 80–100 human LINE-1 elements (L1s) are mobilization-competent, with about 10% of intact L1s being highly active or “hot” [4]. Non-autonomous SINEs such as the human Alu or SVA elements retrotranspose by co-opting proteins encoded by LINEs [1].
Figure 1. Comparative genomic landscape of a retrovirus, endogenous retrovirus and LINE element.
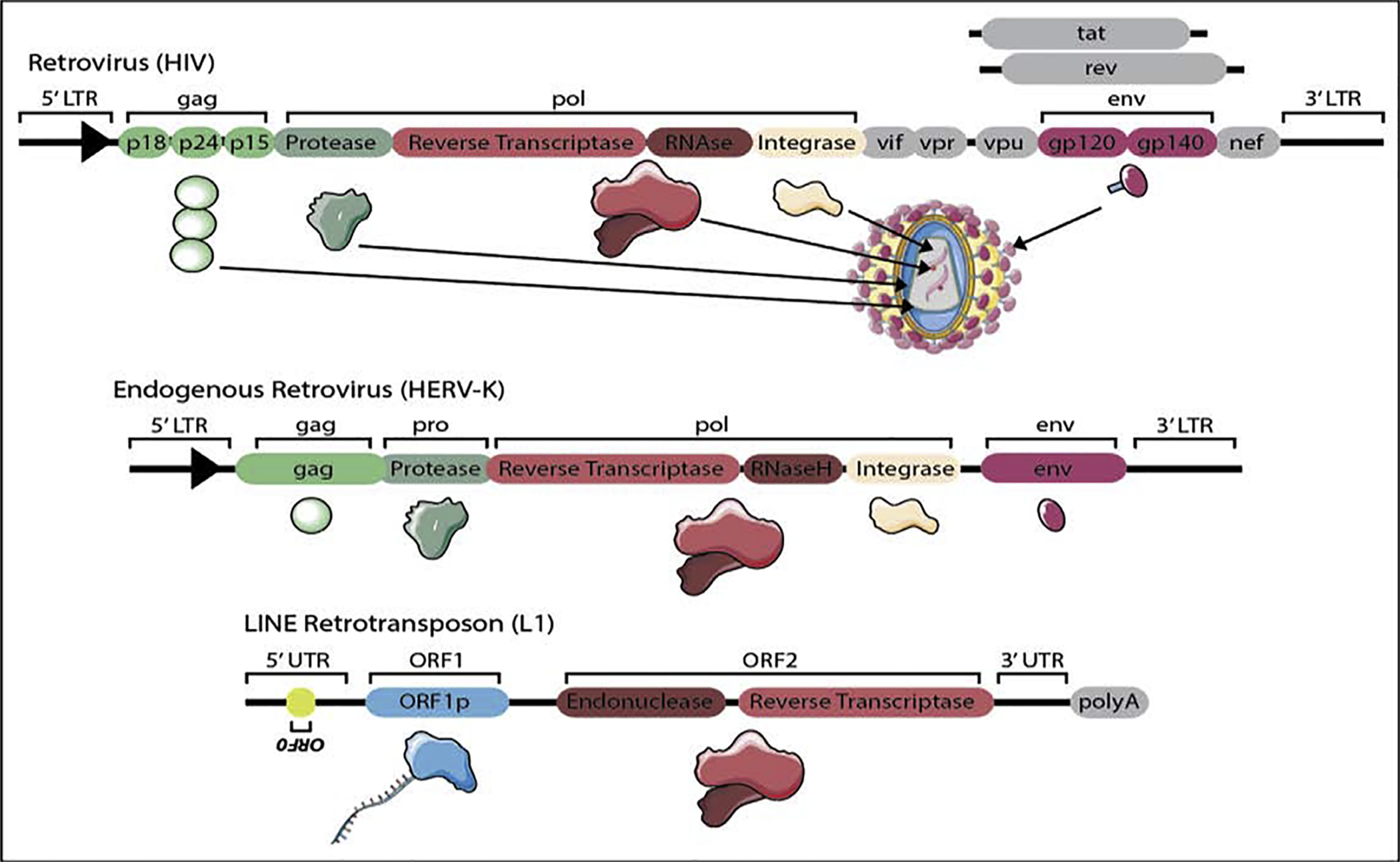
HIV is illustrated as an example of an intact retrovirus, HERV-K is illustrated as an example of an intact endogenous retrovirus, and L1 is illustrated as an example of an intact LINE. Protein products that share functional similarity among the retrovirus, LTR and LINE are represented by the same shape and color.
