

The dimethyl sulfoxide solvate of 2,3,5,6-tetrafluoro-1,4-diiodobenzene is polymorphic; one polymorph undergoes a phase transformation on cooling, associated with re-orientation of the dimethyl sulfoxide molecules.
Keywords: halogen bonding, polymorphism, variable temperature, crystal structure, phase transformation, PIXEL
Abstract
A new polymorph (form II) is reported for the 1:1 dimethyl sulfoxide solvate of 2,3,5,6-tetrafluoro-1,4-diiodobenzene (TFDIB·DMSO or C6F4I2·C2H6SO). The structure is similar to that of a previously reported polymorph (form I) [Britton (2003 ▸). Acta Cryst. E59, o1332–o1333], containing layers of TFDIB molecules with DMSO molecules between, accepting I⋯O halogen bonds from two TFDIB molecules. Re-examination of form I over the temperature range 300–120 K shows that it undergoes a phase transformation around 220 K, where the DMSO molecules undergo re-orientation and become ordered. The unit cell expands by ca 0.5 Å along the c axis and contracts by ca 1.0 Å along the a axis, and the space-group symmetry is reduced from Pnma to P212121. Refinement of form I against data collected at 220 K captures the (average) structure of the crystal prior to the phase transformation, with the DMSO molecules showing four distinct disorder components, corresponding to an overlay of the 297 and 120 K structures. Assessment of the intermolecular interaction energies using the PIXEL method indicates that the various orientations of the DMSO molecules have very similar total interaction energies with the molecules of the TFDIB framework. The phase transformation is driven by interactions between DMSO molecules, whereby re-orientation at lower temperature yields significantly closer and more stabilizing interactions between neighbouring DMSO molecules, which lock in an ordered arrangement along the shortened a axis.
Introduction
The molecule 2,3,5,6-tetrafluoro-1,4-diiodobenzene (TFDIB) is a common halogen-bond donor (Metrangolo & Resnati, 2001 ▸; Cavallo et al., 2016 ▸). The work described in this article originated from a cocrystal screening, where TFDIB was combined with a series of potential halogen-bond acceptors in dimethyl sulfoxide (DMSO) solution. Crystals of TFDIB·DMSO (see Scheme) were quite commonly obtained from these experiments, some of which were found to be different from a previously reported crystal structure at 297 K [Britton, 2003 ▸; Cambridge Structural Database (CSD; Groom et al., 2016 ▸) refcode IKIFOX]. We refer to the previously reported structure (IKIFOX) as form I and the newly obtained polymorph as form II. The structures are similar and we suspected at first that form II might have arisen from a phase transformation on cooling of form I in the N2 cryostream during single-crystal data collection. We therefore obtained crystals of form I and measured them at various temperatures. We did not find any transformation of form I to form II, but instead observed re-orientation of the DMSO molecules in form I to give a further new structure measured at 120 K. We describe herein the various crystal structures of TFDIB·DMSO and the application of dispersion-corrected DFT and PIXEL calculations (Gavezzotti, 2002 ▸, 2003 ▸, 2011 ▸) to examine the DMSO re-orientation on cooling of form I.
Experimental
Synthesis and crystallization
Crystals of forms I and II were produced during a sequence of attempted cocrystallization experiments. TFDIB and an anticipated coformer were dissolved in DMSO, and crystals were produced by vapour diffusion of water into the solution under ambient conditions. Form I (CSD refcode IKIFOX; Britton, 2003 ▸) was obtained frequently, while crystals of form II were obtained specifically from a 2:3 mixture of TFDIB and melamine (C3H6N6). The structure of form II was measured at 180 K, whereby the crystals were plunged directly from ambient conditions into a cold N2 stream. Similar treatment of form I resulted in cracking and the loss of single crystallinity. Analysis of form I was therefore made by placing the crystal initially into the N2 stream at room temperature, followed by slow cooling as described in §3.2.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1 ▸. Determination of the structure of form II at 180 K was straightforward. The DMSO molecule in the asymmetric unit is situated with its internal mirror plane on the crystallographic mirror plane at x, ,z in the space group Pnma (Fig. 1 ▸). The S atom is split into two atomic sites within the mirror plane, with refined site occupancies of 0.424 (5) and 0.576 (5).
