

Abstract
Extracellular vesicles (EVs), such as exosomes, can mediate long-distance communication between cells by delivering biomolecular cargo. It is speculated that EVs undergo back-fusion at multivesicular bodies (MVBs) in recipient cells to release their functional cargo. However, direct evidence is lacking. Tracing the cellular uptake of EVs with high resolution as well as acquiring direct evidence for the release of EV cargo is challenging mainly because of technical limitations. Here, we developed an analytical methodology, combining state-of-the-art molecular tools and correlative light and electron microscopy, to identify the intracellular site for EV cargo release. GFP was loaded inside EVs through the expression of GFP-CD63, a fusion of GFP to the cytosolic tail of CD63, in EV producer cells. In addition, we genetically engineered a cell line which expresses anti-GFP fluobody that specifically recognizes the EV cargo (GFP). Incubation of anti-GFP fluobody-expressing cells with GFP-CD63 EVs resulted in the formation of fluobody punctae, designating cytosolic exposure of GFP. Endosomal damage was not observed in EV acceptor cells. Ultrastructural analysis of the underlying structures at GFP/fluobody double-positive punctae demonstrated that EV cargo release occurs from endosomes/lysosomes. Finally, we show that neutralization of endosomal pH and cholesterol accumulation in endosomes leads to blockage of EV cargo exposure. In conclusion, we report that a fraction of internalized EVs fuse with the limiting membrane of endosomes/lysosomes in an acidification-dependent manner, which results in EV cargo exposure to the cell cytosol.
Keywords: extracellular vesicles, endosomes, nanobody, endosomal escape, cargo delivery, correlative microscopy
Secreted factors, including messenger molecules and extracellular vesicles, allow long-distance communication between mammalian cells. Extracellular vesicles (EVs), including exosomes, microvesicles, and apoptotic bodies, carry biomolecules (lipids, proteins, nucleic acids) and impart phenotypic changes in recipient cells. EVs have been reported to play a role in a wide variety of processes within the human body, including immune response, neurodegenerative disease pathogenesis, viral dissemination, and tumor formation and metastasis.1−8 Next to their role in cell–cell communication, EVs show promise as biological drug delivery vehicles.9−11 Multiple types of EVs exist, which are first categorized on the basis of their biogenesis: (i) Microvesicles and (ii) apoptotic bodies are generated by plasma membrane outward budding, whereas (iii) exosomes are created by endosome membrane invagination, resulting in intraluminal vesicles within multivesicular bodies (MVBs). Subsequent fusion of MVBs with the plasma membrane results in exosome release from cells. EVs can enter cells in the local or distant environment,1,12−18via fusion and/or endocytosis.19−24
Different mechanisms for EV cargo release in recipient cells have been proposed, including (i) fusion with the plasma membrane,19,20 (ii) kiss and run fusion with the endoplasmic reticulum,21 (iii) fusion with the endosome membrane,22 and (iv) endosomal rupture (Figure 1).22,25,26 Although fusion of EVs with the plasma membrane of recipient cells has been proposed as a mechanism for content release,19,20 endocytosis is the major pathway of EV uptake.21−24 Escape of the EV content from the endosomal confinement is then a requirement for its functionality, as it needs to access cytoplasmic targets in the host cell, such as the RNA-induced silencing complex (RISC) machinery for miRNAs. Possible mechanisms for cargo release of EVs from endosomes include endosomal lysis, endosomal permeabilization, and membrane fusion between EV and endosomal membrane.27
Figure 1.
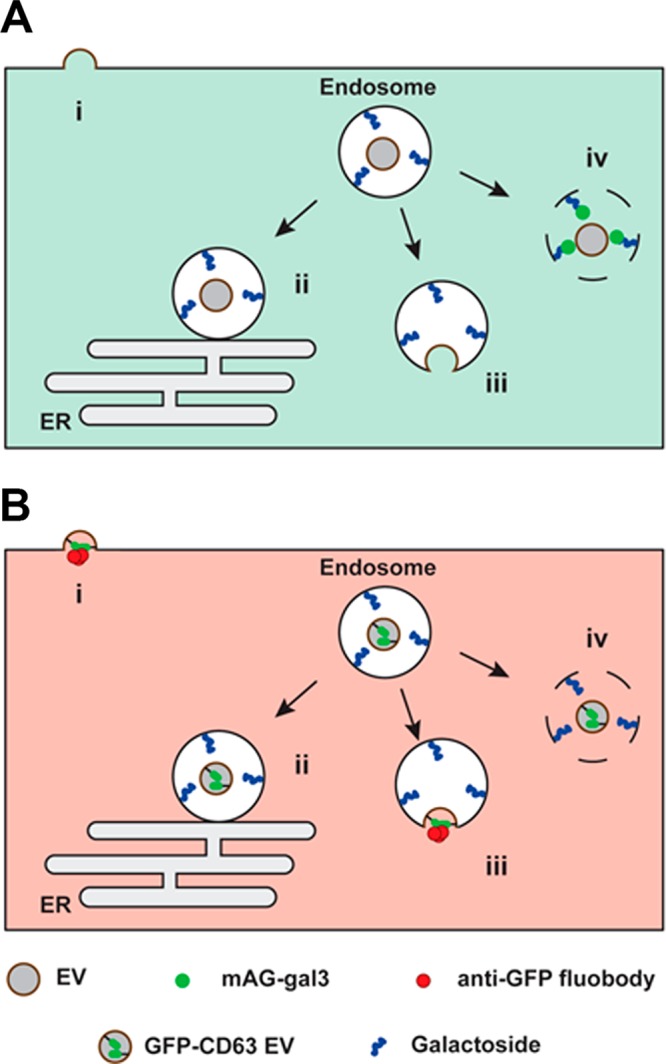
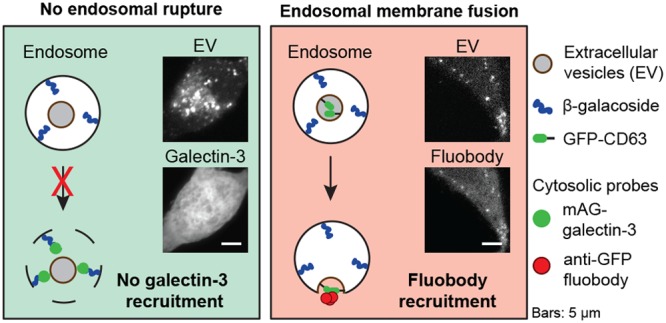
Experimental setup to elucidate the intracellular site of EV-cargo release. EVs interacting with recipient cells can release their cargo via: (i) direct fusion with the plasma membrane; (ii) kiss and run fusion with the endoplasmic reticulum; (iii) fusion with the endosome membrane; and (iv) endosomal rupture. (A) In cells engineered to cytosolically express monomeric azami-green galectin-3 fusion protein (mAG-gal3), mAG-gal3 punctae formation only occurs in case of endosomal rupture (iv). (B) In cells engineered to cytosolically express mCherry-tagged anti-GFP fluobody, mCherry punctae formation only occurs in case of fusion of GFP-CD63 EVs (i.e., membrane-bound GFP inside the EV) with the plasma membrane (i) or endosome membrane (iii).
The incidence of cytosolic delivery of EV cargo has been largely indirectly inferred from functional studies in which, for example, EV-mediated delivery of miRNA was shown to result in altered gene expression.15,28−32 However, direct evidence for EV content release into the cytosol of recipient cells is lacking. Here, we employed state-of-the-art molecular tools with correlative light and electron microscopy (CLEM)33 to detect endosomal lysis and identify the underlying ultrastructure of intracellular sites of EV cargo exposure to the cell cytosol, following EV uptake in HEK293T cells. To this end, fluorescent GFP-CD63 EVs were generated, carrying GFP at the interior EV membrane. Cytosolic expression of anti-GFP fluobody,34 an mCherry-tagged anti-GFP single-domain antibody (nanobody),35,36 in recipient cells was exploited to detect EV cargo exposure to the cytosol (Figure 1).
Results and Discussion
Extracellular Vesicles Are Internalized via Endocytosis
In order to study the processing of exogenously added EVs in mammalian cells by fluorescence light microscopy (LM), a stable GFP-CD63 HEK293T cell line was generated for the production of fluorescently labeled EVs. In GFP-CD63 HEK293T cells, GFP fluorescence showed cell surface staining and a punctate staining pattern consistent with the cytoplasmic distribution of endosomes (Figure S1A), which corresponds with the localization of endogenous CD63.37 EVs were isolated by differential centrifugation of the conditioned cell culture medium, with final ultracentrifugation at 100,000×g (small EVs). Following isolation, both wild-type (WT) and GFP-CD63 EVs showed cup-shaped vesicular morphology and a diameter of 100–150 nm, by electron microscopic investigation (Figure S1B). WT and GFP-CD63 EVs displayed a similar extent of enrichment of EV marker proteins and low levels of the Golgi protein golgin-97, an EV negative marker, in comparison to the respective parent producer cells (Figure S1C). Furthermore, size distribution analysis using dynamic light scattering confirmed the similar size of WT and GFP-CD63 EVs and also their surface charge (ζ-potential) was shown to be identical (Figure S1D–F). Hence, GFP-CD63 expression did not alter morphology nor size or surface charge of the EVs. Therefore, GFP-CD63 EVs were considered similar to WT EVs and were further used in the study.
