

Abstract
Transcription factors have roles at focal points in signaling pathways, controlling many normal cellular processes such as cell growth and proliferation, metabolism, apoptosis, immune responses, and differentiation. Their activity is frequently deregulated in disease and targeting this class of proteins is a major focus of interest. However, the structural disorder and lack of binding pockets have made design of small molecules for transcription factors challenging. Here, we review some of the most recent developments for small molecule inhibitors of transcription factors emphasized in James Darnell’s vision 17 years ago. We also discuss the progress so far on transcription factors recently nominated by genome-scale loss of function screens from the cancer dependency map project.
Keywords: transcription factor, chemical probe, Stat, NF-ĸB, Myc, Myb
Targeting transcription factors
Transcription factors, or proteins that bind DNA to regulate transcription, are frequently aberrant in disease. In 2002, James Darnell argued that targeting transcription factors overactive in diseases such as in human cancers provides the most direct strategy for therapeutics [1]. This class of proteins contains fewer possible targets and multiple signaling pathways can converge on the same transcription factor, which exists at focal points in signaling pathways [1]. However, this class of proteins is also challenging to target. These proteins lack enzymatic activity and unlike enzymes with active sites, they also lack obvious binding pockets for small molecule design. Instead, their activity depends on association with other proteins and these interactions occur over large surfaces that generally contact at multiple points and lack hydrophobic folds [2,3]. Thus, modulation of transcription factor activity requires disruption of DNA-protein or protein-protein interactions [4]. Additionally, many transcription factors exhibit conformational plasticity as they engage in a variety of biomolecular interactions and may be disordered when isolated from their cognate binding partners, presenting further challenges in developing chemical probes (Box 1) [5–7].
Box 1. Chemical Probes.
Chemical probes are small molecules that specifically modulate a protein of interest to allow interrogation of its function in vitro, in cells and/or in animals. These compounds provide an orthogonal tool to CRISPR and RNAi to study the roles of proteins of interest, and they have advantages in their ability to rapidly and reversibly affect that protein with tunable dose-responsive and temporal effects [88]. With characteristics such as high potency, known mechanism of action, activity in cellular contexts, easy accessibility [11], and potential to modulate specific functions of a protein [89], chemical probes are valuable resources in scientific research and in some cases, have potential for translational applications. However, the high bar for a chemical probe combined with challenges in targeting transcription factors translates to a low number of high-quality chemical probes for this class of proteins.
There are many strategies to regulate transcription with small molecules, including recruitment of E3 ubiquitin ligases to hijack proteasomal degradation [8,9], and targeting nuclear hormone receptors and upstream proteins [10]. This review will survey some of the most recent updates on inhibitors of transcription factors highlighted by Darnell in 2002 [1], with many developments focused on perturbing protein-protein interactions. Examples will include inhibitors of the signal transducer and activator of transcription (Stat) family, NF-ĸB, and Myc with a focus on small molecules that most closely fit the requirements of a chemical probe [11]. We will conclude with emerging targets recently nominated or reiterated by the cancer dependency map project (DepMap) (https://depmap.org/portal), a combination of 501 genome-scale loss of function screens to discovery 426 dependencies in a wide variety of human cancer cell lines [12]. For one of the classes of marker dependency pairs with dependency related to elevated expression, 45% of this class included transcription factors. This dataset nominates a variety of transcription factors including Myb for which chemical probes need to be developed. Many of these transcription factors have lineage-specific dependencies and chemical probes will help clarify their potential as therapeutics targets and facilitate the trend toward personalized medicines for cancer [12] (see Clinician’s Corner).
Box 2. Clinician’s Corner.
Transcription factors are located in a central node in signaling pathways to regulate transcription of target genes. Many transcription factors are master regulators, controlling a host of different cellular processes such as cell growth and proliferation, metabolism, apoptosis, immune responses, and differentiation.
Aberrant transcription factor activity, often due to elevated levels or translocation of a transcription factor, is associated with a majority of human cancers. In these cancers, high transcription factor levels can drive expression of target genes that enable tumor cells to grow rapidly and proliferate.
Many proof of concept experiments have suggested various transcription factors as promising therapeutic targets. In the phenomenon of oncogene addiction, cancer cells can become dependent on an oncogene for survival. Inhibition of transcription factor activity can lead to selective killing of cancer cells compared to normal cells. However, targeting transcription factors has traditionally been challenging due to disordered structures and the necessity to modulate large protein-protein or protein-DNA interfaces. Rules for rational drug design do not exist for transcription factors and this class has often been regarded as “undruggable.”
Thus, screens of large compound libraries are typically performed to discover small molecules with desirable properties. These screens may generally select compounds that inhibit transcriptional activity or viable cell counts, or may be designed to home in on a specific mechanism of action such as inhibition of a protein-protein interaction or post-translational modification. Some of the major challenges facing researchers are understanding mechanism of action and target identification. For example, screens for compounds that inhibit the transcription factor activity or have a specific mechanism of action in solution may in fact be acting through an off-target in cells.
If the target protein of a small molecule is known, one of the most exciting advances toward clinical application is the development of a degrader by linking the small molecule to a molecule that recruits an E3 ubiquitin ligase. These degraders catalyze loss of target protein and the first of these degraders to enter clinical trials is currently in phase I (NCT03888612)I.
The toolbox of small molecules to inhibit various transcription factors has been vastly expanded over the last 17 years, and as our ability to understand transcriptional signatures and transient protein-protein interactions improves, this toolbox will continue to grow. The trend in the research community has been that we are closer and closer to making this “undruggable” class of proteins chemically tractable, if not druggable.
Small molecule inhibitors of the Stat family
The Stat family of proteins includes seven members of cytoplasmic transcription factors that are activated via phosphorylation by Janus kinases (JAKs), receptors with kinase activity, or non-receptor kinases. Upon phosphorylation, Stat proteins dimerize through SH2 interactions. Following importin binding, the phosphorylated Stat proteins translocate to the nucleus and activate transcription to regulate – in the case of Stats 1, 3 and 5 – genes involved with cell cycle, survival and angiogenesis (Figure 1) [1,13,14]. Stats3 and 5 are overactive in many human cancer cell lines as well as primary tumors and have been shown to demonstrate oncogene addiction [15,16].
