

Abstract
Recognition that coronary blood flow is tightly coupled with myocardial metabolism has been appreciated for well over half a century. However, exactly how coronary microvascular resistance is tightly coupled with myocardial oxygen consumption (MV̇o2) remains one of the most highly contested mysteries of the coronary circulation to this day. Understanding the mechanisms responsible for local metabolic control of coronary blood flow has been confounded by continued debate regarding both anticipated experimental outcomes and data interpretation. For a number of years, coronary venous Po2 has been generally accepted as a measure of myocardial tissue oxygenation and thus the classically proposed error signal for the generation of vasodilator metabolites in the heart. However, interpretation of changes in coronary venous Po2 relative to MV̇o2 are quite nuanced, inherently circular in nature, and subject to confounding influences that remain largely unaccounted for. The purpose of this review is to highlight difficulties in interpreting the complex interrelationship between key coronary outcome variables and the arguments that emerge from prior studies performed during exercise, hemodilution, hypoxemia, and alterations in perfusion pressure. Furthermore, potential paths forward are proposed to help to facilitate further dialogue and study to ultimately unravel what has become the Gordian knot of the coronary circulation.
Keywords: coronary circulation, coronary venous Po2, local metabolic control, myocardial oxygen consumption
INTRODUCTION
Mythology provides the legend of Gordius, who upon becoming king of Phrygia, dedicated his chariot to Zeus and fastened it to a pole with an impossible knot. An oracle predicted that whoever could disentangle the knot would be the future king of Asia. Many tried and failed, until an impatient Alexander the Great took out his sword and solved the conundrum by merely slicing through the knot. The “Gordian knot” has thus come to symbolize a complex and impossible problem to solve. This metaphor is applicable to the coronary circulation where coronary blood flow is inextricably tied to myocardial metabolism as perfusion not only dictates the substrate supply for, but remains primarily dependent on, the level of oxidative phosphorylation (9, 27, 47). The fundamental coupling between coronary flow and metabolism is essential in a highly metabolically active tissue such as the heart, where baseline levels of oxygen extraction typically exceed 70% (28, 40, 105). It is within these basal constraints that local metabolic mechanisms are proposed to predominate to ensure that alterations in myocardial oxygen consumption (MV̇o2) are balanced by commensurate changes in myocardial oxygen delivery (Fig. 1A) such that cardiac contractile function and output are adequately maintained across a wide range of (patho-)physiologic perturbations (106). Exactly how coronary microvascular resistance is precisely coupled with the underlying level of MV̇o2 remains one of the most highly contested mysteries of the coronary circulation to this day.
Fig. 1.

Classic relationships between coronary blood flow and myocardial oxygen consumption (A), coronary blood flow and coronary venous Po2 (B), and coronary venous Po2 and myocardial oxygen consumption (C) in dogs.
The central tenet of the local metabolic hypothesis of coronary blood flow control proposes that production of vasodilator metabolites corresponds with reductions in myocardial tissue Po2 and thus acts in a negative feedback manner to preserve tissue oxygenation within normal physiological limits (23, 27, 40, 47). Accordingly, the ability to assess how myocardial tissue Po2 is influenced by variations in key determinants of myocardial perfusion (i.e., MV̇o2, arterial oxygen levels and coronary perfusion pressure) is essential for the delineation of mechanisms responsible for local metabolic control of coronary blood flow. To this end, coronary venous Po2 has been proposed, and generally accepted, to directly represent underlying changes in myocardial oxygenation (23, 40, 58). This theory is directly supported by data that demonstrate that coronary blood flow is progressively increased as coronary venous Po2 (i.e., myocardial tissue Po2) falls below a critical threshold value of ~18–20 mmHg in conscious dogs at rest and during exercise induced increases in MV̇o2 (Fig. 1B) (106). As such, changes in coronary venous Po2 relative to MV̇o2 have been utilized as a means to directly assess the balance between coronary blood flow and MV̇o2 (Fig. 1C) (27, 28, 105, 106). Within this paradigm, if increases in coronary blood flow perfectly match increases in MV̇o2, coronary venous Po2 remains unchanged as cardiac workload is elevated. Alternatively, if/when a pathway that contributes to local metabolic coronary vasodilation is inhibited, the impaired flow response results in reductions in myocardial oxygenation and enhanced oxygen extraction, both of which act to significantly steepen the relationship between coronary venous Po2 and MV̇o2 (27, 47).
Herein begins the circular reasoning that is inherent to the coupling between coronary blood flow and myocardial metabolism (Fig. 2). First, it is undisputed that MV̇o2 is the primary determinant of coronary blood flow. However, it must be recognized that inhibition of any mechanism that contributes to metabolism-mediated increases in coronary flow will diminish myocardial oxygen delivery. Furthermore, if/when this reduction is severe enough, MV̇o2 (the principal factor driving the flow response) will be decreased commensurate with the degree of fall in oxygen delivery (47, 105). Second, coronary venous Po2 simultaneously represents the proposed “error signal” responsible for the production of local metabolites (i.e., myocardial tissue Po2) as well as the resultant of the overall balance between myocardial oxygen supply and consumption (106). While this is a logical extension of any variable controlled by a negative feedback system, conclusions regarding changes (or lack thereof) in coronary venous Po2 can prove quite challenging given that a perfect balance between coronary flow and MV̇o2 (Fig. 1C) is not consistent with reductions in tissue Po2 driving the response (Fig. 1B). Lastly, interpretation of changes in coronary venous Po2 is also confounded by the fact that increases in myocardial oxygen extraction reduce coronary venous Po2, thus leaving one to question whether alterations in coronary venous Po2 are driven primarily by changes in myocardial oxygen extraction, myocardial tissue Po2, or perhaps both. Taken together, the dynamic interplay between these essential elements of coronary control continues to generate much debate regarding the most appropriate means by which to assess the balance between coronary blood flow and MV̇o2. Below we consider several situations that highlight the difficulties in interpreting this complex interrelation and the arguments that emerge from the different conclusions that can be drawn from these scenarios. Finally, we consider potential paths forward to help resolve this physiologic Gordian knot by proposing updated criteria of local metabolic mechanisms that seek to simplify analyses and downplay constraints of continued overreliance on interpretation of changes in coronary venous Po2. This review is principally focused on short-term adaptations in coronary blood flow (i.e., acute intervention studies) but not on chronic changes to exercise, pressure, or flow and does not aim to provide a detailed discussion of individual metabolites that have been proposed to contribute to metabolic coronary vasodilation. For these aspects, readers are referred to several comprehensive reviews by Duncker et al. (27, 30), Canty et al. (14), and Goodwill et al. (47).
Fig. 2.
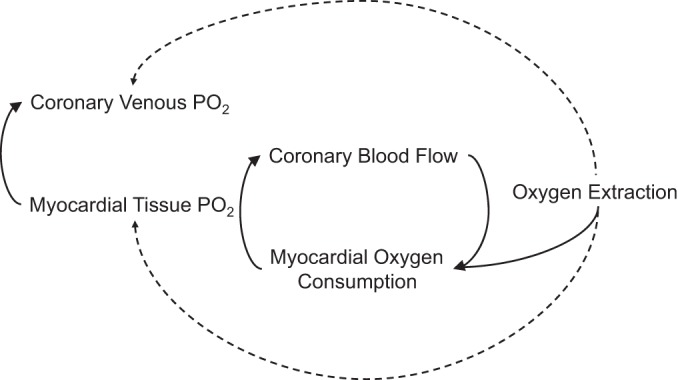
Schematic representation of the interrelations between the primary variables proposed to modulate local metabolic control of coronary blood flow.
DIFFICULTIES WITH ASSESSMENT AND INTERPRETATION OF LOCAL METABOLIC CONTROL
There is a substantial amount of evidence that the relationship between coronary blood flow and MV̇o2 is linear and extremely consistent across many species, including mice (84), rats (41), dogs (3), swine (70), horses (77), and humans (53) (Fig. 3; heart rate used as index of MV̇o2) [see Duncker et al. (27–29) for more detailed review]. However, it should be appreciated that reported normalized values of coronary blood flow (per gram myocardium perfused) in mice are substantially (up to an order of magnitude) higher compared with other (larger) species (84). Nonetheless, within this highly conserved consistency it is reasonable to surmise that disruption of any pathway that contributes to balance between oxygen delivery and metabolism should produce proportional reductions in the slope of this relationship. However, practical application of this premise is flawed for a few reasons. Initially, it is important to recognize that MV̇o2 itself is typically not measured but calculated from the Fick principle, i.e., coronary blood flow times myocardial oxygen extraction (arterial minus coronary venous oxygen content) (47). As such, plotting coronary blood flow (or coronary venous Po2) relative to MV̇o2 results in a certain degree of redundancy and raises questions of validity as specific variables are plotted relative to themselves (57). Next, the relationship between coronary blood flow and MV̇o2 operates near the maximum level of oxygen extraction, and thus the slope of this relationship cannot be appreciably decreased within these tight physiologic constraints. Consequently, alterations in the slope of the coronary blood flow versus MV̇o2 relationship cannot be utilized as a means to reliably assess the contribution of specific mechanisms of local metabolic coronary vasodilation [see Goodwill et al. (47) for more in depth review]. Thus the field has largely relied on assessment of changes in coronary venous Po2 relative to MV̇o2 as a means to interpret the overall balance between myocardial oxygen delivery and metabolism (27, 47, 58, 105, 106).
Fig. 3.