Upon incubation of WT HEK293T cells with GFP-CD63 EVs, a punctate staining pattern was observed throughout the cytosol by LM, suggesting the involvement of endocytosis in EV uptake by cells (Figure 2A). Indeed, inhibition of endocytosis through the use of the dynamin inhibitor dynasore38 resulted in a decrease in EV uptake (Figure S2A). In addition, EV uptake was inhibited at a nonpermissive temperature (4 °C) for endocytosis (Figure S2B). Taking a CLEM approach allowed for the identification of the ultrastructure of the GFP-positive spots by EM (Figure 2B,C), revealing the presence of GFP-CD63 EVs in membranous compartments, that is, endosomes (Figure 2C and Figure S3). To confirm the presence of GFP-CD63 EVs within these endosomal structures, GFP was immunolabeled and detected with a secondary antibody conjugated to QD655. Indeed, the endosomes that were identified by EM (Figure 2C) and appeared positive for GFP by LM examination (Figure 2B) were also found positive for GFP after immunolabeling (Figure 2D). Taken together, the findings demonstrate that GFP-CD63 EVs are taken up by HEK293T cells via endocytosis. Of note, not all compartments that were positive for GFP in the CLEM image stained positive for GFP upon immunolabeling. This can be explained by the low efficiency of EM immunolabeling in general.39
Figure 2.

Exogenously added EVs localize in membrane-bound compartments in HEK293T acceptor cells. (A) HEK293T cells incubated for 12 h with GFP-CD63 EVs show a punctate staining pattern in the cytosol (scale bars, 10 μm). (B) Correlative light (green) and EM (greyscale) microscopy for ultrastructural analysis of the internalized EVs (GFP punctae) of the boxed area in (A) (scale bars, 5 μm). (C) Underlying ultrastructures of areas 1–3 in (B) reveal vesicular structures. Additional snap shots available in Supplementary Figure 3B. (Scale bars, 0.2 μm.) (D) Structures given in (C) are labeled with QDs (indicated by arrowhead) following anti-GFP immunolabeling, confirming the presence of GFP-CD63 EVs within the vesicular structures (scale bars, 0.2 μm). All EM data sets at full resolution are available viawww.nanotomy.org.
Extracellular Vesicles Do Not Induce Endosomal Permeabilization
Following endocytosis of EVs by HEK293T cells, a prerequisite for delivery of their cargo to the cell cytosol is their escape from endosomal confinement. To address whether EVs permeabilize the endosomal membrane to escape from endosomes, a galectin-3-based assay was used. For this purpose, HEK293T cells were transduced to express monomeric azami green-tagged galectin-3 (mAG-gal3) in their cytosol. mAG-gal3 identifies damaged endosomes by binding to β-galactosides that are present at the luminal leaflet of endosomes, which results in the formation of mAG-gal3 punctae (Figure 3A).40 To address whether mAG-gal3 punctae (green) are formed upon incubation of HEK293T cells with EVs, mAG-gal3 HEK293T cells were incubated with red fluorescent CD63-RFP EVs (Figure S4).
Figure 3.

EVs do not induce endosomal permeabilization. (A) Cartoon illustrating the galectin-3-based assay to detect endosomal permeabilization. HEK293T cells are engineered to cytosolically express monomeric azami-green galectin-3 fusion protein (mAG-gal3). Upon endosomal permeabilization, galactoside residues at the inner leaflet of the endosome are accessible to mAG-gal3, resulting in mAG-gal3 accumulation in the endosome and puncta formation. (B) Fluorescence images of mAG-gal3 expressing HEK293T cells untreated (control), treated with CD63-RFP EVs (EV) and transfected with Lipofectamine (Lipo) (t = 12 h). Red, EVs; green, mAG-gal3; blue, nucleus (scale bars, 10 μm). (C) Quantification of CD63-RFP EV uptake (red line) and mAG-gal3 punctae formation (green line) in cells over time (error bars represent SD, n = 3, ≥ 36 cells analyzed per time point). (D) Quantification of mAG-gal3 punctae upon 12 h treatment of mAG-Gal3 HEK293T cells with EVs and Lipofectamine-based lipoplexes. mAG-gal3 punctae only appear in cells with lipoplex treatment (n = 3; ≥ 45 cells analyzed per condition; **P < 0.01, ***P < 0.001, ns; not significant, ANOVA Tukey’s post hoc test).
A diffuse cytosolic green fluorescence without punctae was observed in mAG-gal3 expressing HEK293T cells in the presence of CD63-RFP EVs for 12 h, which was similar to in untreated cells (Figure 3B). As a positive control, cells were incubated with Lipofectamine-based lipoplexes, which are known to disrupt endosomes.40 Indeed, multiple mAG-gal3 punctae were formed in cells treated with lipoplexes (Figure 3B, quantified in D). To exclude the possibility that mAG-gal3 punctae were present at earlier time points but disappeared during the prolonged incubation time, mAG-gal3 cells were incubated with RFP-tagged EVs for 2, 4, 8, and 12 h (Figure S5), after which EV uptake and mAG-gal3 punctae were quantified. Significant uptake of EVs by mAG-gal3 HEK293T cells was seen after 2 h of incubation. A plateau in EV uptake was reached after 4 h incubation, showing 66 ± 9 RFP-positive spots per cell, indicating that EVs were efficiently internalized by mAG-gal3 cells (Figure 3C, red line). However, a very limited number of galectin punctae was observed at all of the investigated time points (Figure 3C, green line), which was similar to the number of punctae in untreated (control) cells (Figure 3D). Moreover, no colocalization was observed between CD63-RFP EVs and mAG-gal3 punctae at any of the investigated time points. Live cell imaging of mAG-gal3 HEK293T cells incubated with CD63-RFP EVs for 4 h confirmed the absence of mAG-gal3 punctae formation, indicating that such events did not remain undetected because of a transient nature of the mAG-gal3 punctae, nor that punctae were absent due to fixation-induced artifacts41 (Movie S1 and still images in Figure S7). A five-times increase in the EV concentration with the mAG-gal3 cells did not result in mAG-gal3 punctae formation (Figure S6). Collectively, the data show that internalized EVs do not permeabilize endosomes in HEK293T cells.
Extracellular Vesicles Release Their Cargo from Endosomes/Lysosomes
Next, we considered the involvement of nondisruptive EV-endosome membrane fusion in the unloading of EV cargo. To visualize EV cargo release into the cell cytosol, we used a fluorescent membrane-bound cargo, because a soluble cargo would rapidly dilute in the cytosol, resulting in a fast loss of signal, precluding further investigation by means of CLEM. Specifically, GFP-CD63 EVs with GFP present at the N-terminal end of CD63, that is, inside the EVs, were used (Figure S4A). First, the presence of GFP at the inside of EVs was verified. To this end, GFP-CD63 EVs were immuno-stained with anti-GFP antibody and secondary antibody conjugated to gold without prior permeabilization. WT EVs were used as a negative control, and EVs with GFP fused to the extracellular loop of CD63 (outGFP-CD63 EVs) were used as a positive control. OutGFP-CD63 EVs showed a positive signal in immunostaining, whereas WT and GFP-CD63 EVs showed no signal (Figure S4D). These data confirm that GFP when fused at the N-terminal end of CD63 is not accessible to the anti-GFP antibody and thus present at the inside of the EVs.