Based on the potentially catastrophic consequence of retrotransposition in the germline, there is a tendency to emphasize the destructive nature of retrotransposons. While this view is well-justified in many contexts, it is important to keep in mind that retrotransposons provide abundant regulatory sequences for host genomes, mediate innate immunity, and drive evolution [5]. Nevertheless, cells have developed mechanisms to keep potentially deleterious retrotransposon activation in check (Fig. 2). First, retrotransposon transcription is regulated at the epigenetic level based on the location of many retrotransposons in highly condensed heterochromatin. Second, retrotransposons are subject to post-transcriptional silencing by two classes of small RNA, endogenous small-interfering RNAs (esiRNAs) [6] and PIWI-interacting RNAs (piRNA) [7], which also have effects on promoter methylation [8]. Long-thought to be germline-specific, piRNAs are now well-appreciated components of somatic tissues, including adult brain [9–12].
Figure 2. Two cellular layers of transposable element control.
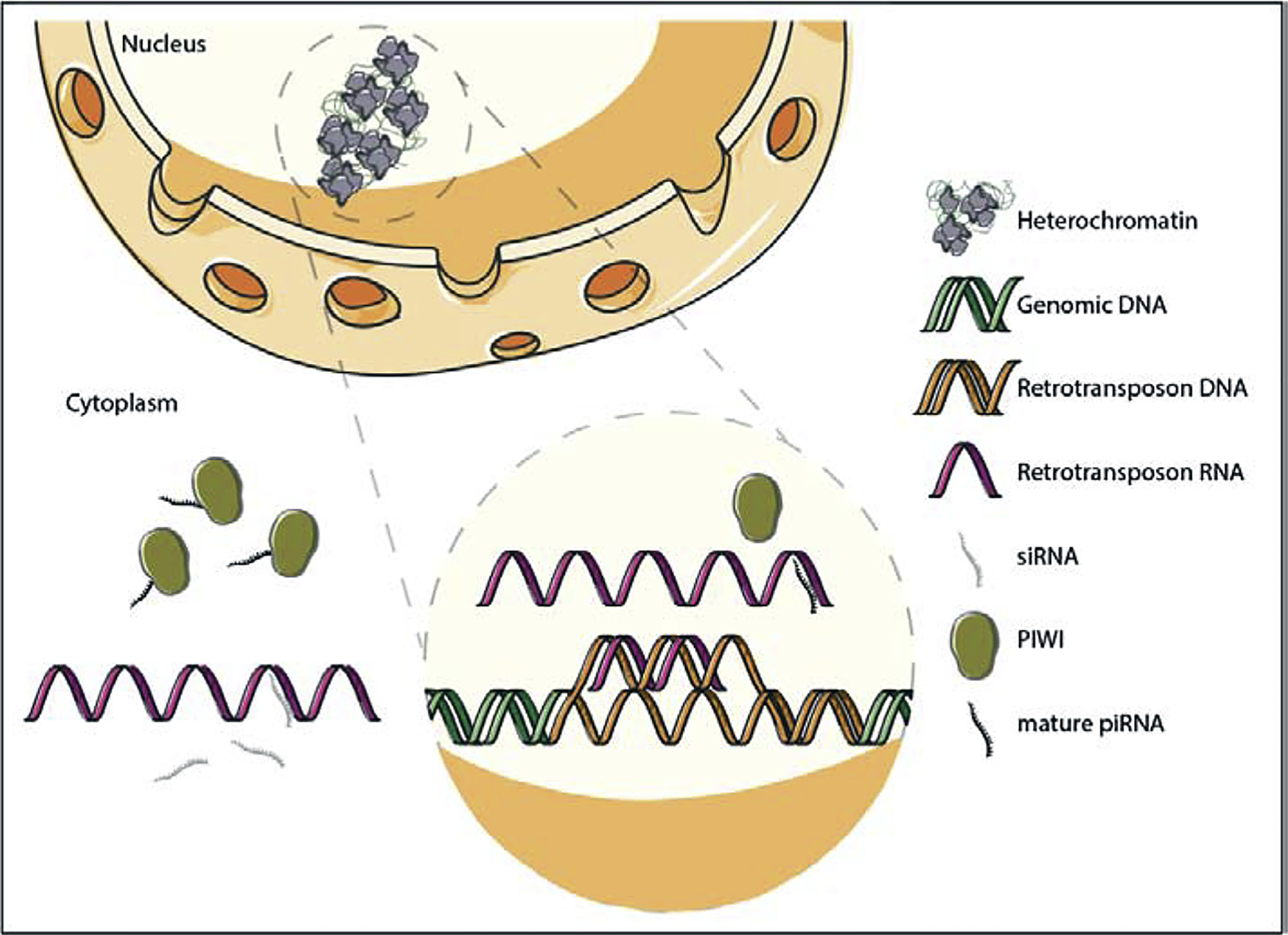
Under normal conditions in somatic cells, many transposable elements are transcriptionally silenced by heterochromatin. Post-transcriptionally, retrotransposons are subject to nuclear and cytoplasmic degradation by piRNAs and siRNAs, respectively.
