

Abstract
Although hypoxia is detrimental to most cell types, it aids survival of progenitor cells and is associated with diseases like cancer and pulmonary hypertension in humans. Therefore, understanding the underlying mechanisms that promote survival of progenitor cells in hypoxia and then developing novel therapies to stop their growth in hypoxia-associated human diseases is important. Here we demonstrate that the proliferation and growth of human CD133+ progenitor cells, which contribute to tumorigenesis and the development of pulmonary hypertension, are increased when cultured under hypoxic conditions. Furthermore, glucose-6-phosphate dehydrogenase (G6PD) activity was increased threefold in hypoxic CD133+ cells. The increased G6PD activity was required for CD133+ cell proliferation, and their growth was arrested by G6PD inhibition or knockdown. G6PD activity upregulated expression of HIF1α, cyclin A, and phospho-histone H3, thereby promoting CD133+ cell dedifferentiation and self-renewal and altering cell cycle regulation. When CD133+ cells were cocultured across a porous membrane from pulmonary artery smooth muscle cells (PASMCs), G6PD-dependent H2O2 production and release by PASMCs recruited CD133+ cells to the membrane, where they attached and expressed smooth muscle markers (α-actin and SM22α). Inhibition of G6PD reduced smooth muscle marker expression in CD133+ cells under normoxia but not hypoxia. In vivo, CD133+ cells colocalized with G6PD+ cells in the perivascular region of lungs from rats with hypoxia-induced pulmonary hypertension. Finally, inhibition of G6PD by dehydroepiandrosterone in pulmonary arterial hypertensive rats nearly abolished CD133+ cell accumulation around pulmonary arteries and the formation of occlusive lesions. These observations suggest G6PD plays a key role in increasing hypoxia-induced CD133+ cell survival in hypertensive lungs that differentiate to smooth muscle cells and contribute to pulmonary arterial remodeling during development of pulmonary hypertension.
Keywords: pentose phosphate pathway, pulmonary hypertension, Notch, HIF, cyclin A, phospho-histone H3, cell cycle, dedifferentiation, phenotype, pulmonary artery smooth muscle cells
molecular oxygen is essential for most life on Earth, functioning as the final electron acceptor during the oxidation of glucose to produce energy in the form of ATP. Ironically, hypoxia promotes the survival and expansion capacity of embryonic stem cells, cancer stem cells (16, 40), and neuronal crest stem cells (30, 44), as well as the differentiation potential of bone marrow-derived CD133+ progenitor cells (33, 40).
CD133+ progenitor cells are capable of differentiating into hematopoietic, endothelial, smooth muscle, and neuronal cell types. They play a role in tissue genesis during gestation (35) and in the angiogenesis that promotes wound healing (4). CD133+ and bone marrow-derived stem cells also contribute to tumorigenesis (39, 47, 54) and pathogenic remodeling that reduces conductance in coronary (49) and pulmonary arteries (13, 28, 48, 51), thereby contributing to chronic hypoxia-induced pulmonary hypertension (23, 41). In response to hypoxia, circulating bone marrow-derived CD34+/CD133+/Flk+ and c-kit+ cells are recruited to the adventitial, medial or intimal compartments, where they assume mesenchymal or smooth muscle cell-like characteristics (29, 38, 43). In vitro, these cells acquire the morphology and phenotype of the cells with which they are cocultured (11). For example, CD133+ cells cocultured with isolated human pulmonary arterial segments migrate through the intima and differentiate into smooth muscle cells. More interestingly, transplantation of bone marrow-derived CD133+ progenitor cells from patients with pulmonary arterial hypertension (PAH) into mice resulted in angioproliferative pulmonary vascular remodeling, right ventricular failure, and death (3). However, the molecular mechanisms that underlie the expansion and differentiation of hypoxic CD133+ progenitor cells remain unclear.
Self-renewal of progenitor cells is mediated by Wnt-Notch (4, 22, 36) and TGF-β-SMAD signaling pathways (25, 50), and NADPH oxidase (NOX)-derived reactive oxygen species (ROS) are also involved in the differentiation of stem/progenitor cells to endothelial and smooth muscle cells (26). For example, mobilization of endothelial progenitor cells is dependent on NOX2-derived ROS (9, 33, 37). In addition, TGF-β upregulates glucose-6-phosphate dehydrogenase (G6PD) expression in fetal brown adipocytes and increases levels of NOX4-derived ROS (45). Because production of superoxide anion from NOX is dependent on G6PD-derived NADPH (18, 20), we hypothesized that increases in G6PD-dependent NADPH redox and NOX-derived ROS act in concert to mediate hypoxia-induced CD133+ cell expansion and differentiation. Our findings indicate that G6PD activity promotes 1) hypoxia-induced CD133+ cell hyperproliferation via a TGF-β- and ROS-independent pathway, and 2) the expression of smooth muscle contractile proteins in CD133+ cells cocultured with pulmonary artery smooth muscle cells (PASMCs). Furthermore, oral administration of dehydroepiandrosterone (DHEA), an endogenous 17α-ketosteroid that blocks G6PD activity (19), to PAH rats for 5 wk inhibited accumulation of CD133+ cells around their pulmonary arteries and reduced the formation of occlusive neointimal and plexiform lesions.
MATERIALS AND METHODS
Chemicals.
The TGF-β receptor blocker SB525334 [6-(2-(1,1-dimethylethyl)-5-(6-methyl-2-pyridinyl)-1H-imidazol-4-yl)quinoxaline] was purchased from Selleckchem (Houston, TX). All other chemicals were from Sigma Aldrich (St. Louis, MO), unless indicated otherwise.
CD133+ cell culture.
Human CD133+ progenitor cells were purchased from PromoCell and expanded in hematopoietic progenitor cell expansion medium (PromoCell) supplemented with Cytokine mix E (PromoCell) containing human thrombopoietin, stem cell factor, flt-3 ligand, and IL-3. Cells were expanded for 1 wk in 75-cm2 flasks (Corning, Corning, NY) at 37°C in a humidified CO2 incubator before use.
Cell coculture.
CD133+ cells were labeled with a nontransferable dye, Alexa Fluor 594-conjugated AcLDL (10 μg/ml Ac-LDL A594, Life Technologies, Grand Island, NY) at 37°C and seeded (2,800 cells/Transwell) onto membranes (0.4-μm pores) in Transwell inserts in a 12-well plate (Corning). Forty-eight hours prior to seeding the CD133+ cells, PASMCs were seeded on the reverse side of the Transwell membrane at a density of 50,000 cells/Transwell insert. After seeding the CD133+ cells, the coculture setup was incubated under normoxia or hypoxia for 8 days. CD133+ cells cultured alone served as a negative control for coculture. PASMCs cultured on Transwell inserts without CD133+ cells served as a positive control for smooth muscle phenotype markers.