,z in the space group Pnma (Fig. 1 ▸). The S atom is split into two atomic sites within the mirror plane, with refined site occupancies of 0.424 (5) and 0.576 (5).
Table 1. Experimental details.
For all structures: C6F4I2·C2H6OS, M r = 479.99, Z = 4. Experiments were carried out with Mo Kα radiation using a Nonius KappaCCD diffractometer. Absorption was corrected for by multi-scan methods (SORTAV; Blessing, 1995 ▸). H-atom parameters were constrained.
| Form II | Form I (120 K) | Form I (220 K) | |
|---|---|---|---|
| Crystal data | |||
| Crystal system, space group | Orthorhombic, P n m a | Orthorhombic, P212121 | Orthorhombic, P n m a |
| Temperature (K) | 180 | 120 | 220 |
| a, b, c (Å) | 12.8308 (6), 21.3307 (12), 4.6463 (2) | 10.6731 (2), 18.0023 (5), 6.5470 (2) | 11.6799 (4), 18.2664 (8), 6.0984 (2) |
| V (Å3) | 1271.65 (11) | 1257.94 (5) | 1301.09 (8) |
| μ (mm−1) | 5.14 | 5.19 | 5.02 |
| Crystal size (mm) | 0.14 × 0.14 × 0.14 | 0.12 × 0.10 × 0.10 | 0.12 × 0.10 × 0.10 |
| Data collection | |||
| T min, T max | 0.411, 0.464 | 0.475, 0.599 | 0.541, 0.611 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 7999, 1463, 952 | 11512, 2834, 2141 | 8062, 1520, 948 |
| R int | 0.049 | 0.089 | 0.057 |
| (sin θ/λ)max (Å−1) | 0.649 | 0.649 | 0.650 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.033, 0.086, 1.01 | 0.043, 0.078, 0.99 | 0.038, 0.082, 1.06 |
| No. of reflections | 1463 | 2834 | 1520 |
| No. of parameters | 83 | 148 | 112 |
| No. of restraints | 0 | 0 | 71 |
| Δρmax, Δρmin (e Å−3) | 1.01, −1.27 | 0.91, −1.18 | 0.62, −0.69 |
| Absolute structure | – | Refined as an inversion twin. | – |
| Absolute structure parameter | – | 0.19 (5) | – |
Figure 1.

The molecular structure of form II at 180 K, with displacement ellipsoids at the 50% probability level for non-H atoms. The site-occupancy factors for atoms S1 and S1A are 0.424 (5) and 0.576 (5), respectively. Only the major component is shown as connected. [Symmetry codes: (i) −x + 1, −y + 1, −z; (ii) x, −y +  , z.]
, z.]
For form I at 120 K, the space group is clearly P212121, with the DMSO molecule ordered on a general equivalent position and with no significant residual electron density in the vicinity of the molecule (Fig. 2 ▸). The structure was refined as an inversion twin with the Flack parameter converging to 0.19 (5). The applied unit-cell setting and origin (placing the 21 screw axes at x,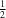 ,; 0,y,0; ,0,z) are nonstandard for the space group P212121 [Hall symbol: P 2ac 2n], but chosen to maintain the relationship with the form I structure at 297 K (IKIFOX) in its standard setting of Pnma.
,; 0,y,0; ,0,z) are nonstandard for the space group P212121 [Hall symbol: P 2ac 2n], but chosen to maintain the relationship with the form I structure at 297 K (IKIFOX) in its standard setting of Pnma.
Figure 2.