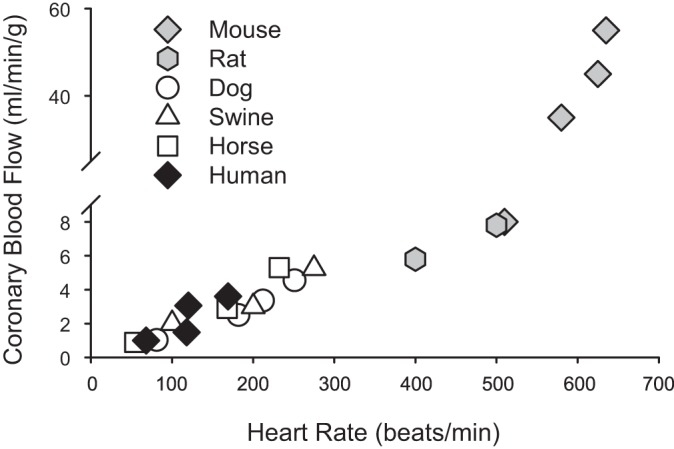
Relationship between coronary blood flow and heart rate [index of myocardial oxygen consumption (MV̇o2)] across multiple species: mouse (84), rat (41), dog (3), swine (70), horse (77), and human (53).
Interpretation of Cause Versus Effect Across Conditions and Species
Classically, studies in conscious, instrumented dogs reliably demonstrate significant reductions in coronary venous Po2 with exercise-induced increases in MV̇o2 (Fig. 4) (27, 47, 49, 106). These data, which represent global, timed averaged snapshots of these variables, support the hypothesis that reductions in myocardial tissue Po2 (oxygen-sensing mechanisms) are responsible for, or at least significantly contribute to, the production of local metabolites and increases in coronary blood flow (Fig. 4A) (52, 111). However, it is evident that these reductions in coronary venous Po2 in dogs (typically from ~20 mmHg at rest to ~15 mmHg with exercise) are not linear and characteristically plateau as MV̇o2 increases above ~150 μl O2·min·−1·g−1 (Fig. 4B). This response lies in stark contrast with data from swine (43, 54, 81) and humans (53, 55), which demonstrate relatively modest changes (~0–1 mmHg) in coronary venous Po2 with approximately two- to fourfold increases in MV̇o2; i.e., relationship between coronary venous Po2 and MV̇o2 remains essentially flat (Fig. 4B). One reason for these discrepant results is the involvement of different mechanisms/pathways that modulate coronary responses to increased MV̇o2 in a species-dependent manner. In particular, prior studies of autonomic control of coronary blood flow in dogs determined that inhibition of α-adrenergic receptors significantly reduces the slope of the relationship between coronary venous Po2 and MV̇o2 (27, 47, 52). In contrast, α-adrenergic blockade has no effect on coronary venous Po2 at rest or during increases in MV̇o2 in swine (31, 96). Thus paradoxical α-adrenergic vasoconstriction appears to largely account for the distinct relationship between coronary venous Po2 and MV̇o2 reported in dogs (Fig. 4B). Taken together, these findings collectively argue against a requisite role for myocardial tissue Po2 as the error signal for local metabolic vasodilation.
Fig. 4.

Relationship between coronary blood flow and coronary venous Po2 (A) and coronary venous Po2 and myocardial oxygen consumption (B) across multiple species: dog (38, 51, 110), swine (43, 54, 81), and human (53, 55).
The discrepancies outlined above also relate to the more recent proposal of the adenine-nucleotide hypothesis (38, 51). This relatively new paradigm postulates that erythrocytes act as oxygen sensors in coronary capillary beds, wherein hemoglobin saturation declines and results in the release of ATP with subsequent activation of endothelial purinergic receptors and the initiation/propagation of a retrograde conducted vasodilator response (49). While there is evidence to support this hypothesis (38, 51, 87), the effective relationship between coronary blood flow and coronary venous hemoglobin saturation (Fig. 5) produces qualitatively similar responses to that of coronary venous Po2 across species (Fig. 4). Here again, data from exercising dog studies are most consistent with this postulate as reductions in coronary venous hemoglobin saturation of up to 10–12% are associated with an ~65% increase in coronary venous plasma ATP concentration and a two- to threefold increase in coronary blood flow (38, 51). In contrast, markedly higher slopes are observed in swine (43, 81) and humans (36, 55), with 1–5% reductions in coronary venous hemoglobin saturation corresponding with two- to fourfold increases in coronary blood flow (Fig. 5). Given these findings, and the fact that the unique relationship in dogs is essentially abolished by inhibition of α-adrenergic receptors (27, 47, 52), it seems unlikely that such modest changes in hemoglobin saturation are the primary sensor responsible for linking myocardial oxygen delivery with metabolism. Furthermore, questions and inconsistencies regarding interpretation of changes in coronary venous Po2 are also relevant to reported alterations in coronary venous hemoglobin saturation by virtue of their essential physiologic relationship.
Fig. 5.
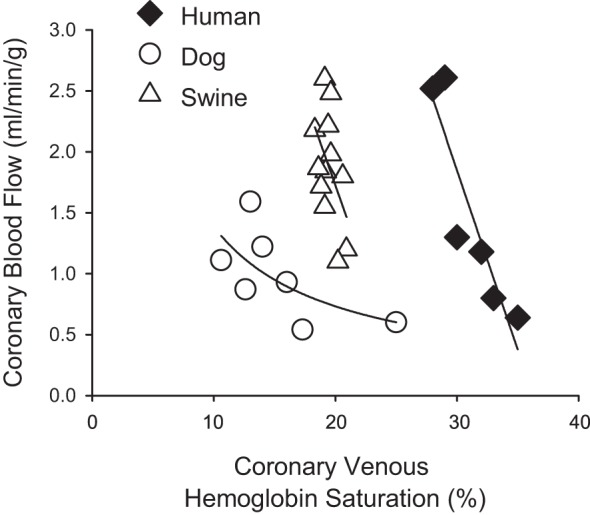
Relationship between coronary blood flow and coronary venous hemoglobin saturation across multiple species: dog (38, 51), swine (43, 81), and human (36, 55).
On the surface, reasons to question whether measurements of coronary venous Po2 reflect a given level of myocardial tissue oxygenation are few. Although evidence of arteriovenous shunting and countercurrent exchange in the heart has been documented (6, 16, 24, 92), diffusional shunting of oxygen is quite limited and impeded by hemoglobin binding (63), i.e., consistent with very low values of coronary venous Po2 reported across multiple species (27, 40, 47). However, it is important to appreciate that species differences in coronary vascular structural patterns exist (5, 42, 44, 60, 62) and that reported values of coronary venous Po2 (and other primary response variables) represent global steady-state values that may not necessarily reflect the highly dynamic processes that correspond with oscillatory (20, 94, 112, 119) and transmurally heterogeneous variations (27, 104, 105) across the coronary circulation. These phenomena lead to issues with variability in coronary response data both within and between studies, which are further complicated by inconsistencies in the measurements themselves. While recognition of these confounding influences is critical, evaluation of static measures of key coronary response variables clearly provides insight into the regulation of coronary blood flow in response to a variety of patho-physiologic perturbations. Nevertheless, interpretation of changes in these variables, such as coronary venous Po2, can be quite nuanced. This point is illustrated by comparing coronary responses to anemia versus hypoxemia. Numerous studies have demonstrated that increases in coronary blood flow to reductions in hematocrit (hemodilution) (18, 19, 46, 65, 76, 113) or arterial Po2 (hypoxemia) (37, 45, 56, 75, 82, 114, 115) are quite similar (Fig. 6A) and that these increases in flow are sufficient to maintain overall levels of myocardial oxygen delivery (Fig. 6B). Despite these comparable effects, which reportedly occur over relatively similar ranges of MV̇o2 (~50–125 μl O2·min·−1·g−1), coronary venous Po2 consistently falls in response to hypoxemia (Y = 0.94x + 6.4; r = 0.75) yet remains unchanged in response to hemodilution (Y = 0.14x + 20.5; r = 0.28; Fig. 6C; P value for slope < 0.01). Such divergent responses have been demonstrated across a wide variety of species, including dogs (18, 19, 37, 56, 76, 82, 114), lambs (74), swine (45, 65, 68, 113), and humans (46). Based on the apparent preservation of oxygen delivery at comparable levels of MV̇o2 in both cases, it seems unlikely that the reduction of coronary venous Po2 in response to hypoxemia represents a progressive decline of myocardial tissue oxygenation. While these divergent responses implicate the presence of distinct vasodilator mechanisms, underlying differences between viscosity, shifts in oxy-hemoglobin dissociation curve, and/or diffusion properties must also be acknowledged (76, 116). Interestingly, data from the Feigl laboratory (7, 95) demonstrate discrepant, nonlinear relationships between myocardial oxygenation (assessed by mean myoglobin saturation) and venous Po2 in isolated, buffer-perfused guinea pig hearts in the absence and presence of erythrocytes (5% hematocrit). Thus, while it is readily apparent that the heart possesses exquisitely sensitive oxygen-sensing mechanisms that act to maintain adequate myocardial oxygen delivery, exactly how these pathways are sensed and regulated remains poorly understood [see Duncker et al. (27) and Goodwill et al. (47) for more comprehensive review].
Fig. 6.

Relationship between coronary blood flow (A), myocardial oxygen delivery (B), coronary venous Po2 (C) relative to arterial oxygen content in response to hemodilution (solid line; Refs. 18, 19, 46, 65, 76, 113) and hypoxemia (dashed line; Refs. 37, 45, 56, 74, 82, 114, 115). Arterial oxygen content was used to normalize the level of oxygen deficit in each study.
