

Abstract
Heart failure, a major cause of morbidity and mortality in patients with pulmonary arterial hypertension (PAH), is an outcome of complex biochemical processes. In this study, we determined changes in microRNAs (miRs) in the right and left ventricles of normal and PAH rats. Using an unbiased quantitative miR microarray analysis, we found 1) miR-21-5p, miR-31-5 and 3p, miR-140-5 and 3p, miR-208b-3p, miR-221-3p, miR-222-3p, miR-702-3p, and miR-1298 were upregulated (>2-fold; P < 0.05) in the right ventricle (RV) of PAH compared with normal rats; 2) miR-31-5 and 3p, and miR-208b-3p were upregulated (>2-fold; P < 0.05) in the left ventricle plus septum (LV+S) of PAH compared with normal rats; 3) miR-187-5p, miR-208a-3p, and miR-877 were downregulated (>2-fold; P < 0.05) in the RV of PAH compared with normal rats; and 4) no miRs were up- or downregulated with >2-fold in LV+S compared with RV of PAH and normal. Upregulation of miR-140 and miR-31 in the hypertrophic RV was further confirmed by quantitative PCR. Interestingly, compared with control rats, expression of mitofusin-1 (MFN1), a mitochondrial fusion protein that regulates apoptosis, and which is a direct target of miR-140, was reduced in the RV relative to LV+S of PAH rats. We found a correlation between increased miR-140 and decreased MFN1 expression in the hypertrophic RV. Our results also demonstrated that upregulation of miR-140 and downregulation of MFN1 correlated with increased RV systolic pressure and hypertrophy. These results suggest that miR-140 and MFN1 play a role in the pathogenesis of PAH-associated RV dysfunction.
Listen to this article's corresponding podcast at http://ajpheart.podbean.com/e/mir140-and-right-heart-hypertrophy/.
Keywords: miR, heart, hypertrophy, heart failure, pulmonary, hypertension, lungs, rats
NEW & NOTEWORTHY
We show that in an experimental model of severe pulmonary arterial hypertension, miR-140 is significantly upregulated in the hypertrophic right ventricle, and its target protein mitofusin-1 is significantly downregulated. The increase in miR-140 is significantly correlated with increase in right ventricular systolic pressure and right ventricle hypertrophy.
the pathophysiology of pulmonary arterial hypertension (PAH) is heterogeneous. In PAH, pulmonary vascular resistance and arterial pressure are increased, and the right ventricle (RV) is hypertrophied in response to increased afterload/pressure overload. Increased afterload in PAH leads to myocardial dysfunction, tricuspid regurgitation with progressive annular dilatation, and ultimately RV failure (67).
Heart failure, a major cause of mortality in PAH patients (4, 5), is a complex syndrome. In the failing hearts, oxidative stress is increased and the rate of cardiomyocyte apoptotic and autophagic death is augmented (6, 49, 51, 60, 62). Activation of these processes causes myocardial stiffening, dilatation, and loss of contractility (49, 51). However, the mechanisms that regulate apoptosis and autophagy in this complex pathology are unclear and are an area under investigation.
MicroRNAs (miRs) are a class of small noncoding RNAs that regulate gene expression by either inhibiting mRNA translation or promoting mRNA degradation (8, 29). The miR profile is altered in various left heart cardiomyopathies compared with nonfailing hearts (8, 14, 66). Several studies have shown that miRs are involved in the pathogenesis of left heart failure (7, 9, 24, 66), and that manipulation of miR expression is a promising therapeutic target (8, 24). In this regard, very little work has been done to evaluate the profile of miRs in the right and left ventricles, and the roles of various miRs in the pathogenesis of PAH-associated heart failure remains elusive. Also, whether increased afterload or unknown factors released from diseased lungs mediate right and left heart failure in PAH is still unclear. We postulated that the miR profile in the hypertrophic RV, induced by increased afterload, would be different from that in the left ventricle (LV) in Sugen5416/hypoxia/normoxia (Su/Hx/Nx)-induced PAH rats, as well as that in the RV of normotensive rats. Therefore, the objective of this study was to determine miR profiles and validate the expression of their targets in the RV vs. LV of control vs. PAH rats.
METHODS
Animal model of pulmonary arterial hypertension.
All animal procedures were approved by the Institutional Animal Care and Use Committee. Adult male Sprague-Dawley rats (200–250 g) were injected with 20 mg/kg Sugen-5416 (Cayman Chemical, Ann Arbor, MI) subcutaneously and exposed to normobaric hypoxia (10% O2) for 3 wk followed by exposure to normoxia (21% O2) for an additional 5 and 10 wk to establish 8-wk and 13-wk Su/Hx/Nx PAH as described previously (1, 63). At 8 and 13 wk, hemodynamic measurements were performed. After hemodynamic measurements, the heart was harvested and RV free wall was carefully separated from LV plus septal wall (LV+S). The ratio of RV to LV+S mass (Fulton's index) was measured to assess RV hypertrophy. Immediately after this measurement, RV and LV+S were snap frozen in liquid nitrogen.
Hemodynamic.