The molecular structure of form I at 120 K, with displacement ellipsoids at the 50% probability level for non-H atoms.
For the refinement of form I after cooling to 220 K, the DMSO molecule was modelled in four orientations (Fig. 3 ▸). Two orientations are similar to those in form II, with the internal mirror plane of the DMSO molecule coincident with the mirror plane at x,,z in the space group Pnma, and with the S atom split into two atomic sites with refined site occupancies of 0.356 (3) and 0.191 (3). A further orientation is defined with the S atom out of the mirror plane with a refined site occupancy of 0.226 (2), giving two further orientations of the DMSO molecule. The site occupancies of the three refined components were tightly restrained to sum to unity (using SUMP in SHELXL; Sheldrick, 2015b
▸) and restraints were applied to all S—O, S—C and C⋯C distances. All non-H atoms were refined with anisotropic displacement parameters, restrained to resemble isotropic behaviour (ISOR in SHELXL). An extinction coefficient was refined. In all structures, the H atoms of the DMSO molecule were placed in idealized positions, with U
iso(H) = 1.5U
eq(C). The methyl groups were not permitted to rotate around their local threefold axes, since this prevented convergence of the refinement. The structure and refinement details are presented in Table 1 ▸.
Figure 3.

The molecular structure of form I at 220 K, with displacement ellipsoids at the 50% probability level. H atoms have been omitted from the disordered DMSO molecule for clarity, and only the major component is shown as connected. The site-occupancy factors for the DMSO components containing atom S1, S1A and S1B are 0.356 (3), 0.191 (3) and 0.226 (2), respectively. [Symmetry codes: (i) −x + 1, −y + 1, −z − 1; (ii) x, −y + , z.]
Computational details
The crystal structures were energy-minimized with dispersion-corrected density functional theory (DFT-D) using the CASTEP module (Clark et al., 2005 ▸) in Materials Studio (Accelrys, 2011 ▸). The PBE functional (Perdew et al., 1996 ▸) was applied with a plane-wave cut-off energy of 520 eV, in combination with the Grimme semi-empirical dispersion correction (Grimme, 2006 ▸). The structures in the space group Pnma were reduced to the space group P212121 to allow the definition of complete molecules, so all optimizations were carried out in P212121. The unit-cell parameters were constrained to the experimental values. For the disordered structures, models were built containing the various individual DMSO components and optimized separately. The DFT-D-optimized structures were used as input for the PIXEL module of the CSP package (Gavezzotti, 2002 ▸, 2003 ▸, 2011 ▸) to examine the energies of the pairwise intermolecular interactions. The calculated interaction energies are estimated to have accuracy within the range ca ±3 kJ mol−1.
Results and discussion
Structure of form II
Both form I and form II adopt structures with a layered arrangement of TFDIB molecules in the (020) planes (Fig. 4 ▸). The DMSO molecules occupy sites between these layers. The difference between forms I and II reveals some flexibility in the structure of the TFDIB layers within the crystalline state. Taking one layer and looking side-on to the molecules [projecting onto the (110) planes], form II shows an approximately perpendicular arrangement of molecules, while form I shows a smaller angle between the molecular planes (Fig. 5 ▸). This difference is reflected in the unit-cell parameters (Table 1 ▸), particularly in the substantially shorter c axis for form II. The sites occupied by the DMSO molecules between the TFDIB layers in form II are substantially similar to those in form I, as described in §3.2. The DMSO molecules lie on the crystallographic mirror planes at x,,z and x, ,z, accepting I⋯O halogen bonds from two TFDIB molecules either side of the mirror plane [I1⋯O1 = 2.847 (2) Å]. Disorder is present in the manner described for form I at 297 K (§3.2), with the DMSO molecules adopting orientations A and B with refined site occupancies of 0.576 (5) and 0.424 (5), respectively.