Local Metabolic Control in Response to Changes in Coronary Perfusion Pressure
The coronary circulation has the intrinsic ability to maintain blood flow relatively constant over a wide range of perfusion pressures (1). This autoregulatory capacity is especially crucial in times of coronary stenosis, where hypoperfusion can lead to rapid reductions in cardiac function and myocardial injury (39, 59). While this phenomenon is distinctly different from a pure metabolic stimulus (e.g., exercise) in that it involves changes in perfusion pressure and myocardial ischemia, the most prominent and accepted theory to explain coronary pressure-flow autoregulation centers around local metabolic regulation of coronary vascular resistance (23, 39). In this context, the local metabolic hypothesis proposes that myocardial oxygen tension determines the coronary autoregulatory capacity by increasing the production of vasodilator metabolites as perfusion pressure is reduced. This contention is supported by the classic study of Dole and Nuno (23), which demonstrated that coronary venous Po2 decreases with coronary perfusion pressure and that autoregulatory gain [equal to
where a value of 1 represents perfect autoregulation] is dependent on normal physiologic levels of myocardial tissue Po2, i.e., coronary venous Po2 < 32 mmHg. This association between coronary venous Po2 and perfusion pressure has been demonstrated by a number of laboratories in dogs (4, 100) and swine (11, 64). Thus there is substantial evidence to support that mechanisms closely associated with reductions in coronary venous Po2 are directly associated with decreases in coronary microvascular resistance in response to changes in perfusion pressure and/or hypoxemia (25). However, whether the reduction in coronary venous Po2 reflects a cause of autoregulatory behavior or a consequence of lowered perfusion pressure and augmented myocardial oxygen extraction remains unclear.
To examine the extent to which coronary autoregulatory behavior is contingent on normal physiologic levels of coronary venous Po2, Kiel et al. (64) examined coronary responses to changes in perfusion pressure (140–40 mmHg) in the absence and presence of euvolemic anemia (50% reduction in hematocrit) and infusion of dobutamine to augment MV̇o2. Hemodilution was utilized in this study as reductions in hematocrit diminish coronary vasomotor tone with little/no change in coronary venous Po2 (17, 65, 76). Consistent with prior studies supporting local metabolic control mechanism in the autoregulatory response (4, 23), autoregulatory gain (determined by changes in coronary flow in response to 20 mmHg increments at pressures ranging from 120 to 60 mmHg) remains relatively constant over coronary venous Po2 values ranging from ~30 to ~10 mmHg (Fig. 7B; Y = −0.01x + 0.6; r = 0.24). In contrast, autoregulatory capacity significantly decreased (P value for slope = 0.03) over a similar range of coronary venous Po2 values in the presence of hemodilution and dobutamine (Fig. 7B; Y = 0.03x −0.7; r = 0.55). These findings indicate that the local metabolic hypothesis is not sufficient to explain autoregulatory behavior. Interestingly, additional data from the study of Kiel et al. (64) suggest that the primary mechanism of autoregulation could be more myogenic in origin, as coronary zero flow pressure (index of overall vascular tone) was highly predictive of changes in flow and autoregulatory gain. Nevertheless, this conclusion, along with those for other scenarios such as exercise, hemodilution, and hypoxemia, rests on the general assumption that has been applied to coronary studies for decades; i.e., coronary venous Po2 is a valid measure of myocardial Po2. However, each of these examples highlight clear inconsistencies that not only obscure interpretation but provide reasons to question whether this is truly always the case or not (21).
Fig. 7.

Relationship between coronary blood flow and coronary perfusion pressure (A) and autoregulatory gain (change in coronary flow over 20-mmHg increments at pressures ranging from 120 to 60 mmHg) relative to coronary venous Po2 (B). Responses are plotted in the absence (solid line) and presence of euvoluemic hemodilution (dashed line) (~50% reduction in hematocrit) plus dobutamine (increase heart rate ~75–100% above baseline levels). Data from Kiel et al. (64).
Additional Considerations in Interpreting Potential Local Metabolic Mechanisms
Traditional interpretation of coronary response variables has held that inhibition of a factor that has a sustained influence on coronary microvascular resistance will produce a reduction in coronary venous Po2 (or venous saturation) across a comparable range of MV̇o2, with little/no effect on coronary blood flow (27, 28, 47, 49, 51, 105, 107). One classic example of this are studies that have demonstrated a parallel downward shift in the relationship between coronary venous Po2 and MV̇o2 following the inhibition of nitric oxide synthase (Fig. 8B). These studies have largely concluded that nitric oxide exerts a modest, tonic vasodilator influence in the coronary circulation but that it is not required for local metabolic coronary vasodilation (2, 26, 61, 80, 98, 109). However, this downward shift is associated with a relatively modest increase in the relationship between coronary blood flow and MV̇o2 (Fig. 8A). One hypothesis to explain these divergent responses is that reductions in coronary venous Po2 following nitric oxide synthesis inhibition reflect an augmented error signal for metabolic vasodilation such that the magnitude of exercise-mediated coronary vasodilation is slightly augmented (51). Another hypothesis that has not been readily considered is that nitric oxide itself has been demonstrated to be an enhancer of oxygen transfer from erythrocytes, i.e., produces a right shift of the oxygen-hemoglobin dissociation curve (66, 67). Consequently, inhibition of nitric oxide production would be postulated to result in a leftward shift of the dissociation curve, thereby providing a potential explanation for a reduction in coronary venous Po2 in the absence of any reduction in coronary blood flow or MV̇o2. Another example of such an effect involves the disruption of adenosine receptor signaling, which has been proposed as a target of sickle cell disease as adenosine augments 2,3-diphophoglycerate (2,3 DPG) production from erythrocytes (120). Thus nonselective inhibition of adenosine receptors with antagonists such as 8-phenyltheophylline (8-PT) would also be predicted to produce a leftward shift of the oxyhemoglobin dissociation curve. Such a consequence could indeed explain the paradoxical findings that 8-PT has no effect on coronary blood flow yet produces a parallel downward shift of the relationship between coronary venous Po2 and MV̇o2 (32, 61, 73, 110) at the same time as measured values of adenosine are reported to be below the threshold value necessary for vasodilation to occur (110). Accordingly, more rigorous consideration of potential influences of antagonists on hemoglobin binding affinity is warranted before definitive conclusions regarding the role of specific pathways can be made.
Fig. 8.

Relationship between coronary blood flow (A) and coronary venous Po2 (B) vs. myocardial oxygen consumption in conscious instrumented dogs at rest and during graded treadmill exercise. Responses are plotted in the absence and presence of nitric oxide synthesis inhibition with N-nitro-l-arginine. Data replotted from Tune et al. (109).
Data that chronic alterations in oxygen transport in the coronary circulation can influence coronary response variables have also been documented. In particular, prior studies in exercise trained animals suggest that augmented levels of myocardial oxygen extraction, hence lower values of coronary venous Po2 (117), are likely related to training-induced increases in the capillary transport capacity (69, 71, 72, 85, 118). Conversely, reductions in coronary venous Po2 in obese (10, 13, 97, 103) and diabetic, dyslipidemic (8, 99) swine have been shown to be directly associated with reductions in capillary density (rarefaction) and overall reductions in myocardial lactate consumption (13). The presence of capillary rarefaction in chronic disease would act to reduce oxygen extraction capacity such that reductions in coronary venous Po2 likely reflect impaired myocardial oxygenation. Regardless, potential alterations in the coronary circulation should be duly considered when interpreting changes in coronary venous Po2 in the setting of chronic disease states.
APPROACHES TO ASSESS LOCAL METABOLIC CONTROL OF CORONARY BLOOD FLOW
The ability to resolve what has been the inextricable Gordian knot of the coronary circulation lies first in the general agreement of the most appropriate means by which to assess the question. Unfortunately, this essential component remains contested as members of the field continue to debate the paradox of whether coronary venous Po2 (Fig. 4) or hemoglobin saturation (Fig. 5) represents the error signal (cause) for metabolic vasodilation (51, 87) versus the consequence of an imposed imbalance between flow and metabolism (27, 28, 47, 106) or somehow both the beginning and end of the knot itself. Accordingly, it has been essentially impossible to arrive at any consensus when differences in anticipated experimental outcomes and data interpretation persist. Below we propose a potential, more straightforward path for assessing and interpreting the contribution of local metabolic mechanisms based on essential underlying experimental findings.
Fundamental a Priori Prediction
When considering how to determine whether a specific factor or pathway contributes to local metabolic coronary vasodilation, the most fundamental assumption that can be made, a priori, is that inhibition of the factor (or pathway) should diminish metabolic-mediated increases in coronary blood flow. As outlined above, such an effect cannot manifest as a marked reduction in the slope of the relationship between coronary blood flow and MV̇o2, but there is evidence that this response results in a commensurate decrease in MV̇o2 itself. There are two primary examples of this in the literature. First, studies in both dogs (93) and swine (10, 48) have demonstrated that inhibition of voltage-gated K+ (KV) channels diminishes coronary blood flow (Fig. 9A) and MV̇o2 (Fig. 9B), during cardiac pacing, catecholamine infusion, and exercise-mediated increases in myocardial metabolism. Interestingly, despite baseline reductions in coronary venous Po2, these changes were not accompanied by progressive reductions in venous Po2 as cardiac workload was elevated (Fig. 9C). Alternatively, studies in mice lacking smooth muscle KV1.5 channels demonstrated marked reductions in the relationship between myocardial blood flow and cardiac work, diminished myocardial tissue Po2, and cardiac contractile dysfunction (84). There are several potential explanations for the more modest effect of KV channel blockade in large animals, including partial antagonism with pharmacologic inhibition, steady-state measurements in the large animal studies versus dynamic state measurements in the mouse studies, and/or that myocardial Po2 and coronary venous Po2 are distinctly different variables. Regardless, the truncation of the MV̇o2 response adheres to the law of mass balance and fits with the primary expectation of antagonist effect; i.e., reduction of the overall flow response limits the ability to increase MV̇o2. For these reasons, pathways that converge on KV channels are proposed to be involved in local metabolic control of coronary blood flow (47, 105). However, corresponding reductions in coronary flow and MV̇o2 elicit a “chicken or egg” argument as to whether the response is mediated by increases in coronary microvascular resistance versus primary reductions in myocardial metabolism.