Hemodynamic measurements were performed as described previously (41). Briefly, at the end of the experiment protocol, rats were anesthetized by 4% isoflurane, and 1–2% isoflurane was used to maintain anesthesia for the entire duration of the surgery and data acquisition. Body temperature of the animal during the surgery was maintained with a heating pad. The right jugular vein was then carefully exposed, and a liquid-filled catheter was inserted and pushed into the RV to measure right ventricular systolic pressure (RVSP) as described previously (41).
MicroRNA microarrays.
Snapped frozen RV and LV+S tissue samples were homogenized in 700 μl QIAzol (Qiagen, No. 79306) as described previously (41). MiRs were extracted from homogenized tissues by using the miRNeasy Mini Kit (Qiagen, No. 217994). Total RNA concentration and quality were determined by absorbance at 260/280 nm. Total RNA quality was also determined by intact 28S and 18S rRNA and their ratios (28S/18S) of ∼2:1, and miR expression profiling was then performed by using 0.5–2 μg total RNA on miRCURYLNA microRNA Arrays (v.11.0) by Exiqon. The array contained >1,700 locked nucleic acid modified oligonucleotide capture probes covering most of the known human, mouse, and rat miR sequences at the time of the assay. Samples of RNA from each animal within a treatment group were labeled with Hy3 dye. A pooled sample of RNA from each animal was labeled with Hy5 dye and used as a common reference standard for interslide normalization. Intraslide normalization of expression data was performed by locally weighted scatterplot smoothing.
MicroRNA expression by quantitative PCR.
MiR analysis was performed by quantitative PCR (qPCR) as previously described (11). Briefly, total RNA was extracted from the RV and LV+S with a Qiagen miRNeasy kit (No. 217004). Input RNA quality and concentration were measured on NanoDrop and cDNA was prepared by miR-specific TaqMan miR assays (Applied Biosystems, Foster City, CA). qPCR was performed in triplicates with TaqMan Universal PCR Master mix (No. 4324018). The primers for the miR-140 (Assay ID No. 001187), miR-31 (Assay ID No. 000185), and U6 (Assay ID No. 001973) were purchased from Thermo Fisher Scientific/TaqMan. Results were normalized to U6, and relative expression of microRNA was determined by ΔΔCt method as described previously (38).
Cell culture.
H9c2 cells were maintained under 5% CO2 at 37°C in Dulbecco's modified Eagle's medium supplemented with l-glutamine, 4.5 g/l glucose (GIBCO, No. 11995-065), and 10% fetal bovine serum (GIBCO, No. 10082-147). Cells at ∼70% confluence were subcultured weekly with 0.05% 100 trypsin-EDTA (GIBCO, No. 25300-054, Thermo Fischer Scientific, Grand Island, NY). About 3 × 105 cells per well were plated in a 6-well plate. Next day, it was transfected with miRIDIAN microRNA Mimics, rno-miR-140-5pmimic (50nM) (Dharmacon, No. C-320700-00) or Mimic Negative (Scramble) Control (50nM) (Dharmacon, No. CN-001000-01). Forty-eight hours posttransfection, cells were washed three times with 1X PBS and harvested by using NP-40 lysis buffer (50 mmol/l Tris·HCl pH 7.4, 150 mmol/l NaCl, 0.5% NP-40) containing protease and phosphatase inhibitors (1X) (Roche Life Sciences).
Mitochondrial DNA quantification.
Total DNA was extracted from H9c2 cells after 48 h of transfection with miRIDIAN microRNA Mimics, rno-miR-140-5p mimic (Dharmacon, No. C-320700-00), or Mimic Negative (Scramble) Control (Dharmacon, No. CN-001000-01) with DNeasy Blood & Tissue Kit (Qiagen, No. 69504) following manufacturer's protocol. Mitochondrial DNA (mtDNA) copy number relative to nuclear DNA (nucDNA) copy number was quantified by qPCR using the equation: relative mtDNA content = 2 × 2ΔCT, where CT = (nucDNA CT − mtDNA CT) as described previously (53). To quantify the amount of mtDNA, the primer set located within the mitochondrial 16s rRNA region was used, which are 5′-TGTGGAATTAGTTGTGTGTGTAAG-3′ Tm=59 and 5′-AGGTGAAAAGCCTATCGACCTTG-3′ Tm = 62. To quantify the amount of nuclear DNA, the primer set located within the actin region was used, which are 5′-GCGGTGACCATAGCCCTCTTT-3′ Tm=61 and 5′-TGCCACTCCCAAAGTAAAGGGTCA-3′ Tm=62.
Mitochondrial membrane potential measurement.
Mitochondrial membrane potential (Δψm) was measured by Fluorescence-activated cell sorting (FACS) by using JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazolocarbo-cyanine iodide) dye. It exhibits potential-dependent accumulation in mitochondria, indicated by formation of J-aggregates in energized mitochondria emitting red fluorescence (∼590 nm). This is spectrally distinguishable from dye monomers emitting green (∼488–529 nm) fluorescence. Mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio (55). For FACS study, H9c2 cells were transfected with miR-140 mimic or scramble control in the presence and absence of endothelin-1 (ET1) (10 nM) for 48 h, followed by incubating them with 1 mM JC-1 for 30 min at 37°C. Cells were washed with 1X PBS, trypsinized, and transferred to a centrifuge tube. Cells were then washed twice in 1X PBS, and analyzed with a Guava EasyCyte Mini (Millipore, Billerica, MA) flow cytometer. The relative Δψm was expressed as the ratio of the red-to-green fluorescence.