,z, accepting I⋯O halogen bonds from two TFDIB molecules either side of the mirror plane [I1⋯O1 = 2.847 (2) Å]. Disorder is present in the manner described for form I at 297 K (§3.2), with the DMSO molecules adopting orientations A and B with refined site occupancies of 0.576 (5) and 0.424 (5), respectively.
Figure 4.

The form I and II structures, viewed along the c axis, showing layers of TFDIB molecules in the (020) planes. The arrangement of TFDIB molecules in form I is closely comparable at 297 and 120 K (the 120 K structure is shown).
Figure 5.

A single layer of TFDIB molecules, looking side-on to the molecules [projecting approximately onto the (110) planes]. Form I shows a smaller angle between the molecular planes and has a longer c axis. The arrangement of TFDIB molecules in form I is closely comparable at both 297 and 120 K (the 120 K structure is shown).
Temperature-dependent structure of form I
The previously-reported structure of form I at 297 K (Britton, 2003 ▸; CSD refcode IKIFOX) exhibits two orientations of the DMSO molecules (labelled A and B), as illustrated in Fig. 6 ▸. We obtained an identical disorder model in our own refinements at 300 K (not reported). Each DMSO molecule lies on a mirror plane in a pocket between eight TFDIB molecules. The position of the O atom is approximately consistent in both orientations, acting as an acceptor for I⋯O halogen bonds from two TFDIB molecules (I⋯O ≃ 2.80–2.90 Å). In orientation A, the S—CH3 bond vectors point approximately perpendicular to the planes of two TFDIB molecules. In orientation B, the S—CH3 bonds lie closer to parallel to the TFDIB planes (Fig. 6 ▸). Thus, the DMSO molecules are ‘anchored’ by the I⋯O halogen bonds, but the S—CH3 bond vectors can point either perpendicular or parallel to the neighbouring TFDIB rings. In the structure reported by Britton (2003 ▸), the refined site occupancies for A and B were 0.620 (17) and 0.380 (17), respectively.
Figure 6.

Two orientations (A and B) of the DMSO molecule in the form I structure at 297 K (Britton, 2003 ▸). The molecules occupy a site between eight TFDIB molecules. The arrows indicate the directions of the S—CH3 bond vectors, i.e. perpendicular (A) or parallel (B) to the planes of the two TFDIB molecules at the top in the front plane.
On cooling of form I to 220 K, the unit cell and positions of the TFDIB molecules remain comparable to those at 297 K and both DMSO orientations A and B remain present. However, new peaks arise in the electron density corresponding to further orientations of the DMSO molecules. At first it was difficult to unravel this disorder, but the situation became clear after the structure was determined at 120 K. The disorder at 220 K corresponds to four DMSO orientations, comprising A, B and two new (symmetry-related) orientations described below for the 120 K structure. A significant feature of the form I structure at 220 K is that its unit-cell parameters and TFDIB positions remain comparable to those at 297 K (Britton, 2003 ▸). Thus, cooling of the structure from 297 to 220 K causes some re-orientation of the DMSO molecules, but the crystal does not yet appear to have undergone any phase transformation.