While much focus is placed on the consequences and frequency of de novo retrotransposon insertions into the genome (i.e. retrotransposon “jumping”), the RNAs, protein products, and episomal DNA generated from retrotransposons can also affect cellular function. For example, double stranded RNAs formed via bidirectional transcription of retrotransposons can induce an interferon response through the RNA-sensing innate immune network [13–15], and ERV-encoded proteins can drive autoimmunity [16] and motor neuron disease [17].
Evidence for transposable element mobilization in the human brain
A seminal paper from Muotri, Gage and colleagues used a fluorescent reporter of retrotransposon mobilization to determine that human L1 can undergo complete retrotransposition when expressed in either cultured rat neural precursor cells or mouse brain [18]. This study was the first to suggest that retrotransposition can generate genomic diversity among neurons. Based on the copy-and-paste mechanism of retrotransposons, in which a single mobilization event increases the DNA copy number of a mobilizing retrotransposon by one copy, a later study detected increased DNA copy number of L1, Alu, and SVA elements in the human brain when compared to liver or heart from the same donor, and estimated the frequency of L1 mobilization at about 80 insertions per neuron [19]. While these findings were consistent with retrotransposon activation in the adult brain, both genomic and episomal retrotransposon DNA can contribute to total DNA copy number. Faulkner and colleagues thus later identified integration sites of novel somatic insertions of L1, Alu, and SVA in the hippocampus and caudate nucleus of the adult human brain [20], establishing that complete retrotransposition, including insertion of retrotransposon DNA into genomic DNA, occurs in the adult human brain.
Estimations of the occurrence and frequency of retrotransposition based on genomic sequencing continue to be an outstanding debate in the field. Results are highly dependent on the method used for genome amplification, library preparation and sequencing platform, as well as how results are analyzed. Current estimates based on single-nucleus DNA sequencing range from <0.6–16.3 L1 mobilization events per neuron in a neurotypical brain [21,22]. Waddell and colleagues have recently called the frequency of retrotransposon mobilization in the brain into question, concluding that the majority of putative de novo transposition events identified by genomic sequencing result from chimeric artifacts formed during library preparation for whole genome sequencing [23]. A general consensus on the best approach for DNA sequencing-based detection of transposition is currently lacking but very much needed.
One interpretation of these studies is that the frequency of mobilization may be fairly low in a neurotypical human brain and thus potentially of little consequence to normal brain functioning. Several of these studies, however, report that retrotransposons selectively insert into genes associated with neuronal function [18,20,22,24], which may increase the impact of relatively rare mobilization events. Even in the absence of complete retrotransposition, retrotransposons generate RNAs, protein, DNA damage, and episomal DNA copies that are known to affect cellular function [25].
Transposable element activation in ALS/FTD
The earliest clues pointing toward involvement of retrotransposons in a neurodegenerative disorder came from multiple studies reporting high reverse transcriptase activity in serum of patients with ALS [26–28]. Having ruled out exogenous retroviral infection, later detection of elevated HERV-K-derived pol transcripts and reverse transcriptase protein in postmortem ALS brain samples [29] led to the conclusion that ALS is associated with activation of endogenous retrovirus rather than exogenous viral infection.
Investigation into a direct association between HERV activation and neurodegeneration demonstrated that expression of the env domain of HERV-K causes retraction and beading of neurites in cultured human neurons. In mice, transgenic expression of env induces a progressive motor phenotype and motor cortex degeneration, reduced synaptic activity of pyramidal neurons, dendritic spine abnormalities, nucleolar dysfunction, and DNA damage [17], suggesting that aberrant expression of HERV-K encoded protein is sufficient to induce neurotoxicity.
Depletion of TAR DNA-binding protein 43 (TDP-43) from the nucleus and accumulation of TDP-43 in the cytoplasm of neurons and glia is a hallmark pathology of ALS and FTD [30], that also appears in 24–70% of Alzheimer’s disease cases [31]. In post-mortem brains of patients with ALS, Nath and colleagues found that HERV-K reverse transcriptase is elevated in neurons harboring cytoplasmic TDP-43 inclusions [29]. Based on TDP-43 immunoprecipitation and RNA sequencing from healthy rat, mouse and human brain, TDP-43 was found to bind directly to a UGUGU pentamer motif present in multiple families of transposable element-encoded RNAs, including SINEs, LINEs and ERVs [32]. The direct binding between TDP-43 and transposable element transcripts is reduced in post-mortem brains of patients with FTD and correlates with increased transcript levels of retrotransposons that have lost TDP-43 binding. Mouse models of TDP-43 overexpression (thought to act as a dominant-negative) as well as TDP-43 knockdown are associated with elevated retrotransposon transcript levels [32], pointing toward a causal association between dysfunctional TDP-43 and loss of retrotransposon silencing. Taken together, these studies suggest that a physiological function of TDP-43 is to silence retrotransposons in neurotypical brains, and that retrotransposon silencing is compromised in ALS and, potentially, other TDP-43 proteinopathies.