Protein extraction and Western blotting.
Tissue samples were pulverized in liquid nitrogen and added to NP-40 lysis buffer (50 mmol/l Tris·HCl pH 7.4, 150 mmol/l NaCl, 0.5% NP-40) containing protease and phosphatase inhibitors (1X) protease and phosphatase inhibitors (Roche Life Sciences). This was followed by centrifugation at 12,000 rpm for 15 min at 4°C, and then the supernatants were collected, discarding the pellets. These supernatants were used to obtain protein (20 μg) for Western blot analysis and to measure protein content. After mixing with 5X loading buffer, the protein samples were boiled for 5 min and then subjected to 10% SDS-PAGE; transferred to nitrocellulose membranes which were blocked by incubation in blocking buffer (5% Bovine Serum Antigen); followed by incubation with specific primary antibodies, mitofusin-1 (MFN1) (1:1,000, No. ab57602, Abcam), tubulin-1α (TUBA1A;) (1:1,000, No. sc5286, Santa Cruz), c-kit (1:1,000, No. sc168, Santa Cruz), and GAPDH (1:1,000, No. 2118S, Cell Signaling), washed; and incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz). Signals were visualized by chemiluminescent detection (SuperSignal West Pico Chemiluminescent Substrate, Thermo scientific). GAPDH was used as loading control, and protein levels were analyzed by densitometric analysis with ImageJ software.
Statistical analysis.
Values are means ± SE of the number of samples (n) from different animals. Statistical analysis was performed with a paired Student's t-test, and a one-way ANOVA with Bonferroni correction was used for comparing multiple groups. P < 0.05 was used to establish statistical significance.
RESULTS
RVSP and RV/LV+S are increased in PAH rats.
Right heart catheterization results indicate that RVSP was elevated in 8- and 13-wk Su/Hx/Nx PAH rats compared with normal rats (Fig. 1A). RV hypertrophy, measured as RV/LV+S, was similarly increased in the PAH rats (Fig. 1B). However, the RV hypertrophy was slightly lower in the 13-wk than in the 8-wk PAH rats (Fig. 1B), as observed previously by Toba et al. (64). RVSP in the control and PAH rats was positively correlated with the respective RV/LV+S. Consistent with our previous studies (2, 49), the PAH rats were not in dilated cardiomyopathy stage at either 8 or 13 wk.
Fig. 1.

Right ventricular systolic pressure and Fulton's index are increased in Su/Hx/Nx rats. A: a measure of right ventricle systolic pressure (RVSP) in normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. B: a measure of right ventricular (RV) hypertrophy (Fulton's index) determined by the RV to left ventricle plus septum (LV+S) ratio in normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. C and D: Pearson correlation between RVSP and RV hypertrophy (Fulton's index). *P < 0.05 vs. normoxic control, #P < 0.05 vs. 8-wk Su/Hx/Nx. Data are means ± SE (n = 4).
miR-140 and miR-31 are elevated in the hypertrophic RV of PAH rats.
The degree of RV hypertrophy was greater at 8-wk than at 13-wk in PAH rats (Fig. 1B). Therefore, to investigate the miR signature of the hypertrophic RV, a microRNA microarray was performed in the RV of 8-wk PAH and normal rats. From an unbiased quantitative analysis, we found that a total of 304 miRs were either up- or downregulated in the hypertrophic RV. Out of these miRs, miR-21-5p, miR-31a-5p, miR-31a-3p, miR-140-5p, miR-140-3p, miR-208b-3p, miR-221-3p, miR-222-3p, miR-702-3p, and miR-1298 were upregulated (>1 logFC; P < 0.05), and miR-187-5p, miR-208a-3p, and miR-877 were downregulated (>1 logFC; P < 0.05) (Fig. 2A).
Fig. 2.

MiroRNA-140 is elevated in RV Su/Hx/Nx rats compared with RV normoxic rats. A: a subset of microRNA determined by unbiased microRNA microarrays that are differentially regulated more than 1 logFC between normal right ventricle (Nx RV) and hypertrophied RV at 8-wk Su/Hx/Nx PAH rats. B: qPCR validation of increase in miR-140 in the RV of the Su/Hx/Nx PAH rat. C: Pearson correlation between RVSP and miR-140 in the RV of 8-wk Su/Hx/Nx PAH rats. D: Pearson correlation between RV hypertrophy (Fulton's index) and miR-140 in the RV of 8-wk Su/Hx/Nx PAH rats. E: Pearson correlation between RVSP and miR-140 in the RV of 13-wk Su/Hx/Nx PAH rats. F: Pearson correlation between RV hypertrophy (Fulton's index) and miR-140 in the RV of 13-wk Su/Hx/Nx PAH rats. *P < 0.05 vs. normoxic control, #P < 0.05 vs. 8-wk Su/Hx/Nx. Data are means ± SE (n = 3–4).
miR-140, which was upregulated in the hypertrophic RV (Fig. 2A), directly targets mitofusin-1 (MFN1) protein, one of the important mitochondrial fusion proteins that modifies cell function and plays a role in cardiomyocyte death (35). Although a recent study indicated a role for miR-140 in the pathogenesis of PAH (54), very little is known about the role of miR-140 in the PAH-associated hypertrophic RV. Furthermore, in the microRNA microarray analysis, miR-140 was the second highly upregulated microRNA (Fig. 2A). Therefore, we confirmed that the levels of miR-140 were elevated in hypertrophic RV by QPCR (Fig. 2B). However, the level of miR-140 was significantly lower in 13-wk PAH RV than in 8-wk PAH RV (Fig. 2B). Interestingly, increased miR-140 levels in the RV positively correlated with the elevated RVSP (Fig. 2, C and E) and the increased RV hypertrophy (Fig. 2, D and F).