After cooling the crystal slowly (ca 1 K min−1) to 120 K, the structure changes clearly from the 297 and 220 K structures. The unit cell expands by ca 0.5 Å along the c axis and contracts by ca 1.0 Å along the a axis. Looking side-on to the TFDIB molecules in one layer shows only a very subtle change compared to the 297 K structure (Fig. 7 ▸). However, the DMSO molecules are ordered and the space-group symmetry is reduced to P212121. The DMSO orientation (labelled C, Fig. 8 ▸) retains essentially the same O-atom position, anchored by the I⋯O halogen bonds [I1⋯O1i = 2.874 (7) Å and I4⋯O1ii = 2.871 (7) Å; symmetry codes: (i) x, y, z − 1; (ii) −x, y −  , −z + 1]. Compared to orientations A and B (Fig. 6 ▸), the molecule rotates approximately around its S—O bond. One of the S—CH3 bond vectors retains a position comparable to orientation A, with a ‘perpendicular’ approach to the face of the neighbouring TFDIB molecule. The other adopts a new position pointing approximately along the c axis, between TFDIB molecules. Although the symmetry of the structure is clearly reduced to P212121, the TFDIB molecules retain the effective mirror symmetry of the Pnma structure, so that two locally equivalent DMSO orientations can be envisaged, with the S—CH3 bond pointing towards the face of either TFDIB molecule related by the local mirror symmetry (Fig. 8 ▸). Neighbouring DMSO molecules along the a axis alternate in this respect (visible in Fig. 4 ▸), and the two additional components seen in the 220 K structure correspond to an overlay of these two orientations. It appears from the partial observation of orientation C at 220 K that some degree of DMSO reorientation can be tolerated within the ‘high-temperature’ TFDIB framework in form I, but the DMSO re-orientation ultimately drives the phase transformation to the ordered ‘low-temperature’ structure.
, −z + 1]. Compared to orientations A and B (Fig. 6 ▸), the molecule rotates approximately around its S—O bond. One of the S—CH3 bond vectors retains a position comparable to orientation A, with a ‘perpendicular’ approach to the face of the neighbouring TFDIB molecule. The other adopts a new position pointing approximately along the c axis, between TFDIB molecules. Although the symmetry of the structure is clearly reduced to P212121, the TFDIB molecules retain the effective mirror symmetry of the Pnma structure, so that two locally equivalent DMSO orientations can be envisaged, with the S—CH3 bond pointing towards the face of either TFDIB molecule related by the local mirror symmetry (Fig. 8 ▸). Neighbouring DMSO molecules along the a axis alternate in this respect (visible in Fig. 4 ▸), and the two additional components seen in the 220 K structure correspond to an overlay of these two orientations. It appears from the partial observation of orientation C at 220 K that some degree of DMSO reorientation can be tolerated within the ‘high-temperature’ TFDIB framework in form I, but the DMSO re-orientation ultimately drives the phase transformation to the ordered ‘low-temperature’ structure.
Figure 7.

Overlay of a single layer of TFDIB molecules [projecting approximately onto the (110) planes] in the form I structure at 297 K (red; Britton, 2003 ▸) and 120 K (blue). The change in the unit-cell parameters is clear, but the positions of the TFDIB molecules change only very slightly.
Figure 8.

Orientation C for the DMSO molecule in the form I structure at 120 K. The arrows indicate the directions of the S—CH3 bond vectors: one is equivalent to orientation A (Fig. 6 ▸), while one is distinct, pointing between TFDIP molecules. Due to the local mirror symmetry, two locally equivalent orientations are possible for the DMSO molecule, pointing either to the left or to the right in the diagram; these orientations alternate for neighbouring molecules along the c axis (Fig. 4 ▸).
Additional temperature-dependent measurements were made to examine the unit-cell parameters in the region of the phase transformation. A crystal of form I was cooled from 300 to 200 K at a rate of ca 2 K min−1, with the unit cell determined at 10 K intervals. The unit-cell volume (Fig. 9 ▸ a) shows an approximately linear decrease over the range 300→230 K, but a clear change of gradient occurs between 230 and 220 K, suggesting that reorientation of the DMSO molecules begins to take place significantly around this temperature. Clear discontinuities are evident for both the a and the c axes (Fig. 9 ▸ b) between the measurements made at 220 K (resembling the 297 K structure) and 210 (resembling the 120 K structure), suggesting that the reorientation is largely complete by 210 K. Hence, the disordered structure at 220 K captures the (average) structure of the crystal mid-transformation.
Figure 9.

Variation in (a) the unit-cell volume (•) and (b) the a (□) and c (filled □) axis lengths over the temperature range 300→200 K for form I. Error bars (where visible) are drawn at ±(3 × s.u.). The clear discontinuities in the axis lengths correspond to the phase transformation from the ‘high-temperature’ TFDIB framework to the ‘low-temperature’ framework.
