

An efficient synthesis of 1-arylisochromeno[3,4-d][1,2,3]triazol-5(1H)-ones is reported, along with the structures of four examples and the structure of a ring-opened transesterification product.
Keywords: synthesis, heterocyclic compounds, isocoumarins, triazoles, crystal structure, molecular conformation, hydrogen bonding, supramolecular assembly
Abstract
An efficient synthesis of 1-arylisochromeno[3,4-d][1,2,3]triazol-5(1H)-ones, involving the diazotization of 3-amino-4-arylamino-1H-isochromen-1-ones in weakly acidic solution, has been developed and the spectroscopic characterization and crystal structures of four examples are reported. The molecules of 1-phenylisochromeno[3,4-d][1,2,3]triazol-5(1H)-one, C15H9N3O2, (I), are linked into sheets by a combination of C—H⋯N and C—H⋯O hydrogen bonds, while the structures of 1-(2-methylphenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, C16H11N3O2, (II), and 1-(3-chlorophenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, C15H8ClN3O2, (III), each contain just one hydrogen bond which links the molecules into simple chains, which are further linked into sheets by π-stacking interactions in (II) but not in (III). In the structure of 1-(4-chlorophenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, (IV), isomeric with (III), a combination of C—H⋯O and C—H⋯π(arene) hydrogen bonds links the molecules into sheets. When compound (II) was exposed to a strong acid in methanol, quantitative conversion occurred to give the ring-opened transesterification product methyl 2-[4-hydroxy-1-(2-methylphenyl)-1H-1,2,3-triazol-5-yl]benzoate, C17H15N3O3, (V), where the molecules are linked by paired O—H⋯O hydrogen bonds to form centrosymmetric dimers.
Introduction
Isocoumarins are an important building block in synthetic medicinal chemistry because they have shown interesting bioactivities, for example, as anticoagulants (Oweida et al., 1990 ▸), as herbicides (Zhang et al., 2016 ▸) and as insecticides (Qadeer et al., 2007 ▸). In order to gain access to compounds of this type in a straightforward way, a synthetic route has been developed using reactions between 2-formylbenzoic acid, hydrogen cyanide and anilines to yield N-aryldiaminoisocoumarins (Opatz & Ferenc, 2005 ▸). We have reported the structures of several compounds of this type (Vicentes et al., 2013 ▸) and, more recently using such compounds as precursors, we have developed the synthesis of a new heterocyclic system, namely fused imidazoloisocoumarins, as part of an exploration of possible synergies between the imidazole and isocoumarin pharmacophores (Rodríguez et al., 2017 ▸).
The bioactivity of hybrid systems containing the 1,2,3-triazole unit has been reviewed recently (Xu et al., 2019 ▸) and, with this in mind, we have now developed an efficient synthesis of 1-arylisochromeno[3,4-d][1,2,3]triazol-5(1H)-ones starting from the same N-aryldiaminoisocoumarins as were used in the synthesis of imidazoloisocoumarins (Rodríguez et al., 2017 ▸). Thus, we now report the synthesis and spectroscopic characterization, and the molecular and supramolecular structures of four representative examples, namely, 1-phenylisochromeno[3,4-d][1,2,3]triazol-5(1H)-one, (I), 1-(2-methylphenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, (II), 1-(3-chlorophenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, (III), and 1-(4-chlorophenyl)isochromeno[3,4-d][1,2,3]triazol-5(1H)-one, (IV), carrying substituents at different positions in the pendent aryl group, along with those of a transesterification product, namely, methyl 2-[4-hydroxy-1-(2-methylphenyl)-1H-1,2,3-triazol-5-yl]benzoate, (V). Compounds (I)–(IV) were prepared by reaction of sodium nitrite in acetic acid with the corresponding 3-amino-4-arylamino-1H-isochromen-1-ones (A) (see Scheme 1); the precursors of type (A) having aryl = phenyl, 2-methylphenyl or 4-chlorophenyl were prepared (Scheme 1) as reported previously (Rodríguez et al., 2017 ▸), and the new analogue having aryl = 3-chlorophenyl was prepared in the same way. The conversion of the precursors of type (A) to the products (I)–(IV) proceeds via the diazonium intermediate (B) (Scheme 1), which itself undergoes an intramolecular cyclization to form the triazolo ring. It is important to stress here the necessity of using a weak acid, here acetic acid, in the diazotization of (A) to form (B), as isocoumarins often readily undergo ring opening in the presence of strong acids. To confirm this, a sample of compound (II) was stirred in methanol in the presence of aqueous hydrochloric acid, resulting in a quantitative conversion of (II) to ester (V).