Then, HEK293T cells were engineered to express an anti-GFP nanobody fused to mCherry (anti-GFP fluobody) in the cytosol. Upon fusion of GFP-CD63 EVs with the endosomal membrane, GFP that is present inside the EVs would become exposed to the cytoplasm. Consequently, the cytosolic anti-GFP fluobody would identify such a fusion event (Figure 4A). Upon the incubation of fluobody-expressing HEK293T cells with GFP-CD63 EVs, mCherry punctae were formed, designating the cytosolic exposure of EV cargo (Figure 4B). Fluobody punctae were present in nearly all cells (94% ± 4.4%; Figure S8) after 12 h. In addition, maximum colocalization (yellow) between EVs (green) and fluobody punctae (red) was seen after 12 h of incubation, when 24% ± 1.2% of GFP spots colocalized with mCherry punctae (Figure 4B, C). Importantly, mCherry punctae always colocalized with GFP spots, validating the specificity of the anti-GFP fluobody. Moreover, incubation of fluobody-expressing cells with WT EVs revealed the absence of mCherry punctae, confirming the specificity of mCherry puncta formation toward GFP-CD63 EVs (Figure S9). Of note, the maximum EV/fluobody colocalization at t = 12 h does not mean that maximum cargo exposure occurs at this time point. It more likely reflects persistence over time of the structures from where release has taken place. Next, to identify the underlying ultrastructure at the sites of colocalization between GFP-CD63 EVs and fluobody punctae, CLEM was performed. To maximize the chance of detecting sites of EV-fluobody colocalization, mCherry-fluobody HEK293T cells that were incubated with GFP-CD63 EVs for 12 h were investigated. Ultrastructural analysis revealed the presence of late endosomes/MVBs and lysosomes at the intracellular sites of colocalization between EVs and fluobody punctae (Figure 4D,E). This implies that EV cargo is released from late endosomes and lysosomes and/or that EV cargo is released from (early) endosomes that subsequently undergo maturation. To confirm the identity of the endosomal structures as revealed by CLEM investigation, fluobody-expressing cells were incubated with GFP-CD63 EVs for 12 h and immunostained for LAMP1, a marker for both late endosomes/MVBs and lysosomes. The extent of colocalization between EV/fluobody spots and LAMP1 was 43 ± 13% (Figure 4F). Altogether, the data demonstrate that a fraction of internalized EVs undergoes fusion with endosomes and/or lysosomes, resulting in EV cargo exposure to the cell cytosol.
Figure 4.

EV cargo release occurs from endosomes. (A) Cartoon illustrating the experimental design to identify EV cargo exposure to the cell cytosol. When GFP-CD63 EVs in endosomes undergo fusion with the endosome membrane in anti-GFP fluobody (mCherry) HEK293T cells, the anti-GFP fluobody can access and recognize the GFP at the EV interior, resulting in formation of mCherry punctae. (B) Fluorescence images of anti-GFP fluobody (mCherry) HEK293T cells incubated with GFP-CD63 EVs for 4, 8, and 12 h. Yellow punctae represent colocalization. Green, EV; red, fluobody (scale bars, 10 μm). (C) EV uptake (green line) and colocalization with fluobody punctae (yellow line). Colocalization of GFP and mCherry indicate EV cargo exposure to the cell cytosol (error bars represent SD, n = 3, ≥ 36 cells analyzed per time point). (D) Correlative light (red + green) and EM (greyscale) microscopy of anti-GFP fluobody (mCherry) cells incubated with GFP-CD63 EVs for 12 h. Numbers 1–4 indicate areas of red and green (yellow) colocalization (scale bars, 2 μm). (E) The underlying ultrastructure of intracellular sites of EV cargo exposure to the cell cytosol (areas 1–4 in D). Membrane-bound structures containing numerous ILVs (1, 3, and 4) represent MVBs, while structures with electron-dense interior (2) represent lysosomes (scale bars, 0.2 μm). Complete data set at maximum resolution is available at www.nanotomy.org. (F) Immunostaining for the late endosome/lysosome marker LAMP1 in anti-GFP fluobody cells incubated for 12 h with GFP-CD63 EVs. Green, EV; red, fluobody; 633, antibody staining color-coded blue (scale bars, 10 μm).
Neutral pH and Elevated Cholesterol in Endosomes Block EV Content Release from Endosomes
Viral fusion with endosomes has been shown to be pH and cholesterol dependent.42 To address whether these factors also control EV content release from endosomes, two metabolic inhibitors, Bafilomycin A1 (BafA1) and U18666A, were used. BafA1 is a V-ATPase inhibitor that prevents the acidification of endosomes during endosomal maturation.43 In the presence of BafA1, fluobody punctae formation was almost completely abolished in anti-GFP fluobody (mCherry)-expressing cells that were treated with EVs (Figure 5A). The overlap between GFP-CD63 EVs and fluobody punctae decreased 9-to-10-fold as compared to untreated (control) cells (Figure 5B). Importantly, treatment with BafA1 did not affect cell viability and EV internalization (Figure S10A–B), while it effectively prevented endosomal acidification (Figure S10C). Treatment of cells with U18666A, an inhibitor of lysosomal cholesterol export, has been shown to trigger cholesterol accumulation in late endosomes and lysosomes.44,45 Incubation of anti-GFP fluobody HEK293T cells with EVs in the presence of U18666A resulted in a reduction in fluobody punctae compared to incubation in the absence of U18666A (Figure 5A), while the colocalization between fluobody punctae and GFP-CD63 EVs showed a 5-to-6-fold decrease (Figure 5B). Treatment of HEK293T cells with U18666A resulted in enlarged LAMP1-positive compartments and cholesterol accumulation in endosomes (Figure S10D–E, respectively). Collectively, the data indicate that EV-endosome membrane fusion is inhibited by neutralization of endosomal pH and cholesterol accumulation in endosomes, which prevents EV content release from endosomes into the cytosol.
Figure 5.

EV cargo release is blocked by treatment of acceptor cells with the V-ATPase inhibitor Bafilomycin A1 or U18666A, an inhibitor of cholesterol export from late endosomes/lysosomes. (A) Anti-GFP fluobody (mCherry) expressing cells were incubated for 12 h with GFP-CD63 EVs in the absence and presence of BafA1 or U18666A. Note that colocalization (yellow) between EVs (green) and fluobody punctae (red) is largely absent in BafA1 and U18666A treated cells. Green, EV; red, fluobody; blue, nucleus (scale bars, 10 μm). (B) Quantification of colocalization of GFP-CD63 EVs and fluobody punctae after 12 h incubation in the absence and presence of BafA1 or U18666A. Number of GFP/mCherry double-positive spots in control cells is set at 100% (error bars indicate SD, n = 3, ≥ 24 cells analyzed per condition; *P < 0.05, **P < 0.001, two-tailed Student’s t-test).
Conclusions
Extracellular vesicles (EVs), including exosomes, enable (long-distance) cell–cell communication. Their potential use for disease diagnosis and drug delivery is extensively investigated. However, the mechanism of EV-mediated cargo transfer between cells remains largely obscure. Different sites for EV cargo release in acceptor cells have been proposed, including (i) the plasma membrane,19 (ii) the endosome,22,25 and (iii) the endoplasmic reticulum.21 In this study, an analytical methodology was developed to visualize EV uptake and cargo release in acceptor cells, in order to identify the intracellular site for EV cargo release (Figure 1). Using correlative light and electron microscopy (CLEM), a combination of fluorescence microscopy and EM, we reveal the ultrastructural context of cellular structures containing fluorescently labeled EVs (Figure 2). EV localization in endosomes and lysosomes confirmed their uptake by endocytosis in line with hitherto reported studies.21−24
Escape from endosomal confinement is necessary for EVs to expose their cargo to the cytosol. Studying the fate of EV cargoes has been challenging owing to the low quantities of encapsulated cargo molecules.18,46 Despite the development of tools to enhance EV loading efficiency, direct proof for cargo release is lacking.23,24 Recently, EV cargo release from endosomes was examined by the use of GFP-carrying EVs labeled with a quenching concentration of R18.22 Dequenching of the R18 probe at the level of late endosomes/MVBs revealed membrane interaction between exosomes and late endosomes/MVBs, indicative for cargo release at late endosomes/MVBs. However, dequenching of R18 shows dilution of exosomal lipids and cannot distinguish endosomal rupture from membrane fusion. Moreover, R18 dilution does not demonstrate cargo exposure to the cytosol, and release of GFP content from endosomes into the cytosol was not detected in the study,22 possibly because release of soluble GFP cargo remains undetected because of its rapid dilution in the cytosol. In the present study, to increase the chance of detecting EV cargo exposure to the cytosol of acceptor cells, a membrane-bound EV cargo (i.e., GFP-CD63) was used. Formation of mCherry punctae in mCherry-tagged anti-GFP nanobody-expressing HEK293T cells upon their exposure to GFP-CD63 EVs revealed GFP exposure, that is, EV cargo delivery, to the cytosol (Figure 4B,C). Examination of the underlying ultrastructure at GFP/mCherry double-positive punctae revealed late endosomes/MVBs and lysosomes (Figure 4D,E). This finding was corroborated by a positive immunostaining for the late endosome/MVB and lysosome marker LAMP1 at the double-positive punctae (Figure 4F). Of note, mCherry punctae were never detected at the plasma membrane, suggesting that EV fusion with the plasma membrane (Figure 1B(i)) does not occur. However, taking into consideration the possibility of rapid diffusion of lipids and proteins at the plasma membrane, a failure to capture fluobody punctae at the plasma membrane cannot be ruled out. Therefore, with the presented data, we do not rule out the possibility of direct fusion of EVs with the plasma membrane.