A recent study suggests that TDP-43 dysfunction may cause complete L1 transposition in human neurons. Using neuronal nuclei isolated from ALS/FTD human brain, Lee and colleagues found that neuronal nuclei with low levels of TDP-43 have increased L1 DNA copy number compared to nuclei with high levels of nuclear TDP-43. While increased L1 DNA copy number may simply reflect episomal L1 DNA, accompanying experiments reveal that knockdown of TDP-43 is sufficient to induce active L1 mobilization in HeLa cells [33]. Similarly, a previous study reports that wild-type TDP-43 suppresses L1 mobilization in HEK293T cells [34]. A study by Chang and Dubnau [35] recently reported that transgenic expression of human TDP-43 in glia of the adult Drosophila brain induces transposition based on the newly developed gypsy-CLEVR [36] reporter of de novo transposon insertions. Interestingly, the authors find that TDP-43-associated retrotransposon activation in glia negatively affects survival of surrounding neurons, suggesting that retrotransposon activation contributes to both cell autonomous and non-cell autonomous forms of toxicity.
Despite the significant evidence connecting ALS/FTD and TDP-43 to retrotransposon activation, two recent studies failed to detect differential expression of HERV-K encoded gag, pol, or env transcripts in post-mortem brain samples from sporadic ALS [37,38]. Greater insight into the discrepancy among studies may be gleaned from a recent machine learning approach developed by Hammell and colleagues, who find that retrotransposon activation occurs in a distinct subset of ALS patients that exhibit signatures of TDP-43 dysfunction [39]. In addition, two recent publications report that increased retrotransposon transcript levels are limited to cases of ALS associated with C9orf72 [34,40], a GGGGCC repeat expansion in a non-coding region of chromosome 9 open reading frame. C9orf72 expansion is the most common genetic abnormality in ALS, and was not included as a biological variable in studies that failed to detect HERV-K activation in ALS [37,38]. Indeed, a mouse model of C9orf72-associated toxicity features elevated transcript levels of many classes of repeat elements, including LINEs, LTRs, and SINEs [41]. A summary of the retrotransposons reported as dysregulated in brain, CSF or plasma from patients with neurodegenerative disorders is provided in Table 1.
Table 1. Retrotransposons reported as dysregulated in brain, cerebrospinal fluid (CSF) or plasma from patients with neurodegenerative disorders.
A comprehensive summary of the retrotransposons identified in human neurodegenerative diseases, including retrotransposon subtype. Font in blue: transposable element-derived transcripts and protein detected in human brain tissue, grey: transposable element-derived transcripts detected in human cerebral spinal fluid, and red: transposable element activity detected in human serum. Refer to references for full details of each study indicated.
| Disease | Increased | Decreased | ||
|---|---|---|---|---|
| Transposable Element | Subfamily | Transposable Element | Subfamily | |
| Alzheimer’s Disease (AD) | ERV | ERV17, ERV9, ERVH48I, ERVK22I, ERVKC4, ERVL
[11] ERVFc_1 [46] |
||
| LINE1 | L1PA7_5 [11] L1MB4_5 [46] |
|||
| LTR | LTR12C, LTR14 [11] LTR77, PRIMA4_LTR [46] |
|||
| SINE | AluYh9, AluYc5, AluSp [46] | SINE | AluYa5, AluYb8, AluYc1, AluYi6 [10] | |
| Other | SVA_B, SVA_C, TIGGER2 [11] THER2, PB1D11 [46] |
|||
| Progressive Supranuclear Palsy (PSP) | ERV | ERV17, ERV9, ERVH48I, ERVK22I, ERVKC4, ERVL, ERVH, ERVP71A_I [11] | ||
| LINE1 | L1PB2c [11] | |||
| LTR | LTR12C, LTR14 [11] | LTR14A [10] | ||
| SINE | AluYa5, AluYb8, AluYc1, AluYi6, AluSp, AluY, AluYd8, AluYe5, AluYg6, AluYk11, AluYk12 [10] | |||
| Amyotrophic Lateral Sclerosis (ALS) | ERV | ERVK pol and RT protein
[30] ERV-K gag, pol, env transcripts and env protein [18] |
||
| LINE1 | L1MA9 [40] | |||
| LTR | LTR2, LTR70, MER21B, MER51C [40] | |||
| SINE | AluYk12, AluYa5, FRAM [40] | |||
| Other | Reverse transcriptase activity [27–29] | |||
| Frontotemporal lobar degeneration (FTD) | LINE1 | L1MA9 [40] | ||
| LTR | LTR2, LTR70, MER21B, MER51C [40] | |||
| SINE | AluYk12, AluYa5, FRAM [40] | |||
| Sporadic Creutzfeldt-Jakob Disease (sCJD) | ERV | ERV-W, ERV-T, ERV-FRD, ERV- L, ERV-9 [56] | ||
Mechanism
Several lines of evidence suggest that TDP-43 binds directly to retrotransposon transcripts in human, cell, and Drosophila-based systems [32,39,42]. Binding of TDP-43 to retrotransposon transcripts is thought to aid in retrotransposon silencing, as TDP-43 knockdown increases almost all expressed retrotransposon transcripts in neuroblastoma cells [39]. Consistent with these findings, expression of TDP-43 in either glia or neurons of the Drosophila mushroom body increases retrotransposon transcript levels and reduces siRNA-mediated transcript clearance [42]. A recent ATAC-seq analysis of neuronal nuclei isolated from patients with ALS reveals that heterochromatin is decondensed in nuclei lacking TDP-43 and that L1 elements are particularly affected, suggesting that disrupted heterochromatin-mediated silencing may be an additional mechanism contributing to retrotransposon activation in TDP-43 proteinopathy [33].