Figure 1. Inhibitors of the Stat family of transcription factors.
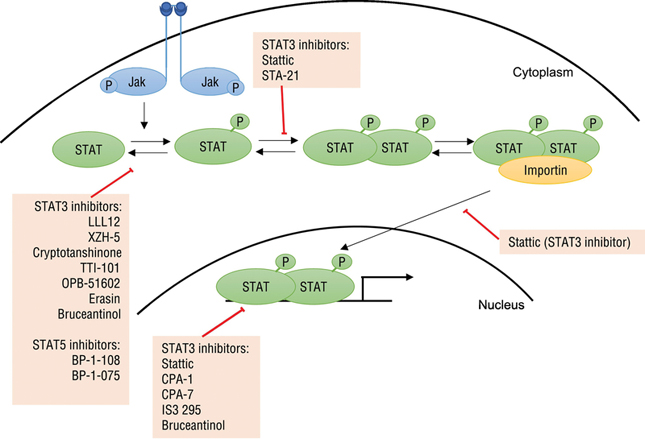
Upon stimulation, kinases such as JAKs phosphorylate Stat proteins, which can then dimerize via SH2 domains. Binding to importin allows translocation of phosphorylated Stat dimers into the nucleus, where they bind DNA and regulate transcription. Inhibitors of the Stat family of transcription factors have been found to affect various aspects of this pathway directly or indirectly.
Many small molecule inhibitors specific for Stats3 and 5 have previously been reviewed [13]. From fluorescence polarization screens of a 17,298 compound library for inhibition of binding between a peptide with a phosphotyrosine and the Stat3 SH2 domain, 144 compounds were prioritized. From these hits, Stattic was found to inhibit translocation of Stat3 into the nucleus and DNA binding of phosphorylated Stat3. Treatment with Stattic (10 μM) also led to apoptosis specifically in Stat3-dependent breast cancer cell lines [17]. STA-21, which was discovered from a virtual screen, inhibited Stat3 dimerization, DNA binding and growth of breast cancer cell lines with overactive Stat3 activity [18]. Many other inhibitors – such as LLL12 [19], XZH-5 [20], cryptotanshinone [21] and analogues of curcumin [22–24] – inhibit the phosphorylation of Stat3 with most IC50 values in a variety of cancer cell lines ranging from 0.16 μM to ~15 μM. Other compounds such as CPA-1, CPA-7 [25], and IS3 295 [26] target at the nucleus to prevent Stat3 binding to the DNA. Among the most potent Stat5 inhibitors, BP-1–108 and BP-1–075 inhibited Stat5 phosphorylation, downregulated Stat5 target genes and caused apoptosis of human leukemia cell lines [27].
Recently, inhibition of Stat proteins is becoming a more promising strategy. TTI-101 is a Stat3 inhibitor that binds the SH2 domain of Stat3 to inhibit phosphorylation, activation and subsequent translocation of Stat3 into the nucleus. This potential drug is currently in Phase 1 clinical trials (NCT03195699)II. Treatment with OPB-51602, a drug tested in phase I clinical trials (NCT01184807)III, resulted in decreases in Stat3 phosphorylation in peripheral blood mononuclear cells and a partial response in two of 37 patients with solid tumors refractory to the current standard of care [28]. Bruceantinol, a compound that was found from a panel of quassinoids to inhibit DNA binding of Stat3 with an IC50 of 2.4 pM, suppressed phosphorylation of Stat3 and downstream Stat3-dependent gene expression through reverse-phase protein array and immunoblot analyses. Moreover, bruceantinol treatment of a mouse model bearing Stat3-expressing HCT116 xenografts resulted in inhibition of tumor growth without general toxicity effects [29]. Another more recent Stat3 inhibitor, Erasin, was discovered using fluorescence polarization along with molecular docking models to evaluate synthesized compounds. This compound inhibited Stat3 phosphorylation in HepG2 cells dose-dependently without significantly affecting Stat1 or Stat5 phosphorylation, and induced apoptosis in breast cancer cell lines and non-small cell lung cancer cells with overactive Stat3 without affecting cancer cells that do not constitutively express Stat3. Interestingly, Erasin could kill HCC-827 cells with acquired resistance to Erlotinib, an inhibitor of the upstream EGF receptor [30]. In support of Darnell’s vision, this example suggests that targeting a focal point in a signaling pathway may be a viable strategy to overcome resistance mechanisms that arise from targeting an upstream pathway.
Inhibitors of NF-ĸB activity
Similar to the Stat family of proteins, NF-ĸB transcription factors control many cellular processes such as cell growth, apoptosis, angiogenesis and immune responses [31,32]. In the canonical pathway, NF-ĸB dimers (most commonly, p65/p50 heterodimers) reside in the cytoplasm in association with IĸB inhibitor proteins. Upon stimulation of the pathway, these inhibitor proteins are phosphorylated by IĸB kinase (IKK) complexes, ubiquitinated and degraded. The free NF-ĸB can then translocate into the nucleus and regulate gene expression [33] (Figure 2). As this pathway is often constitutively active in disease, these proteins have been a major target of interest and over 750 inhibitors have been developed and reviewed, including small molecules that inhibit IKK, IĸB phosphorylation and IĸB degradation, as well as compounds that are more direct by inhibiting NF-ĸB translocation, DNA binding of NF-ĸB and transactivation [31,32].
Figure 2. Modulators of NF-ĸB activity.