Hypoxia treatment.
CD133+ progenitor cells cultured in 25-cm2 flasks (Corning), (n = 6 flasks/condition) were incubated for 72 h in a hypoxic chamber (InvivO2 300, Ruskin Technology) under 3% O2 and 5% CO2, or under normoxia at 21% O2 and 5% CO2.
Adenovirus preparation.
We developed adenoviral vectors to deliver shRNA into cultured CD133+ cells. Briefly, a G6PD-specific shRNA gene sequence (CGGAAACGUCGUACACUUtt) that specifically and efficaciously downregulated G6PD (based on our preliminary results) and a scrambled sequence (negative control) were custom cloned by GeneScript in an adenoviral vector under the H1 promoter to drive short hairpin (sh) RNA expression. To monitor transfection efficiency, the vector also carried a green fluorescent protein (GFP) marker (coral GPF, cGFP) under the control of the CMV promoter. These vector-based shRNAs were packaged in adenoviruses by Welgen Laboratories. Stocks of adenoviral vector (3 × 1010–13 pfu) encoding the G6PD or scrambled shRNA were diluted threefold (1012 pfu) and used for transfecting cultured CD133+ cells.
Immunohistochemistry.
Paraffin-embedded lung sections from rats left untreated (normoxia) or subjected to 5 wk of hypoxia were deparaffinized and placed in 1× citrate buffer. The endogenous peroxidase activity was then suppressed by use of 3% H2O2, and nonspecific binding was blocked with blocking serum (Vectastain Universal Elite ABC kit, Vector Laboratories, Burlingame, CA). The slides were next incubated with primary antibodies, anti-G6PD (1:300; Santa Cruz, CA) and anti-CD133 (1:300; Santa Cruz, CA), overnight at 4°C. Secondary antibody incubation was for 1 h at room temperature and was followed by incubation for 30 min with avidin-biotin complex. Finally, the slides were developed by use of diaminobenzidine. Nuclei were stained with hematoxylin.
Immunofluorescent staining.
CD133+ cells on Transwell membranes were fixed in 3.7% paraformaldehyde for 30 min at 37°C and then blocked with 0.5% BSA. PASMCs on the reverse side of the Transwell membranes were wiped off with a moist tissue, after which the membranes were cut out and incubated with anti-α-actin and anti-SM22α (Sigma Aldrich) overnight at 4°C. They were then washed with 1× TBP (0.5% BSA, 0.2% Triton X-100 in 1× PBS), incubated with secondary antibody (Alexa Fluor 488-conjugated anti-mouse and anti-rabbit, Life Technologies) for 1 h at room temperature, and washed again with 1× TBP. Nuclei were stained with DAPI (1 μg/ml), after which the Transwell membranes were mounted on slides with DAKO mounting medium (DAKO, Carpinteria, CA) and examined via a Nikon-A1 confocal microscope.
Western blot analysis.
Cells were collected by centrifugation at 240 g, after which the protein was extracted from the cell pellets by use of Nonidet P-40 (NP-40) lysis buffer [50 mmol/l Tris·HCl (pH 7.4), 150 mmol/l NaCl, 0.5% NP-40, 100 mmol/l PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 200 mmol/l pepstatin]. Samples (35 μg) were then loaded and run on SDS-PAGE gels, transferred to nitrocellulose membranes, exposed to primary and secondary antibodies, and detected by ECL on autoradiography film.
G6PD activity.
G6PD activity was measured in the protein extracts by estimating the reduction of NADP+ to NADPH (17). NADPH fluorescence was detected at 340 nm (excitation) and 460 nm (emission) by use of an Flx800 microplate fluorescence detector (BioTek Instruments, Winooski, VT).
Cell cytotoxicity.
Necrotic cell death was assessed based on LDH release from cells into the culture medium. LDH assays were performed according to the manufacturer's instructions with LDH-cytotoxicity assay kits from Biovision (Milpitas, CA).
Superoxide assay.
Protein extracted from CD133+ cells was treated with lucigenin (5 μM) in black 96-well plates, after which chemiluminescence was measured with a microplate reader (BioTek Instruments). Readings were normalized to the protein contents of corresponding samples.
Hypoxia-induced pulmonary hypertension and pulmonary arterial hypertension.
To test whether CD133+ cells contribute to pulmonary vascular remodeling we used both moderate hypoxia-induced pulmonary hypertension and severe Sugen5416/hypoxia/normoxia (SU/Hx/Nx)-induced rat PAH models in which, respectively, medial thickening and occlusive neointimal lesions develop. Adult male Sprague-Dawley rats weighing 180 to 220 g were made pulmonary hypertensive by established methods. Severe PAH was induced as described previously (1). Briefly, rats were injected subcutaneously with SU5416 (20 mg/kg) and exposed to normobaric hypoxia (10% O2) for 3 wk. They were returned to normoxia (21% O2) for an additional 5 wk (total 8 wk after SU5416 injection). DHEA was administered to rats in food (1% of daily diet; Teklad Custom Research Diet) for a period of 5 wk (weeks 3 to 8). The dosage of DHEA was based from previous studies (32). The number of CD133+ cells, around 10 vessels (50–200 um diameter), per whole left lobe cross section (5 sections/rat) including the hilum from three groups of rats (6 rats/group) were counted by investigators who were unaware of the source of the sections. The number of CD133+ cells was normalized for the total number of perivascular cells. Staining for CD133 was done with a Vectastain kit. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of South Alabama. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Statistical analysis.
Values are presented as means ± SE. ANOVA and post hoc Student's t-tests were used for analysis. Values of P < 0.05 were considered significant. In all cases, the number of experimental determinations (n) was equal to the number of rats from which pulmonary arteries were harvested for this study.
RESULTS
CD133+ cell numbers increase under hypoxia.