Endosomal permeabilization as the mechanism of EV cargo release was excluded, because of the absence of mAG punctae in EV-treated HEK293T acceptor cells expressing cytosolic mAG-gal3 (Figure 3), specifying EV-endosome membrane fusion as a mechanism behind EV cargo delivery to acceptor cells. Of note, only a fraction of the EVs that were internalized by cells exposed their cargo to the cytoplasm, varying from 10% after 2 h to 24.5% after 12 h of incubation. This shows that functional delivery of EV cargo is limited. The design of EVs with a higher propensity to fuse with (endosomal) membranes will facilitate the advancement of improved EV-based therapeutics. Such approaches may include insertion of cell penetrating peptides (CPPs), (viral) fusogenic peptides/proteins, and cationic lipids and are currently under investigation.47−51 Moreover, surface functionalization of EVs to alter their tissue tropism will enhance cell specificity and prevent off-target effects. Combining the advantages of EVs, that is, a biological delivery system, and synthetic delivery systems provides opportunities to improve target specificity, safety, and efficiency in drug delivery.51−55
EV entry by endocytosis and subsequent cargo release via membrane fusion suggests that EVs exploit mechanisms akin to certain viruses.19,56 These viruses exploit a low pH-induced change in the tertiary structure of viral envelope proteins to induce fusion with acidified endosomal compartments.42 A reduction in EV cargo delivery to cytosol after inhibition of endosomal acidification by Bafilomycin A1 showed that EVs respond to low pH for undergoing membrane fusion with endosomal membranes (Figure 5). Further investigation is required to see if low pH can alter membrane characteristics of the internalized EVs, rendering them susceptible for membrane fusion. Importantly, at different stages of maturation, endosomes differ not only in their luminal pH but also in lipid and protein composition.57,58 Anionic lipids in late endosomes have been shown to act as cofactors for fusion of viruses with the endosomal membrane.59 Likewise, the endosomal escape of genetic cargo mediated by synthetic gene delivery vectors was shown to occur from maturing endosomes,60 initiating discussion on the involvement of a specific class of anionic lipids in mediating endosomal escape.61,62 U18666A treatment is well-known to trigger an accumulation of cholesterol and the anionic lipid LBPA in maturing endosomes.63,64 In the present study, a reduction in the number of fluobody punctae upon treatment of anti-GFP fluobody expressing HEK293T cells with GFP-CD63 EVs in the presence of U18666A (Figure 5) revealed that EV cargo release was significantly inhibited in the presence of the cholesterol transport inhibitor U18666A, even though endosomal compartments in U18666A-treated cells were acidic in nature, as indicated by the presence of Lysotracker (Figure S10). This may suggest that the lipid composition of the endosomal membrane plays a role in EV-endosome membrane fusion.
Taken together, using a sophisticated approach that combines CLEM, cytosolic expression of mAG-gal3 that detects endosomal damage, and cytosolic expression of a fluorescently tagged probe that recognizes EV cargo, we provide experimental proof for the exposure of EV cargo to the cytosol via fusion of EVs with endosomes/lysosomes. This fusion event provides a possible target for treatment and/or prevention of tumor progression, viral pathogenesis, and neurodegenerative diseases where EVs have been implicated as key mediators.2,4−6,65 Blocking EV cargo release by manipulating the EV-endosome membrane fusion process may assist in preventing or decelerating disease pathogenesis. However, a safe disposal of the EVs, for example, through lysosomal degradation, is required in addition to the prevention of intracellular EV cargo release. For instance, treatment of cells with U18666A, which prevents exosome-endosome membrane fusion and consequently intracellular cargo release, has been shown to stimulate exosome secretion by cells.65 This would unwantedly increase the risk of spreading the disease-causing entities. Indeed, inhibition of exosome secretion by bacteria-infected macrophages was shown to inhibit sepsis-induced inflammation and cardiac dysfunction.66 Therefore, prevention of exosome secretion by “diseased” cells and/or inhibition of exosome internalization by surrounding cells may offer safer alternatives to inhibition of EV cargo release in order to prevent EV-mediated spreading of disease.
Methods
Plasmids
N-Flag-Apex2-emGFP-CD63 (a gift from Nicole C. Meisner-Kober)21 was amplified adding BsmBI site at both the ends of the sequence. Next, the amplified segment was inserted into an entry vector pENTR1A (a gift from Eric Campeau and Paul Kaufman; Addgene, plasmid# 17398; http://n2t.net/addgene:17398; PRID:Addgene_17398)67 by golden gate assembly method.68 The plasmid obtained was then recombined with pLenti-CMV-Puro-DEST (a gift from Eric Campeau and Paul Kaufman; Addgene, plasmid# 17452; http://n2t.net/addgene:17452; PRID:Addgene_17452)67 using a gateway LR clonase enzyme (ThermoFisher Scientific, 11791100) to achieve the expression vector pLenti-CMV-N-Flag-APEX2-emGFP-CD63-Puro-DEST. For GFP display at the surface of EVs, emGFP was inserted at the second extracellular loop of CD63 following a reported strategy, to generate pLenti-CMV-N-CD63-emGFP-CD63-C-Puro-DEST.69 To generate the expression vector pLenti-CMV-CD63-mRFP using the same cloning strategy, CD63 was amplified from N-Flag-Apex2-emGFP-CD63, and mRFP sequence was amplified from Lamp1-mRFP plasmid (a gift from Walther Mothes; Addgene, plasmid# 1817; http://n2t.net/addgene:1817; PRID:Addgene_1817).70 mAzami-Green (mAG)-galectin 3 (Gal3) plasmid was a gift from Niels Geijsen (Addgene, plasmid# 62734; http://n2t.net/addgene:62734; PRID:Addgene_62734).71 The secretion signal peptide sequence in the mCherry-APEX2-anti-GFP FLIPPER-body vector72 was removed during PCR, and pLenti-CMV-mCherry-APEX2-anti-GFP FLIPPER-body-Puro-DEST was generated using aforementioned golden gate and gateway cloning strategy. In this study, the APEX2 in the FLIPPER-body was not used, and therefore the probe is called fluobody in the text. Full sequences of used proteins are provided in the Supporting Information.
Generation of EV Producer and Acceptor HEK293T Cell Lines and Cell Culture
The EV producer cell lines (GFP-CD63 and CD63-RFP) and acceptor cell lines (mCherry anti-GFP fluobody and galectin-3 Azami-Green) were created via lentiviral transduction followed by Puromycin (Sigma P8833) selection at 1 μg/mL. HEK293T cells were cultured in DMEM (Gibco 41965-039) supplemented with 10% fetal bovine serum (FBS, Bodinco, 5010) and 1% penicillin-streptomycin sulfate (Gibco, 15140-122) at 37 °C under 5% CO2. For LM imaging, acceptor cells were seeded on glass coverslips coated (VWR, 631-0150) with poly l-lysine (Sigma, P-2636). For CLEM imaging, acceptor cells were seeded on glass bottom Petri dishes (Greiner, 627870). The Petri dishes were, before cell seeding, sputter coated with 2 nm palladium/gold (LeicaSCD050) after which a pattern was made to enable to relocate the ROI in EM as defined by LM.
Preparation of Exosome-Depleted Medium
DMEM containing 10% FBS was centrifuged at 110,000×g for 16 h at 4 °C to deplete the fetal bovine serum-derived EVs. The resulting supernatant was filter sterilized through a 0.2 μm filter (Millipore) and stored at 4 °C.
EV Isolation
EV-producer cells were seeded in T162 flasks (Corning), and 15 mL exosome-depleted medium was added when cells reached ∼40% confluence. 48 h later, medium was collected and EVs were isolated by sequential centrifugation (Table S1). Please note that the EV fraction obtained after ultracentrifugation at 100,000×g represents small EVs. The final pellet was resuspended in 50 μL of PBS, and protein concentration was measured with DC protein assay kit (Bio-Rad, 5000114). EV concentrations are given as total EV protein weight per volume (μg/mL). Of note, besides EVs, other proteinaceous medium components may have become pelleted by sequential centrifugation, which may have caused an overestimation of the EV protein content.