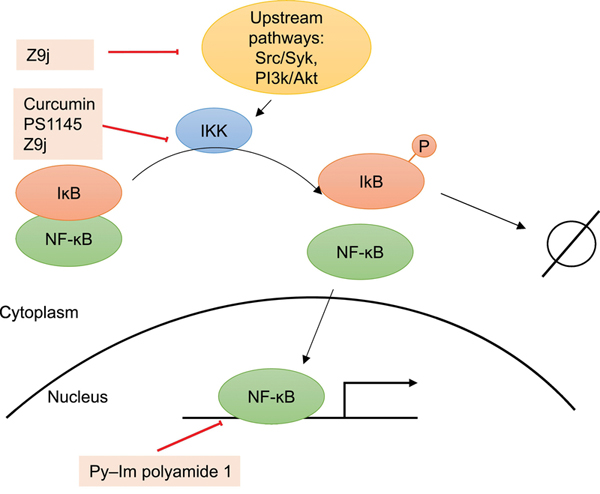
NF-ĸB is inactivated by association with IĸB proteins. Upon stimulation, IKK complexes phosphorylate IĸB proteins, leading to their degradation and allowing NF-ĸB to translocate to the nucleus and activate transcription. Inhibitors of NF-ĸB activity have been discovered to act directly or indirectly at various aspects of this pathway.
Since these reviews were published, a variety of additional NF-ĸB inhibitors have been discovered. Curcumin, a compound that was found to inhibit IKK activity [34] and Stat3 phosphorylation via direct interaction with Cys259 of Stat3 [35,36], decreased NF-ĸB activity and cancer stem cell populations specifically in sensitive cell lines. Gene set enrichment analysis suggested enrichment of histone deacetylase (HDAC) I and II targets in curcumin-sensitive cells compared to untreated cells, and Connectivity Map analysis further revealed HDAC inhibitors as the compounds with the highest connectivity scores to curcumin [37]. In fact, treatment with both curcumin and an HDAC inhibitor significantly reduced colony and sphere formation of curcumin-resistant cells, suggesting potential for combinatory treatments for patients with liver cancer [37]. In another study, hits from a cell-based screen for inhibitors of Toll-like receptor 7 (TLR7) or TLR7-dependent activation of NF-ĸB were prioritized based on potency and their structural similarity to each other. Following structure activity relationship studies on these hits, Z9j was discovered as an analogue with an IC50 of 0.26 μM for its effect on NF-ĸB activation. This compound appeared to inhibit IKK as well as upstream pathways such as Src/Syk and PI3K/Akt to affect NF-ĸB activation, although there may be other ways that the compound indirectly affects NF-ĸB activity [38]. Another compound acting upstream of NF-ĸB, iNUB, was found from a screen of a natural compound library to inhibit the interaction between IKKγ and ubiquitin [39]. Treatment with this compound decreased NF-ĸB activity following TNFα stimulation, reduced expression of NF-ĸB target genes and selectively killed lymphoma cells addicted to high levels of NF-ĸB signaling at 20 μM [39]. Finally, by binding the minor groove of DNA at promoters of NF-ĸB target genes, Py-Im polyamide 1 inhibits the DNA binding ability of NF-ĸB and reduces expression of target genes [40]. Genome-wide comparison with an established IKK inhibitor (PS1145) [41] showed similar effects on a group of genes along with distinct regulation of other genes, suggesting that small molecules modulating different points of the NF-ĸB signaling pathway can potentially be used to answer different biological questions [40].
Modulators of Oncogenic Myc and Obligate Partner Max
The transcription factor Myc is deregulated in most of human cancers [42]. Inhibition of Myc in transgenic mouse models of Myc addicted cancers [43,44] and utilization of the dominant negative Omomyc [45,46] suggest that Myc is a promising therapeutic target. Myc forms a heterodimer with its obligate partner Max to bind to DNA at E-box sequences and regulate transcription (Figure 3) [47,48]. Thus, many research groups have focused on inhibiting this Myc/Max interaction, which involves a large protein-protein interface stretching an area of 3,206 Å2 [3,49]. Many of the resulting compounds were thus discovered from mechanism-specific assays focused on this Myc/Max interaction such as fluorescence resonance energy transfer (FRET) assays, yeast two-hybrid assays, and DNA binding assays, and were found to inhibit c-Myc/Max dimerization, block Myc-dependent oncogenic chicken embryo fibroblast transformation, and inhibit Myc-dependent transcription and cell proliferation [50,51]. These molecules – including 10058-F4 [52] and Mycro3 [53] - have previously been reviewed [50,51] and some of the more potent IC50s are in the 10 to 40 μM range in a variety of cell-based assays. A more recent addition to the toolbox of Myc/Max disrupters is sAJM589 [54], which was discovered using a protein-fragment complementation assay (PCA). This compound displayed potent effects on viable cell levels (with an IC50 of 1.9 μM in P493–6, an engineered B cell line with a Tet off system for Myc), affected transcription in a manner to mimic Myc depletion, and decreased levels of Myc protein possibly due to increased ubiquitin-mediated degradation [54].
Figure 3. Modulators of Myc-driven transcription.
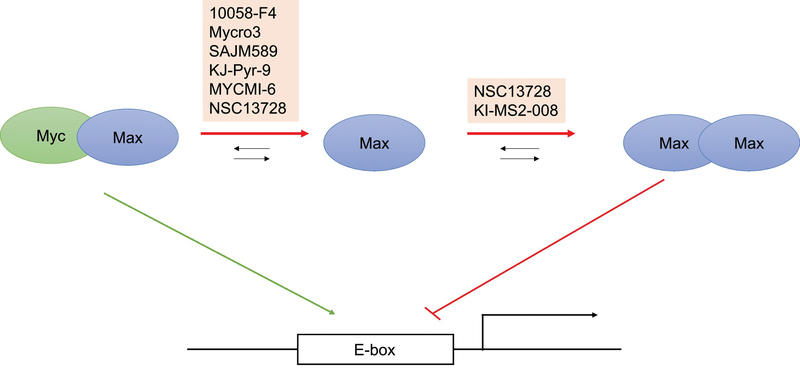
A simplified diagram of Myc-driven transcription in which Myc/Max dimers generally activate transcription, while Max/Max homodimers attenuate Myc-driven transcription. While the majority of inhibitors of Myc-driven transcription disrupt the Myc/Max heterodimer, NSC13728 (which also has roles in affecting Myc/Max heterodimers) and KI-MS2–008 stabilize the Max homodimer to indirectly attenuate Myc activity.