A remarkable property of CD133+ cells is that they self-renew and differentiate. To determine the effect of hypoxia, we compared CD133+ cell numbers/self-renewal between cells cultured under hypoxia (Hx, 3% O2, 5% CO2) or normoxia (Nx, 21% O2, 5% CO2). Both expression and cell numbers of CD133 were increased in cultures exposed to hypoxia (Fig. 1, A and B), as did cell size (Fig. 1C). To determine whether the observed increases in cell number and size reflected a reduction in the incidence of cell death, we used LDH-cell cytotoxicity assays to assess necrotic cell death and found no difference between normoxic and hypoxic cells (data not shown). CD133+ cells apparently thrive under hypoxic conditions.
Fig. 1.

Immunoblots showing an increase in CD133 expression under hypoxia (Hx) (A), which correlated with increases in cell number (B) and cell size (C). Nx, normoxia.
Increased G6PD activity contributes to increased cell growth in hypoxia.
DHEA and 6-aminonicotinamide (6AN), both known to inhibit G6PD (19), suppressed hypoxia-induced increases in G6PD activity in CD133+ cells (Fig. 2A). This inhibition of G6PD also suppressed the hypoxia-induced increases in cell numbers (Fig. 2B) and cell size (Fig. 2C) and tended to increase cytotoxicity (Fig. 2D). This suggests G6PD activity makes a significant contribution to cell growth. We then further confirmed the positive effect of G6PD activity on CD133+ cell growth by knocking down G6PD expression under hypoxic conditions using an adenoviral vector (1013 pfu) harboring a GFP-tagged shRNA targeting G6PD (Fig. 2, E and F). Both G6PD activity and cell numbers were reduced in CD133+ cells transfected with G6PD-shRNA but were unaffected in cells transfected with a vector harboring GFP alone (Fig. 2, G and H).
Fig. 2.
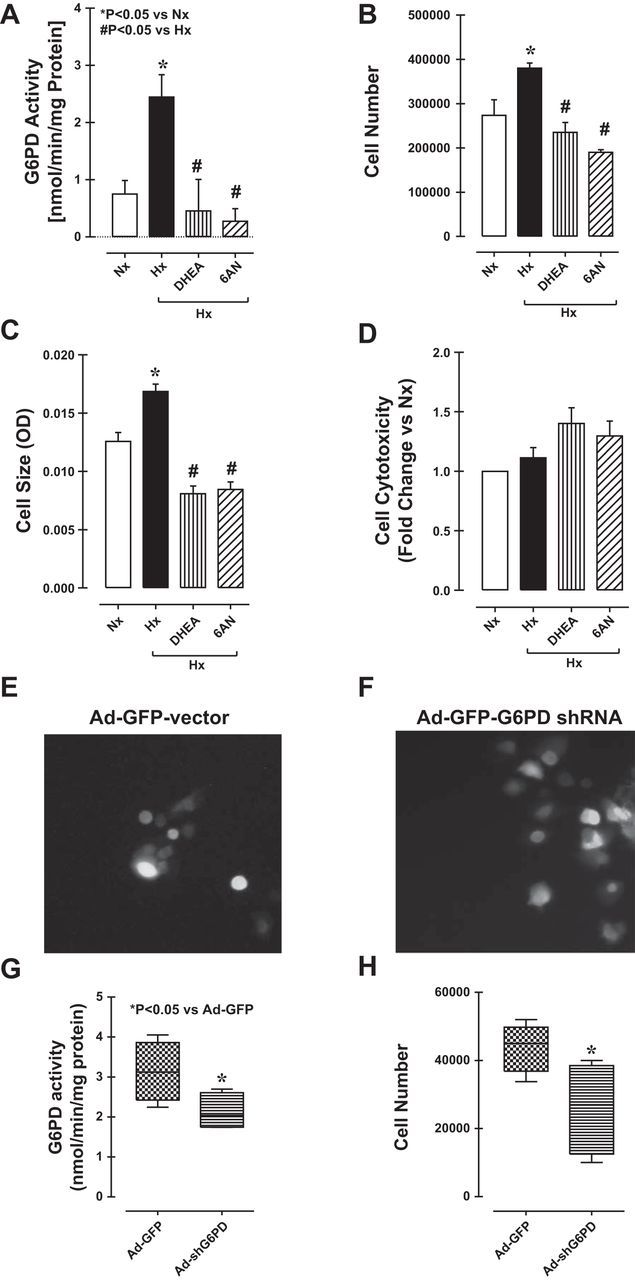
A: hypoxia increased glucose-6-phosphate dehydrogenase (G6PD) activity in CD133+ cells, but the effect was suppressed by dehydroepiandrosterone (DHEA) and 6-aminonicotinamide (6AN). Inhibition of G6PD with DHEA or 6AN also suppressed hypoxia-induced increases in cell number (B) and cell size (C) but slightly increased the incidence of necrotic cell death (D). Fluorescence micrographs showing CD133+ cells transfected with an adenoviral vector encoding green fluorescent protein (GFP) with G6PD-shRNA or GFP alone (E and F). G: G6PD activity was reduced in cells transfected with G6PD shRNA. H: transfection with G6PD shRNA also reduced CD133+ cell numbers.
TGF-β and ROS signaling do not contribute to increased CD133+ cell numbers in hypoxia.
TGF-β signaling is important for self-renewal of stem cells (25), which is mediated via activation of downstream SMAD2/3 signaling (50). To determine whether the same pathway is involved in the self-renewal of CD133+ cells under hypoxia, we treated the cells with or without the TGF-β receptor blocker 6-(2-(1,1-dimethylethyl)-5-(6-methyl-2-pyridinyl)-1H-imidazol-4-yl)quinoxaline (SB525334; 1, 5 and 10 μM; Selleckchem, Houston, TX) (6). SB525334 (1 μM) reduced phosphorylation of SMAD2 but not SMAD3 in CD133+ cells (Fig. 3, A and B). Under normoxia, SB525334 (1 μM) effectively inhibited CD133+ cell growth, but it had no effect on the increase in cell numbers caused by hypoxia (Fig. 3C). In addition, lucigenin chemiluminescence assays showed that ROS (superoxide) levels declined under hypoxia and that inhibition of TGF-β signaling had no effect on superoxide levels under normoxia or hypoxia, compared with untreated controls (Fig. 3D). On the other hand, G6PD activity increased under hypoxia, and SB525344 treatment further increased that activity (Fig. 3E).
Fig. 3.

A and B: SB525334 (1 μM) decreased phosphorylation of SMAD2 but not SMAD3. C: inhibition of TGF-β signaling by treatment with SB525334 for 72 h reduced cell numbers under normoxia (Nx) but not hypoxia (Hx). D: hypoxia reduced superoxide levels in CD133+ cells, as measured by lucigenin chemiluminescence, but SB525334 treatment had no effect on superoxide levels under hypoxia or normoxia. E: G6PD activity was increased in CD133+ cells exposed to hypoxia for 72 h, and that effect was enhanced by SB525334 treatment.