Experimental
Synthesis and crystallization
The known precursors of type (A) (see Scheme 1) having Ar = C6H5, 2-CH3C6H4 and 4-ClC6H4 were prepared as described previously (Rodríguez et al., 2017 ▸); the new analogue having Ar = 3-ClC6H4 was prepared following the same procedure. Analytical data for 3-amino-4-(3-chloroanilino)-1H-isochromen-1-one: yellow solid, yield 71%, m.p. 451–452 K; IR (ATR, cm−1): 3456, 3319, 2922, 1701, 1592, 1474, 1306, 1089, 767, 679; NMR [CDCl3, the numbering of the chlorophenyl ring follows that for compound (III)]: δ(1H) 8.15 (dd, J = 8.0, 0.7 Hz, 1H, H8), 7.53 (t, J = 7.6 Hz, 1H, H6), 7.20 (t, J = 7.6 Hz, 1H, H7), 7.16 (d, J = 8.1 Hz, 1H, H5), 7.10 (t, J = 8.0 Hz, 1H, H15), 6.76 (dd, J = 7.9, 1.1 Hz, 1H, H14), 6.65 (t, J = 2.0 Hz, 1H, H12), 6.56 (dd, J = 8.2, 1.6 Hz, 1H, H16), 4.85 (s, 1H, NH), 4.57 (s, 2H, NH2); δ(13C) 160.64 (CO), 154.78 (C3), 147.59 (C11), 140.40 (C4A), 135.65 (C13), 135.52 (C6), 130.86 (C15), 130.56 (C8), 124.23 (C7), 119.74 (C5), 119.29 (C14), 116.15 (C8A), 113.24 (C12), 111.59 (C16), 92.19 (C4); MS (EI, 70 eV): m/z (%) 285.9 (12) [M]+, 259.94 (31), 257.93 (100), 177.97 (16), 148.92 (20), 129.93 (21), 110.89 (17), 103.92 (17); HRMS (ESI–QTOF) found 287.0582, C15H11 35ClN2O2 requires for [M + H]+ 287.0578.
For the synthesis of compounds (I)–(IV), sodium nitrite (153 mg, 2.22 mmol) was added to a suspension of the appropriate precursor (A) [1.09 mmol; 275 mg for (I), 290 mg for (II) and 313 mg for each of (III) and (IV)] in acetic acid (1.0 ml) and the resulting mixture was then stirred at ambient temperature for 5 min. The resulting solid precipitate was collected by filtration and washed with an aqueous solution of sodium hydrogen carbonate (10% w/v) and then with water. The crude solid products were purified by column chromatography on silica gel 60 (0.040–0.063 mm) using dichloromethane as eluent.
Analytical data for compound (I), colourless solid, yield 77%, m.p. 433–434 K; IR (ATR, cm−1): 3065, 1736, 1622, 1493, 1208, 1019, 763, 715; NMR (CDCl3): δ(1H) 8.46 (dd, J = 7.5, 1.6 Hz, 1H, H6), 7.72–7.55 (m, 7H, H7, H8, H12, H13, H14, H15, H16), 7.29 (dd, J = 7.8, 1.0 Hz, 1H, H9); δ(13C) 160.18 (CO), 154.65 (C3A), 136.89 (C11), 135.41 (C8), 132.84 (C6), 131.24 (C14), 130.23 (C13, C15), 129.79 (C7), 126.27 (C9A), 126.11 (C12, C16), 121.25 (C9), 120.57 (C9B), 115.26 (C5A); MS (EI, 70 eV): m/z (%) 262.98 (2) [M]+, 223.99 (35), 178.98 (100), 178.00 (29), 148.93 (36), 104.94 (23), 76.95 (35); HRMS (ESI–QTOF) found 264.0768, C15H9N3O3 requires for [M + H]+ 264.0768.