Transposable element activation in tauopathy
We have recently identified transposable element activation as a novel driver of neuronal death in tauopathies [11], a group of age-related neurodegenerative disorders that are pathologically defined by deposits of tau protein in the brain [43]. RNA-seq analysis of brain lysates from post-mortem human controls, late-stage Alzheimer’s disease and progressive supranuclear palsy, a “primary” tauopathy, revealed elevated levels of specific L1, HERV, and SVA transcripts, and decreased levels of Alu family members [11]. Coincident with our work, Shulman and colleagues reported a significant association between decreased cognitive performance in the year prior to death and elevation of specific HERV subfamilies in human Alzheimer’s disease brain, as well as an association between tau tangle burden and increased transcript levels of select L1 and HERV elements [44]. It is currently unknown if the increase in L1 and HERV transcripts translates to an increase in L1 and HERV-encoded protein, or if retrotransposition frequency is increased in human Alzheimer’s disease and associated tauopathies compared to neurotypical aged controls. Of note, a recent quantitative PCR-based approach failed to detect increased DNA copy number of active L1 elements in late-stage postmortem human Alzheimer’s disease frontal cortex compared to controls [45].
Mechanism
Experiments in Drosophila provide greater mechanistic insight into transposable element mobilization and the cell biology mediating transposable element activation in tauopathy. We have reported that pan-neuronal transgenic expression of human tau in Drosophila disrupts two arms of transposable element control – heterochromatin and piRNA-mediated retrotransposon silencing [11]. Consistent with the increase in DNA copy number of select retrotransposons in heads of tau transgenic Drosophila [44], we demonstrated active retrotransposition as a consequence of human tau in neurons of the adult Drosophila brain. We found that genetic manipulation of retrotransposon regulatory machinery modifies tau-induced neurodegeneration, which suggests a causal link between transposable element dysregulation and neurodegeneration. Together with reports of heterochromatin relaxation [46,47] and piRNA dysregulation [10,48] in post-mortem human Alzheimer’s disease brain, these studies suggest that tau-induced heterochromatin decondensation, piwi/piRNA dysregulation and consequent transposable element activation is a novel, conserved driver of neurodegeneration in tauopathy. We do not think that tau-induced transposable element activation requires formation of large tau aggregates, as the Drosophila models that feature heterochromatin decondensation [46], piRNA depletion, and transposable element activation [11] produce disease-associated forms of phosphorylated tau and exhibit progressive neurodegeneration but do not produce insoluble tau species [49].
Transposing elements into treatment
Based on the similarities between exogenous retroviruses and ERVs, numerous studies have investigated the therapeutic efficacy of anti-retroviral medications, including nucleoside analog reverse transcriptase inhibitors (NRTIs), to prevent transposable element expression and mobilization [11,50–52]. Current clinical trials for anti-retroviral therapy in the context of neurodegeneration include the “Lighthouse” study, an open label, multi-center study to investigate the safety and tolerability of Triumeq, a combination anti-retroviral therapy (dolutegravir, abacavir, and lamivudine) in ALS patients (Clinical Trials ID NCT02868580).
We originally became interested in the utility of lamivudine (3TC) [53], an NRTI that is FDA-approved for HIV and Hepatitis B, to suppress tau-induced retrotransposition based on studies reporting that lamivudine suppresses L1 retrotransposition at concentrations within the standard dosing range for HIV [50,54,55]. Similarly, we found that lamivudine suppresses tau-induced retrotransposition and consequent neurodegeneration in Drosophila [11]. Lamivudine is also reported to suppress L1-induced interferon-1 (IFN-1) activation and the senescence-associated secretory phenotype in vitro and in vivo [51], suggesting that, beyond their effects on dampening retrotransposition, NRTIs limit production of retrotransposon-derived RNA or protein products.