DFT-D and PIXEL calculations
To investigate the energetics of the associated intermolecular interactions, models were constructed containing the various DMSO components and optimized using dispersion-corrected DFT calculations (DFT-D; see Experimental, §2). The purpose of the DFT-D step is to produce a model with an optimized representation of the disordered solvent molecules, where the geometry from the X-ray refinement is likely to be less well defined. The pairwise interactions in the optimized structures were analysed using the PIXEL approach (Gavezzotti, 2002 ▸, 2003 ▸, 2011 ▸). The principal interest is the total interaction energy between the DMSO molecules and its neighbours for orientations A, B and C in the form I structure. In each structure, the pairwise interactions sorted by centroid–centroid distance show a clear set of eight DMSO–TFDIB interactions, as indicated in Figs. 6 ▸ and 8 ▸, with no other DMSO–TFDIB interactions having significant interaction energy. Similarly, each structure shows a clear set of six significant DMSO–DMSO interactions, which are directly comparable between the structures. Table 2 ▸ shows the sums of the total energies for these sets of interactions. It is evident that the total interaction energy between DMSO and the TFDIB framework changes little between orientations A, B and C. Orientation B appears slightly favoured over orientation A in the form I structure at 297 K.1 For orientation C, however, the calculations give a clear indication: orientation C is favoured on account of significantly more stabilizing interactions between the DMSO molecules. In particular, the interaction between DMSO molecules along the a axis (Fig. 4 ▸) has a centroid–centroid distance ca 0.5 Å shorter than any other DMSO–DMSO interaction and is particularly stabilizing (E tot = −17.3 kJ mol−1). It appears that this interaction drives the ordering of the DMSO molecules, resulting in the ca 1.0 Å contraction of the a axis and symmetry reduction to the space group P212121. The DMSO reorientation takes place within a TFDIB framework that is clearly flexible, as evidenced by the existence of the three closely-related framework structures reported herein, and with little consequence for the total energy of the DMSO–TFDIB interactions.
Table 2. Total intermolecular interaction energies (kJ mol−1) involving the DMSO molecules in form I, calculated using the PIXEL method, applied to the DFT-D-optimized structures.
| Structure | Disorder component | Refinement temperature (K) | a (Å) | b (Å) | c (Å) | E tot DMSO–TFDIB | E tot DMSO–DMSO |
|---|---|---|---|---|---|---|---|
| Form I | A | 297 | 11.819 | 18.418 | 6.075 | −107.4 | −25.0 |
| Form I | B | 297 | 11.819 | 18.418 | 6.075 | −112.0 | −27.4 |
| Form I | C | 120 | 10.673 | 18.002 | 6.547 | −109.8 | −44.6 |
Conclusion
The existence of TFDIB·DMSO form II and the variation of the form I structure as a function of temperature shows that the layered arrangement of TFDIB molecules can exhibit significant flexibility in the crystalline state. This flexibility accommodates several orientations for the DMSO molecules between the layers, with apparently little variation in the DMSO–TFDIB interaction energies. The DMSO molecules are consistently anchored by accepting I⋯O halogen bonds, but their orientation can vary relative to the TFDIB molecules and relative to each other. The PIXEL calculations suggest no clear preference for orientations A or B in form I, consistent with the observed disorder in the structure at 297 K, but they show clearly why orientation C is preferred in the structure at 120 K. Optimization of the interactions between neighbouring DMSO molecules locks in an ordered arrangement, which accounts for the observed changes in the unit-cell parameters and space group on cooling of form I below ca 220 K. The applied combination of temperature-dependent X-ray diffraction measurements and intermolecular energy calculations provides a clear picture of the temperature-dependent phase transformation in this case
Supplementary Material
Crystal structure: contains datablock(s) Form_II, Form_I_120K, Form_I_220K, global. DOI: 10.1107/S2053229620005690/sk3750sup1.cif
Structure factors: contains datablock(s) Form_II. DOI: 10.1107/S2053229620005690/sk3750Form_IIsup2.hkl
Structure factors: contains datablock(s) Form_I_120K. DOI: 10.1107/S2053229620005690/sk3750Form_I_120Ksup3.hkl
Structure factors: contains datablock(s) Form_I_220K. DOI: 10.1107/S2053229620005690/sk3750Form_I_220Ksup4.hkl
Supporting information file. DOI: 10.1107/S2053229620005690/sk3750Form_I_120Ksup5.cml
DFT-D-optimized structures. DOI: 10.1107/S2053229620005690/sk3750sup6.txt
Acknowledgments
The cocrystallization study that led to this work was carried out by CLT under the supervision of Professor Stuart Clarke (Department of Chemistry, University of Cambridge).