Fig. 9.

Coronary blood flow (A), myocardial oxygen consumption (B), and coronary venous Po2 (C) at rest and during exercise in the absence and presence of the KV channel inhibitor 4-aminopyridine (4-AP) in conscious instrumented swine. Data from Berwick et al. (10). *P < 0.05 vs. control.
Another case consistent with this axiom are studies that have examined redundant and compensatory mechanisms invoked in response to the inhibition of ATP-sensitive K+ (KATP) channels. These studies reliably show that inhibition of KATP channels with glibenclamide produces reductions in coronary blood flow and MV̇o2 at rest and during increases in metabolism in dogs (33, 88, 89) as well as swine (78, 79). Furthermore, studies by the Bache laboratory (34, 35, 61) demonstrate that the addition of adenosine receptor (8-PT) and nitric oxide synthesis [N-nitro-l-arginine (LNNA)] inhibitors results in further progressive reductions in the coronary blood flow response to graded treadmill exercise (Fig. 10A). These reductions in coronary flow were accompanied by decreases in cardiac contractile function (systolic wall thickening) and MV̇o2 (Fig. 10B) (34, 35, 61), as would be expected with impairments in myocardial oxygen delivery. These data indicate reductions in coronary flow produced by glibenclamide were sufficient to induce compensatory increases in myocardial adenosine release and in the contribution of nitric oxide to exercise-induced increases in coronary blood flow. Importantly, subsequent experiments by the Bache laboratory (35, 61) directly addressed the chicken or egg conundrum by demonstrating that the restoration of coronary blood flow to normal, untreated control values (with sodium nitroprusside, which by itself had no effect on regional contractile function) in the presence of glibenclamide and 8-PT (35) completely restored values of MV̇o2 (Fig. 10C) and systolic wall thickening to normal levels under baseline resting conditions (Fig. 10D). These data are critical in confirming that the reductions in MV̇o2 in the presence of glibenclamide and 8PT were related to deficits in coronary blood flow and not to a direct negative inotropic effect or to inhibition of mitochondrial respiration. These data are further corroborated by the lack of effect of the mitochondrial selective KATP channel blocker 5-hydroxydecanoate (15). Taken together, these findings provide direct evidence that inhibition of a specific pathway that contributes to the regulation of coronary blood flow diminishes the coronary flow response and is accompanied by proportional (and correctable) reductions in MV̇o2. Further support for this contention can be found in prior studies designed to test the adenosine hypothesis, in that inhibition of adenosine signaling was shown to diminish exercise-induced increases in coronary blood flow only in the presence of a coronary stenosis (73). In other words, reductions in coronary blood flow are indeed evident when a specific factor involved in the response is inhibited.
Fig. 10.

Coronary blood flow (A) and myocardial oxygen consumption (B) at rest and during exercise (Ex) in the absence and presence of the KATP channel inhibitor glibenclamide + the adenosine receptor blocker 8-phenytheophylline (8-PT) ± the nitric oxide synthase inhibitor [N-nitro-l-arginine (LNNA)] in conscious instrumented dogs. Myocardial oxygen consumption (C) and systolic wall thickening (D) vs. coronary blood flow with and without glibenclamide (Glib) + 8-PT and the vasodilator sodium nitroprusside (SNP) under baseline resting conditions [data from Duncker et al. (35) and Ishibashi et al. (61)]. *P < 0.05 vs. control. †P < 0.05 vs. glibenclamide + 8PT.
It is also important to consider the extent to which the attenuation of the coronary flow and MV̇o2 response influences coronary venous Po2. Specifically, while inhibition of either KV or KATP channels alone produces reductions in resting coronary venous Po2, the classically predicted steepening (clockwise rotation shown in Fig. 1C) of the relationship between coronary venous Po2 and MV̇o2 is not readily apparent in either dog (34, 35, 93) or swine (10, 48) studies (Fig. 11). We submit that the lessening of coronary venous Po2 (truncated, parallel shift) reflects the consequence of antagonist-mediated decreases in myocardial oxygen delivery-metabolism balance. However, cases of the steepening of the relationship between coronary venous Po2 and MV̇o2 have been documented in the presence of combined KATP channel (high-dose, intracoronary glibenclamide), adenosine receptor, and nitric oxide synthesis inhibition (61) as well as in chronic disease conditions such as obesity-metabolic syndrome (10). Such progressive increases in myocardial oxygen extraction (diminished delivery-metabolism balance) appear to only occur under conditions in which coronary blood flow is substantially diminished, typically ≥ 40% at higher levels of exercise. As proposed above, findings of truncated, parallel shifts versus steepened slopes are likely dependent on the degree of pharmacologic antagonism and/or the level of physiologic compensation. Regardless, we propose that antagonist-induced reductions in coronary blood flow and MV̇o2 (correctable with vasodilator infusion), along with accompanied truncation and reductions in indexes of the balance between coronary blood flow and myocardial metabolism, are the most fundamental experimental findings that must be observed to satisfy an obligatory role for any factor or pathway in local metabolic coronary vasodilation.
Fig. 11.

Coronary venous Po2 vs. myocardial oxygen consumption at rest and during exercise in the absence and presence of the Kv channel blocker 4-aminopyridine [4-AP; data from Berwick et al. (10); A] and the KATP channel inhibitor glibenclamide [data from Duncker et al. (35); B].
Updated Criteria for Defining Local Metabolic Mechanisms
In 1983, Feigl proposed criteria for the classification of specific factors as local “metabolites” (40). These standards, a modification of Koch’s classic postulates, include the necessary biochemical machinery for the production and release of the metabolite to be present, along with appropriate mechanisms for inactivation and/or reuptake. Further experimental requirements are that the metabolite is quantifiably released under the appropriate conditions, that administration of the metabolite mimics the desired physiologic response, and that inhibition of the metabolite has effects consistent with the hypothesis. Without a doubt, differences in opinion of what finding(s) constitute the predicted inhibitory effects have significantly clouded interpretation and conclusions regarding mechanisms of local metabolic control to this point. To this end, we propose the following updates to the criteria for defining mechanisms of local metabolic control:
-
1.
Quantitative studies demonstrate that increases in MV̇o2 correspond with increases in metabolite concentration (release). Such studies include direct measures via placement of myocardial electrodes, microdialysis probes, and/or collection of coronary venous blood. However, limitations of these approaches including myocardial damage and the potential for rapidly degraded substances to not appear in venous blood should be recognized.
-
2.
Physiologically relevant levels of the released metabolite are sufficient to produce coronary vasodilation in a concentration-dependent manner. Experiments are needed to validate that physiologic concentrations of the metabolite are sufficient to produce coronary vasodilation. Recognition that intracoronary administration of the metabolite (or related agonist) does not specifically equate with myocardial-dependent release and responses should be appreciated.
-
3.
Inhibition of metabolite production and/or receptor signaling diminishes coronary blood flow at rest and/or in response to increases in MV̇o2. A requisite role of a metabolite in the control of coronary microvascular resistance is demonstrated by dose-dependent reductions in coronary blood flow in response to metabolite blockade. Furthermore, if the effect of the metabolite is only present at rest, inhibition will diminish baseline coronary blood flow but not the overall increase (delta) of coronary response to increases in MV̇o2. Alternatively, if the metabolite contributes only during increases in MV̇o2, then inhibition will not influence baseline flow but will diminish MV̇o2-mediated increases in flow. If the metabolite involvement is at rest and during increases in MV̇o2, then inhibition will decrease coronary flow and rest and in response to augmented MV̇o2.
-
4.
Reductions in coronary blood flow produced by inhibition of the metabolite correspond with commensurate decreases in MV̇o2 (and indexes of cardiac function) and indexes of the balance between coronary blood flow and metabolism. Findings with regard to effects of metabolite inhibition on coronary blood flow demonstrate proportionate reductions in MV̇o2 and indexes of cardiac function. Collectively, the data should not violate mass balance and inhibition results in the truncation of the relationships of coronary blood flow and coronary venous Po2 relative to MV̇o2.
-
5.
Corresponding reductions of MV̇o2 (and indexes of cardiac function) are reversed by vasodilator-mediated restoration of coronary blood flow to normal (untreated) levels. Studies to demonstrate that reductions in MV̇o2 and cardiac function produced by metabolite inhibition are related to deficits in myocardial oxygen delivery and not to a direct negative inotropic effect or to an impairment in mitochondrial respiration, are imperative.
Despite well over 50 years of dedicated research in this area, no study has documented evidence to support that any specific metabolite fulfills these criteria. Furthermore, there are only a couple factors, most notably H2O2 (90, 91, 93) and ATP (adenine nucleotides) (38, 50, 51, 87), that satisfy some of these criteria and merit further consideration. Interestingly, these metabolites have been shown to converge on K+ channels (22); however, definitive data to establish causation are presently lacking [see Duncker et al. (27) and Goodwill et al. (47) for more comprehensive review]. Another mechanism that should be recognized is that of the dual role of catecholamines, which contribute to both arms of the delivery/metabolism equation in mediating β-adrenergic vasodilation and increases in heart rate and contractility (27, 47). This pathway is proposed to occur without an error signal (feedforward or open-loop control) and thus could explain how coronary venous Po2 is maintained over a wide range of MV̇o2 (Fig. 4). However, precisely how autonomic β-adrenergic pathways work with proposed parallel oxygen-sensing pathways remains to be determined. Importantly, the criteria outlined above are appropriate for both feedback and feedforward pathways. Additionally, while the presence of alternative parallel and/or redundant pathways that can act in concert or in a compensatory manner must be appreciated (34, 35, 61, 78, 108), there is ample evidence to support the assumption of a (correctable) truncation of the coronary flow/MV̇o2 response when a specific pathway that contributes to the regulation of coronary blood flow is inhibited (10, 35, 61, 93) (see Fig. 10). We propose that the application of the updated criteria above, which appear particularly suited for short-term studies using acute interventions, will help to facilitate interpretation and understanding of mechanisms responsible for metabolic control of coronary blood flow.