Concluding thoughts
Despite continued controversy in transposable element biology and computational analyses, the studies described make a strong case that retrotransposition and retrotransposon-derived RNA and protein products are involved in human neurodegenerative disorders and are potentially pharmacologically targetable. We speculate that activation of endogenous retroviruses contributes to the anti-viral response in Alzheimer’s disease that has been attributed to exogenous viral infection [56]. Continued studies focusing on the prevalence of retrotransposition in human neurodegenerative disorders, toxicity of retrotransposon-derived RNA and protein products, the contribution of retrotransposon activation to inflammation and anti-viral responses, and the utility of anti-retroviral therapy will allow us to better understand the darker half of our genome and its involvement in neurodegenerative biology.
Highlights.
Elevated levels of retrotransposon-encoded RNAs and protein products are present in several human neurodegenerative disorders.
Pathogenic forms of TDP43 lose their ability to directly bind and silence retrotransposon transcripts
Pathogenic forms of TDP43 may contribute to retrotransposon activation by disrupting heterochromatin-mediated silencing mechanisms
Pathogenic forms of tau drive retrotransposon activation by disrupting piRNA- and heterochromatin-mediated silencing mechanisms.
Use of reverse transcriptase inhibitors to suppress retrotransposition provide a potential therapeutic strategy for patients with ALS/FTD and tauopathy.
Acknowledgements
The figures in this review were prepared using media adapted from and publicly available through Sevier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License (https://smart.servier.com/). This work was supported by the Rainwater Foundation, the Owens Foundation, and NIH (1 RF1 NS112931).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Nothing declared
References
- 1.Slotkin RK, Martienssen R: Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 2007, 8:272–285. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, Fitzhugh W, et al. : Initial sequencing and analysis of the human genome. Nature 2001, 409:860–921. [DOI] [PubMed] [Google Scholar]
- 3.Tam OH, Ostrow LW, Gale Hammell M: Diseases of the nERVous system: Retrotransposon activity in neurodegenerative disease. Mob DNA 2019, 10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV., Kazazian HH: Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 2003, 100:5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuong EB, Elde NC, Feschotte C: Regulatory activities of transposable elements: From conflicts to benefits. Nat Rev Genet 2017, 18:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. : Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453:539–543. [DOI] [PubMed] [Google Scholar]
- 7.Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ: Discrete Small RNA-Generating Loci as Master Regulators of Transposon Activity in Drosophila. Cell 2007, 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- 8.Goodier JL: Restricting retrotransposons: A review. Mob DNA 2016, doi: 10.1186/s13100-016-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EJ, Banerjee S, Zhou H, Jammalamadaka A, Arcila M, Manjunath BS, Kosik KS: Identification of piRNAs in the central nervous system. RNA 2011, 17:1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu W, Guo X, Lin X, Yang Q, Zhang W, Zhang Y, Zuo L, Zhu Y, Li CSR, Ma C, et al. : Transcriptome-wide piRNA profiling in human brains of Alzheimer’s disease. Neurobiol Aging 2017, 57:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **11.Sun W, Samimi H, Gamez M, Zare H, Frost B: Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci 2018, 21:1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using Drosophila models of tauopathy, this study reported that pathogenic tau drives retrotransposition in the adult brain. Mechanistically, the authors report that loss of heterochromatin condensation and a disruption of piwi/piRNA retrotransposon silencing mediates tau-induced transposable element dysregulation. The authors additionally identify transposable elements that are differentially expressed in post-mortem brains of human Alzheimer’s Disease and progressive supranuclear palsy.
- *12.Perera BPU, Tsai ZTY, Colwell ML, Jones TR, Goodrich JM, Wang K, Sartor MA, Faulk C, Dolinoy DC: Somatic expression of piRNA and associated machinery in the mouse identifies short, tissue-specific piRNA. Epigenetics 2019, 5:504–521. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports presence of PIWIL1, PIWIL2 and PIWIL4 protein in the adult mouse brain. Based on small RNA-seq, the authors find that hippocampus has the highest piRNA expression among the tissues tested (cortex, kidney, liver).