Despite success in cell culture experiments, many Myc/Max interaction disrupters failed to exhibit efficacy in vivo. To our knowledge, the first small molecule inhibiting the Myc/Max interaction in vivo without first pre-treating tumor cells is KJ-Pyr-9 [55]. This molecule was identified as the most soluble in water out of four small molecules effective in a fluorescence polarization screen and an assay to test Myc-induced oncogenic transformation in chicken embryo fibroblasts. It displayed specificity for Myc and N-Myc compared to other oncoproteins such as v-Src, v-Jun and an H1047R mutant of PI3K. The compound inhibited proliferation of P493–6 cells and downregulated Myc-regulated gene expression. It also suppressed the growth of an MDA-MB-231 xenograft in mice treated with daily intraperitoneal (IP) injections at 10 mg/kg [55]. The potential of Myc/Max disrupters to display in vivo efficacy was also seen with Mycro3, which promoted cancer cell apoptosis, decreased cell proliferation and led to tumor shrinkage of mutant KRAS-driven pancreatic ductal adenocarcinoma when moribund mice were dosed daily with 100 mg/kg Mycro3 [56]. Additionally, an example overcoming obstacles delivering Myc/Max inhibitors was observed with a prodrug version of 10058-F4 encapsulated in nanoparticles targeting integrin-expressing multiple myeloma cells [57]. The use of a prodrug allowed stabilization and encapsulation of the compound in the hydrophobic portion of the nanoparticles and prevented early release during delivery. The original inhibitor 10058-F4 without nanoparticle-aided delivery did not improve survival of mice and this study was the first to suggest that Myc/Max inhibitors previously limited by poor bioavailability or pharmacokinetics could be effective when coupled with targeted delivery [57]. Most recently, MYCMI-6 was discovered as another Myc/Max disrupter from a cell-based screen of nearly 2000 compounds based on bimolecular fluorescence complementation [58]. The compound bound the basic helix-loop-helix leucine zipper domain of Myc with a Kd of 1.6 μM as measured by surface plasmon resonance (SPR), decreased tumor cell growth with IC50s down to 0.5 μM and inhibited Myc-driven transcription. In a mouse model of N-Myc amplified neuroblastoma, daily IP injections at 20 mg/kg increased apoptosis of tumor cells, reduced proliferation and microvasculature, and exhibited on-target effects at the tumor [58].
In addition to forming heterodimers with Myc, Max can also form dimers with itself, Mga and a family of Mxd proteins [59]. An alternative strategy pioneered by Vogt’s group involves stabilization of the Max homodimer to attenuate Myc-driven transcription by reducing the amount of Max available to bind Myc [60]. By conducting virtual ligand screens on the full structures of Myc/Max and Max/Max dimers, this group identified three sites that contained 85% of the compounds predicted to bind Myc/Max and Max/Max dimers. The lead compound NSC13728 bound to one of these binding sites, which in contrast to the other two sites, allowed for specificity for Max/Max dimers over Myc/Max dimers. NSC13728 was observed to stabilize the Max/Max homodimer in FRET and analytical ultracentrifugation experiments, and was also found to directly inhibit the Myc/Max interaction in co-immunoprecipitation, enzyme-linked immunosorbent assay (ELISA) and SPR studies. It decreased the proliferation of Q8 cells – a cell line transformed by v-myc, decreased Myc-mediated oncogenic transformation of chicken embryonic fibroblasts with an IC50 of 3 μM, and lowered Myc-mediated transcription in HEK293T cells [60]. Additional support of stabilizing the Max/Max homodimer as an alternative strategy to inhibit Myc transcriptional activity came from the discovery of KI-MS2–008 [61]. KI-MS2–008 was discovered as a Max-binding small molecule that inhibited Myc transcriptional activity through small molecule microarray screens of purified recombinant Max and dual luciferase-based reporter assays of Myc transcriptional activity. This compound was observed to stabilize the Max homodimer in in vitro studies, while having no effect on the biophysical interaction between Myc and Max. Treatment with the compound led to a decrease in c-Myc protein levels in P493–6 cells, as well as global changes in the transcriptome with specific decreases in Myc-regulated genes, and decreases in Myc binding and increases in Max binding at the promoters of Myc occupied genes. At relatively low doses of 0.06 mg/kg and 0.24 mg/kg, treatment with KI-MS2–008 resulted in suppression of T-cell acute lymphoblastic leukemia and hepatocellular carcinoma in mouse models [61]. As this compound did not directly disrupt the Myc/Max interaction, it provides even stronger evidence for the alternative strategy to stabilize the Max/Max homodimer proposed by the Vogt lab. With the extensive network in which Max is involved, it immediately follows that stabilization of Max/Mxd or Max/Mga heterodimers could also offer potential strategies for Myc inhibition.
Myb – an acute myeloid leukemia (AML) dependency reinforced by DepMap data
The master regulator Myb has been found to be a dependency in acute myeloid leukemia (AML) [62] and more recently, Myb was identified as a top dependency specifically in AML from analysis of DepMap data focused on lineage-specific targeting of master transcription factors to mitigate off-target effects [12]. In various cancers such as AML, breast cancers and colon cancers, overexpression of Myb can drive proliferation of tumor cells [63–65]. Proof of concept experiments with peptides showed that squelching of Taf12 or disrupting the interaction between Myb and CREB-binding protein/p300 could perturb Myb activity and exhibit efficacy in mouse models of leukemia [66,67]. While there are small molecules that inhibit Myb activity, development of small molecule probes that directly and specifically inhibit Myb would provide additional value for studying Myb in cancer.