G6PD inhibition reduces expression of HIF1-α and cell cycle proteins.
Because Notch-HIF1α signaling mediates stem cell self-renewal and is required to maintain stem cells in a dedifferentiated state under hypoxia (22), we assessed the levels of HIF1α expression in CD133+ cells cultured under normoxia or hypoxia. We found that HIF1α expression increased under hypoxia was reduced by G6PD inhibition with 6AN (Fig. 4). Moreover, cyclin A and phospho-histone H3 that promote cell proliferation were also elevated in hypoxia and inhibition of G6PD suppressed this increase (Fig. 4).
Fig. 4.
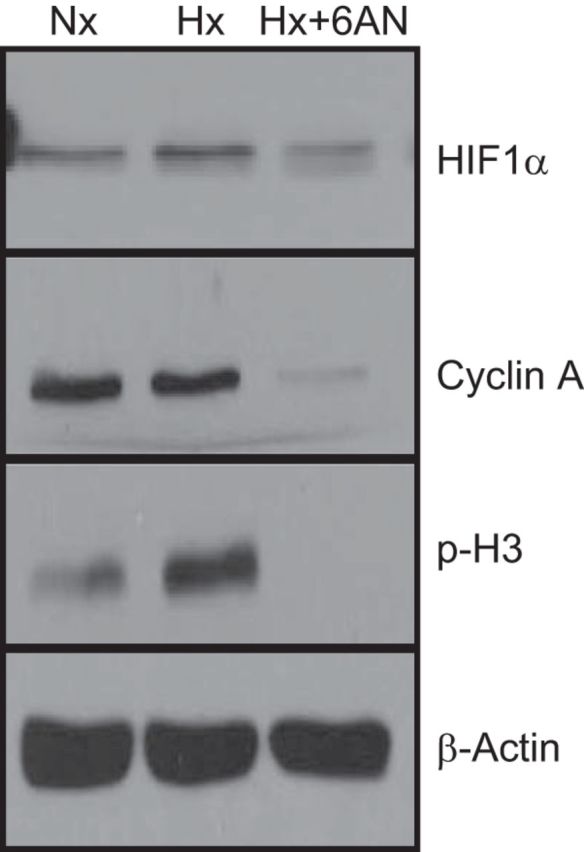
Western blot analysis CD133+ cells exposed to normoxia or hypoxia, with or without 6AN treatment. Inhibition of G6PD with 6AN reduced expression of HIF1-α, cyclin A, and phospho-histone H3 (H3). A representative Western blot of 4 different experiments is shown.
Differentiation of CD133+ cells into smooth muscle-like cells is influenced by G6PD-dependent signaling.
To determine whether G6PD-dependent signaling influences differentiation of CD133+ to PASM-like cells, we cocultured CD133+ cells and PASMCs in a Transwell chamber on opposite sides of a porous membrane (0.4-μm pores) that allowed transmembrane movement of small molecules but not cells (Fig. 5A). We then examined the expression of smooth muscle cell phenotype markers in the CD133+ cells. Immunofluorescent staining showed that CD133+ cells expressed the smooth muscle markers SM22α and α-actin when cocultured with PASMCs but remained undifferentiated in suspension when cultured alone for the same period (Fig. 5, B and C). In addition, significantly more (7.5-fold) CD133+ cells were attached to the membrane after 8 days of coculture with PASMCs than were attached after culture alone for the same period (Fig. 5E). Our next goal was to determine whether PASMC released G6PD-dependent factors that affected the recruitment and differentiation of CD133 cells. Inhibiting G6PD in PASMCs cultured in the lower chamber (see schematic; Fig. 5A) using 6AN or DHEA reduced the number of CD133+ cells attached to the membrane and prevented their expression of SM22α (Fig. 5C). Consequently, the SM22α+-to-total cell ratio was reduced by inhibition of G6PD (Fig. 5, C and F) but the CD133+-to-total cell ratio was unaffected (data not shown). Interestingly, the SM22α+-to-total cell ratio was not reduced by inhibition of G6PD in cocultures exposed to hypoxia (Fig. 5, D and F).
Fig. 5.
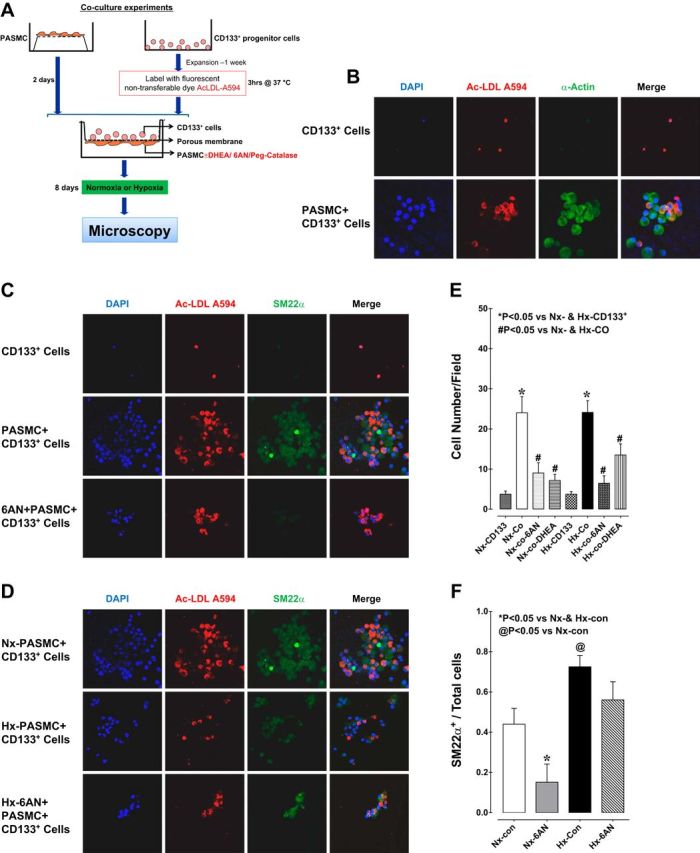
A: schematic representation of the protocol used for coculture of CD133+ cells and pulmonary artery smooth muscle cells (PASMCs) in Transwells under hypoxia or normoxia. Initially, PASMCs were seeded onto the underside of porous Transwell membranes (0.4-μm pores) that allow transmembrane movement of small molecules but not cells. After 2 days, CD133+ cells labeled with Alexa Fluor 594-conjugated Ac-LDL were seeded into the Transwells in the upper chamber. The coculture (Co) setup was then incubated under normoxia or hypoxia for 8 days. G6PD inhibitors (6AN/DHEA) or catalase were added 3 days prior to the end of incubation to PASMCs in the lower chamber. The cells were then fixed, PASMCs were removed, and the CD133+ cells were immunostained and imaged. B: top and bottom, respectively, show staining of CD133+ cells cultured alone or with PASMCs. α-Actin is stained green, CD133+ cells are stained red, and nuclei are stained blue (DAPI). CD133+ cells expressed α-actin only when cocultured with PASMCs. C: CD133+ cells were cultured for 8 days alone (top), with PASMCs (middle), or with PASMCs in the presence of 6AN (bottom). D: CD133+ cells were cocultured for 8 days with PASMCs under normoxia (top), hypoxia (middle), or hypoxia in the presence of 6AN (bottom). 6AN reduced the numbers of cells attached to the membrane and the numbers of SM22α+ cells. E and F: coculture increased the numbers of CD133+ cells attached to the membrane and the numbers of cells expressing SM22α+. These effects were blocked by 6AN.