- 13.Buhu S, Pardoll DM, Makarov V, Rote NS, Slamon DJ, Merghoub T, Strick R, Wolchok JD, Chan TA, Zahnow CA, et al. : Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162:974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuellar TL, Herzner A-M, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D, et al. : Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol 2017, 11:3535–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. : DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162:961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volkman HE, Stetson DB: The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol 2014, 15:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, Geldern G Von, Johnson K, et al. : Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015, 7:307ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muotri AR, Chu VT, Marchetto MCN, Deng W, Moran JV., Gage FH : Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435:903–910. [DOI] [PubMed] [Google Scholar]
- 19.Coufal NG, Garcia-perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, Shea KSO, Moran JV, Gage FH: L1 Retrotransposition in Human Neural Progenitor Cells. Nature 2009, 460:1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. : Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479:534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann HS, Parker JJ, Atabay KD, Gilmore EC, Poduri A, et al. : Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151:483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sánchez-Luque FJ, Bodea GO, Ewing AD, Salvador-Palomeque C, van der Knaap MS, Brennan PM, et al. : Ubiquitous L1 Mosaicism in Hippocampal Neurons. Cell 2015, 161:228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treiber CD, Waddell S: Resolving the prevalence of somatic transposition in Drosophila. Elife 2017, 6:e28297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perrat PN, DasGupta S, Wang J, Theurkauf W, Weng Z, Rosbash M, Waddell S: Transposition-driven genomic heterogeneity in the Drosophila brain. Science (80- ) 2013, 340:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faulkner GJ, Billon V: L1 retrotransposition in the soma: A field jumping ahead. Mob DNA 2018, 9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele AJ, Al-Chalabi A, Ferrante K, Cudkowicz ME, Brown RH, Garson JA: Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 2005, 64:454–458. [DOI] [PubMed] [Google Scholar]
- 27.MacGowan DJL, Scelsa SN, Imperato TE, Liu KN, Baron P, Polsky B: A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 2007, 68:1944–1946. [DOI] [PubMed] [Google Scholar]
- 28.McCormick AL, Brown RH, Cudkowicz ME, Al-Chalabi A, Garson JA: Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 2008, 70:278–283. [DOI] [PubMed] [Google Scholar]
- 29.Douville R, Liu J, Rothstein J, Nath A: Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 2011, 69:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. : Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (80- ) 2006, 314:130–133. [DOI] [PubMed] [Google Scholar]
- 31.Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW: Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 2016, 131:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Jin Y, Prazak L, Hammell M, Dubnau J: Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS One 2012, 7:e44099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **33.Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB: Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep 2019, 27:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that loss of nuclear TDP-43 is associated with increased chromatin accessibility of LINEs based on ATAC-seq of neuronal nuclei isolated from human ALS brain. TDP-43 negative nuclei were found to have elevated L1 DNA content compared to TDP-43 positive nuclei. In HeLa cells, TDP-43 knockdown was found to activate a GFP-based reporter of L1 transposition.
- **34.Pereira GC, Sanchez L, Schaughency PM, Rubio-Roldán A, Choi JA, Planet E, Batra R, Turelli P, Trono D, Ostrow LW, et al. : Properties of LINE-1 proteins and repeat element expression in the context of amyotrophic lateral sclerosis. Mob DNA 2018, 9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that TDP-43 overexpression limits L1 mobilization in cultured cells, and disease-associated mutations in TDP-43 disrupt L1 silencing. Analysis of publicly available RNA-seq data reveales that differential expression of retrotransposons is limited to C9orf72-associated cases of ALS, as the authors found minimal altered expression between controls and sporadic ALS cases.
- 35.Chang YH, Dubnau J: The Gypsy Endogenous Retrovirus Drives Non-Cell-Autonomous Propagation in a Drosophila TDP-43 Model of Neurodegeneration. Curr Biol 2019, 29:3135–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **36.Chang YH, Keegan RM, Prazak L, Dubnau J: Cellular labeling of endogenous retrovirus replication (CLEVR) reveals de novo insertions of the gypsy retrotransposable element in cell culture and in both neurons and glial cells of aging fruit flies. PLoS Biol 2019, 17:e3000278. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that transgenic expression of human TDP-43 in glia of the adult Drosophila brain induces transposition. TDP-43-associated retrotransposon activation in glia negatively affects survival of surrounding neurons, suggesting that retrotransposon activation contributes to both cell autonomous and non-cell autonomous forms of toxicity.
- **37.Mayer J, Harz C, Sanchez L, Pereira GC, Maldener E, Heras SR, Ostrow LW, Ravits J, Batra R, Meese E, et al. : Transcriptional profiling of HERV-K(HML-2) in amyotrophic lateral sclerosis and potential implications for expression of HML-2 proteins. Mol Neurodegener 2018, 13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, HERV-K(HML-2) transcript levels were quantified in brain and spinal cord tissue from patients with ALS versus controls. In conflict with previous studies, HERV-K(HML-2) transcripts were not found to differ across samples, and the authors detected fragments of HERV-K-derived env protein rather than full length env reported in previous studies. Overall, this study does not support involvement of HERV dysregulation in ALS.
- **38.Garson JA, Usher L, Al-Chalabi A, Huggett J, Day, McCormick AL: Quantitative analysis of human endogenous retrovirus-K transcripts in postmortem premotor cortex fails to confirm elevated expression of HERV-K RNA in amyotrophic lateral sclerosis. Acta Neuropathol Commun 2019, 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors of this study quantified HERV-K RNA levels in the premotor cortex of human ALS samples versus controls using RT-PCR. Similar to Mayer et al [37], differences in the levels of HERV-K transcripts for env and pol did not differ between samples from ALS patients and neurotypical controls.