The first example of a small molecule inhibitor of Myb activity was discovered from a screen of 30 sesquiterpene lactones using a fluorescence-based reporter of Myb activity. The compound mexicanin-I significantly inhibited Myb activity as well as Myb target gene expression at 1–3 μM and suppressed proliferation of human leukemia cell lines without affecting Myb expression levels [68,69]. After additional compounds were screened using this Myb reporter assay, two compounds – helenalin acetate and goyazensolide – were observed to inhibit Myb activity with IC50 values of 0.6 to 0.7 μM [70], although helenalin acetate was later found to bind and target full-length C/EBPβ [71], a cooperative transcription factor of Myb in myeloid cells [72]. Additionally, the triterpenoid Celastrol was discovered from this screen to inhibit Myb activity with an IC50 of 0.85 μM [73]. The compound disrupted the interaction between the transactivation domain of Myb and p300’s KIX domain at 1 μM, decreased Myb target gene expression, and caused differentiation of HL60 cells in response to Myb inhibition. In a mouse model of AML, Celastrol treatment extended survival of mice to 10 weeks from 4 weeks [73]. Additional studies including reporter assays and mammalian two-hybrid experiments revealed that Celastrol also inhibited C/EBPβ activity in a Myb-independent manner and disrupted the interaction between C/EBPβ and p300’s Taz2 domain via Cys1789 or Cys1790 [74]. Similar results disrupting the Myb – p300 interaction, decreasing Myb target gene expression and causing differentiation of HL60 cells were observed with naphthoquinones [75].
Another cell-based screen for Myb inhibitors utilized HEK293 cells with a Tet-On system for mutant Myb with increased transactivation activity [76]. After screening a library of 120 natural compounds at 5 μM, two compounds – toyocamycin and teniposide – were found to inhibit Myb activity in a dose dependent manner with submicromolar concentrations as low as ~30 nM. While teniposide is a known topoisomerase II inhibitor, it also inhibited the activity of both mutant and wild-type Myb, and caused differentiation of a promyelocytic leukemia cell line in a dose-dependent manner through Myb inhibition. Teniposide additionally decreased Myb protein levels in AML cells via proteasome-mediated degradation [76].
Mebendazole was discovered to drive global transcriptional changes most resembling a c-MYB signature out of a library of 1309 CMAP drugs [77,78]. In cell culture, it inhibited viability (IC50 values between 0.07 and 0.26 μM) and colony formation of human AML cell lines at 1.25 μM, and decreased c-Myb protein levels in AML cell lines after 6 hours of treatment at concentrations as low as 1.1 μM. Additional studies suggested that mebendazole causes c-Myb to be degraded by the proteasome likely through dissociation of c-Myb from the Hsp70/Hsc70 chaperone complex, generally without affecting c-MYB gene expression. Finally, mebendazole administered orally extended life expectancy of NSG mice transplanted with THP1 cells [77].
Additional transcription factor targets nominated by DepMap data
To our knowledge, there are no small molecule probes available for a number of other targets nominated by the dependency map project such as ESR1, TFAP2C, SPDEF, FOXA1 [79], and LYL1. Other targets have inhibitors, but no direct chemical probes. For example, the glucocorticoid dexamethasone dose-dependently inhibited GATA-3 transcriptional activity, but did not inhibit GATA-3 binding to DNA [80]. Metformin treatment led to decreases in N-cadherin protein levels and sarcosphere numbers to mimic shRNA-mediated knockdown of SATB2 in osteosarcoma cells [81]. BET bromodomain inhibitors such as JQ1, MA4–022-1, and MA4–022-2 could be used to decrease expression and protein levels of HOXB13, resulting in suppression of tumor cell growth in castration-resistant prostate cancers [82]. Antisense oligonucleotide targeting of IRF4 suggest its promise as a therapeutic target in multiple myeloma [83], while IRF4 siRNA suggests a role for inhibiting IRF4 to improve liver transplant efficiency [84]. Small molecules 10-E-09, 12-P-16 and 13-I-18 decreased IRF4 protein levels in myeloma cells and decreased viable cell levels of a variety of myeloma cells compared to IRF4 negative cells [85]. As mentioned previously, many cancer types were dependent on specific transcription factors nominated by the dependency map project [12]. Indeed, there is a general trend toward higher specificity of dependence within specific lineages of cancer compared to other cancer types for these new transcription factor targets (Figure 4) [12,86]. Establishing chemical probes for these transcription factors may lead to drugs and strategies that have fewer side effects due to the specificity of targeting limited tissues.
Figure 4.
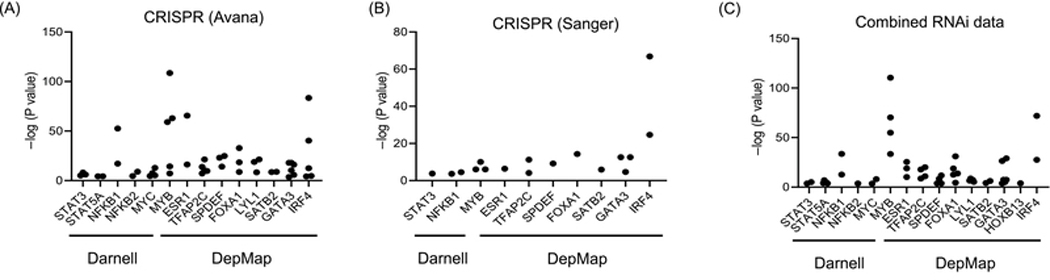
Significance of tissue specificity for targets nominated by the dependency map project. –log (P values) for P values computed between cancer cell lines of a specific type and all other cancer cell lines using empirical Bayes statistics. Data points were selected in October 2019 from specific types that were statistically different and more dependent than the rest of the cancer cell lines, using (A) CRISPR (Avana) data, (B) CRISPR (Sanger) data and (C) combined RNAi data from the Broad, Novartis, and Marcotte.