H2O2 acts as a chemotactic factor that recruits CD133+ cells to PASMCs.
Because G6PD-derived NADPH fuels production of NOX-dependent ROS (12, 42), we tested whether H2O2 was involved in the observed recruitment of CD133+ cells to the Transwell membrane in the presence of PASMCs. When we treated the PASMCs in the lower chamber with pegylated catalase (100 μg/ml), a H2O2 scavenger (Fig. 6A, bright-field; Fig. 6B, DAPI-stained nuclei of CD133+ cells), fewer CD133+ cells attached to the membrane (Fig. 6C). By contrast, attachment of CD133+ cells cultured alone was stimulated by H2O2 (50 μmol/l; Fig. 6D) added to the lower chamber. This suggests that the PASMC-related recruitment of CD133+ cells was mediated, at least partly, by H2O2.
Fig. 6.

Bright-field images (A) and DAPI-stained nuclei (B) of CD133+ cells attached to Transwell membranes in the presence and absence of catalase (Cat). C: addition of catalase in the coculture reduced the numbers of CD133+ cells attaching to membrane under hypoxia. D: bright-field images and DAPI stained nuclei of CD133+ cells attached to Transwell membranes in the presence of H2O2. Peg, pegylated catalase.
G6PD and CD133 colocalize in the pulmonary arterial adventitia in hypoxic rats.
When we used immunohistochemistry to examine the distributions of G6PD and CD133 in consecutive sections of paraffin-embedded lungs from rats exposed to normoxia or hypoxia for 3 wk, we found that cells positive for CD133 and G6PD colocalize in the adventitia (or perivascular space) of pulmonary arteries from chronically hypoxic lungs (Fig. 7A, arrows). By contrast, the normoxic control lungs were negative for CD133. The pulmonary wall thickness, determined as medial wall thickness-to-diameter ratio, increased by 1.92-fold in lungs of hypoxic compared with normoxic control rats. Concurrently, right ventricle-to-left ventricle + septum ratio increased (normoxia: 0.169 ± 0.008 and hypoxia: 0.240 ± 0.009; P < 0.05) in hypoxic rats was reduced by DHEA treatment (0.148 ± 0.005; P < 0.05 vs. hypoxia).
Fig. 7.
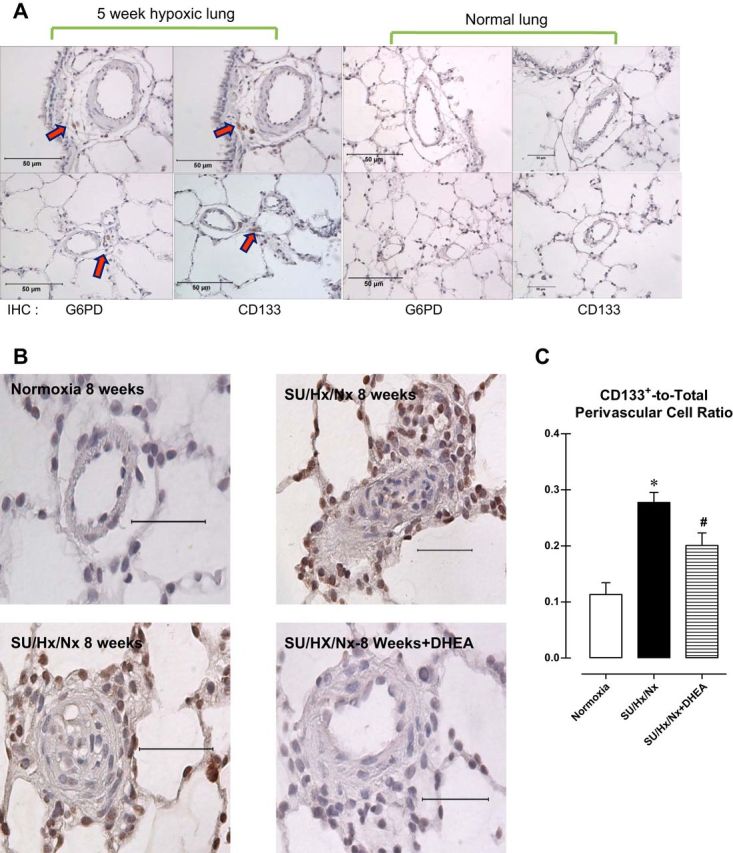
A: immunohistochemical staining of sections of rat lung exposed to normoxia or hypoxia for 5 wk. Arrows indicate cells positive for G6PD and CD133+. B: immunohistochemical staining for CD133 in pulmonary arteries from a normotensive rat (top left), pulmonary arterial hypertensive rat (SU/Hx/Nx 8 wk: top right and bottom left), and pulmonary arterial hypertensive rat treated with DHEA (bottom right). C: summary data showing CD133+ cell accumulation in perivascular regions was decreased by DHEA treatment.
DHEA reduces CD133+ cell numbers and plexiform lesions in the lungs of PAH rats.