Since miR-31 regulates the migration of endothelial progenitor cells to form microvascular tubes (68), we also confirmed elevated miR-31 levels in the hypertrophic RV of PAH rats by qPCR (Fig. 3A). A positive correlation (P < 0.05) was found between the increased miR-31 levels and elevated RVSP (Fig. 3, B and D) and RV hypertrophy (Fig. 3, C and E).
Fig. 3.

MiroRNA-31 is elevated in RV Su/Hx/Nx rats compared with RV normoxic rats. A: qPCR validation of increase in miR-31 in the RV of the Su/Hx/Nx PAH rat. B: Pearson correlation between RVSP and miR-31 in the RV of 8-wk Su/Hx/Nx PAH rats. C: Pearson correlation between RV hypertrophy (Fulton's index) and miR-31 in the RV of 8-wk Su/Hx/Nx PAH rats. D: Pearson correlation between RVSP and miR-31 in the RV of 13-wk Su/Hx/Nx PAH rats. E: Pearson correlation between RV hypertrophy (Fulton's index) and miR-31 in the RV of 13-wk Su/Hx/Nx PAH rats. *P < 0.05 vs. normoxic control. Data are means ± SE (n = 4).
Expression of MFN1 is downregulated in the hypertrophic RV of PAH rats.
Since Mfn1 gene is a direct target of miR-140 (35) (Fig. 4A), we investigated the expression levels of MFN1 in RV and LV of normal and PAH rats. We first tested the effects of a miR-140 mimic on MFN1 expression in cultured cells. Transfection of 50 nM miR-140 mimic in H9c2 cells decreased 1) expression of MFN1 by ∼30% (Fig. 4, C and D) and 2) mitochondrial DNA content by ∼32% (Fig. 4E). However, transfection of miR-140 mimic did not change mitochondrial potential either in the presence or absence of ET1 (10 nM; Fig. 4F). In the hypertrophic RV of 8-wk PAH rats, the expression of MFN1 was significantly deceased (Fig. 5, A and B). However, the expression of MFN1 in the RV of 13-wk PAH rats was not different from that in the normal RV (Fig. 5, A and B). Importantly, at 8 wk of PAH the elevated RVSP, RV hypertrophy, and increased miR-140 positively correlated with the decreased MFN1 expression (Fig. 5, C–E). However, at 13 wk of PAH, the elevated RVSP, RV hypertrophy, and increase in miR-140 levels did not correlate with the change in MFN1 expression (Fig. 5, F–H).
Fig. 4.

MiR-140 targets 3′UTR of MFN1 and decreases the expression of Mfn1 in cardiomyocytes. A: a predicted consequential pairing of target region (top) position 476–483 of MFN-1 3′UTR and miR-140 (bottom) obtained from targetscan.org. B: conserved target sequence (red box) for miR-140 in position 476–483 of MFN1 3′UTR among different species. C: representative Western blot showing the decreased levels of mitofusin-1 (MFN1) proteins in H9c2 cells after transfection of meridian mimic for miR-140-5p. D: quantitative determination of relative expression of mitofusin-1 (MFN1) in H9c2 cells after transfection of meridian mimic for miR-140-5p E: relative mitochondrial DNA copy number in H9c2 cells after transfection of meridian mimic for miR-140-5p. F: relative mitochondrial membrane potential (Δψm) expressed as a ratio of J-aggregates (Red) to monomers (Green), in H9c2 cells after transfection of meridian mimic for miR-140-5p or scramble control with and without enthodelin-1 (ET1) treatment. *P < 0.05 vs. control, #P < 0.05 vs. scramble control. Data are means ± SE (n = 4).
Fig. 5.

Expression of MFN1 is downregulated in RV Su/Hx/Nx rats compared with RV normoxic rats. A: representative Western blot of MFN1 from RV and LV of normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. Blots were extracted from different lanes of the same gel to compare side by side. B: quantitative determination of relative expression of MFN1 in RV normalized by LV-MFN1. C: Spearman correlation between RVSP and the relative expression levels of MFN1 in RV of 8-wk Su/Hx/Nx PAH rats. D: Spearman correlation between RV hypertrophy and the relative expression levels of MFN1 in RV of 8-wk Su/Hx/Nx PAH rats. E: Pearson correlation between miR-140 levels and the relative expression levels of MFN1 in RV of 8-wk Su/Hx/Nx PAH rats. F: Spearman correlation between RVSP and the relative expression levels of MFN1 in RV of 13-wk Su/Hx/Nx PAH rats. G: Spearman correlation between RV hypertrophy and the relative expression levels of MFN1 in RV of 13-wk Su/Hx/Nx PAH rats. H: Pearson correlation between miR-140 levels and the relative expression levels of MFN1 in RV of 13-wk Su/Hx/Nx PAH rats. *P < 0.05 vs. normoxic control, #P < 0.05 vs. 8-wk Su/Hx/Nx. Data are means ± SE (n = 3).