- **39.Tam OH, Rozhkov NV., Shaw R, Kim D, Hubbard I, Fennessey S, Propp N, Phatnani H, Kwan J, Sareen D, et al. : Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep 2019, 29:1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]; Based on a machine-learning driven analysis of RNA-seq data across two cohorts (NYGC ALS Consortium and UCSD), this study demonstrates that ALS gene expression profiles segregate into three subtypes. 20% of patient brains fall into the “ALS-Transposable Element” subtypes, which is characterized by retrotransposon elevation and transcriptional signatures of TDP-43 dysfunction, further supporting a key role of TDP-43 in retrotransposon regulation.
- 40.Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, Ebbert MTW, van Blitterswijk M, Zhang YJ, Jansen-West K, et al. : Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 2017, 26:3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **41.Zhang Y-J, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, O’Raw AD, Pickles SR, Prudencio M, Carlomagno Y, et al. : Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363:eaav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that C9orf72-associated poly(PR) localizes to heterochromatin in mouse models and human ALS/FTD, and causes epigenetic changes. Patient samples and cell models of poly(PR) had increases in dsRNA that were a consequence of heterochromatin decondensation. The authors hypothesize that poly(PR) disrupts HP1α liquid phases, causing changes in lamin structure, nuclear invaginations and further HP1α depletion, which permits repetitive element expression and dsRNA formation.
- 42.Krug L, Chatterjee N, Borges-Monroy R, Hearn S, Liao WW, Morrill K, Prazak L, Rozhkov N, Theodorou D, Hammell M, et al. : Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet 2017, 13:e1006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arendt T, Stieler JT, Holzer M: Tau and tauopathies. Brain Res Bull 2016, 126:238–292. [DOI] [PubMed] [Google Scholar]
- **44.Guo C, Jeong HH, Hsieh YC, Klein HU, Bennett DA, De Jager PL, Liu Z, Shulman JM: Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep 2018, 5:2874–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports a significant association between tau tangle burden and differential expression of several retrotransposons in human Alzheimer’s disease brain tissue, including specific L1 and HERV family members. The authors report reduced levels of H3K9Ac at HERV-Fc1 loci in Alzheimer’s disease brains, consistent with heterochromatin decondensation. Drosophila models of tauopathy are reported to have elevated transcript levels and DNA copy number of several retrotransposons.
- 45.Protasova MS, Gusev FE, Grigorenko AP, Kuznetsova IL, Rogaev EI, Andreeva TV.: Quantitative analysis of L1-retrotransposons in Alzheimer’s disease and aging. Biochem 2017, 82:962–971. [DOI] [PubMed] [Google Scholar]
- 46.Frost B, Hemberg M, Lewis J, Feany MB: Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci 2014, 17:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *47.Klein HU, McCabe C, Gjoneska E, Sullivan SE, Kaskow BJ, Tang A, Smith RV., Xu J, Pfenning AR, Bernstein BE, et al. : Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat Neurosci 2019, 22:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]; Consistent with the findings of Frost et al. [46], this study reports a significant association between tau burden and loss of heterochromatin-mediated silencing in post-mortem human cortex. In addition, the authors report that tau expression in iPSC-derived neurons is sufficient to induce widespread epigenetic changes.
- 48.Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B: Small RNA sequencing revealed dysregulated piRNAs in Alzheimer’s disease and their probable role in pathogenesis. Mol Biosyst 2017, 13:565–576. [DOI] [PubMed] [Google Scholar]
- 49.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB: Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science (80- ) 2001, 293:711–714. [DOI] [PubMed] [Google Scholar]
- 50.Jones RB, Garrison KE, Wong JC, Duan EH, Nixon DF, Ostrowski MA: Nucleoside Analogue Reverse Transcriptase Inhibitors Differentially Inhibit Human LINE-1 Retrotransposition. PLoS One 2008, 3:e1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *51.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. : L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that L1 activation and subsequent interferon response is a phenotype of cell senescence. In particular, the authors find that episomal DNA produced from L1 induced during senescence triggers the interferon response, which is reduced upon treatment with the reverse transcriptase inhibitor lamivudine.
- 52.Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, Garcia M, Hartnett DA, Burhenn L, Neretti N, et al. : Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci 2016, 113:11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coates JA, Cammack N, Jenkinson HJ, Jowett AJ, Jowett MI, Pearson BA, Penn CR, Rouse PL, Viner KC, Cameron JM: (−)-2’-deoxy-3’-thiacytidine is a potent, highly selective inhibitor of human immunodeficiency virus type 1 and type 2 replication in vitro. Antimicrob Agents Chemother 1992, 36:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dai L, Huang Q, Boeke JD: Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem 2011, 12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, Macia A, Crow YJ, Muotri AR: Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, et al. : Microbes and Alzheimer’s disease. J Alzheimer’s Dis 2016, 51:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]