CD133+ cells have been implicated in the pathogenesis of PAH. Because these cells accumulate around the abluminal side of the pulmonary arteries in hypertensive rat and human lungs, we speculated that they participate in the pathogenesis of the occlusive neointimal and plexiform lesions associated with the PAH. Immunohistochemical analysis of sections of paraffin-embedded lungs from rats exposed to SU5416+hypoxia for 3 wk plus reexposure to normoxia for 5 wk (SU/Hx/Nx 8 wk) revealed that the numbers of CD133+ cells, mainly around occlusive lesions, were observed in lungs from hypertensive rats (Fig. 7B). Because increased G6PD activity contributes to increased CD133+ proliferation (Fig. 2), we speculated that inhibiting G6PD could decrease CD133+ cell number and reduce occlusive lesion formation. Treating the PAH rats for 5 wk (from week 3 through week 8) with DHEA (1% of total food intake), which blocks G6PD activity in CD133+ cells (Fig. 3A) and other cell types, including vascular smooth muscle cells (10), decreased the accumulation of CD133+ cells (Fig. 7, B and C). Coincidently, DHEA decreased wall thickness (in μl/min normoxia: 0.066 ± 0.014; SU/Hx/Nx: 0.128 ± 0.021; SU/Hx/Nx+DHEA: 0.068 ± 0.012; P < 0.05 normoxia vs. SU/Hx/Nx and SU/Hx/Nx vs. SU/Hx/Nx+DHEA) and increased cardiac output (normoxia: 34,460 ± 3,725; SU/Hx/Nx: 21,133 ± 2,502; SU/Hx/Nx: 29,101 ± 1,805; P < 0.05 normoxia vs. SU/Hx/Nx and SU/Hx/Nx vs. SU/Hx/Nx+DHEA).
DISCUSSION
Pluripotent CD133+ cells give rise to hematopoietic, endothelial, smooth muscle, liver, and neuronal cells. They also can acquire the phenotypes of the cells they grow with and participate in organogenesis, tissue repair, and wound healing and have been implicated in oncogenesis and the pathogenesis of coronary artery disease. They thrive and expand under hypoxia, and so contribute to the pathogenesis of hypoxia-associated diseases like cancer and PAH. However, the signaling pathways that lead to CD133+ cell growth under hypoxia remain obscure. In the present study, we 1) identified G6PD as a pivotal mediator involved in the proliferation and differentiation of CD133+ cells, 2) described a G6PD-associated pathway via which proliferation and differentiation of CD133+ cells appear to be increased under hypoxia, and 3) demonstrated that inhibition of G6PD reduced both CD133+ cell numbers and occlusive lesions in the lungs of PAH rats.
Among the products generated from glucose catabolism is glucose-6-phosphate, which generates electron donors that reduce molecular oxygen to water and produce energy in the mitochondrial respiratory chain. And when the cell has surplus energy, glucose-6-phosphate is shunted into the pentose phosphate pathway to produce NADPH, which is required to protect the cell from oxidative damage and to synthesize fatty acids and cholesterol, as well as the ribose required for de novo synthesis of RNA and DNA. The pentose phosphate pathway is also oxygen sensitive. In coronary arterial smooth muscle, the pentose phosphate pathway activity is decreased by reductions in Po2 (21), but in the pulmonary arterial smooth muscle (10, 18) and in solid tumors (15) the activity is increased by hypoxia. Similarly, we found that G6PD activity is enhanced in hypoxic CD133+ cells, compared with normoxic cells, and inhibition or knockdown of G6PD reduces the hyperproliferation of CD133+ cells seen under hypoxia. Consistent with that finding, increased activity in the pentose phosphate pathway appears to promote growth in other cell types (14, 52). Moreover, changes in cellular energy metabolism and hypoxia promote proliferation and differentiation of neural crest and central nervous system (CNS) stem cell population (30, 44). Collectively, these findings suggest that adaptive metabolic changes mediate the increases in stem cell proliferation induced by hypoxia and that the pentose phosphate pathway plays a key role. We observed that hypoxia had no effect on the incidence of apoptosis in CD133+ cells (data not shown); nonetheless, both G6PD inhibition and knockdown reduced cell numbers without significantly increasing the incidence of necrotic cell death. This finding is consistent with the notion that G6PD plays a role in regulating the growth and death of cells (7) and demonstrates for the first time that activation of G6PD is crucial for mediating sustained CD133+ cell proliferation under hypoxia.
TGF-β upregulates G6PD expression and increases levels of NOX4-derived ROS (45), which in turn stimulate G6PD activity in a variety of cell types, including erythrocytes, hepatocytes, and smooth muscle myocytes. We found that TGF-β receptor blockade significantly reduced CD133+ cell numbers under normoxia but not hypoxia. This suggests the TGF-β pathway does not mediate proliferation of CD133+ cells under hypoxia and that G6PD does not play a role in TGF-β-mediated self-renewal of CD133+ cells under normoxia. In addition, our observation that TGF-β receptor blockade did not affect ROS production in CD133+ cells under either normoxia or hypoxia suggests that TGF-β-associated signaling does not mediate ROS generation in CD133+ cells. Indeed, ROS production was reduced in hypoxic CD133+ cells. This indicates that neither hypoxia-evoked G6PD overactivation nor hyperproliferation of CD133+ cells was mediated by TGF-β/SMAD or ROS signaling. This is not unprecedented, as oxidative stress is known to reduce stem cell growth (5). Moreover, we found that inhibition of G6PD stalled cell growth and reduced hypoxia-induced expression of HIF1α, phospho-histone H3 and cyclin A. Under hypoxia, Notch interacts with HIF1α, recruiting it to Notch-responsive promoters, and prevents differentiation of C2C12 and neural crest stem cells (22). G6PD-derived NADPH stabilizes HIF1α in human mesangial cells (34) and activates NADPH-dependent thioredoxin reductase that promotes HIF-responsive element (HRE) activity in rat PASMCs (46). Taken together with those findings, our results suggest that hypoxia-induced activation of G6PD and elevation of G6PD-derived NADPH promotes CD133+ cell growth, perhaps through upregulation of cell cycle proteins and HIF1α associated with Notch pathways. Besides HIF-Notch pathway the role of other HIF-dependent transcription factors (ETS, Oct3/4, and SOX) that activate CD133 promoter activity (24, 27, 31), in hypoxia-induced growth/self-renewal of CD133+ cells cannot be ruled out.