SUMMARY AND IMPLICATIONS
The local metabolic hypothesis of blood flow control remains one of the primary questions of coronary physiologists to this day. However, discerning the mechanisms responsible for equilibrating alterations in coronary microvascular resistance with underlying changes in MV̇o2 has been confounded by the continued debate around both anticipated experimental outcomes and data interpretation. For a number of years, coronary venous Po2 has been generally accepted to represent a measure of myocardial tissue oxygenation (12, 27, 28, 40, 47, 58, 105, 106) and thus the classically proposed error signal for the generation of vasodilator metabolites in the heart. Yet, evidence from the coronary literature across species indicates that reductions in coronary venous Po2 (tissue oxygenation) are not required for coupling coronary flow with MV̇o2 in response to exercise (Fig. 4), reductions in arterial oxygenation (Fig. 6), or changes in perfusion pressure (Fig. 7). Similar questions regarding hemoglobin saturation serving as the primary sensor for linking myocardial oxygen delivery with metabolism also remain. Together these findings importantly argue against the original formulation of the metabolic hypothesis and implicate the presence and involvement of alternative oxygen- or metabolic (e.g., ADP/ATP ratio)-sensing mechanisms within the vasculature and/or myocardium that are capable of maintaining myocardial oxygen delivery/metabolism balance in response to a variety of (patho-)physiologic perturbations. Nevertheless, interpretation of changes in coronary venous Po2 (or coronary venous hemoglobin saturation) relative to MV̇o2 is quite nuanced, inherently circular in nature, and subject to the largely unaccounted for potential for confounding alterations in oxy-hemoglobin dissociation and/or myocardial diffusion properties. Accordingly, a more straightforward path for assessing and interpreting the contribution of local metabolic mechanisms is greatly needed to begin to better understand this fundamental physiologic phenomenon.
We propose that the simplest a priori prediction that can be made is that inhibition of a factor or pathway that contributes to local metabolic control should attenuate the coronary flow response relative to the untreated control response. Prior studies demonstrate that this is indeed the case to the extent that imposed reductions in myocardial oxygen delivery result in commensurate, yet correctable decreases in MV̇o2 (Figs. 9 and 10). Such effects are importantly accompanied by anticipated reductions in coronary venous Po2 (Fig. 11) and are evident in scenarios that involve activation of compensatory vasodilator pathways (Fig. 10). We submit that decreases in coronary venous Po2 (or coronary venous hemoglobin saturation) in the absence of clear reductions in coronary blood flow and MV̇o2 (Fig. 8) are not consistent with a requisite role for that factor (pathway) in local metabolic coronary vasodilation. While alternative interpretations certainly remain, we are hopeful that the proposed updated criteria will help facilitate further dialogue and study and assist in better understanding what remains the Gordian knot of the coronary circulation. Given that impaired coronary microvascular function (in the absence of overt atherosclerosis) is now recognized to be a powerful, independent predictor of cardiac morbidity and mortality (83, 86, 101, 102), delineation of the precise mechanisms responsible for the dynamic regulation of coronary blood flow in health and disease is more important than ever.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-136386, Dutch Heart Foundation Grants 2000D038 and 2000D042, and The Netherlands CardioVascular Research Initiative (with support of the Dutch Heart Foundation Grants CVON2014-11 (RECONNECT). Additional support was provided by the use of resources and facilities at the Indiana University School of Medicine (Indianapolis, IN) and the Harry S. Truman Memorial Veteran’s Hospital (Columbia, MO).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.T., A.K., H.E.B., S.B.B., D.M., and D.J.D. analyzed data; J.D.T., A.G.G., A.K., H.E.B., S.B.B., D.M., and D.J.D. interpreted results of experiments; J.D.T. and A.G.G. prepared figures; J.D.T. and D.J.D. drafted manuscript; J.D.T., A.G.G., A.K., H.E.B., S.B.B., D.M., and D.J.D. edited and revised manuscript; J.D.T., A.G.G., A.K., H.E.B., S.B.B., D.M., and D.J.D. approved final version of manuscript.
REFERENCES
- 1.Alella A, Williams FL, Bolene-Williams C, Katz LN. Interrelation between cardiac oxygen consumption and coronary blood flow. Am J Physiol 183: 570–582, 1955. doi: 10.1152/ajplegacy.1955.183.3.570. [DOI] [PubMed] [Google Scholar]
- 2.Altman JD, Kinn J, Duncker DJ, Bache RJ. Effect of inhibition of nitric oxide formation on coronary blood flow during exercise in the dog. Cardiovasc Res 28: 119–124, 1994. doi: 10.1093/cvr/28.1.119. [DOI] [PubMed] [Google Scholar]
- 3.Bache RJ, Vrobel TR, Ring WS, Emery RW, Andersen RW. Regional myocardial blood flow during exercise in dogs with chronic left ventricular hypertrophy. Circ Res 48: 76–87, 1981. doi: 10.1161/01.RES.48.1.76. [DOI] [PubMed] [Google Scholar]
- 4.Bai XJ, Iwamoto T, Williams AG Jr, Fan WL, Downey HF. Coronary pressure-flow autoregulation protects myocardium from pressure-induced changes in oxygen consumption. Am J Physiol Heart Circ Physiol 266: H2359–H2368, 1994. doi: 10.1152/ajpheart.1994.266.6.H2359. [DOI] [PubMed] [Google Scholar]
- 5.Bassingthwaighte JB, Yipintsoi T, Harvey RB. Microvasculature of the dog left ventricular myocardium. Microvasc Res 7: 229–249, 1974. doi: 10.1016/0026-2862(74)90008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassingthwaighte JB, Yipintsoi T, Knopp TJ. Diffusional arteriovenous shunting in the heart. Microvasc Res 28: 233–253, 1984. doi: 10.1016/0026-2862(84)90020-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beard DA, Schenkman KA, Feigl EO. Myocardial oxygenation in isolated hearts predicted by an anatomically realistic microvascular transport model. Am J Physiol Heart Circ Physiol 285: H1826–H1836, 2003. doi: 10.1152/ajpheart.00380.2003. [DOI] [PubMed] [Google Scholar]
- 8.Bender SB, de Beer VJ, Tharp DL, Bowles DK, Laughlin MH, Merkus D, Duncker DJ. Severe familial hypercholesterolemia impairs the regulation of coronary blood flow and oxygen supply during exercise. Basic Res Cardiol 111: 61, 2016. doi: 10.1007/s00395-016-0579-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berne RM. Regulation of coronary blood flow. Physiol Rev 44: 1–29, 1964. doi: 10.1152/physrev.1964.44.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD. Contribution of voltage-dependent K+ channels to metabolic control of coronary blood flow. J Mol Cell Cardiol 52: 912–919, 2012. doi: 10.1016/j.yjmcc.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berwick ZC, Moberly SP, Kohr MC, Morrical EB, Kurian MM, Dick GM, Tune JD. Contribution of voltage-dependent K+ and Ca2+ channels to coronary pressure-flow autoregulation. Basic Res Cardiol 107: 264, 2012. doi: 10.1007/s00395-012-0264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borbouse L, Dick GM, Asano S, Bender SB, Dincer UD, Payne GA, Neeb ZP, Bratz IN, Sturek M, Tune JD. Impaired function of coronary BKCa channels in metabolic syndrome. Am J Physiol Heart Circ Physiol 297: H1629–H1637, 2009. doi: 10.1152/ajpheart.00466.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borbouse L, Dick GM, Payne GA, Payne BD, Svendsen MC, Neeb ZP, Alloosh M, Bratz IN, Sturek M, Tune JD. Contribution of BKCa channels to local metabolic coronary vasodilation: Effects of metabolic syndrome. Am J Physiol Heart Circ Physiol 298: H966–H973, 2010. doi: 10.1152/ajpheart.00876.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canty JM Jr, Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol 52: 822–831, 2012. doi: 10.1016/j.yjmcc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Traverse JH, Zhang J, Bache RJ. Selective blockade of mitochondrial KATP channels does not impair myocardial oxygen consumption. Am J Physiol Heart Circ Physiol 281: H738–H744, 2001. doi: 10.1152/ajpheart.2001.281.2.H738. [DOI] [PubMed] [Google Scholar]
- 16.Crystal GJ, Boatwright RB, Downey HF, Bashour FA. Shunting of microspheres across the canine coronary circulation. Am J Physiol Heart Circ Physiol 236: H7–H12, 1979. doi: 10.1152/ajpheart.1979.236.1.H7. [DOI] [PubMed] [Google Scholar]