Concluding Remarks
Overall, the toolbox of small molecule probes discovered for transcription factors originally suggested by Darnell – including the Stat family of transcription factors, NF-ĸB and Myc – has been drastically improved. Among the compounds that most closely match the description of a chemical probe [11], there are small molecules that selectively inhibit their target over structurally related proteins with IC50 values equal or more potent than single digit micromolar range. For many of these small molecules, however, one of the major shortcomings is lack of understanding for the precise mechanism of action and/or target identification profile in cells. For example, knowledge of how the small molecule binds the target protein or how selective the compound is in cellular contexts will improve characterization to help meet criteria of a high quality chemical probe. This additional information will allow scientists to study specific functions of a protein of interest, such as disruption of a particular protein-protein interaction rather than general changes such as an overall protein level decrease. As the nuanced differences of individual cancer subtypes become clearer, this knowledge will also be critical in predicting how patients will respond to a given small molecule in the clinical setting.
While this characterization of existing small molecules, and better understanding of the biology and models related to transcription factor targets will improve the toolkit for studying those transcription factors (see Outstanding Questions), DepMap has uncovered a number of dependencies in cancer cell lines in an unbiased manner – many of which are specific to certain tissue types. Moving forward, these additional transcription factors may become potential therapeutic targets of high interest, but many lack tools for effective research. This gap between the knowledge gained from genome-scale screens and capabilities to act on these observations has slowed scientific research [87]. In July 2018, scientists gathered to plan how to generate tools for understudied proteins of high interest. As a result, Target 2035 was established as a global federation with an ambitious vision to create a chemical probe and/or antibody to interrogate the entire proteome through open science [87]. The additional transcription factors nominated and reinforced by DepMap should be prioritized among the first wave of proteins for which chemical probes will be developed under Target 2035.
Outstanding Questions Box.
Which biological functions of transcription factors implicated in disease should we consider more for targeting? Is it functionally or technically more desirable to target a transcription factor directly or to target other proteins that interact closely with the implicated transcription factor (e.g. targeting Myb through C/EBPβ, Myc through Max or Mxd proteins, androgen receptor through CDK9)?
What factors govern sensitivity of a cell line toward a compound? Transcription factors function primarily via protein-protein and protein-DNA interactions. In some scenarios, the ratio between various proteins may be more indicative of the cell state than absolute protein levels. Understanding such factors may be critical in predicting response to an inhibitor.
Which cell line models are most appropriate for each disease when studying aberrant transcription factor activity? Using representative cell lines with natural levels of transcription factors may be key in modeling the disease, rather than using engineered cell lines with overexpressed protein that are commonly used for research. Additionally, transcription factors may regulate different sets of genes depending on cellular context. Thus, use of cell lines that most closely mimic the target disease will be critical to translate research findings to therapeutic applications.
Can current chemical probes or modulators of transcription factors be translated into clinically relevant drugs? Cancer cell dependence on a deregulated transcription factor may provide a therapeutic window in which treatment specifically kills cancer cells, but side effects may still be a major concern for small molecules targeting master regulators. Additionally, while current small molecule inhibitors are useful as research tools, many lack the pharmacological properties to translate into a drug. A handful of transcription factor inhibitors are currently in clinical trials, including inhibitors of protein-protein interactions.
Highlights.
Transcription factors have previously been nominated as targets in disease due to their location in focal points of signaling pathways.
Significant progress has been made for many of these previously suggested transcription factors, including the STAT family, NF-ĸB and Myc, resulting in small molecules inhibiting transcription factor activity in cell culture and in some cases, in vivo.
The cancer dependency map project uncovered many critical proteins in human cancers based on genome-wide loss of function screens, and transcription factors represent a large class of the dependencies. Among these, a number of transcription factors including Myb have limited or no suitable small molecule probes, and chemical probe research on these transcription factors will be a major area of interest moving forward.
Acknowledgments:
We thank Becky Leifer for helpful comments. This work was financially supported by the National Cancer Institute through R01-CA160860, the Leukemia & Lymphoma Society, the Ono Pharma Foundation, and a Koch Institute Graduate Fellowship in Cancer Research.
Footnotes
Declaration of Interests: A.N.K. is a founder of Kronos Bio and a member of its scientific advisory board. A.C. is an employee of Kronos Bio. A.C. and A.N.K. have a patent related to work on KI-MS2-008 that is licensed by Kronos Bio.
Resources:
This study is registered with ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03888612
This study is registered with ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03195699
This study is registered with ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01184807
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Darnell JE (2002) Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749 [DOI] [PubMed] [Google Scholar]
- 2.Berg T. (2008) Inhibition of transcription factors with small organic molecules. Curr. Opin. Chem. Biol 12, 464–471 [DOI] [PubMed] [Google Scholar]
- 3.McKeown MR and Bradner JE (2014) Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koehler AN (2010) A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol 14, 331–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J. et al. (2006) Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsafou K. et al. (2018) Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol 430, 2321–2341 [DOI] [PubMed] [Google Scholar]