Consistent with the fact that CD133+ cells differentiate into the vascular endothelial and smooth muscle cell lineages (8, 53), we found that significantly more (7.5-fold) CD133+ cells expressed SM22α and α-actin when cocultured in Transwells with PASMCs. Like neural crest and CNS stem cells, which respectively differentiate into the sympathoadrenal and dopaminergic lineages under hypoxia (22, 30, 44), the numbers of CD133+ cells attaching to the membrane and expressing smooth muscle cell markers significantly increased in hypoxia compared with normoxia. Inhibition of G6PD in PASMCs by 6AN or DHEA reduced the numbers of CD133+ cells recruited to Transwell membranes as well as the numbers expressing SM22α+ under normoxia but not hypoxia. This likely reflects our finding that G6PD-derived NADPH supports NOX-mediated production of PASMCs ROS/H2O2 (18), which acts as a chemoattractant to recruit CD133+ cells. Collectively, then, these findings suggest that CD133+ cell expansion and differentiation are dependent on a G6PD-activated redox pathway under normoxia (Fig. 8). However, although their expansion under hypoxia is also critically dependent on a G6PD-activated redox pathway, interestingly their differentiation is mediated through a G6PD-independent pathway.
Fig. 8.

Schematic illustration demonstrating a potential role of G6PD in hypoxia-induced CD133+ cell proliferation and differentiation. Hypoxia stimulates G6PD in both CD133+ and PASMCs. Increased G6PD activity and G6PD-derived NADPH redox play a potential role in upregulating HIF1α (and phospho-histone H3 cell cycle-promoting proteins) expression and increasing CD133+ cell proliferation. In addition, G6PD-derived NADPH redox fuels NADPH oxidases (Nox)-derived ROS production in the PASMCs. H2O2 produced by PASMCs facilitate CD133+ attachment to the membrane and differentiation to smooth muscle-like cells that express SM22α and α-actin. Dotted arrow depicts diffusion of H2O2.
In hypoxia-induced pulmonary hypertension, circulating progenitor cells infiltrate the arteries and migrate to the adventitia or media. These progenitor cells signal to resident smooth muscle cells to dedifferentiate and proliferate, or they themselves differentiate into smooth muscle cells, leading to pulmonary arterial hypertrophy and hyperplasia (3, 13, 23, 28, 41, 48, 51). Therefore, to determine whether CD133+ contributes to pulmonary vascular remodeling, we used both moderate hypoxia-induced pulmonary hypertension and severe Sugen5416/hypoxia/normoxia-induced PAH models in which, respectively, medial thickening and occlusive neointimal lesions develop. We observed that CD133+ cell proliferation increased in a G6PD-dependent manner under hypoxia (Fig. 7), leading to the accumulation of CD133+/G6PD+ cells within the lungs of moderate and severe pulmonary hypertensive rats. Treatment with DHEA, a well-tolerated steroid hormone widely used to inhibit G6PD in vivo, reduced the numbers of CD133+ cells around the pulmonary arteries of PAH rats. DHEA treatment also reduced occlusive lesions and RV remodeling (2), which typically are seen in SU/Hx/Nx-treated rat lungs (1).
In summary, our findings indicate that G6PD, the rate-limiting enzyme in the pentose phosphate pathway, plays a fundamental role in hypoxia-induced expansion/growth and differentiation of CD133+ cells. Supporting that finding are the observations that 1) cultured cells hyperproliferate and grow in size under hypoxia; 2) hyperproliferation is mediated by overactivation of G6PD via a TGF-β- and ROS-independent pathway; 3) inhibition or knockdown of G6PD downregulates HIF1α and cell cycle promoting proteins and reduces cell number; and 4) treating PAH rats with DHEA, a G6PD blocker, inhibits pulmonary accumulation of CD133+ cells and reduces remodeling of the pulmonary arteries. We therefore propose that the development of second-generation drugs that potently and selectively block G6PD and the pentose phosphate pathway is a promising approach to the treatment of PAH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.C., S.R.J., and A.A.Z. performed experiments; S.C. and S.A. Gebb analyzed data; S.C., S.A. Gebb, I.F.M., R.S.G., and S.A. Gupte interpreted results of experiments; S.C., S.R.J., and A.A.Z. prepared figures; S.C. and S.A. Gupte drafted manuscript; S.R.J., S.A. Gebb, I.F.M., R.S.G., and S.A. Gupte edited and revised manuscript; I.F.M., R.S.G., and S.A. Gupte conception and design of research; I.F.M., R.S.G., and S.A. Gupte approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for S. Chettimada: Department of Cancer Immunology and AIDS, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA.
Present address for S. R. Joshi, R. Gupte, and S. A. Gupte: Department of Pharmacology and Pulmonary Hypertension Center, New York Medical College, Valhalla, NY.
REFERENCES
- 1.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation : 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol : H1708–H1718, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, Hazen SL, Loyd J, Tuder R, Erzurum SC. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood : 1218–1227, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barcelos LS, Duplaa C, Krankel N, Graiani G, Invernici G, Katare R, Siragusa M, Meloni M, Campesi I, Monica M, Simm A, Campagnolo P, Mangialardi G, Stevanato L, Alessandri G, Emanueli C, Madeddu P. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ Res : 1095–1102, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boregowda SV, Krishnappa V, Chambers JW, Lograsso PV, Lai WT, Ortiz LA, Phinney DG. Atmospheric oxygen inhibits growth and differentiation of marrow-derived mouse mesenchymal stem cells via a p53-dependent mechanism: implications for long-term culture expansion. Stem Cells : 975–987, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyd NL, Nunes SS, Jokinen JD, Krishnan L, Chen Y, Smith KH, Stice SL, Hoying JB. Microvascular mural cell functionality of human embryonic stem cell-derived mesenchymal cells. Tissue Eng Part A : 1537–1548, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchakjian MR, Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol : 715–727, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Chan-Ling T, Dahlstrom JE, Koina ME, McColm JR, Sterling RA, Bean EG, Adamson S, Hughes S, Baxter LC. Evidence of hematopoietic differentiation, vasculogenesis and angiogenesis in the formation of human choroidal blood vessels. Exp Eye Res : 361–376, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Chen CT, Hsu SH, Wei YH. Upregulation of mitochondrial function and antioxidant defense in the differentiation of stem cells. Biochim Biophys Acta : 257–263, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol : L64–L74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diez M, Barbera JA, Ferrer E, Fernandez-Lloris R, Pizarro S, Roca J, Peinado VI. Plasticity of CD133+ cells: role in pulmonary vascular remodeling. Cardiovasc Res : 517–527, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med : 1340–1351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diller GP, Thum T, Wilkins MR, Wharton J. Endothelial progenitor cells in pulmonary arterial hypertension. Trends Cardiovasc Med : 22–29, 2010. [DOI] [PubMed] [Google Scholar]