Analytical data for compound (II), colourless solid, yield 74%, m.p. 439–440 K; IR (ATR, cm−1): 1745, 1622, 1012, 987, 764, 680; NMR (CDCl3): δ(1H) 8.45 (dd, J = 7.1, 2.2 Hz, 1H, H8), 7.66–7.57 (m, 3H, H6, H7, H13), 7.55–7.48 (m, 2H, H14, H15), 7.46 (dd, J = 7.8, 1.6 Hz, 1H, H16), 6.92 (dd, J = 7.0, 2.1 Hz, 1H, H5), 2.10 (s, 3H, CH3); δ(13C) 160.22 (CO), 154.34 (C3A), 135.85 (C11), 135.69 (C8), 132.67 (C6), 131.89 (C14), 131.66 (C13), 129.78 (C7), 127.73 (C16), 127.38 (C15), 126.19 (C9A), 120.63 (C9), 120.46 (C9B), 115.62 (C5A), 17.32 (CH3); MS (EI, 70 eV): m/z (%) 276,97 (2) [M]+, 220.99 (16), 194.02 (15), 193.00 (100), 192.02 (21), 164.97 (22), 88.94 (22); HRMS (ESI–QTOF) found 278.0923, C16H11N3O2 requires for [M + H]+ 278.0924.
Analytical data for compound (III), yellow solid, yield 75%, m.p. 448–449 K; IR (ATR, cm−1): 3072, 2919, 2850, 1748, 1617, 1587, 1010, 885, 867, 783; NMR (CDCl3): δ(1H) 8.46 (dd, J = 7.9, 1.4 Hz, 1H, H6), 7.75–7.60 (m, 5H, H7, H8, H12, H14, H15), 7.56 (ddd, J = 7.7, 1.9, 1.4 Hz, 1H, H16), 7.34 (dd, J = 7.9, 0.6 Hz, 1H, H9); δ(13C) 159.94 (CO), 154.65 (C3A), 137.75 (C11), 136.03 (C13), 135.57 (C8), 132.95 (C6), 131.47 (C15), 131.22 (C14), 130.04 (C7), 126.46 (C12), 125.92 (C9A), 124.23 (C16), 121.20 (C9), 120.57 (C9B), 115.22 (C5A); MS (EI, 70 eV): m/z (%) 296.95 (2) [M]+, 214.96 (29), 213.98 (15), 212.01 (100), 177.98 (47), 150.97 (15), 74.94 (27); HRMS (ESI–QTOF) found 298.0380, C15H8 35ClN3O2 requires for [M + H]+ 298.0378.
Analytical data for compound (IV), pink solid, yield 62%,, m.p. 490–492 K; IR (ATR, cm−1): 3087, 3066, 1740, 1620, 1457, 1217, 1013, 832, 764; NMR (CDCl3) δ(1H) 8.46 (ddd, J = 7.8, 1.5, 0.6 Hz, 1H, H6), 7.72–7.58 (m, 6H, H7, H8, H12, H13, H15, H16), 7.32 (ddd, J = 7.9, 1.3, 0.5 Hz, 1H, H9); δ(13C) 159.98 (CO), 154.69 (C3A), 137.45 (C11), 135.51 (C8), 135.30 (C14), 132.96 (C6), 130.53 (C13, C15), 130.00 (C7), 127.39 (C12, C16), 126.02 (C9A), 121.14 (C9), 120.59 (C9B), 115.25 (C5A); MS (EI, 70 eV): m/z (%) 296.93 (1.4) [M]+, 214.95 (31), 213.96 (15), 212.90 (100), 177.97 (39), 150.96 (14), 110.91 (13), 74.93 (25); HRMS (ESI–QTOF) found 298.0379, C15H8 35ClN3O2 requires for [M + H]+ 298.0378.