SDS-PAGE and Western Blot
SDS-PAGE samples were prepared using an established protocol.73 Briefly, after measuring the protein content of EVs and cell lysates, 30 μg of protein per sample was mixed with Laemmli loading buffer with SDS and protease inhibitors (Roche, 11697498001). Samples were boiled for 5 min at 90 °C, loaded on a 10% SDS-PAGE gel, and subjected to electrophoresis at 100 V for 2 h. After SDS-PAGE, proteins were transferred to a polyvinylidene difluoride membrane (PVDF, Millipore, IPFL00010) and blocked with Odyssey blocking buffer (Li-COR, 927-40000) for 1 h at RT. Blots were incubated overnight with primary antibodies (Table S2) in blocking buffer at 4 °C. Next, blots were washed with 0.1% PBS-Tween 20 and subsequently incubated in secondary antibody for 1 h at RT. They were washed with 0.1% PBS-Tween 20 and imaged with an Odyssey Infrared Imaging system (Li-COR).
Size, Polydispersity, and ζ-Potential of EVs
An EV suspension with a concentration of 10 μg/mL in PBS was loaded into a folded capillary cell (Malvern Instruments DTS1070) and measured on a Zetasizer Nano ZS particle analyzer (Malvern Instruments) using a 633 nm laser. Dynamic light scattering measurements were performed in triplicate. Size and polydispersity were calculated by the cumulant analysis method using Zetasizer software version 7.10. The ζ-potential was determined by measuring the electrophoretic mobility and calculated using the Smoluchowski approximation.
mAG-gal3 Endosomal Permeabilization Assay
mAG-gal3 expressing HEK293T cells were seeded in 24-well plates on glass coverslips at a density of 150 × 103 cells/mL (0.5 mL) and treated the next day with 0.5 mL of 20 μg/mL CD63-RFP EVs for 2, 4, 8, or 12 h in exosome-depleted medium. Lipoplex treatment was used as a positive control. Lipoplexes composed of Lipofectamine 2000 and pDNA were prepared according to the manufacturer’s instructions and incubated with mAG-gal3 cells for 12 h in exosome-depleted medium. Control cells were treated in exosome-depleted medium in the absence of CD63-RFP EVs. The effect of a high(er) concentration of CD63-RFP EVs on endosomal integrity in mAG-Gal3 expressing cells was studied by using 0.5 mL of 100 μg/mL EVs, while keeping the rest of the protocol unchanged. Cells were fixed (4% PFA) and examined for mAG-gal3 punctae formation by fluorescence microscopy (Leica DMI6000B fluorescence microscope (HCX PL FLUOTAR 63×/N.A. 1.25 OIL).
anti-GFP Fluobody EV Cargo Release Assay
HEK293T cells expressing mCherry anti-GFP fluobody were seeded in 24-well plates on glass coverslips at a density of 150 × 103 cells/mL (0.5 mL). The next day, cells were treated with 0.5 mL of 20 μg/mL GFP-CD63 EVs for 2, 4, 8, or 12 h in an exosome-depleted medium. Control cells were treated in exosome-depleted medium in the absence of GFP-CD63 EVs. Cells were fixed, and mCherry fluobody punctae formation was examined by fluorescence microscopy.
Pharmacological Inhibitor, Temperature, and Fluorescent Tracer Treatments of HEK293T Cells
HEK293T cells expressing mCherry anti-GFP fluobody were plated in 24-well plates on glass coverslips at a density of 150 × 103 cells/mL (0.5 mL). The next day, cells were treated with 0.1 μM Bafilomycin A1 (BafA1, Enzo Lifesciences BML-CM110–0100) for 30 min in 0.5 mL of exosome-depleted medium, followed by addition of 20 μg/mL (10 μg per well) EVs in continued presence of the drug for 12 h. Cells were treated with 1 μg/mL U18666A (Merck Millipore, 662015-10MG) for 4 h in 0.5 mL of exosome-depleted medium, followed by addition of 20 μg/mL (0.5 mL) EVs in continued presence of the drug for 12 h. LysoTracker Red DND-99 (Invitrogen, L7528) was added to cells in the inhibitor experiments 1 h before fixation. TopFluor (Bodipy)-cholesterol (Avanti lipids, 810255P) was added (10 μM) to the cells for 1 h before addition of inhibitors. Cells were (pre)treated with 80 μM dynasore (Bioconnect, 2897/10) for 30 min in 0.5 mL of exosome-depleted medium, followed by addition of 20 μg/mL (0.5 mL) EVs in continued presence of the drug for 2 h. To inhibit energy-dependent passage across the plasma membrane, cells were incubated at 4 °C for 30 min and then treated with 0.5 mL of 20 μg/mL GFP-CD63 EVs in ice-cold EV-depleted medium for 1 h before fixation. Control cells were treated similarly at 37 °C before fixation.
MTT Assay
The viability of HEK293T after exposure to pharmacological inhibitors was evaluated by performing a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich #M2128) assay. Two ×104 (0.2 mL) HEK293T cells were seeded in 96-well plates precoated with PLL. Cells were treated in triplicate with 0.1 μM BafA1 and 1 μg/mL U18666A in exosome depleted DMEM (final volume of 0.2 mL), for 21 h. Untreated cells in exosome depleted DMEM were used as a negative control. During the final 3 h of incubation, cells were exposed to 20 μL MTT solution (5 mg/mL in PBS). The medium was removed, and formazan crystals were dissolved in 200 μL of DMSO. Upon complete solubilization of the crystals, the optical density of each well was measured at a wavelength of 520 nm using a microplate spectrophotometer (μQuant, BIO-TEK Instruments inc).
Electron Microscopic Investigation of Isolated EVs
Isolated EVs were fixed in 50 μL of 2% paraformaldehyde (PFA, Merck, 1.04005.1000) prepared in 0.1 M sodium cacodylate buffer pH 7.4 (Sigma, C0250-500g). Four μL of the EV solution was incubated on Formvar-coated 150 meshed copper grids (Electron Microscopy Sciences, 0150-Cu) for 25 min. The grids were rinsed with PBS for 1 min and subsequently incubated with 1% glutaraldehyde (GA, Polysciences, 01909-100) in 0.1 M sodium cacodylate buffer pH 7.4 for 5 min followed by rinsing with Milli-Q 7 times. All steps were performed at RT. For EV immunostaining, grids were incubated for 1 h with primary anti-GFP antibody, rinsed, and incubated for 1 h with a secondary antibody conjugated to 10 nm gold. Next, grids were incubated with 2% uranyl oxalate (pH7; SPI, 02624-AB) for 4 min on ice, briefly rinsed, and incubated for 10 min in methyl cellulose-uranyl acetate (pH 4) on ice. Images were generated by EM (FEI, CM100).
Fixation and Immunolabeling for LM
Cells were fixed for 30 min with 4% PFA in PBS and rinsed with PBS. Prior to immunolabeling, cells were permeabilized (0.2% Tween-20 (Sigma, P1379) in PBS) and treated with blocking solution (1 h; 3% BSA (Sigma, A7906) in PBS). Cells were then incubated with primary antibodies overnight at 4 °C (Table S2) in blocking solution followed by rinsing with PBS. Next, cells were incubated with secondary antibodies in blocking solution and subsequently rinsed with PBS. DAPI (1 μg/mL in PBS; Sigma, D9542) was added for 20 min, followed by rinsing with PBS and mounting of the coverslips using Vectashield (Vector Laboratories, H-1000) onto microscope slides. Images were generated using confocal microscopy (Leica SP8, HC PL APO CS2 63×/N.A. 1.4). All steps were performed at RT unless mentioned otherwise.
Fixation and Imaging for CLEM
An equal volume of 2% PFA and 0.2% GA in 0.1 M sodium cacodylate buffer (pH 7.4) was added to the cells in medium and incubated for 10 min. Cells were then incubated with fresh pure fixative (2% PFA and 0.2% GA) for 30 min and rinsed twice with 0.1 M sodium cacodylate. All steps were performed at RT. Fluorescent images were generated using confocal microscopy (Zeiss LSM 780, Plan-Neofluar 63×/N.A. 1.3 Imm Corr DIC M27 lens) before being processing for EM.
Embedding and EM
Epon EM embedding was performed as described previously.74 The ROI identified by confocal microscopy was traced using a stereo microscope by using the palladium/gold marks. The selected areas were sawn out and trimmed prior to ultrathin sectioning. Serial sections (100 nm) of entire cells were collected on nickel one-hole grids (Electron Microscopy Sciences, 1000-Ni). For stabilization, the grids were pre-irradiated using a TEM (FEI, CM100) at 80 kV. Subsequently, large-scale areas, with 2.5 nm pixel size, were scanned using the STEM detector in a SEM (Zeiss Supra55) at 28 kV.39,74 An overlay was made in Adobe Photoshop. All data sets are available at www.nanotomy.org.