- 14.Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL, Buffa FM, McIntyre A, Harris AL. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab : 751–764, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Frederiks WM, Vizan P, Bosch KS, Vreeling-Sindelarova H, Boren J, Cascante M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. Int J Exp Pathol : 232–240, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR, Gillespie GY. CD133 is a marker of bioenergetic stress in human glioma. PloS One : e3655, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal : 543–558, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem : 19561–19571, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupte SA. Glucose-6-phosphate dehydrogenase: a novel therapeutic target in cardiovascular diseases. Curr Opin Investig Drugs : 993–1000, 2008. [PubMed] [Google Scholar]
- 20.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol : H13–H21, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Gupte SA, Wolin MS. Hypoxia promotes relaxation of bovine coronary arteries through lowering cytosolic NADPH. Am J Physiol Heart Circ Physiol : H2228–H2238, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell : 617–628, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Hayashida K, Fujita J, Miyake Y, Kawada H, Ando K, Ogawa S, Fukuda K. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest : 1793–1798, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Iida H, Suzuki M, Goitsuka R, Ueno H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol : 71–79, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Itoh F, Watabe T, Miyazono K. Roles of TGF-beta family signals in the fate determination of pluripotent stem cells. Semin Cell Dev Biol 2014. [DOI] [PubMed] [Google Scholar]
- 26.Kane NM, Xiao Q, Baker AH, Luo Z, Xu Q, Emanueli C. Pluripotent stem cell differentiation into vascular cells: a novel technology with promises for vascular re(generation). Pharmacol Ther : 29–49, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Lehnus KS, Donovan LK, Huang X, Zhao N, Warr TJ, Pilkington GJ, An Q. CD133 glycosylation is enhanced by hypoxia in cultured glioma stem cells. Int J Oncol : 1011–1017, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Majka SM, Skokan M, Wheeler L, Harral J, Gladson S, Burnham E, Loyd JE, Stenmark KR, Varella-Garcia M, West J. Evidence for cell fusion is absent in vascular lesions associated with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol : L1028–L1039, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, Huertas A, Hammad H, Lambrecht B, Simonneau G, Launay JM, Cohen-Kaminsky S, Humbert M. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med : 116–123, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, Anderson DJ. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell : 499–510, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Ohnishi S, Maehara O, Nakagawa K, Kameya A, Otaki K, Fujita H, Higashi R, Takagi K, Asaka M, Sakamoto N, Kobayashi M, Takeda H. Hypoxia-inducible factors activate CD133 promoter through ETS family transcription factors. PLoS One : e66255, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, Limbird J, Imamura M, Gebb SA, Fagan KA, McMurtry IF. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res : 377–387, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ong LL, Li W, Oldigs JK, Kaminski A, Gerstmayer B, Piechaczek C, Wagner W, Li RK, Ma N, Steinhoff G. Hypoxic/normoxic preconditioning increases endothelial differentiation potential of human bone marrow CD133+ cells. Tissue Eng Part C Methods : 1069–1081, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Osada-Oka M, Hashiba Y, Akiba S, Imaoka S, Sato T. Glucose is necessary for stabilization of hypoxia-inducible factor-1alpha under hypoxia: contribution of the pentose phosphate pathway to this stabilization. FEBS Lett : 3073–3079, 2010. [DOI] [PubMed] [Google Scholar]
- 35.Potgens AJ, Bolte M, Huppertz B, Kaufmann P, Frank HG. Human trophoblast contains an intracellular protein reactive with an antibody against CD133—a novel marker for trophoblast. Placenta : 639–645, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature : 409–414, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Schroder K, Kohnen A, Aicher A, Liehn EA, Buchse T, Stein S, Weber C, Dimmeler S, Brandes RP. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res : 537–544, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Schwarz J. Emerging role of c-kit+ progenitor cells in pulmonary hypertension. Am J Respir Crit Care Med : 5–7, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, Chadburn A, Murphy AJ, Valenzuela DM, Gale NW, Thurston G, Yancopoulos GD, D'Angelica M, Kemeny N, Lyden D, Rafii S. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest : 2111–2120, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF, Park DM. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene : 3949–3959, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J, Prockop DJ. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J : 1226–1236, 2008. [DOI] [PubMed] [Google Scholar]
- 42.Spencer NY, Yan Z, Boudreau RL, Zhang Y, Luo M, Li Q, Tian X, Shah AM, Davisson RL, Davidson B, Banfi B, Engelhardt JF. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J Biol Chem : 8977–8987, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res : 675–691, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci : 7377–7383, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teruel T, Valverde AM, Benito M, Lorenzo M. Transforming growth factor beta 1 induces mitogenesis in fetal rat brown adipocytes. J Cell Physiol : 577–584, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Tipple TE, Nelin V, Gin Y, Rogers L, Nelin L. Thioredoxin-1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation. FASEB J : 873.–10., 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, Rocco G, Pirozzi G. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg : 446–453, 2009. [DOI] [PubMed] [Google Scholar]
- 48.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, Hecker M, Zhu Z, Gehling U, Seeger W, Pepke-Zaba J, Morrell NW. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med : 780–787, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanderheyden M, Vercauteren S, Mansour S, Delrue L, Vandekerckhove B, Heyndrickx GR, Van Haute I, De Bruyne B, Timmermans F, Wijns W, Bartunek J. Time-dependent effects on coronary remodeling and epicardial conductance after intracoronary injection of enriched hematopoietic bone marrow stem cells in patients with previous myocardial infarction. Cell Transplant : 919–925, 2007. [DOI] [PubMed] [Google Scholar]
- 50.Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood : 1250–1256, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Yao W, Firth AL, Sacks RS, Ogawa A, Auger WR, Fedullo PF, Madani MM, Lin GY, Sakakibara N, Thistlethwaite PA, Jamieson SW, Rubin LJ, Yuan JX. Identification of putative endothelial progenitor cells (CD34+CD133+Flk-1+) in endarterectomized tissue of patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol : L870–L878, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science : 975–980, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin L, Ohanyan V, Pung YF, Delucia A, Bailey E, Enrick M, Stevanov K, Kolz CL, Guarini G, Chilian WM. Induction of vascular progenitor cells from endothelial cells stimulates coronary collateral growth. Circ Res : 241–252, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature : 603–607, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]