- 7.Dyson HJ and Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 8.Ottis P. and Crews CM (2017) Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol 12, 892–898 [DOI] [PubMed] [Google Scholar]
- 9.Winter GE et al. (2015) Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bushweller JH (2019) Targeting transcription factors in cancer — from undruggable to reality. Nat. Rev. Cancer DOI: 10.1038/s41568-019-0196-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frye SV (2010) The art of the chemical probe. Nat. Chem. Biol 6, 159–161 [DOI] [PubMed] [Google Scholar]
- 12.Tsherniak A. et al. (2017) Defining a Cancer Dependency Map. Cell 170, 564–576.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furqan M. et al. (2013) STAT inhibitors for cancer therapy. J. Hematol. Oncol.J Hematol Oncol 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Shea JJ et al. (2015) The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med 66, 311–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buettner R. et al. (2002) Activated STAT Signaling in Human Tumors Provides Novel Molecular Targets for Therapeutic Intervention. Clin. Cancer Res 8, 945–954 [PubMed] [Google Scholar]
- 16.Yu H. and Jove R. (2004) The STATs of cancer — new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 [DOI] [PubMed] [Google Scholar]
- 17.Schust J. et al. (2006) Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol 13, 1235–1242 [DOI] [PubMed] [Google Scholar]
- 18.Song H. et al. (2005) A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A 102, 4700–4705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ball S. et al. (2011) The Small Molecule, LLL12, Inhibits STAT3 Phosphorylation and Induces Apoptosis in Medulloblastoma and Glioblastoma Cells. PLoS ONE 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu A. et al. (2011) Novel small molecule, XZH-5, inhibits constitutive and interleukin-6-induced STAT3 phosphorylation in human rhabdomyosarcoma cells. Cancer Sci. 102, 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin D-S et al. (2009) Cryptotanshinone Inhibits Constitutive Signal Transducer and Activator of Transcription 3 Function through Blocking the Dimerization in DU145 Prostate Cancer Cells. Cancer Res. 69, 193–202 [DOI] [PubMed] [Google Scholar]
- 22.Lin L. et al. (2010) A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol. Cancer 9, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin L. et al. (2009) New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 100, 1719–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bill MA et al. (2012) Structurally Modified Curcumin Analogs Inhibit STAT3 Phosphorylation and Promote Apoptosis of Human Renal Cell Carcinoma and Melanoma Cell Lines. PLoS ONE 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turkson J. et al. (2004) Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther 3, 1533–1542 [PubMed] [Google Scholar]
- 26.Turkson J. et al. (2005) A Novel Platinum Compound Inhibits Constitutive Stat3 Signaling and Induces Cell Cycle Arrest and Apoptosis of Malignant Cells. J. Biol. Chem 280, 32979–32988 [DOI] [PubMed] [Google Scholar]
- 27.Page BDG et al. (2012) Small Molecule STAT5-SH2 Domain Inhibitors Exhibit Potent Antileukemia Activity. J. Med. Chem 55, 1047–1055 [DOI] [PubMed] [Google Scholar]
- 28.Wong AL et al. (2015) Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 26, 998–1005 [DOI] [PubMed] [Google Scholar]
- 29.Wei N. et al. (2019) Targeting colon cancer with the novel STAT3 inhibitor bruceantinol. Oncogene 38, 1676–1687 [DOI] [PubMed] [Google Scholar]
- 30.Lis C. et al. (2017) Development of Erasin: a chromone-based STAT3 inhibitor which induces apoptosis in Erlotinib-resistant lung cancer cells. Sci. Rep 7, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilmore TD and Herscovitch M. (2006) Inhibitors of NF- κ B signaling: 785 and counting. Oncogene 25, 6887–6899 [DOI] [PubMed] [Google Scholar]
- 32.Gupta SC et al. (2010) Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta BBA - Gene Regul. Mech 1799, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann A. et al. (2002) The IκB-NF-κB Signaling Module: Temporal Control and Selective Gene Activation. Science 298, 1241–1245 [DOI] [PubMed] [Google Scholar]
- 34.Bharti AC et al. (2003) Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor–κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 101, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 35.Alexandrow MG et al. (2012) Curcumin: a novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. ECP 21, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hahn Y-I et al. (2018) Curcumin interacts directly with the Cysteine 259 residue of STAT3 and induces apoptosis in H- Ras transformed human mammary epithelial cells. Sci. Rep 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquardt JU et al. (2015) Curcumin effectively inhibits oncogenic NF-κB signaling and restrains stemness features in liver cancer. J. Hepatol 63, 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L. et al. (2018) Discovery of Novel Small-Molecule Inhibitors of NF-κB Signaling with Antiinflammatory and Anticancer Properties. J. Med. Chem 61, 5881–5899 [DOI] [PubMed] [Google Scholar]
- 39.Vincendeau M. et al. (2016) Inhibition of Canonical NF-κB Signaling by a Small Molecule Targeting NEMO-Ubiquitin Interaction. Sci. Rep 6, 18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raskatov JA et al. (2012) Modulation of NF-κB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci 109, 1023–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yemelyanov A. et al. (2006) Effects of IKK inhibitor PS1145 on NF- κ B function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene 25, 387–398 [DOI] [PubMed] [Google Scholar]
- 42.Tansey WP (2014) , Mammalian MYC Proteins and Cancer. , New Journal of Science. [Online]. Available: https://www.hindawi.com/journals/njos/2014/757534/. [Accessed: 20-Mar-2019] [Google Scholar]
- 43.Felsher DW (2010) MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell–Intrinsic and Host-Dependent Mechanisms. Genes Cancer 1, 597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Felsher DW and Bishop JM (1999) Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol. Cell 4, 199–207 [DOI] [PubMed] [Google Scholar]
- 45.Soucek L. et al. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soucek L. et al. (2002) Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 62, 3507–3510 [PubMed] [Google Scholar]
- 47.Blackwell TK et al. (1990) Sequence-specific DNA binding by the c-Myc protein. Science 250, 1149–1151 [DOI] [PubMed] [Google Scholar]
- 48.Blackwood EM and Eisenman RN (1991) Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217 [DOI] [PubMed] [Google Scholar]
- 49.Kiessling A. et al. (2006) Selective Inhibition of c-Myc/Max Dimerization and DNA Binding by Small Molecules. Chem. Biol 13, 745–751 [DOI] [PubMed] [Google Scholar]