For the conversion of compound (II) into compound (V), a sample of (II) (2.00 g, 7.22 mmol) and aqueous hydrochloric acid (1 mol dm−3, 1 ml) were added to methanol (9 ml) and the resulting mixture was then stirred for 24 h at ambient temperature. The solvent was removed under reduced pressure and the resulting solid product was washed with an aqueous solution of sodium hydrogen carbonate (10% w/v) and then with water and finally dried in air to provide (V) as a colourless solid in quantitative yield (m.p. 463–464 K). Analytical data: IR (ATR, cm−1): 2982, 2948, 1722, 1623, 1512, 1259, 764, 713; NMR (CDCl3): δ(1H) 10.48 (s, 1H, OH), 7.76 (d, J = 7.7 Hz, 1H, H3), 7.51 (td, J = 7.5, 1.2 Hz, 1H, H5), 7.44 (t, J = 7.2 Hz, 1H, H4), 7.40–7.29 (m, 2H, H213, H214), 7.26 (td, J = 7.7, 1.1 Hz, 1H, H215), 7.23–7.16 (m, 2H, H6, H216), 3.67 (s, 3H, OCH3), 2.02 (s, 3H, CH3); δ(13C) 166.53 (CO), 155.36 (C24), 135.61 (C211), 134.49 (C214), 131.80 (C5), 131.38 (C6), 131.09 (C213), 130.85 (C25), 129.94 (C3), 129.70 (C214), 128.85 (C4), 127.39 (C216), 126.52 (C215), 126.40 (C1), 119.03 (C2), 52.12 (OCH3), 17.07 (CH3); MS (EI, 70 eV): m/z (%) 309.00 (2) [M]+, 239.01 (16), 237.98 (100), 219.99 (21), 193.00 (70), 164.97 (23), 90.96 (30); HRMS (ESI–QTOF) found 310.1186, C17H15N3O2 requires for [M + H]+ 310.1186.
Crystals of compounds (I)–(V) suitable for single-crystal X-ray diffraction were grown by slow evaporation, at ambient temperature and in the presence of air, of solutions in chloroform.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1 ▸. All H atoms were located in difference maps. H atoms bonded to C atoms were subsequently treated as riding atoms in geometrically idealized positions, with C—H = 0.95 (alkenyl and aromatic) or 0.98 Å (CH3) and with U
iso(H) = kU
eq(C), where k = 1.5 for the methyl groups, which were allowed to rotate but not to tilt, and 1.2 for all other H atoms bonded to C atoms. For the H atom bonded to an O atom in compound (V), the atomic coordinates were refined with U
iso(H) = 1.5U
eq(O), giving an O—H distance of 0.90 (2) Å. Several low-angle reflections which had been attenuated by the beam stop were omitted, i.e.
 01 for (II) and 02 for (IV); in addition, one bad outlier reflection, i.e.
01 for (II) and 02 for (IV); in addition, one bad outlier reflection, i.e.
 06, was omitted from the data set for (II) before the final refinements. For several of the refinements, the final analyses of variance showed unexpected values of K = [mean(F
o
2)/mean(F
c
2)] for the groups of the very weakest reflections. Thus, for (III) and (IV), respectively, −0.035 and −0.125 for 312 and 289 reflections in the F
c/F
c(max) ranges 0.000–0.008 and 0.000–0.010, and for (V), 3.550 for 339 reflections in the F
c/F
c(max) range 0.000–0.009; these values are probably statistical artefacts.
06, was omitted from the data set for (II) before the final refinements. For several of the refinements, the final analyses of variance showed unexpected values of K = [mean(F
o
2)/mean(F
c
2)] for the groups of the very weakest reflections. Thus, for (III) and (IV), respectively, −0.035 and −0.125 for 312 and 289 reflections in the F
c/F
c(max) ranges 0.000–0.008 and 0.000–0.010, and for (V), 3.550 for 339 reflections in the F
c/F
c(max) range 0.000–0.009; these values are probably statistical artefacts.
Table 1. Experimental details.
For all structures: Z = 4. Experiments were carried out at 100 K with Mo Kα radiation using a Bruker D8 Venture diffractometer. Absorption was corrected for by multi-scan methods (SADABS; Bruker, 2016 ▸).