Expression of c-kit and TUBA1A are upregulated in the hypertrophic RV of PAH rats.
Because miR-31 is associated with migration of c-kit+ endothelial progenitor cells (27), we investigated the expression levels of c-kit in RV and LV of normal and PAH rats. The c-kit expression was significantly higher in hypertrophic RV of 8-wk PAH rats than in normal RV (Fig. 6, A and B). Interestingly, at 13 wk, the expression level of c-kit in the RV was significantly lower than that in 8-wk PAH RV (Fig. 6, A and B). Importantly, the elevated RVSP, RV hypertrophy, and increased miR-31 positively correlated with increased c-kit expression at 8 wk (Fig. 6, C–E) but not at 13 wk (Fig. 6, F–H).
Fig. 6.

Expression of c-kit is upregulated in RV Su/Hx/Nx rats compared with RV normoxic rats. A: representative Western blot of c-kit from RV and LV of normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. Blots were extracted from different lanes of the same gel to compare side by side. B: quantitative determination of relative expression of c-kit in RV normalized by LV-c-kit. C: Spearman correlation between RVSP and the relative expression levels of c-kit in RV of 8-wk Su/Hx/Nx PAH rats. D: Spearman correlation between RV hypertrophy and the relative expression levels of c-kit in RV of 8-wk Su/Hx/Nx PAH rats. E: Pearson correlation between miR-31 levels and the relative expression levels of c-kit in RV of 8-wk Su/Hx/Nx PAH rats. F: Spearman correlation between RVSP and the relative expression levels of c-kit in RV of 13-wk Su/Hx/Nx PAH rats. G: Spearman correlation between RV hypertrophy and the relative expression levels of c-kit in RV of 13-wk Su/Hx/Nx PAH rats. H: Pearson correlation between miR-31 levels and the relative expression levels of c-kit in RV of 13-wk Su/Hx/Nx PAH rats. *P < 0.05 vs. normoxic control, #P < 0.05 vs. 8-wk Su/Hx/Nx. Data are means ± SE (n = 3).
TUBA1A was also increased in the RV of both 8- and 13-wk PAH vs. control rats (Fig. 7, A and B). Notably, a correlation (P < 0.05) was found between the increased TUBA1A and elevated RVSP and RV hypertrophy (Fig. 7, C–F). The internal control GAPDH was not different between the RV free wall of the PAH vs. the control rats (Fig. 7, G and H).
Fig. 7.

Expression of TUBA1 is upregulated in RV Su/Hx/Nx rats compared with RV normoxic rats. A: representative Western blot of TUBA1 from RV and LV of normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. Blots were extracted from different lanes of the same gel to compare side by side. B: quantitative determination of relative expression of TUBA1 in RV normalized by LV- TUBA1. C: Pearson correlation between RVSP and the relative expression levels of TUBA1 in RV of 8-wk Su/Hx/Nx PAH rats. D: Pearson correlation between RV hypertrophy and the relative expression levels of TUBA1 in RV of 8-wk Su/Hx/Nx PAH rats. E: Pearson correlation between RVSP and the relative expression levels of TUBA1 in RV of 13-wk Su/Hx/Nx PAH rats. F: Pearson correlation between RV hypertrophy and the relative expression levels of TUBA1 in RV of 13-wk Su/Hx/Nx PAH rats. G: quantitative determination of relative expression of GAPDH in RV normalized by LV-GAPDH. H: representative Western blot of GAPDH from RV and LV of normoxic control, 8-wk, and 13-wk Su/Hx/Nx PAH rats. Blots were extracted from different lanes of the same gel to compare side by side. *P < 0.05 vs. normoxic control. Data are means ± SE (n = 3).
MiR-208b is elevated in LV+S of PAH rats.
We also estimated the microRNA expression levels in LV+S of PAH rats. From an unbiased quantitative analysis, we found that miR-208b-3p, miR-31a-5p, and miR-31a-3p were upregulated (Fig. 8) in the LV+S of 8-wk PAH compared with that of the control rats.
Fig. 8.
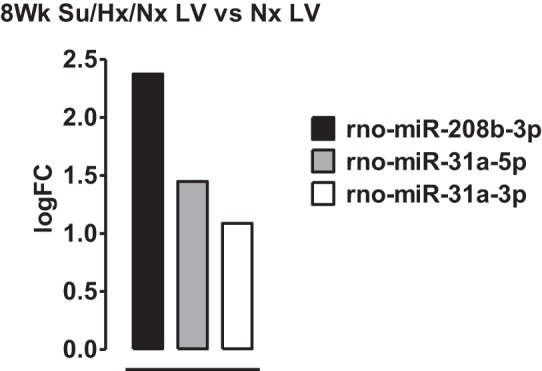
MicroRNA 208b is elevated in LV of Su/Hx/Nx rats compared with LV normoxic rats. A subset of microRNAs determined by unbiased microRNA microarrays that are differentially regulated more than 1 logFC between left ventricle (LV) of normoxic (Nx) and 8-wk Su/Hx/Nx PAH rats.