Post-Embedding Immunolabeling
Thin sections in Epon were etched for 10 min in 1% periodic acid (Merck, 1.00524.0025) in Milli-Q, followed by washes (3 × 2 min Milli-Q) and blocking with 1% BSA in PBS for 30 min. Next, the sections were immunolabeled (anti-GFP, Table S2) for 4 h, washed 2 × 5 min with PBS, and incubated with a biotinylated secondary antibody (1 h). Samples were rinsed (3 × 5 min PBS) and incubated with Quantum dot 655 (QD655)-labeled streptavidin for 1 h. Finally, sections were rinsed (3 × 5 min PBS). Images were generated with 2.5 nm pixel size, using the STEM detector in a SEM (Zeiss Supra55) at 28 kV.
LM Image Analysis
Representative images were selected and cropped. The images were adjusted in Adobe Photoshop by adding a layer to change the levels, which was implemented to all the images to the same extent. Quantification analysis was performed on the recorded z-stacks and analyzed using the software Icy.75 The Colocalizer with binary and excel output plugin was used to determine the number of EVs and fluobody spots and colocalization.
For colocalization studies in cells co-incubated with GFP-CD63 and CD63-RFP EVs, images were first adjusted in brightness/contrast minimum and maximum before colocalization measurements were applied. Brightness/contrast minimum and maximum were set at 2–70 for GFP-CD63, and 1–35 for CD63-RFP.76 Next, Fiji plugin colocalization threshold was used, without the use of a ROI, to obtain the Pearson’s value for the whole image. For colocalization studies in immunostained fluobody-expressing cells, fluobody (red channel) adjustments were set at 11 (min) and 84 (max). Similarly, for LAMP1 (633 channel), brightness/contrast minimum-maximum was set at 0–70. JACoP plugin was used to obtain the Mander’s coefficient.76 Values were calculated after setting a threshold to remove the background to only include fluobody punctae (mCherry channel) or LAMP1 (633 channel) spots.
Live Cell Imaging of mAG-gal3 HEK293T Cells
For live cell imaging, cells were grown on glass bottomed 2-well plates (Lab-TEK chambered coverglass, Thermo Fisher Scientific, Denmark). On the day of the experiment, cells were placed in a DeltaVision Elite microscope equipped with a temperature/CO2 controlled cabinet and automated stage. HEK293T cells expressing mAG-gal3 were selected before addition of CD63-RFP EVs. Experiments were carried out in exosome-depleted medium. Image acquisition took place from 30 to 270 min after the addition of the CD63-RFP EVs to the cells using softWoRx 6 acquisition and integrated deconvolution software (GE Healthcare, Issaquah, WA). The UPLSAPO objective (100× oil, N.A. 1.4, WD 0.12 mm) and GFP/mCherry filter combination were used for image acquisition. Images were further analyzed using Fiji.77
Statistical Analysis
Experiments were performed at n = 3. Data are presented as mean ± SD. The significance of the difference between two independent samples was determined using Student’s t test. Groups were compared using one-way analysis of variance, with Tukey’s post hoc. All the statistical analyses were performed using GraphPad Prism.
Acknowledgments
I.S.Z. is supported by the Dutch Technology Foundation TTW, which is part of The Netherlands Organization for Scientific Research (NWO) and is partly funded by the Ministry of Economic Affairs. Part of the work was funded by The Netherlands organization for scientific research (STW Microscopy Valley 12718, NWO 175-010-2009-023, and ZonMW 91111.006) to BNGG and de Cock-Hadders Stichting to MAdB and B.S.J. B.S.J. received a NAMASTE scholarship funded by the Erasmus Mundus India-EU mobility consortium.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b10033.
Figure S1: Biophysical characterization of WT EVs and GFP-CD63 EVs isolated from HEK293T cells and GFP-CD63 HEK293T cells, respectively. Figure S2: GFP-CD63 EVs are taken up via endocytosis. Figure S3: EVs localize in membrane-bound compartments in HEK293T acceptor cells. Figure S4: GFP-CD63 and CD63-RFP EVs show similar EV characteristics and intracellular localization in recipient cells. Figure S5: CD63-RFP EVs do not induce endosomal permeabilization. Figure S6: Increased EV concentration does not induce endosomal permeabilization. Figure S7: EVs do not induce endosomal permeabilization. Figure S8: Quantification of fraction of mCherry-fluobody-expressing cells that show fluobody punctae after 2, 4, 8, and 12 h incubation with 20 μg/mL CD63-GFP EVs. Figure S9: WT EVs do not induce fluobody punctae in anti-GFP fluobody (mCherry) HEK293T cells. Figure S10: Effects of BafA1 and U18666A metabolic inhibitors on HEK293T cells. Table S1: EV isolation method by means of sequential centrifugation. Table S2: List of antibodies used in the study. Supplementary data: Nucleotide and corresponding amino acid sequence of cDNAs used in this study (PDF)
Movie S1: EVs do not induce endosomal permeabilization (AVI)
Author Contributions
§ These authors contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Raposo G.; Nijman H. W.; Stoorvogel W.; Liejendekker R.; Harding C. V.; Melief C. J.; Geuze H. J. B Lymphocytes Secrete Antigen-Presenting Vesicles. J. Exp. Med. 1996, 183, 1161–1172. 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L.; Regnault A.; Lozier A.; Wolfers J.; Flament C.; Tenza D.; Ricciardi-Castagnoli P.; Raposo G.; Amigorena S. Eradication of Established Murine Tumors Using a Novel Cell-Free Vaccine: Dendritic Cell-Derived Exosomes. Nat. Med. 1998, 4, 594–600. 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- Thery C.; Duban L.; Segura E.; Veron P.; Lantz O.; Amigorena S. Indirect Activation of Naive CD4+ T Cells by Dendritic Cell-Derived Exosomes. Nat. Immunol. 2002, 3, 1156–1162. 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- Wiley R. D.; Gummuluru S. Immature Dendritic Cell-Derived Exosomes Can Mediate HIV-1 Trans Infection. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 738–743. 10.1073/pnas.0507995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fevrier B.; Vilette D.; Archer F.; Loew D.; Faigle W.; Vidal M.; Laude H.; Raposo G. Cells Release Prions in Association with Exosomes. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9683–9688. 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L.; Honsho M.; Zahn T. R.; Keller P.; Geiger K. D.; Verkade P.; Simons K. Alzheimer’s Disease β-Amyloid Peptides Are Released in Association with Exosomes. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11172–11177. 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfers J.; Lozier A.; Raposo G.; Regnault A.; Thery C.; Masurier C.; Flament C.; Pouzieux S.; Faure F.; Tursz T.; Angevin E.; Amigorena S.; Zitvogel L. Tumor-Derived Exosomes Are a Source of Shared Tumor Rejection Antigens for CTL Cross-Priming. Nat. Med. 2001, 7, 297–303. 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- Iero M.; Valenti R.; Huber V.; Filipazzi P.; Parmiani G.; Fais S.; Rivoltini L. Tumour-Released Exosomes and Their Implications in Cancer Immunity. Cell Death Differ. 2008, 15, 80–88. 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- Sterzenbach U.; Putz U.; Low L. H.; Silke J.; Tan S. S.; Howitt J. Engineered Exosomes As Vehicles for Biologically Active Proteins. Mol. Ther. 2017, 25, 1269–1278. 10.1016/j.ymthe.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney M. J.; Klyachko N. L.; Zhao Y.; Gupta R.; Plotnikova E. G.; He Z.; Patel T.; Piroyan A.; Sokolsky M.; Kabanov A. V.; Batrakova E. V. Exosomes As Drug Delivery Vehicles for Parkinson’s Disease Therapy. J. Controlled Release 2015, 207, 18–30. 10.1016/j.jconrel.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y.; Li S.; Song J.; Ji T.; Zhu M.; Anderson G. J.; Wei J.; Nie G. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials 2014, 35, 2383–2390. 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- Colombo M.; Moita C.; van Niel G.; Kowal J.; Vigneron J.; Benaroch P.; Manel N.; Moita L. F.; Thery C.; Raposo G. Analysis of ESCRT Functions in Exosome Biogenesis, Composition and Secretion Highlights the Heterogeneity of Extracellular Vesicles. J. Cell Sci. 2013, 126, 5553–5565. 10.1242/jcs.128868. [DOI] [PubMed] [Google Scholar]