| (I) | (II) | (III) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C15H9N3O2 | C16H11N3O2 | C15H8ClN3O2 |
| M r | 263.25 | 277.28 | 297.69 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/c |
| a, b, c (Å) | 11.6814 (5), 6.4310 (3), 16.0589 (8) | 7.5676 (5), 20.2663 (14), 9.2503 (7) | 11.0993 (8), 12.8996 (9), 9.2234 (6) |
| β (°) | 100.687 (2) | 112.957 (3) | 106.675 (2) |
| V (Å3) | 1185.47 (10) | 1306.33 (16) | 1265.04 (15) |
| μ (mm−1) | 0.10 | 0.10 | 0.31 |
| Crystal size (mm) | 0.20 × 0.12 × 0.06 | 0.28 × 0.17 × 0.16 | 0.20 × 0.12 × 0.05 |
| Data collection | |||
| T min, T max | 0.936, 0.994 | 0.941, 0.985 | 0.895, 0.985 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 26237, 2747, 2400 | 36209, 3242, 2915 | 28194, 2905, 2495 |
| R int | 0.037 | 0.034 | 0.044 |
| (sin θ/λ)max (Å−1) | 0.652 | 0.668 | 0.650 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.036, 0.103, 1.04 | 0.036, 0.098, 1.05 | 0.032, 0.089, 1.08 |
| No. of reflections | 2747 | 3242 | 2905 |
| No. of parameters | 181 | 191 | 190 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.31, −0.21 | 0.31, −0.25 | 0.32, −0.36 |
| (IV) | (V) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C15H8ClN3O2 | C17H15N3O3 |
| M r | 297.69 | 309.32 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c |
| a, b, c (Å) | 16.7331 (12), 5.9676 (4), 13.681 (1) | 11.1518 (5), 9.3143 (4), 14.2417 (6) |
| β (°) | 112.820 (3) | 98.655 (2) |
| V (Å3) | 1259.21 (16) | 1462.46 (11) |
| μ (mm−1) | 0.31 | 0.10 |
| Crystal size (mm) | 0.25 × 0.14 × 0.11 | 0.19 × 0.11 × 0.07 |
| Data collection | ||
| T min, T max | 0.880, 0.967 | 0.960, 0.993 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 34775, 2891, 2413 | 33834, 3360, 3084 |
| R int | 0.056 | 0.036 |
| (sin θ/λ)max (Å−1) | 0.650 | 0.650 |
| Refinement | ||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.034, 0.087, 1.12 | 0.042, 0.098, 1.07 |
| No. of reflections | 2891 | 3360 |
| No. of parameters | 190 | 213 |
| H-atom treatment | H-atom parameters constrained | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.37, −0.26 | 0.34, −0.22 |
Results and discussion
The constitutions of compounds (I)–(V) were all fully established by a combination of high-resolution mass spectrometry (HRMS), IR spectrosopy and 1H and 13C NMR spectroscopy, further confirmed by the structure analyses reported here (Figs. 1 ▸–5 ▸ ▸ ▸ ▸). The HRMS data for (I)–(IV) demonstrate the incorporation of an additional H atom, the IR data show the absence of an NH2 absorption around 3400 cm−1 and the 1H NMR spectra show the absence of signals around δ 4.5–5.0 arising from an amino group; these observations taken together confirm the conversion of the diamino precursors of type (A) (Scheme 1) into the triazolo products (I)–(IV), whose constitutions were fully confirmed by the detailed assignments of the 1H and 13C NMR spectra (see §2.1). Hence, the constitutions of (I)–(IV) show clearly that the anticipated triazolo ring formation has occurred, with the additional N atom arising from the diazotization process; similarly, the constitution of (V) confirms the occurrence of a ring-opening transesterification process.
Figure 1.

The molecular structure of compound (I), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level.
Figure 2.

The molecular structure of compound (II), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level.
Figure 3.

The molecular structure of compound (III), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level.
Figure 4.

The molecular structure of compound (IV), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level.
Figure 5.

The molecular structure of compound (V), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level.
Aside from the orientation of the 2-methyl and 3-chloro substituents in compounds (II) and (III), respectively, the conformations of compounds (I)–(IV) are fairly similar; the dihedral angles between the triazolo ring and the pendent ring (C11–C16) are 65.32 (5), 64.59 (4), 45.48 (8) and 52.32 (9)° in (I)–(IV), respectively. The molecules thus exhibit no internal symmetry and so are conformationally chiral in the crystalline state; the centrosymmetric space groups (Table 1 ▸) confirm that (I)–(IV) have all crystallized as conformational racemates. For all of (I)–(IV), the reference molecules were selected to have the same sign for the torsion angle N2—N1—C11—C12, or N2—N1—C11—C16 in the case of (III). A comparison of the conformation of ester (V) with that of its precursor (II) (Figs. 2 ▸ and 5 ▸) indicates that, in the crystalline state, there appear to have been rotations about both the bonds exocyclic to the triazolo ring in (V), along with a rotation about the bond linking the ester unit to the adjacent aryl ring. The significance of these differences is unclear. The bond lengths in (I)–(V) show no unusual features.