- 50.Berg T. (2011) Small-Molecule Modulators of c-Myc/Max and Max/Max Interactions In Small-Molecule Inhibitors of Protein-Protein Interactions (Vassilev L. and Fry D., eds), pp. 139–149, Springer Berlin Heidelberg [Google Scholar]
- 51.Fletcher S. and Prochownik EV (2015) Small-Molecule Inhibitors of the Myc Oncoprotein. Biochim. Biophys. Acta 1849, 525–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin X. et al. (2003) Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene 22, 6151. [DOI] [PubMed] [Google Scholar]
- 53.Kiessling A. et al. (2007) Selective Inhibition of c-Myc/Max Dimerization by a Pyrazolo[1,5-a]pyrimidine. ChemMedChem 2, 627–630 [DOI] [PubMed] [Google Scholar]
- 54.Choi SH et al. (2017) Targeted Disruption of Myc–Max Oncoprotein Complex by a Small Molecule. ACS Chem. Biol 12, 2715–2719 [DOI] [PubMed] [Google Scholar]
- 55.Hart JR et al. (2014) Inhibitor of MYC identified in a Kröhnke pyridine library. Proc. Natl. Acad. Sci 111, 12556–12561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stellas D. et al. (2014) Therapeutic Effects of an Anti-Myc Drug on Mouse Pancreatic Cancer. JNCI J. Natl. Cancer Inst 106, [DOI] [PubMed] [Google Scholar]
- 57.Soodgupta D. et al. (2015) Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol. Cancer Ther 14, 1286–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castell A. et al. (2018) A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep 8, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conacci-Sorrell M. et al. (2014) An Overview of MYC and Its Interactome. Cold Spring Harb. Perspect. Med 4, a014357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang H. et al. (2009) Stabilizers of the Max Homodimer Identified in Virtual Ligand Screening Inhibit Myc Function. Mol. Pharmacol 76, 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Struntz NB et al. (2019) Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem. Biol DOI: 10.1016/j.chembiol.2019.02.009 [DOI] [PubMed] [Google Scholar]
- 62.Zuber J. et al. (2011) An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 25, 1628–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X. et al. (2018) Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J. Cancer 9, 1259–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pattabiraman DR and Gonda TJ (2013) Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 27, 269–277 [DOI] [PubMed] [Google Scholar]
- 65.Nakano K. et al. (2016) Dysregulation of c-Myb Pathway by Aberrant Expression of Proto-oncogene MYB Provides the Basis for Malignancy in Adult T-cell Leukemia/lymphoma Cells. Clin. Cancer Res 22, 5915–5928 [DOI] [PubMed] [Google Scholar]
- 66.Xu Y. et al. (2018) A TFIID-SAGA Perturbation that Targets MYB and Suppresses Acute Myeloid Leukemia. Cancer Cell 33, 13–28.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramaswamy K. et al. (2018) Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat. Commun 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bujnicki T. et al. (2012) Inhibition of Myb-dependent gene expression by the sesquiterpene lactone mexicanin-I. Leukemia 26, 615–622 [DOI] [PubMed] [Google Scholar]
- 69.Uttarkar S. et al. (2017) Targeting the transcription factor Myb by small-molecule inhibitors. Exp. Hematol 47, 31–35 [DOI] [PubMed] [Google Scholar]
- 70.Schomburg C. et al. (2013) Natural sesquiterpene lactones as inhibitors of Myb-dependent gene expression: Structure–activity relationships. Eur. J. Med. Chem 63, 313–320 [DOI] [PubMed] [Google Scholar]
- 71.Jakobs A. et al. (2016) An isoform-specific C/EBPβ inhibitor targets acute myeloid leukemia cells. Leukemia 30, 1612–1615 [DOI] [PubMed] [Google Scholar]
- 72.Burk O. et al. (1993) Synergistic activation of the chicken mim-1 gene by v-myb and C/EBP transcription factors. EMBO J. 12, 2027–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uttarkar S. et al. (2016) Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood 127, 1173–1182 [DOI] [PubMed] [Google Scholar]
- 74.Coulibaly A. et al. (2018) The natural anti-tumor compound Celastrol targets a Myb-C/EBPβ-p300 transcriptional module implicated in myeloid gene expression. PLOS ONE 13, e0190934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uttarkar S. et al. (2016) Small-Molecule Disruption of the Myb/p300 Cooperation Targets Acute Myeloid Leukemia Cells. Mol. Cancer Ther 15, 2905–2915 [DOI] [PubMed] [Google Scholar]
- 76.Yusenko M. et al. (2018) A novel cell-based screening assay for small-molecule MYB inhibitors identifies podophyllotoxins teniposide and etoposide as inhibitors of MYB activity. Sci. Rep 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walf-Vorderwülbecke V. et al. (2018) Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia 32, 882–889 [DOI] [PubMed] [Google Scholar]
- 78.Lamb J. et al. (2006) The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 313, 1929–1935 [DOI] [PubMed] [Google Scholar]
- 79.Glont S-E et al. (2019) Comprehensive Genomic Analysis Reveals that the Pioneering Function of FOXA1 Is Independent of Hormonal Signaling. Cell Rep. 26, 2558–2565.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liberman AC et al. (2009) Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J. 23, 1558–1571 [DOI] [PubMed] [Google Scholar]
- 81.Xu HY et al. (2017) Metformin reduces SATB2-mediated osteosarcoma stem cell-like phenotype and tumor growth via inhibition of N-cadherin/NF-kB signaling. Eur. Rev. Med. Pharmacol. Sci 21, 4516–4528 [PubMed] [Google Scholar]
- 82.Nerlakanti N. et al. (2018) Targeting the BRD4-HOXB13 Coregulated Transcriptional Networks with Bromodomain-Kinase Inhibitors to Suppress Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther 17, 2796–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou T. et al. (2017) Therapeutic Targeting of Interferon Regulatory Factor 4 with Next Generation Antisense Oligonucleotides Produces Robust In Vivo Antitumor Activity in Preclinical Models of Multiple Myeloma. Blood 130, 3078–3078 [Google Scholar]
- 84.Zhao W. et al. (2015) Inhibition of Interferon Regulatory Factor 4 Attenuates Acute Liver Allograft Rejection in Mice. Scand. J. Immunol 82, 262–268 [DOI] [PubMed] [Google Scholar]
- 85.Vatolin S. et al. (2012) Direct Chromatin PCR (DC-PCR): Hypotonic Conditions Allow Differentiation of Chromatin States during Thermal Cycling. PLoS ONE 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritchie ME et al. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carter AJ et al. (2019) Target 2035: probing the human proteome. Drug Discov. Today DOI: 10.1016/j.drudis.2019.06.020 [DOI] [PubMed] [Google Scholar]
- 88.Arrowsmith CH et al. (2015) The promise and peril of chemical probes. Nat. Chem. Biol 11, 536–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blagg J. and Workman P. (2017) Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 32, 9–25 [DOI] [PMC free article] [PubMed] [Google Scholar]