Expression of miRs in RV vs. LV+S of PAH rats.
Although 304 miRs were differentially regulated in RV, no miRs were up- or downregulated with logFC>1 compared with LV+S of normal and PAH rats.
DISCUSSION
RV remodeling in response to increased afterload is a complex process that involves 1) changes in expression of cytoskeletal proteins in cardiac myocytes, 2) increase in cardiomyocyte size, and 3) proliferation of noncardiomyocytes in the myocardium (12, 13, 58). Emerging studies suggest that miRs play a critical role in regulating expression of proteins that are involved in gene regulation, cell proliferation, and cell apoptosis (8, 29). Accordingly, using an unbiased quantitative miR microarray analysis, we found that the miRs that increase cardiac fibrosis and hypertrophy (miR-21) (10, 15), increase cardiomyocyte apoptosis (miR-140) (35) and myocardial remodeling (miR-208b) (39), attenuate vascular smooth muscle cell proliferation and migration (miR-1298) (26), regulate mesenchymal/embryonic stem cell and endothelial progenitor cell proliferation or migration (miR-31, miR-221/222, and miR-702) (31, 68, 71, 72), and decrease cancer and stem cell apoptosis (miR-31 and miR-702) (17, 73) were augmented in the hypertrophic compared with control RV. In contrast, miRs associated with tumor growth (miR-877) (70), as well as suppression (miR-187) (74), and with cardiac arrhythmia and fibrosis (miR-208a) (45) were decreased in the hypertrophic RV. On the other hand, we found only three miRs (miR-208b, miR-31a-3p, and miR-31a-5p) that were elevated in the LV of PAH rats.
miR-140 directly targets MFN1 and negatively regulates its expression (35). Consistently, we found high miR-140-5p and low MFN1 expression in hypertrophic RV of PAH compared with control rats, as well as a direct correlation between miR-140-5p and low MFN1 expression in hypertrophic RV of PAH rats. Furthermore, miR-140-5p knock-in downregulated MFN1 and mtDNA content in H9C2 cells. In these cells, mitochondrial membrane potential was unaffected by miR-140-5p knock-in in the absence and presence of ET1, which is a well-known mediator of pulmonary hypertension. These results suggest that upregulation of miR-140 potentially decreases mitochondrial biogenesis or damages the mitochondria, but it is unlikely to affect mitochondrial membrane potential. Consistently, studies show that miR-140 plays a role in promoting cardiomyocyte apoptosis by suppressing the expression of MFN1, and knockdown of miR-140 reduces myocardial infarct size (35). Previously, others and we have shown that apoptosis and autophagy/mitophagy are increased in the RV of PAH rats (2, 49) and of mice with pulmonary artery banding (48), suggesting a decline of myocardial function. Mitochondria constantly undergo fusion and fission to maintain organelle homeostasis (28, 30), and abnormality in this phenomenon triggers apoptotic cell death (47). Mitochondrial fission proteins initiate apoptosis, while mitochondrial fusion proteins inhibit apoptosis (59). Downregulation of MFN1 and 2 increases sensitivity to apoptotic stimuli (59). Conversely, MFN1 inhibits mitochondrial fission and apoptotic death of cardiomyocytes (36). It is accepted that apoptosis of cardiac myocytes plays a substantial role in the pathogenesis of dilated cardiomyopathy. In a late-stage dilated cardiomyopathy, ∼80–250 cardiac myocytes per 105 cardiac nuclei undergo apoptosis (23, 46, 56). The use of transgenic mice conditionally expressing active caspase exclusively in the myocardium has shown that very low levels of cardiac myocyte apoptosis (23 cardiac myocytes per 105 cardiac nuclei) are sufficient to cause a lethal, dilated cardiomyopathy (69). Conversely, inhibition of cardiac myocyte apoptosis has been shown to prevent the development of cardiac dilation and contractile dysfunction (69). Therefore, these studies and our current findings taken together suggest that MFN1 downregulated by miR-140 in RV of PAH rats could be a contributor to PAH-associated RV failure.
MiR-31 and c-kit expression were increased in the hypertrophic RV of PAH rats compared with control rats. These findings indirectly suggest that the number of c-kit+ cells was presumably increased in the hypertrophic RV. MiR-31 inhibits apoptosis, stimulates growth-maturation, and promotes migration of c-kit+ endothelial progenitor cells (17, 68). Furthermore there is an association between high levels of miR-31 and c-kit activation (27). Therefore, it can be suggested that miR-31 is increased in the RV of PAH rats as a compensatory mechanism to reduce the decrease in capillary density, which is associated with the failing hearts (2). Alternatively, c-kit+ cells in the mammalian adult heart are endothelial cells (61), telocytes (3), or mast cells (75). Telocytes are involved in regeneration and/or repair of an organ and has been implicated in atrial remodeling (40) and mast cells in cardiac inflammation and myocardial remodeling (21, 34). Thus the accumulation of c-kit+ cells was likely a consequence of the RV hypertrophy in PAH rats, but a contribution to the progressive deterioration of heart function and finally failure cannot be ruled out.