The supramolecular assembly in compounds (I)–(IV) is dominated by contacts of C—H⋯N, C—H⋯O and C—H⋯π(arene) types (Table 2 ▸) and it is therefore worthwhile to specify the criteria under which such interactions are regarded here is structurally significant, or otherwise. Firstly, we discount all C—H⋯N and C—H⋯O contacts in which the D—H⋯A angle is less than 140°, as the interaction energies associated with such contacts are likely to be extremely small (Wood et al., 2009 ▸). Secondly, we discount all contacts involving methyl C—H bonds; these are not only of low acidity, but methyl groups CH3—E are generally undergoing very fast rotation about the C—E bonds, even in the solid state (Riddell & Rogerson, 1996 ▸, 1997 ▸). In particular, for methyl groups bonded to aryl rings, as found in (II) and (V), the rotation of the methyl group relative to the ring is subject to a sixfold rotation barrier, known to be in general extremely low, typically just a few J mol−1 rather than the more typical magnitude of a few kJ mol−1 (Tannenbaum et al., 1956 ▸; Naylor & Wilson, 1957 ▸). Hence, there is just one significant intermolecular C—H⋯X interaction in each of (II), (III) and (V), involving atoms C13, C16 and O24, respectively, as the donors, and two each in (I) and (IV), involving as the donors C8 and C12 in (I), and C7 and C8 in (IV).
Table 2. Hydrogen bonds and short intermolecular contacts (Å, °) for compounds (I)–(V).
Cg1 and Cg2 represent the centroids of the C5A/C6–C9/C9A and C1–C6 rings, respectively.
| Compound | D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|---|
| (I) | C8—H8⋯N3i | 0.95 | 2.52 | 3.4641 (16) | 173 |
| C12—H12⋯O5ii | 0.95 | 2.50 | 3.4136 (16) | 161 | |
| (II) | C7—H7⋯N3iii | 0.95 | 2.55 | 3.2770 (15) | 133 |
| C13—H13⋯O5iv | 0.95 | 2.54 | 3.4083 (16) | 151 | |
| C15—H15⋯O5v | 0.95 | 2.48 | 3.2482 (16) | 138 | |
| (III) | C8—H8⋯N3vi | 0.95 | 2.58 | 3.2465 (19) | 127 |
| C16—H16⋯N2vii | 0.95 | 2.56 | 3.500 (2) | 169 | |
| (IV) | C6—H6⋯O5viii | 0.95 | 2.53 | 3.287 (3) | 137 |
| C8—H8⋯O5ix | 0.95 | 2.57 | 3.578 (2) | 161 | |
| C15—H15⋯N3x | 0.95 | 2.52 | 3.271 (2) | 136 | |
| C7—H7⋯Cg1xi | 0.95 | 2.69 | 3.517 (2) | 146 | |
| (V) | O24—H24⋯N23xii | 0.90 (2) | 1.77 (2) | 2.6721 (15) | 177.5 (19) |
| C8—H8A⋯Cg2xiii | 0.98 | 2.90 | 3.6605 (16) | 135 | |
| C8—H8B⋯Cg2viii | 0.98 | 2.90 | 3.5007 (16) | 120 | |
| C8—H8C⋯O24viii | 0.98 | 2.59 | 3.4004 (19) | 140 | |
| C215—H215⋯O24xiv | 0.95 | 2.57 | 3.2972 (19) | 134 |
Symmetry codes: (i) x +  , −y + , z + ; (ii) −x + , y + , −z + ; (iii) x − 1, y, z − 1; (iv) −x + , y − , −z +
, −y + , z + ; (ii) −x + , y + , −z + ; (iii) x − 1, y, z − 1; (iv) −x + , y − , −z +  ; (v) −x + , y − , −z + ; (vi) x, y, z + 1; (vii) x, −y + , z + ; (viii) −x, −y + 1, −z + 1; (ix) x, −y + , z − ; (x) x, −y +
; (v) −x + , y − , −z + ; (vi) x, y, z + 1; (vii) x, −y + , z + ; (viii) −x, −y + 1, −z + 1; (ix) x, −y + , z − ; (x) x, −y +  , z − ; (xi) −x, y − , −z + ; (xii) −x + 1, −y + 1, −z + 1; (xiii) −x, y − , −z + ; (xiv) x, −y + , z + .
, z − ; (xi) −x, y − , −z + ; (xii) −x + 1, −y + 1, −z + 1; (xiii) −x, y − , −z + ; (xiv) x, −y + , z + .