- 17.Crystal GJ, El-Orbany M, Zhou X, Salem MR, Kim SJ. Hemodilution does not alter the coronary vasodilating effects of endogenous or exogenous nitric oxide. Can J Anaesth 55: 507–514, 2008. doi: 10.1007/BF03016670. [DOI] [PubMed] [Google Scholar]
- 18.Crystal GJ, Ruiz JR, Rooney MW, Salem MR. Regional hemodynamics and oxygen supply during isovolemic hemodilution in the absence and presence of high-grade beta-adrenergic blockade. J Cardiothorac Anesth 2: 772–779, 1988. doi: 10.1016/0888-6296(88)90101-9. [DOI] [PubMed] [Google Scholar]
- 19.Crystal GJ, Salem MR. Myocardial and systemic hemodynamics during isovolemic hemodilution alone and combined with nitroprusside-induced controlled hypotension. Anesth Analg 72: 227–237, 1991. doi: 10.1213/00000539-199102000-00016. [DOI] [PubMed] [Google Scholar]
- 20.Dankelman J, Van der Ploeg CP, Spaan JA. Transients in myocardial O2 consumption after abrupt changes in perfusion pressure in goats. Am J Physiol Heart Circ Physiol 270: H492–H499, 1996. doi: 10.1152/ajpheart.1996.270.2.H492. [DOI] [PubMed] [Google Scholar]
- 21.Deussen A. Mechanisms underlying coronary autoregulation continue to await clarification. Basic Res Cardiol 113: 34, 2018. doi: 10.1007/s00395-018-0693-y. [DOI] [PubMed] [Google Scholar]
- 22.Dick GM, Tune JD. Role of potassium channels in coronary vasodilation. Exp Biol Med (Maywood) 235: 10–22, 2010. doi: 10.1258/ebm.2009.009201. [DOI] [PubMed] [Google Scholar]
- 23.Dole WP, Nuno DW. Myocardial oxygen tension determines the degree and pressure range of coronary autoregulation. Circ Res 59: 202–215, 1986. doi: 10.1161/01.RES.59.2.202. [DOI] [PubMed] [Google Scholar]
- 24.Downey HF, Bashour FA, Jishi B, Parker PE. Arteriovenous shunts in dilated or reperfused canine coronary vasculature. Microvasc Res 17: 22–26, 1979. doi: 10.1016/0026-2862(79)90004-9. [DOI] [PubMed] [Google Scholar]
- 25.Downey HF, Murakami H, Kim SJ. Control of coronary vascular tone during altered myocardial oxygen demand and during altered myocardial oxygen. Hypoxia Med J 3: 120–127, 1998. [Google Scholar]
- 26.Duncker DJ, Bache RJ. Inhibition of nitric oxide production aggravates myocardial hypoperfusion during exercise in the presence of a coronary artery stenosis. Circ Res 74: 629–640, 1994. doi: 10.1161/01.RES.74.4.629. [DOI] [PubMed] [Google Scholar]
- 27.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev 88: 1009–1086, 2008. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 28.Duncker DJ, Bache RJ, Merkus D. Regulation of coronary resistance vessel tone in response to exercise. J Mol Cell Cardiol 52: 802–813, 2012. doi: 10.1016/j.yjmcc.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Duncker DJ, Bache RJ, Merkus D, Laughlin MH. Exercise and the coronary circulation. In: Muscle and Exercise Physiology, edited by Zoladz PJ. Chennai, India: Academic, 2019, p. 467–503. doi: 10.1016/B978-0-12-814593-7.00022-0. [DOI] [Google Scholar]
- 30.Duncker DJ, Koller A, Merkus D, Canty JM Jr. Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis 57: 409–422, 2015. doi: 10.1016/j.pcad.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duncker DJ, Stubenitsky R, Verdouw PD. Autonomic control of vasomotion in the porcine coronary circulation during treadmill exercise: evidence for feed-forward beta-adrenergic control. Circ Res 82: 1312–1322, 1998. doi: 10.1161/01.RES.82.12.1312. [DOI] [PubMed] [Google Scholar]
- 32.Duncker DJ, Stubenitsky R, Verdouw PD. Role of adenosine in the regulation of coronary blood flow in swine at rest and during treadmill exercise. Am J Physiol Heart Circ Physiol 275: H1663–H1672, 1998. doi: 10.1152/ajpheart.1998.275.5.H1663. [DOI] [PubMed] [Google Scholar]
- 33.Duncker DJ, Van Zon NS, Altman JD, Pavek TJ, Bache RJ. Role of K+ATP channels in coronary vasodilation during exercise. Circulation 88: 1245–1253, 1993. doi: 10.1161/01.CIR.88.3.1245. [DOI] [PubMed] [Google Scholar]
- 34.Duncker DJ, van Zon NS, Ishibashi Y, Bache RJ. Role of K+ ATP channels and adenosine in the regulation of coronary blood flow during exercise with normal and restricted coronary blood flow. J Clin Invest 97: 996–1009, 1996. doi: 10.1172/JCI118524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncker DJ, van Zon NS, Pavek TJ, Herrlinger SK, Bache RJ. Endogenous adenosine mediates coronary vasodilation during exercise after K(ATP)+ channel blockade. J Clin Invest 95: 285–295, 1995. doi: 10.1172/JCI117653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edlund A, Sollevi A, Wennmalm A. The role of adenosine and prostacyclin in coronary flow regulation in healthy man. Acta Physiol Scand 135: 39–46, 1989. doi: 10.1111/j.1748-1716.1989.tb08548.x. [DOI] [PubMed] [Google Scholar]
- 37.Erickson HH, Stone HL. Cardiac beta-adrenergic receptors and coronary hemodynamics in the conscious dog during hypoxic hypoxia. Aerosp Med 43: 422–428, 1972. [PubMed] [Google Scholar]
- 38.Farias M 3rd, Gorman MW, Savage MV, Feigl EO. Plasma ATP during exercise: possible role in regulation of coronary blood flow. Am J Physiol Heart Circ Physiol 288: H1586–H1590, 2005. doi: 10.1152/ajpheart.00983.2004. [DOI] [PubMed] [Google Scholar]
- 39.Feigl EO. Coronary autoregulation. J Hypertens Suppl 7: S55–S58, 1989. [PubMed] [Google Scholar]
- 40.Feigl EO. Coronary physiology. Physiol Rev 63: 1–205, 1983. doi: 10.1152/physrev.1983.63.1.1. [DOI] [PubMed] [Google Scholar]
- 41.Flaim SF, Minteer WJ, Clark DP, Zelis R. Cardiovascular response to acute aquatic and treadmill exercise in the untrained rat. J Appl Physiol 46: 302–308, 1979. doi: 10.1152/jappl.1979.46.2.302. [DOI] [PubMed] [Google Scholar]
- 42.Flores NA, Davies RL, Penny WJ, Sheridan DJ. Coronary microangiography in the guinea pig, rabbit and ferret. Int J Cardiol 6: 459–471, 1984. doi: 10.1016/0167-5273(84)90326-7. [DOI] [PubMed] [Google Scholar]
- 43.Gao F, de Beer VJ, Hoekstra M, Xiao C, Duncker DJ, Merkus D. Both β1- and β2-adrenoceptors contribute to feedforward coronary resistance vessel dilation during exercise. Am J Physiol Heart Circ Physiol 298: H921–H929, 2010. doi: 10.1152/ajpheart.00135.2009. [DOI] [PubMed] [Google Scholar]
- 44.Genain MA, Morlet A, Herrtage M, Muresian H, Anselme F, Latremouille C, Laborde F, Behr L, Borenstein N. Comparative anatomy and angiography of the cardiac coronary venous system in four species: human, ovine, porcine, and canine. J Vet Cardiol 20: 33–44, 2018. doi: 10.1016/j.jvc.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Gewirtz H, Olsson RA, Most AS. Role of adenosine in mediating the coronary vasodilative response to acute hypoxia. Cardiovasc Res 21: 81–89, 1987. doi: 10.1093/cvr/21.2.81. [DOI] [PubMed] [Google Scholar]
- 46.Gisselsson L, Rosberg B, Ericsson M. Myocardial blood flow, oxygen uptake and carbon dioxide release of the human heart during hemodilution. Acta Anaesthesiol Scand 26: 589–591, 1982. doi: 10.1111/j.1399-6576.1982.tb01820.x. [DOI] [PubMed] [Google Scholar]
- 47.Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of coronary blood flow. Compr Physiol 7: 321–382, 2017. doi: 10.1002/cphy.c160016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodwill AG, Noblet JN, Sassoon D, Fu L, Kassab GS, Schepers L, Herring BP, Rottgen TS, Tune JD, Dick GM. Critical contribution of KV1 channels to the regulation of coronary blood flow. Basic Res Cardiol 111: 56, 2016. doi: 10.1007/s00395-016-0575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorman MW, Feigl EO. Control of coronary blood flow during exercise. Exerc Sport Sci Rev 40: 37–42, 2012. doi: 10.1097/JES.0b013e3182348cdd. [DOI] [PubMed] [Google Scholar]
- 50.Gorman MW, Ogimoto K, Savage MV, Jacobson KA, Feigl EO. Nucleotide coronary vasodilation in guinea pig hearts. Am J Physiol Heart Circ Physiol 285: H1040–H1047, 2003. doi: 10.1152/ajpheart.00981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorman MW, Rooke GA, Savage MV, Jayasekara MP, Jacobson KA, Feigl EO. Adenine nucleotide control of coronary blood flow during exercise. Am J Physiol Heart Circ Physiol 299: H1981–H1989, 2010. doi: 10.1152/ajpheart.00611.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorman MW, Tune JD, Richmond KN, Feigl EO. Feedforward sympathetic coronary vasodilation in exercising dogs. J Appl Physiol (1985) 89: 1892–1902, 2000. doi: 10.1152/jappl.2000.89.5.1892. [DOI] [PubMed] [Google Scholar]