Footnotes
The fact that this is not immediately consistent with the site occupancies reported by Britton (2003 ▸) might be attributed to several factors: (i) the accuracy of the calculations; (ii) the calculations are static and do not consider entropic contributions that must become relevant at real temperatures; (iii) the DMSO orientations may be established during crystal growth, and not solely determined by their relative interaction energies.
References
- Accelrys (2011). Materials Studio. Accelrys Software Inc., San Diego, CA, USA.
- Blessing, R. H. (1995). Acta Cryst. A51, 33–38. [DOI] [PubMed]
- Britton, D. (2003). Acta Cryst. E59, o1332–o1333.
- Cavallo, G., Metrangolo, P., Milani, R., Pilati, T., Priimagi, A., Resnati, G. & Terraneo, G. (2016). Chem. Rev. 116, 2478–2601. [DOI] [PMC free article] [PubMed]
- Clark, S. J., Segall, M. D., Pickard, C. J., Hasnip, P. J., Probert, M. J., Refson, K. & Payne, M. C. (2005). Z. Kristallogr. 220, 567–570.
- Gavezzotti, A. (2002). J. Phys. Chem. B, 106, 4145–4154.
- Gavezzotti, A. (2003). J. Phys. Chem. B, 107, 2344–2353.
- Gavezzotti, A. (2011). New J. Chem. 35, 1360–1389.
- Grimme, S. (2006). J. Comput. Chem. 27, 1787–1799. [DOI] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Macrae, C. F., Sovago, I., Cottrell, S. J., Galek, P. T. A., McCabe, P., Pidcock, E., Platings, M., Shields, G. P., Stevens, J. S., Towler, M. & Wood, P. A. (2020). J. Appl. Cryst. 53, 226–235. [DOI] [PMC free article] [PubMed]
- Metrangolo, P. & Resnati, G. (2001). Chem. Eur. J. 7, 2511–2519. [DOI] [PubMed]
- Nonius (1998). COLLECT. Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Perdew, J. P., Burke, K. & Ernzerhof, M. (1996). Phys. Rev. Lett. 77, 3865–3868. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. C71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. A71, 3–8.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) Form_II, Form_I_120K, Form_I_220K, global. DOI: 10.1107/S2053229620005690/sk3750sup1.cif
Structure factors: contains datablock(s) Form_II. DOI: 10.1107/S2053229620005690/sk3750Form_IIsup2.hkl
Structure factors: contains datablock(s) Form_I_120K. DOI: 10.1107/S2053229620005690/sk3750Form_I_120Ksup3.hkl
Structure factors: contains datablock(s) Form_I_220K. DOI: 10.1107/S2053229620005690/sk3750Form_I_220Ksup4.hkl
Supporting information file. DOI: 10.1107/S2053229620005690/sk3750Form_I_120Ksup5.cml
DFT-D-optimized structures. DOI: 10.1107/S2053229620005690/sk3750sup6.txt