The supramolecular assembly in compound (I) is mediated by two hydrogen bonds, one each of the C—H⋯N and C—H⋯O types (Table 2 ▸). Molecules which are related by an n-glide plane are linked by the C—H⋯N hydrogen bond to form a C(7) (Etter, 1990 ▸; Etter et al., 1990 ▸; Bernstein et al., 1995 ▸) chain running parallel to the [101] direction, while molecules which are related by a 21 screw axis are linked by a C—H⋯O hydrogen bond to form a C(9) chain running parallel to the [010] direction. The combination of these two chain motifs generates a sheet lying parallel to (10) and built of  (28) rings (Fig. 6 ▸).
(28) rings (Fig. 6 ▸).
Figure 6.

Part of the crystal structure of compound (I), showing the formation of a hydrogen-bonded sheet of (28) rings parallel to (10). Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms not involved in the motifs shown have been omitted.
For compound (II), a single C—H⋯O hydrogen bond links molecules which are related by a 21 screw axis to form a C(10) chain running parallel to the [010] direction (Fig. 7 ▸), and chains of this type are linked by two π–π stacking interactions, both involving the fused carbocyclic ring, which together generate a π-stacked chain running parallel to [100] (Fig. 8 ▸). The combination of these two motifs generates a sheet lying parallel to (001). There is again just one hydrogen bond in the structure of compound (III), this time of the C—H⋯N type, linking molecules which are related by a c-glide plane to form a C(5) chain running parallel to the [001] direction (Fig. 9 ▸), but here there are no direction-specific interactions between adjacent chains.
Figure 7.

Part of the crystal structure of compound (II), showing the formation of a hydrogen-bonded C(10) chain parallel to [010]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms not involved in the motifs shown have been omitted.
Figure 8.

Part of the crystal structure of compound (II), showing the formation of a π-stacked chain parallel to [100]. For the sake of clarity, H atoms have all been omitted. O atoms marked with an asterisk (*), a hash (#), a dollar sign ($) or an ampersand (&) are at the symmetry positions (−x + 1, −y + 1, −z + 1), (−x, −y + 1, −z + 1), (x + 1, y, z) and (x − 1, y, z), respectively.
Figure 9.

Part of the crystal structure of compound (III), showing the formation of a hydrogen-bonded C(5) chain parallel to [001]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms not involved in the motif shown have been omitted.
The assembly in compound (IV) is built from a combination of C—H⋯O and C—H⋯π(arene) hydrogen bonds (Table 2 ▸). The C—H⋯O hydrogen bond links molecules which are related by a c-glide plane to form a C(7) chain running parallel to the [001] direction (Fig. 10 ▸). By contrast, molecules which are related by a 21 screw axis are linked by the C—H⋯π(arene) hydrogen bond to form a chain running parallel to the [010] direction (Fig. 11 ▸), and the combination of these two chain motifs generates a sheet lying parallel to (100).
Figure 10.

Part of the crystal structure of compound (IV), showing the formation of a hydrogen-bonded C(7) chain parallel to [001]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms not involved in the motif shown have been omitted.
Figure 11.

Part of the crystal structure of compound (IV), showing the formation of a hydrogen-bonded chain parallel to [010]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms not involved in the motif shown have been omitted.
Paired O—H⋯O hydrogen bonds link inversion-related pairs of molecules of (V) to form a cyclic centrosymmetric 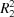 (8) dimer (Fig. 12 ▸), but there are no direction-specific interactions between adjacent dimer units.
(8) dimer (Fig. 12 ▸), but there are no direction-specific interactions between adjacent dimer units.
Figure 12.

Part of the crystal structure of compound (V), showing the formation of a centrosymmetric (8) dimer. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms bonded to C atoms have all been omitted. Atoms marked with an asterisk (*) are at the symmetry position (−x + 1, −y + 1, −z + 1).
Thus, minor variations in the substituent on the pendent aryl ring in compounds (I)–(IV) are associated with significant changes in the pattern of supramolecular assembly. Whereas for the unsubstituted parent compound (I), the molecules are linked into hydrogen-bonded sheets by a combination of C—H⋯N and C—H⋯O hydrogen bonds, the sheet formation in 4-chloro derivative (IV) is based on a combination of C—H⋯O and C—H⋯π(arene) hydrogen bonds. In each of the methyl compound (II) and the 3-chloro compound (III), a single hydrogen bond, of the C—H⋯O and C—H⋯N types, respectively, links the molecules into simple chains; these chains form π-stacked sheets in (II), but not in (III).