- Lai C. P.; Breakefield X. O. Role of Exosomes/Microvesicles in the Nervous System and Use in Emerging Therapies. Front. Physiol. 2012, 3, 228. 10.3389/fphys.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H.; Aleckovic M.; Lavotshkin S.; Matei I.; Costa-Silva B.; Moreno-Bueno G.; Hergueta-Redondo M.; Williams C.; Garcia-Santos G.; Ghajar C.; Nitadori-Hoshino A.; Hoffman C.; Badal K.; Garcia B. A.; Callahan M. K.; Yuan J.; Martins V. R.; Skog J.; Kaplan R. N.; Brady M. S.; et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells Toward a Pro-Metastatic Phenotype Through MET. Nat. Med. 2012, 18, 883–891. 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H.; Ekstrom K.; Bossios A.; Sjostrand M.; Lee J. J.; Lotvall J. O. Exosome-Mediated Transfer of mRNAs and microRNAs Is a Novel Mechanism of Genetic Exchange Between Cells. Nat. Cell Biol. 2007, 9, 654–659. 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Ratajczak J.; Miekus K.; Kucia M.; Zhang J.; Reca R.; Dvorak P.; Ratajczak M. Z. Embryonic Stem Cell-Derived Microvesicles Reprogram Hematopoietic Progenitors: Evidence for Horizontal Transfer of mRNA and Protein Delivery. Leukemia 2006, 20, 847–856. 10.1038/sj.leu.2404132. [DOI] [PubMed] [Google Scholar]
- Edgar J. R. Q&A: What Are Exosomes, Exactly?. BMC Biol. 2016, 14, 46. 10.1186/s12915-016-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camussi G.; Deregibus M. C.; Bruno S.; Cantaluppi V.; Biancone L. Exosomes/Microvesicles As a Mechanism of Cell-to-Cell Communication. Kidney Int. 2010, 78, 838–848. 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- Parolini I.; Federici C.; Raggi C.; Lugini L.; Palleschi S.; De Milito A.; Coscia C.; Iessi E.; Logozzi M.; Molinari A.; Colone M.; Tatti M.; Sargiacomo M.; Fais S. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecalvo A.; Larregina A. T.; Shufesky W. J.; Stolz D. B.; Sullivan M. L.; Karlsson J. M.; Baty C. J.; Gibson G. A.; Erdos G.; Wang Z.; Milosevic J.; Tkacheva O. A.; Divito S. J.; Jordan R.; Lyons-Weiler J.; Watkins S. C.; Morelli A. E. Mechanism of Transfer of Functional microRNAs Between Mouse Dendritic Cells Via Exosomes. Blood 2012, 119, 756–766. 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusermann W.; Hean J.; Trojer D.; Steib E.; von Bueren S.; Graff-Meyer A.; Genoud C.; Martin K.; Pizzato N.; Voshol J.; Morrissey D. V.; Andaloussi S. E.; Wood M. J.; Meisner-Kober N. C. Exosomes Surf on Filopodia to Enter Cells at Endocytic Hot Spots, Traffic Within Endosomes, and Are Targeted to the ER. J. Cell Biol. 2016, 213, 173–184. 10.1083/jcb.201506084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z.; Qiao Y.; Li X.; Chen J.; Ding J.; Bai L.; Shen F.; Shi B.; Liu J.; Peng L.; Li J.; Yuan Z. Exosomes Exploit the Virus Entry Machinery and Pathway to Transmit Alpha Interferon-Induced Antiviral Activity. J. Virol. 2018, 92, e01578 10.1128/JVI.01578-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung M. E.; Leonard J. N. A Platform for Actively Loading Cargo RNA to Elucidate Limiting Steps in EV-Mediated Delivery. J. Extracell. Vesicles 2016, 5, 31027. 10.3402/jev.v5.31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. P.; Kim E. Y.; Badr C. E.; Weissleder R.; Mempel T. R.; Tannous B. A.; Breakefield X. O. Visualization and Tracking of Tumour Extracellular Vesicle Delivery and RNA Translation Using Multiplexed Reporters. Nat. Commun. 2015, 6, 7029. 10.1038/ncomms8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Conde I.; Shrimpton C. N.; Thiagarajan P.; Lopez J. A. Tissue-Factor-Bearing Microvesicles Arise from Lipid Rafts and Fuse with Activated Platelets to Initiate Coagulation. Blood 2005, 106, 1604–1611. 10.1182/blood-2004-03-1095. [DOI] [PubMed] [Google Scholar]
- Mathieu M.; Martin-Jaular L.; Lavieu G.; Thery C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat. Cell Biol. 2019, 21, 9–17. 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- Stewart M. P.; Lorenz A.; Dahlman J.; Sahay G. Challenges in Carrier-Mediated Intracellular Delivery: Moving Beyond Endosomal Barriers. Wiley Interdiscip. Rev. Nanomed Nanobiotechnol. 2016, 8, 465–478. 10.1002/wnan.1377. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wang H.; Xia Y.; Yan F.; Lu Y. Therapeutic Potential of Mesenchymal Cell-Derived miRNA-150–5p-Expressing Exosomes in Rheumatoid Arthritis Mediated by the Modulation of MMP14 and VEGF. J. Immunol. 2018, 201, 2472–2482. 10.4049/jimmunol.1800304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usman W. M.; Pham T. C.; Kwok Y. Y.; Vu L. T.; Ma V.; Peng B.; Chan Y. S.; Wei L.; Chin S. M.; Azad A.; He A. B.; Leung A. Y. H.; Yang M.; Shyh-Chang N.; Cho W. C.; Shi J.; Le M. T. N. Efficient RNA Drug Delivery Using Red Blood Cell Extracellular Vesicles. Nat. Commun. 2018, 9, 2359. 10.1038/s41467-018-04791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang X.; Chen X.; Wang L.; Yang G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis After Ischemia. Mol. Ther.--Nucleic Acids 2017, 7, 278–287. 10.1016/j.omtn.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S.; Takanashi M.; Sudo K.; Ueda S.; Ishikawa A.; Matsuyama N.; Fujita K.; Mizutani T.; Ohgi T.; Ochiya T.; Gotoh N.; Kuroda M. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor microRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Li L.; Yu J.; Zhu D.; Zhang Y.; Li X.; Gu H.; Zhang C. Y.; Zen K. Microvesicle-Mediated Delivery of Transforming Growth Factor β1 siRNA for the Suppression of Tumor Growth in Mice. Biomaterials 2014, 35, 4390–4400. 10.1016/j.biomaterials.2014.02.003. [DOI] [PubMed] [Google Scholar]
- de Boer P.; Hoogenboom J. P.; Giepmans B. N. Correlated Light and Electron Microscopy: Ultrastructure Lights Up!. Nat. Methods 2015, 12, 503–513. 10.1038/nmeth.3400. [DOI] [PubMed] [Google Scholar]
- Rothbauer U.; Zolghadr K.; Tillib S.; Nowak D.; Schermelleh L.; Gahl A.; Backmann N.; Conrath K.; Muyldermans S.; Cardoso M. C.; Leonhardt H. Targeting and Tracing Antigens in Live Cells with Fluorescent Nanobodies. Nat. Methods 2006, 3, 887–889. 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman C.; Atarhouch T.; Muyldermans S.; Robinson G.; Hammers C.; Songa E. B.; Bendahman N.; Hammers R. Naturally Occurring Antibodies Devoid of Light Chains. Nature 1993, 363, 446–448. 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- Helma J.; Cardoso M. C.; Muyldermans S.; Leonhardt H. Nanobodies and Recombinant Binders in Cell Biology. J. Cell Biol. 2015, 209, 633–644. 10.1083/jcb.201409074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols M. S.; Klumperman J. Trafficking and Function of the Tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. 10.1016/j.yexcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Macia E.; Ehrlich M.; Massol R.; Boucrot E.; Brunner C.; Kirchhausen T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Kuipers J.; de Boer P.; Giepmans B. N. Scanning EM of Non-Heavy Metal Stained Biosamples: Large-Field of View, High Contrast and Highly Efficient Immunolabeling. Exp. Cell Res. 2015, 337, 202–207. 10.1016/j.yexcr.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Wittrup A.; Ai A.; Liu X.; Hamar P.; Trifonova R.; Charisse K.; Manoharan M.; Kirchhausen T.; Lieberman J. Visualizing Lipid-Formulated siRNA Release from Endosomes and Target Gene Knockdown. Nat. Biotechnol. 2015, 33, 870–876. 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell U.; Kuipers J.; Giepmans B. N. EpCAM Proteolysis: New Fragments with Distinct Functions?. Biosci. Rep. 2013, 33, e00030 10.1042/BSR20120128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P.; Helenius A. Endocytosis of Viruses and Bacteria. Cold Spring Harbor Perspect. Biol. 2014, 6, a016972 10.1101/cshperspect.a016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weert A. W.; Dunn K. W.; Gueze H. J.; Maxfield F. R.; Stoorvogel W. Transport from Late Endosomes to Lysosomes, but Not Sorting of Integral Membrane Proteins in Endosomes, Depends on the Vacuolar Proton Pump. J. Cell Biol. 1995, 130, 821–834. 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobo K.; Le Blanc I.; Luyet P. P.; Fivaz M.; Ferguson C.; Parton R. G.; Gruenberg J.; van der Goot F. G. Late Endosomal Cholesterol Accumulation Leads to Impaired Intra-Endosomal Trafficking. PLoS One 2007, 2, e851 10.1371/journal.pone.0000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgner F.; Ren H.; Medvedev R.; Ploen D.; Himmelsbach K.; Boller K.; Hildt E. The Intracellular Cholesterol Transport Inhibitor U18666A Inhibits the Exosome-Dependent Release of Mature Hepatitis C Virus. J. Virol. 2016, 90, 11181–11196. 10.1128/JVI.01053-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zong S.; Wang Z.; Lu J.; Zhu D.; Zhang Y.; Zhang R.; Cui Y. Visualization and Intracellular Dynamic Tracking of Exosomes and Exosomal miRNAs Using Single Molecule Localization Microscopy. Nanoscale 2018, 10, 5154–5162. 10.1039/C7NR08800K. [DOI] [PubMed] [Google Scholar]