- 53.Grubbström J, Berglund B, Kaijser L. Myocardial blood flow and lactate metabolism at rest and during exercise with reduced arterial oxygen content. Acta Physiol Scand 142: 467–474, 1991. doi: 10.1111/j.1748-1716.1991.tb09181.x. [DOI] [PubMed] [Google Scholar]
- 54.Haitsma DB, Bac D, Raja N, Boomsma F, Verdouw PD, Duncker DJ. Minimal impairment of myocardial blood flow responses to exercise in the remodeled left ventricle early after myocardial infarction, despite significant hemodynamic and neurohumoral alterations. Cardiovasc Res 52: 417–428, 2001. doi: 10.1016/S0008-6363(01)00426-6. [DOI] [PubMed] [Google Scholar]
- 55.Heiss HW, Barmeyer J, Wink K, Hell G, Cerny FJ, Keul J, Reindell H. Studies on the regulation of myocardial blood flow in man. I: Training effects on blood flow and metabolism of the healthy heart at rest and during standardized heavy exercise. Basic Res Cardiol 71: 658–675, 1976. doi: 10.1007/BF01906411. [DOI] [PubMed] [Google Scholar]
- 56.Herrmann SC, Feigl EO. Adrenergic blockade blunts adenosine concentration and coronary vasodilation during hypoxia. Circ Res 70: 1203–1216, 1992. doi: 10.1161/01.RES.70.6.1203. [DOI] [PubMed] [Google Scholar]
- 57.Heusch G. Reprint of: the paradox of α-adrenergic coronary vasoconstriction revisited. J Mol Cell Cardiol 52: 832–839, 2012. doi: 10.1016/j.yjmcc.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 58.Heyndrickx GR, Vilaine JP, Moerman EJ, Leusen I. Role of prejunctional alpha 2-adrenergic receptors in the regulation of myocardial performance during exercise in conscious dogs. Circ Res 54: 683–693, 1984. doi: 10.1161/01.RES.54.6.683. [DOI] [PubMed] [Google Scholar]
- 59.Hoffman JI, Spaan JA. Pressure-flow relations in coronary circulation. Physiol Rev 70: 331–390, 1990. doi: 10.1152/physrev.1990.70.2.331. [DOI] [PubMed] [Google Scholar]
- 60.Howe BB, Fehn PA, Pensinger RR. Comparative anatomical studies of the coronary arteries of canine and porcine hearts. I. Free ventricular walls. Acta Anat (Basel) 71: 13–21, 1968. doi: 10.1159/000143165. [DOI] [PubMed] [Google Scholar]
- 61.Ishibashi Y, Duncker DJ, Zhang J, Bache RJ. ATP-sensitive K+ channels, adenosine, and nitric oxide-mediated mechanisms account for coronary vasodilation during exercise. Circ Res 82: 346–359, 1998. doi: 10.1161/01.RES.82.3.346. [DOI] [PubMed] [Google Scholar]
- 62.Johnson NP, Kirkeeide RL, Gould KL. Coronary anatomy to predict physiology: fundamental limits. Circ Cardiovasc Imaging 6: 817–832, 2013. doi: 10.1161/CIRCIMAGING.113.000373. [DOI] [PubMed] [Google Scholar]
- 63.Katz SA, Feigl EO. Little carbon dioxide diffusional shunting in coronary circulation. Am J Physiol Heart Circ Physiol 253: H614–H625, 1987. doi: 10.1152/ajpheart.1987.253.3.H614. [DOI] [PubMed] [Google Scholar]
- 64.Kiel AM, Goodwill AG, Baker HE, Dick GM, Tune JD. Local metabolic hypothesis is not sufficient to explain coronary autoregulatory behavior. Basic Res Cardiol 113: 33, 2018. doi: 10.1007/s00395-018-0691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kiel AM, Goodwill AG, Noblet JN, Barnard AL, Sassoon DJ, Tune JD. Regulation of myocardial oxygen delivery in response to graded reductions in hematocrit: role of K+ channels. Basic Res Cardiol 112: 65, 2017. doi: 10.1007/s00395-017-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kosaka H, Seiyama A. Elevation of oxygen release by nitroglycerin without an increase in blood flow in the hepatic microcirculation. Nat Med 3: 456–459, 1997. doi: 10.1038/nm0497-456. [DOI] [PubMed] [Google Scholar]
- 67.Kosaka H, Seiyama A. Physiological role of nitric oxide as an enhancer of oxygen transfer from erythrocytes to tissues. Biochem Biophys Res Commun 218: 749–752, 1996. doi: 10.1006/bbrc.1996.0133. [DOI] [PubMed] [Google Scholar]
- 68.Kuo L, Davis MJ, Chilian WM. Longitudinal gradients for endothelium-dependent and -independent vascular responses in the coronary microcirculation. Circulation 92: 518–525, 1995. doi: 10.1161/01.CIR.92.3.518. [DOI] [PubMed] [Google Scholar]
- 69.Laughlin MH, Bowles DK, Duncker DJ. The coronary circulation in exercise training. Am J Physiol Heart Circ Physiol 302: H10–H23, 2012. doi: 10.1152/ajpheart.00574.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laughlin MH, Muller JM. Vasoconstrictor responses of coronary resistance arteries in exercise-trained pigs. J Appl Physiol (1985) 84: 884–889, 1998. doi: 10.1152/jappl.1998.84.3.884. [DOI] [PubMed] [Google Scholar]
- 71.Laughlin MH, Overholser KA, Bhatte MJ. Exercise training increases coronary transport reserve in miniature swine. J Appl Physiol (1985) 67: 1140–1149, 1989. doi: 10.1152/jappl.1989.67.3.1140. [DOI] [PubMed] [Google Scholar]
- 72.Laughlin MH, Tomanek RJ. Myocardial capillarity and maximal capillary diffusion capacity in exercise-trained dogs. J Appl Physiol (1985) 63: 1481–1486, 1987. doi: 10.1152/jappl.1987.63.4.1481. [DOI] [PubMed] [Google Scholar]
- 73.Laxson DD, Homans DC, Bache RJ. Inhibition of adenosine-mediated coronary vasodilation exacerbates myocardial ischemia during exercise. Am J Physiol Heart Circ Physiol 265: H1471–H1477, 1993. doi: 10.1152/ajpheart.1993.265.5.H1471. [DOI] [PubMed] [Google Scholar]
- 74.Lee JC, Halloran KH, Taylor JF, Downing SE. Coronary flow and myocardial metabolism in newborn lambs: effects of hypoxia and acidemia. Am J Physiol 224: 1381–1387, 1973. doi: 10.1152/ajplegacy.1973.224.6.1381. [DOI] [PubMed] [Google Scholar]
- 75.Lee SC, Mallet RT, Shizukuda Y, Williams AG Jr, Downey HF. Canine coronary vasodepressor responses to hypoxia are attenuated but not abolished by 8-phenyltheophylline. Am J Physiol Heart Circ Physiol 262: H955–H960, 1992. doi: 10.1152/ajpheart.1992.262.4.H955. [DOI] [PubMed] [Google Scholar]
- 76.Levy PS, Kim SJ, Eckel PK, Chavez R, Ismail EF, Gould SA, Ramez Salem M, Crystal GJ. Limit to cardiac compensation during acute isovolemic hemodilution: influence of coronary stenosis. Am J Physiol Heart Circ Physiol 265: H340–H349, 1993. doi: 10.1152/ajpheart.1993.265.1.H340. [DOI] [PubMed] [Google Scholar]
- 77.Manohar M. Left ventricular oxygen extraction during submaximal and maximal exertion in ponies. J Physiol 404: 547–556, 1988. doi: 10.1113/jphysiol.1988.sp017305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merkus D, Haitsma DB, Fung TY, Assen YJ, Verdouw PD, Duncker DJ. Coronary blood flow regulation in exercising swine involves parallel rather than redundant vasodilator pathways. Am J Physiol Heart Circ Physiol 285: H424–H433, 2003. doi: 10.1152/ajpheart.00916.2002. [DOI] [PubMed] [Google Scholar]
- 79.Merkus D, Houweling B, van Vliet M, Duncker DJ. Contribution of KATP+ channels to coronary vasomotor tone regulation is enhanced in exercising swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol 288: H1306–H1313, 2005. doi: 10.1152/ajpheart.00631.2004. [DOI] [PubMed] [Google Scholar]
- 80.Merkus D, Houweling B, Zarbanoui A, Duncker DJ. Interaction between prostanoids and nitric oxide in regulation of systemic, pulmonary, and coronary vascular tone in exercising swine. Am J Physiol Heart Circ Physiol 286: H1114–H1123, 2004. doi: 10.1152/ajpheart.00477.2003. [DOI] [PubMed] [Google Scholar]
- 81.Merkus D, Sorop O, Houweling B, Boomsma F, van den Meiracker AH, Duncker DJ. NO and prostanoids blunt endothelin-mediated coronary vasoconstrictor influence in exercising swine. Am J Physiol Heart Circ Physiol 291: H2075–H2081, 2006. doi: 10.1152/ajpheart.01109.2005. [DOI] [PubMed] [Google Scholar]
- 82.Merrill GF, Downey HF, Jones CE. Adenosine deaminase attenuates canine coronary vasodilation during systemic hypoxia. Am J Physiol Heart Circ Physiol 250: H579–H583, 1986. doi: 10.1152/ajpheart.1986.250.4.H579. [DOI] [PubMed] [Google Scholar]
- 83.Murthy VL, Naya M, Foster CR, Gaber M, Hainer J, Klein J, Dorbala S, Blankstein R, Di Carli MF. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 126: 1858–1868, 2012. doi: 10.1161/CIRCULATIONAHA.112.120402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohanyan V, Yin L, Bardakjian R, Kolz C, Enrick M, Hakobyan T, Kmetz J, Bratz I, Luli J, Nagane M, Khan N, Hou H, Kuppusamy P, Graham J, Fu FK, Janota D, Oyewumi MO, Logan S, Lindner JR, Chilian WM. Requisite role of Kv1.5 channels in coronary metabolic dilation. Circ Res 117: 612–621, 2015. doi: 10.1161/CIRCRESAHA.115.306642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Overholser KA, Laughlin MH, Bhatte MJ. Exercise training-induced increase in coronary transport capacity. Med Sci Sports Exerc 26: 1239–1244, 1994. doi: 10.1249/00005768-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 86.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 87.Pradhan RK, Feigl EO, Gorman MW, Brengelmann GL, Beard DA. Open-loop (feed-forward) and feedback control of coronary blood flow during exercise, cardiac pacing, and pressure changes. Am J Physiol Heart Circ Physiol 310: H1683–H1694, 2016. doi: 10.1152/ajpheart.00663.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Richmond KN, Tune JD, Gorman MW, Feigl EO. Role of KATP+ channels and adenosine in the control of coronary blood flow during exercise. J Appl Physiol (1985) 89: 529–536, 2000. doi: 10.1152/jappl.2000.89.2.529. [DOI] [PubMed] [Google Scholar]