In summary, therefore, we have developed a simple and efficient route to new 1-arylisochromeno[3,4-d][1,2,3]triazol-5(1H)-ones, with full spectroscopic and structural characterization of four examples, which show that small changes in substituents are associated with substantial changes in the patterns of supramolecular aggregation, and we have demonstrated the necessity of using only a weak acid in the synthesis, along with the spectroscopic and structural characterization of a ring-opened derivative.
Supplementary Material
Crystal structure: contains datablock(s) global, I, II, III, IV, V. DOI: 10.1107/S2053229620003757/sk3747sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2053229620003757/sk3747Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2053229620003757/sk3747IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2053229620003757/sk3747IIIsup4.hkl
Structure factors: contains datablock(s) IV. DOI: 10.1107/S2053229620003757/sk3747IVsup5.hkl
Structure factors: contains datablock(s) V. DOI: 10.1107/S2053229620003757/sk3747Vsup6.hkl
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747Isup7.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IIsup8.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IIIsup9.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IVsup10.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747Vsup11.cml
Acknowledgments
The authors thank ‘Centro de Instrumentación Científico-Técnica of Universidad de Jaén’ for data collection. The authors thank Universidad de Ciencias Aplicadas y Ambientales (UDCA), Universidad Nacional de Colombia, the Consejería de Innovación, Ciencia y Empresa (Junta de Andalucía, Spain) and the Universidad de Jaén for financial support.
References
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Bruker (2016). SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2018). APEX3 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Etter, M. C. (1990). Acc. Chem. Res. 23, 120–126.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Naylor, R. E. & Wilson, E. B. (1957). J. Chem. Phys. 26, 1057–1060.
- Opatz, T. & Ferenc, D. (2005). Eur. J. Org. Chem. 2005, 817–821.
- Oweida, S. W., Ku, D. N., Lumsden, A. B., Kam, C. M. & Powers, J. C. (1990). Thromb. Res. 58, 191–197. [DOI] [PubMed]
- Qadeer, G., Rama, N. H., Fan, Z. J., Liu, B. & Liu, X. F. (2007). J. Braz. Chem. Soc. 18, 1176–1182.
- Riddell, F. G. & Rogerson, M. (1996). J. Chem. Soc. Perkin Trans. 2, pp. 493–504.
- Riddell, F. G. & Rogerson, M. (1997). J. Chem. Soc. Perkin Trans. 2, pp. 249–256.
- Rodríguez, R., Vicentes, D. E., Cobo, J. & Nogueras, M. (2017). Tetrahedron Lett. 58, 1487–1489.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spek, A. L. (2020). Acta Cryst. E76, 1–11. [DOI] [PMC free article] [PubMed]
- Tannenbaum, E., Myers, R. J. & Gwinn, W. D. (1956). J. Chem. Phys. 25, 42–47.
- Vicentes, D. E., Rodríguez, R., Cobo, J. & Glidewell, C. (2013). Acta Cryst. C69, 770–773. [DOI] [PubMed]
- Wood, P. A., Allen, F. H. & Pidcock, E. (2009). CrystEngComm, 11, 1563–1571.
- Xu, Z., Zhao, S. J. & Liu, Y. (2019). Eur. J. Med. Chem. 183, 111700. [DOI] [PubMed]
- Zhang, Z., Huo, J. Q., Dong, H. J., Shi, J. M. & Zhang, J. L. (2016). Asian J. Chem. 28, 666–668.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I, II, III, IV, V. DOI: 10.1107/S2053229620003757/sk3747sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2053229620003757/sk3747Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2053229620003757/sk3747IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2053229620003757/sk3747IIIsup4.hkl
Structure factors: contains datablock(s) IV. DOI: 10.1107/S2053229620003757/sk3747IVsup5.hkl
Structure factors: contains datablock(s) V. DOI: 10.1107/S2053229620003757/sk3747Vsup6.hkl
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747Isup7.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IIsup8.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IIIsup9.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747IVsup10.cml
Supporting information file. DOI: 10.1107/S2053229620003757/sk3747Vsup11.cml