Expression of miR-208b and miR-208a, correlates with the expression of MYH7 and MYH6, respectively (50). A switch from MYH6 to MYH7 in myocardium is an established marker of cardiomyopathy (16, 25, 32, 44, 65). Increased MYH7 expression in the myocardium also contributes to left heart failure (25, 33, 50). Notably, in the PAH rats, miR-208b was upregulated in RV and LV+S, while miR-208a was downregulated in the RV. This differential expression of miR-208a and miR-208b indicates that a switch in MYH6 to MYH7 occurred in both RV and LV in 8-wk PAH rats. While a recent study did not find miR-208b in the failing RV of the Sugen/hypoxia model of PAH (18), miR-208b and MYH7 have been reported to be upregulated in the mouse model of PA banding-induced RV failure (50). Clearly, the miR-208 that is involved in the normal functioning of myocardium was dysregulated in the PAH RV and LV+S. Therefore, elevated miR-208b and decreased miR-208a, signature miRs of heart failure, suggest right and left hearts were failing in the experimental model of Su/Hx/Nx-induced PAH. We also found a twofold increase of TUBA1A expression in the hypertrophic RV. Expression of TUBA1A and TUBA1B is increased in the hypertrophic and failing hearts (19, 57, 58), augmented in metabolic cardiomyopathies (22), and implicated in contributing to contractile dysfunction (19, 57).
Cardiac fibrosis, hypertrophy, and abnormal heart rhythm contribute to heart failure (52). Loss of vascular smooth muscle cell function or growth and apoptosis of endothelial cells also contribute to heart failure (20, 37, 43). A recent study showed that miR-1298 inhibits connexin-43 expression and vascular smooth muscle cell proliferation and migration (26). Connexin-43, a gap junction protein, is required for maintenance of the normal cardiac rhythm, regulation of vascular tone, and endothelial function (42). Since miR-1298 increased in the hypertrophic RV but not in the LV, we suggest that miR-1298-mediated decrease of connexin-43 could be one of the causes of arrhythmia and impaired coronary artery function in the right heart of PAH rats.
In summary, we have described the dynamic changes in the miR signatures in the RV and LV+S of the angioproliferative model of severe PAH. Of importance, we have shown that miR-140 is upregulated and that its target protein MFN1 is downregulated in the hypertrophic RV of PAH rats. Moreover, the increase in miR-140 is positively correlated with the increases in RVSP and RV hypertrophy, and the decrease in the expression of MFN1 correlated well with the increase in RVSP. These changes in miR-140 and MFN1 clearly suggest that apoptosis of cardiac myocytes plays a potential role in the pathogenesis of PAH-induced RV failure.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.R.J. and S.A.G. conception and design of research; S.R.J., V.D., and S.G. performed experiments; S.R.J., V.D., S.G., J.G.E., I.F.M., and S.A.G. analyzed data; S.R.J., V.D., S.G., J.G.E., I.F.M., and S.A.G. interpreted results of experiments; S.R.J., V.D., and S.G. prepared figures; S.R.J. and S.A.G. drafted manuscript; S.R.J., V.D., S.G., J.G.E., I.F.M., and S.A.G. edited and revised manuscript; S.R.J., V.D., S.G., J.G.E., I.F.M., and S.A.G. approved final version of manuscript.
REFERENCES
- 1.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation : 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol : H1708–H1718, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bei Y, Zhou Q, Fu S, Lv D, Chen P, Chen Y, Wang F, Xiao J. Cardiac telocytes and fibroblasts in primary culture: different morphologies and immunophenotypes. PLoS One : e0115991, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest : 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Bronicki RA, Baden HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med : S15–22, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld C, Kasanmoentalib S, Visser FC, Ylstra B, Simonides WS. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics : 314–323, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest : 2772–2786, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callis TE, Wang DZ. Taking microRNAs to heart. Trends Mol Med : 254–260, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW 2nd Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med : 613–618, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Cavarretta E, Condorelli G. miR-21 and cardiac fibrosis: another brick in the wall? Eur Heart J : 2139–2141, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG, Gupte SA. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol : H214–H224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper G. Cardiocyte cytoskeleton in hypertrophied myocardium. Heart Fail Rev : 187–201, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Cooper G. Cytoskeletal networks and the regulation of cardiac contractility: microtubules, hypertrophy, and cardiac dysfunction. Am J Physiol Heart Circ Physiol : H1003–H1014, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circ Res : 724–732, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res : 563–572, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Dillmann W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail Rev : 125–132, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong Z, Zhong Z, Yang L, Wang S, Gong Z. MicroRNA-31 inhibits cisplatin-induced apoptosis in non-small cell lung cancer cells by regulating the drug transporter ABCB9. Cancer Lett : 249–257, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol : 1239–1247, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eble DM, Spinale FG. Contractile and cytoskeletal content, structure, and mRNA levels with tachycardia-induced cardiomyopathy. Am J Physiol Heart Circ Physiol : H2426–H2439, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol : 1983–1992, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Fairweather D, Frisancho-Kiss S. Mast cells and inflammatory heart disease: potential drug targets. Cardiovasc Hematol Disord Drug Targets : 80–90, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Gaikwad AB, Sayyed SG, Lichtnekert J, Tikoo K, Anders HJ. Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am J Pathol : 1079–1083, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA, Kajstura J, Anversa P. Myocyte death in the failing human heart is gender dependent. Circ Res : 856–866, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Han M, Toli J, Abdellatif M. MicroRNAs in the cardiovascular system. Curr Opin Cardiol : 181–189, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Herron TJ, McDonald KS. Small amounts of α-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res : 1150–1152, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Hu W, Wang M, Yin H, Yao C, He Q, Yin L, Zhang C, Li W, Chang G, Wang S. MicroRNA-1298 is regulated by DNA methylation and affects vascular smooth muscle cell function by targeting connexin 43. Cardiovasc Res : 534–545, 2015. [DOI] [PubMed] [Google Scholar]