- Nakase I.; Noguchi K.; Aoki A.; Takatani-Nakase T.; Fujii I.; Futaki S. Arginine-Rich Cell-Penetrating Peptide-Modified Extracellular Vesicles for Active Macropinocytosis Induction and Efficient Intracellular Delivery. Sci. Rep. 2017, 7, 1991. 10.1038/s41598-017-02014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I.; Futaki S. Combined Treatment with a pH-Sensitive Fusogenic Peptide and Cationic Lipids Achieves Enhanced Cytosolic Delivery of Exosomes. Sci. Rep. 2015, 5, 10112. 10.1038/srep10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Lee H.; Goh U.; Kim J.; Jeong M.; Lee J.; Park J. H. Cellular Engineering with Membrane Fusogenic Liposomes to Produce Functionalized Extracellular Vesicles. ACS Appl. Mater. Interfaces 2016, 8, 6790–6795. 10.1021/acsami.6b01315. [DOI] [PubMed] [Google Scholar]
- Mangeot P. E.; Dollet S.; Girard M.; Ciancia C.; Joly S.; Peschanski M.; Lotteau V. Protein Transfer into Human Cells by VSV-G-Induced Nanovesicles. Mol. Ther. 2011, 19, 1656–1666. 10.1038/mt.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y. T.; Umezaki K.; Sawada S.; Mukai S. A.; Sasaki Y.; Harada N.; Shiku H.; Akiyoshi K. Engineering Hybrid Exosomes by Membrane Fusion with Liposomes. Sci. Rep. 2016, 6, 21933. 10.1038/srep21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Zang X.; Wang M.; Li Z.; Qiao M.; Hu H.; Chen D. Exosome-Based Nanocarriers As Bio-Inspired and Versatile Vehicles for Drug Delivery: Recent Advances and Challenges. J. Mater. Chem. B 2019, 7, 2421–2433. 10.1039/C9TB00170K. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Wu J.; Gu W.; Huang Y.; Tong Z.; Huang L.; Tan J. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. (Weinh) 2018, 5, 1700611. 10.1002/advs.201700611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin A. J.; Mager I.; de Jong O. G.; Varela M. A.; Schiffelers R. M.; El Andaloussi S.; Wood M. J. A.; Vader P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. 10.1016/j.ymthe.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meel R.; Fens M. H.; Vader P.; van Solinge W. W.; Eniola-Adefeso O.; Schiffelers R. M. Extracellular Vesicles As Drug Delivery Systems: Lessons from the Liposome Field. J. Controlled Release 2014, 195, 72–85. 10.1016/j.jconrel.2014.07.049. [DOI] [PubMed] [Google Scholar]
- van Dongen H. M.; Masoumi N.; Witwer K. W.; Pegtel D. M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. 2016, 80, 369–386. 10.1128/MMBR.00063-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Beuchat M. H.; Chevallier J.; Makino A.; Mayran N.; Escola J. M.; Lebrand C.; Cosson P.; Kobayashi T.; Gruenberg J. Separation and Characterization of Late Endosomal Membrane Domains. J. Biol. Chem. 2002, 277, 32157–32164. 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- Huotari J.; Helenius A. Endosome Maturation. EMBO J. 2011, 30, 3481–3500. 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva E.; Yang S. T.; Melikov K.; Pourmal S.; Chernomordik L. V. Dengue Virus Ensures Its Fusion in Late Endosomes Using Compartment-Specific Lipids. PLoS Pathog. 2010, 6, e1001131 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ur Rehman Z.; Hoekstra D.; Zuhorn I. S. Protein Kinase A Inhibition Modulates the Intracellular Routing of Gene Delivery Vehicles in HeLa Cells, Leading to Productive Transfection. J. Controlled Release 2011, 156, 76–84. 10.1016/j.jconrel.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Durymanov M.; Reineke J. Non-Viral Delivery of Nucleic Acids: Insight into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9, 971. 10.3389/fphar.2018.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degors I. M. S.; Wang C.; Rehman Z. U.; Zuhorn I. S. Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors. Acc. Chem. Res. 2019, 52, 1750–1760. 10.1021/acs.accounts.9b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum A. I.; Zhang G.; Warren J. D.; Maxfield F. R. Endocytosis of Beta-Cyclodextrins Is Responsible for Cholesterol Reduction in Niemann-Pick Type C Mutant Cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5477–5482. 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta-Geijo M. A.; Chiappi M.; Galindo I.; Barrado-Gil L.; Munoz-Moreno R.; Carrascosa J. L.; Alonso C. Cholesterol Flux Is Required for Endosomal Progression of African Swine Fever Virions During the Initial Establishment of Infection. J. Virol. 2016, 90, 1534–1543. 10.1128/JVI.02694-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss K.; Goebel C.; Runz H.; Mobius W.; Weiss S.; Feussner I.; Simons M.; Schneider A. Exosome Secretion Ameliorates Lysosomal Storage of Cholesterol in Niemann-Pick Type C Disease. J. Biol. Chem. 2010, 285, 26279–26288. 10.1074/jbc.M110.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essandoh K.; Yang L.; Wang X.; Huang W.; Qin D.; Hao J.; Wang Y.; Zingarelli B.; Peng T.; Fan G. C. Blockade of Exosome Generation with GW4869 Dampens the Sepsis-Induced Inflammation and Cardiac Dysfunction. Biochim. Biophys. Acta, Mol. Basis Dis. 2015, 1852, 2362–2371. 10.1016/j.bbadis.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E.; Ruhl V. E.; Rodier F.; Smith C. L.; Rahmberg B. L.; Fuss J. O.; Campisi J.; Yaswen P.; Cooper P. K.; Kaufman P. D. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One 2009, 4, e6529 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C.; Kandzia R.; Marillonnet S. A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PLoS One 2008, 3, e3647 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney Z.; Losacco J.; McDevitt S.; Zhang Z.; Lu B. Development of Exosome Surface Display Technology in Living Human Cells. Biochem. Biophys. Res. Commun. 2016, 472, 53–59. 10.1016/j.bbrc.2016.02.058. [DOI] [PubMed] [Google Scholar]
- Sherer N. M.; Lehmann M. J.; Jimenez-Soto L. F.; Ingmundson A.; Horner S. M.; Cicchetti G.; Allen P. G.; Pypaert M.; Cunningham J. M.; Mothes W. Visualization of Retroviral Replication in Living Cells Reveals Budding into Multivesicular Bodies. Traffic 2003, 4, 785–801. 10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- D’Astolfo D. S.; Pagliero R. J.; Pras A.; Karthaus W. R.; Clevers H.; Prasad V.; Lebbink R. J.; Rehmann H.; Geijsen N. Efficient Intracellular Delivery of Native Proteins. Cell 2015, 161, 674–690. 10.1016/j.cell.2015.03.028. [DOI] [PubMed] [Google Scholar]
- de Beer M. A.; Kuipers J.; van Bergen en Henegouwen P. M. P.; Giepmans B. N. G. A Small Protein Probe for Correlated Microscopy of Endogenous Proteins. Histochem. Cell Biol. 2018, 149, 261–268. 10.1007/s00418-018-1632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C.; Amigorena S.; Raposo G.; Clayton A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- Kuipers J.; Kalicharan R. D.; Wolters A. H.; van Ham T. J.; Giepmans B. N. Large-Scale Scanning Transmission Electron Microscopy (Nanotomy) of Healthy and Injured Zebrafish Brain. J. Visualized Exp. 2016, 111, e53635 10.3791/53635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaumont F.; Dallongeville S.; Chenouard N.; Herve N.; Pop S.; Provoost T.; Meas-Yedid V.; Pankajakshan P.; Lecomte T.; Le Montagner Y.; Lagache T.; Dufour A.; Olivo-Marin J. C. Icy: An Open Bioimage Informatics Platform for Extended Reproducible Research. Nat. Methods 2012, 9, 690–696. 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
- Bolte S.; Cordelières F. P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J. Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.