- 89.Richmond KN, Tune JD, Gorman MW, Feigl EO. Role of KATP+ channels in local metabolic coronary vasodilation. Am J Physiol Heart Circ Physiol 277: H2115–H2123, 1999. doi: 10.1152/ajpheart.1999.277.6.H2115. [DOI] [PubMed] [Google Scholar]
- 90.Rogers PA, Chilian WM, Bratz IN, Bryan RM Jr, Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol 292: H1404–H1411, 2007. doi: 10.1152/ajpheart.00696.2006. [DOI] [PubMed] [Google Scholar]
- 91.Rogers PA, Dick GM, Knudson JD, Focardi M, Bratz IN, Swafford AN Jr, Saitoh S, Tune JD, Chilian WM. H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K+ channels. Am J Physiol Heart Circ Physiol 291: H2473–H2482, 2006. doi: 10.1152/ajpheart.00172.2006. [DOI] [PubMed] [Google Scholar]
- 92.Roth AC, Feigl EO. Diffusional shunting in the canine myocardium. Circ Res 48: 470–480, 1981. doi: 10.1161/01.RES.48.4.470. [DOI] [PubMed] [Google Scholar]
- 93.Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26: 2614–2621, 2006. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 94.Samaha FF, Heineman FW, Ince C, Fleming J, Balaban RS. ATP-sensitive potassium channel is essential to maintain basal coronary vascular tone in vivo. Am J Physiol Cell Physiol 262: C1220–C1227, 1992. doi: 10.1152/ajpcell.1992.262.5.C1220. [DOI] [PubMed] [Google Scholar]
- 95.Schenkman KA, Beard DA, Ciesielski WA, Feigl EO. Comparison of buffer and red blood cell perfusion of guinea pig heart oxygenation. Am J Physiol Heart Circ Physiol 285: H1819–H1825, 2003. doi: 10.1152/ajpheart.00383.2003. [DOI] [PubMed] [Google Scholar]
- 96.Schulz R, Oudiz RJ, Guth BD, Heusch G. Minimal alpha 1- and alpha 2-adrenoceptor-mediated coronary vasoconstriction in the anaesthetized swine. Naunyn Schmiedebergs Arch Pharmacol 342: 422–428, 1990. doi: 10.1007/BF00169459. [DOI] [PubMed] [Google Scholar]
- 97.Setty S, Sun W, Tune JD. Coronary blood flow regulation in the prediabetic metabolic syndrome. Basic Res Cardiol 98: 416–423, 2003. doi: 10.1007/s00395-003-0418-7. [DOI] [PubMed] [Google Scholar]
- 98.Shen W, Lundborg M, Wang J, Stewart JM, Xu X, Ochoa M, Hintze TH. Role of EDRF in the regulation of regional blood flow and vascular resistance at rest and during exercise in conscious dogs. J Appl Physiol (1985) 77: 165–172, 1994. doi: 10.1152/jappl.1994.77.1.165. [DOI] [PubMed] [Google Scholar]
- 99.Sorop O, Heinonen I, van Kranenburg M, van de Wouw J, de Beer VJ, Nguyen IT, Octavia Y, van Duin RW, Stam K, van Geuns RJ, Wielopolski PA, Krestin GP, van den Meiracker AH, Verjans R, van Bilsen M, Danser AH, Paulus WJ, Cheng C, Linke WA, Joles JA, Verhaar MC, van der Velden J, Merkus D, Duncker DJ. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res 114: 954–964, 2018. doi: 10.1093/cvr/cvy038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stepp DW, Kroll K, Feigl EO. KATP+ channels and adenosine are not necessary for coronary autoregulation. Am J Physiol Heart Circ Physiol 273: H1299–H1308, 1997. doi: 10.1152/ajpheart.1997.273.3.H1299. [DOI] [PubMed] [Google Scholar]
- 101.Taqueti VR, Everett BM, Murthy VL, Gaber M, Foster CR, Hainer J, Blankstein R, Dorbala S, Di Carli MF. Interaction of impaired coronary flow reserve and cardiomyocyte injury on adverse cardiovascular outcomes in patients without overt coronary artery disease. Circulation 131: 528–535, 2015. doi: 10.1161/CIRCULATIONAHA.114.009716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taqueti VR, Solomon SD, Shah AM, Desai AS, Groarke JD, Osborne MT, Hainer J, Bibbo CF, Dorbala S, Blankstein R, Di Carli MF. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J 39: 840–849, 2018. doi: 10.1093/eurheartj/ehx721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol (1985) 113: 1128–1140, 2012. doi: 10.1152/japplphysiol.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trzeciakowski J, Chilian WM. Chaotic behavior of the coronary circulation. Med Biol Eng Comput 46: 433–442, 2008. doi: 10.1007/s11517-008-0329-8. [DOI] [PubMed] [Google Scholar]
- 105.Tune JD. Coronary Circulation. Williston, VT: Morgan & Claypool Life Sciences, 2014. [Google Scholar]
- 106.Tune JD, Gorman MW, Feigl EO. Matching coronary blood flow to myocardial oxygen consumption. J Appl Physiol (1985) 97: 404–415, 2004. doi: 10.1152/japplphysiol.01345.2003. [DOI] [PubMed] [Google Scholar]
- 107.Tune JD, Richmond KN, Gorman MW, Feigl EO. Control of coronary blood flow during exercise. Exp Biol Med (Maywood) 227: 238–250, 2002. doi: 10.1177/153537020222700404. [DOI] [PubMed] [Google Scholar]
- 108.Tune JD, Richmond KN, Gorman MW, Feigl EO. KATP+ channels, nitric oxide, and adenosine are not required for local metabolic coronary vasodilation. Am J Physiol Heart Circ Physiol 280: H868–H875, 2001. doi: 10.1152/ajpheart.2001.280.2.H868. [DOI] [PubMed] [Google Scholar]
- 109.Tune JD, Richmond KN, Gorman MW, Feigl EO. Role of nitric oxide and adenosine in control of coronary blood flow in exercising dogs. Circulation 101: 2942–2948, 2000. doi: 10.1161/01.CIR.101.25.2942. [DOI] [PubMed] [Google Scholar]
- 110.Tune JD, Richmond KN, Gorman MW, Olsson RA, Feigl EO. Adenosine is not responsible for local metabolic control of coronary blood flow in dogs during exercise. Am J Physiol Heart Circ Physiol 278: H74–H84, 2000. doi: 10.1152/ajpheart.2000.278.1.H74. [DOI] [PubMed] [Google Scholar]
- 111.Tune JD, Yeh C, Setty S, Zong P, Downey HF. Coronary blood flow control is impaired at rest and during exercise in conscious diabetic dogs. Basic Res Cardiol 97: 248–257, 2002. doi: 10.1007/s003950200018. [DOI] [PubMed] [Google Scholar]
- 112.Van der Ploeg CP, Dankelman J, Spaan JA. Heart rate affects the dependency of myocardial oxygen consumption on flow in goats. Heart Vessels 10: 258–265, 1995. doi: 10.1007/BF01744905. [DOI] [PubMed] [Google Scholar]
- 113.Van Woerkens EC, Trouwborst A, Duncker DJ, Koning MM, Boomsma F, Verdouw PD. Catecholamines and regional hemodynamics during isovolemic hemodilution in anesthetized pigs. J Appl Physiol (1985) 72: 760–769, 1992. doi: 10.1152/jappl.1992.72.2.760. [DOI] [PubMed] [Google Scholar]
- 114.Van Wylen DG, Williams AG Jr, Downey HF. Interstitial purine metabolites and lactate during regional myocardial hypoxia. Cardiovasc Res 27: 1498–1503, 1993. doi: 10.1093/cvr/27.8.1498. [DOI] [PubMed] [Google Scholar]
- 115.Vance JP, Parratt JR, Ledingham IM. The effects of hypoxia on myocardial blood flow and oxygen consumption: negative role of beta adrenoreceptors. Clin Sci 41: 257–273, 1971. doi: 10.1042/cs0410257. [DOI] [PubMed] [Google Scholar]
- 116.von Restorff W, Höfling B, Holtz J, Bassenge E. Effect of increased blood fluidity through hemodilution on coronary circulation at rest and during exercise in dogs. Pflugers Arch 357: 15–24, 1975. doi: 10.1007/BF00584541. [DOI] [PubMed] [Google Scholar]
- 117.von Restorff W, Holtz J, Bassenge E. Exercise induced augmentation of myocardial oxygen extraction in spite of normal coronary dilatory capacity in dogs. Pflugers Arch 372: 181–185, 1977. doi: 10.1007/BF00585334. [DOI] [PubMed] [Google Scholar]
- 118.White FC, Bloor CM, McKirnan MD, Carroll SM. Exercise training in swine promotes growth of arteriolar bed and capillary angiogenesis in heart. J Appl Physiol (1985) 85: 1160–1168, 1998. doi: 10.1152/jappl.1998.85.3.1160. [DOI] [PubMed] [Google Scholar]
- 119.Wong AY, Klassen GA. Vasomotor coronary oscillations: a model to evaluate autoregulation. Basic Res Cardiol 86: 461–475, 1991. doi: 10.1007/BF02190714. [DOI] [PubMed] [Google Scholar]
- 120.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Zhang W, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, Xia Y. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med 17: 79–86, 2011. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]