- 27.Itkin T, Ludin A, Gradus B, Gur-Cohen S, Kalinkovich A, Schajnovitz A, Ovadya Y, Kollet O, Canaani J, Shezen E, Coffin DJ, Enikolopov GN, Berg T, Piacibello W, Hornstein E, Lapidot T. FGF-2 expands murine hematopoietic stem and progenitor cells via proliferation of stromal cells, c-Kit activation, and CXCL12 down-regulation. Blood : 1843–1855, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Jeong SY, Seol DW. The role of mitochondria in apoptosis. BMB Rep : 11–22, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Joshi SR, McLendon JM, Comer BS, Gerthoffer WT. MicroRNAs-control of essential genes: implications for pulmonary vascular disease. Pulm Circ : 357–364, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ : 870–880, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Kim BM, Choi MY. Non-canonical microRNAs miR-320 and miR-702 promote proliferation in Dgcr8-deficient embryonic stem cells. Biochem Biophys Res Commun : 183–189, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinugawa K, Minobe WA, Wood WM, Ridgway EC, Baxter JD, Ribeiro RC, Tawadrous MF, Lowes BA, Long CS, Bristow MR. Signaling pathways responsible for fetal gene induction in the failing human heart: evidence for altered thyroid hormone receptor gene expression. Circulation : 1089–1094, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol : H801–H812, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Levick SP, Melendez GC, Plante E, McLarty JL, Brower GL, Janicki JS. Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res : 12–19, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Li Y, Jiao J, Wang J, Qin D, Li P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol : 1788–1799, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Zhou J, Li Y, Qin D, Li P. Mitochondrial fission controls DNA fragmentation by regulating endonuclease G. Free Radic Biol Med : 622–631, 2010. [DOI] [PubMed] [Google Scholar]
- 37.Lighthouse JK, Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol : 52–60, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods : 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Lv P, Zhou M, He J, Meng W, Ma X, Dong S, Meng X, Zhao X, Wang X, He F. Circulating miR-208b and miR-34a are associated with left ventricular remodeling after acute myocardial infarction. Int J Mol Sci : 5774–5788, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandache E, Gherghiceanu M, Macarie C, Kostin S, Popescu LM. Telocytes in human isolated atrial amyloidosis: ultrastructural remodelling. J Cell Mol Med : 2739–2747, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLendon JM, Joshi SR, Sparks J, Matar M, Fewell JG, Abe K, Oka M, McMurtry IF, Gerthoffer WT. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J Control Release : 67–75, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michela P, Velia V, Aldo P, Ada P. Role of connexin 43 in cardiovascular diseases. Eur J Pharmacol : 71–76, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Moore-Morris T, Cattaneo P, Puceat M, Evans SM. Origins of cardiac fibroblasts. J Mol Cell Cardiol : 1–5, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ojamaa K, Kenessey A, Shenoy R, Klein I. Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am J Physiol Endocrinol Metab : E1319–E1324, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Oliveira-Carvalho V, Carvalho VO, Bocchi EA. The emerging role of miR-208a in the heart. DNA Cell Biol : 8–12, 2012. [DOI] [PubMed] [Google Scholar]
- 46.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med : 1131–1141, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Perfettini JL, Roumier T, Kroemer G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol : 179–183, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Qipshidze N, Tyagi N, Metreveli N, Lominadze D, Tyagi SC. Autophagy mechanism of right ventricular remodeling in murine model of pulmonary artery constriction. Am J Physiol Heart Circ Physiol : H688–H696, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, Gupte SA. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension : 1266–1274, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC, Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics : 562–575, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Redout EM, van der Toorn A, Zuidwijk MJ, van de Kolk CW, van Echteld CJ, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am J Physiol Heart Circ Physiol : H1038–H1047, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm : 848–856, 2009. [DOI] [PubMed] [Google Scholar]
- 53.Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, Meyer JN. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol : 23–38, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest : 2495–2508, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salido M, Gonzalez JL, Vilches J. Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol Cancer Ther : 1292–1299, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Saraste A, Pulkki K, Kallajoki M, Heikkila P, Laine P, Mattila S, Nieminen MS, Parvinen M, Voipio-Pulkki LM. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Invest : 380–386, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Sequeira V, Nijenkamp LL, Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta : 700–722, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Stones R, Benoist D, Peckham M, White E. Microtubule proliferation in right ventricular myocytes of rats with monocrotaline-induced pulmonary hypertension. J Mol Cell Cardiol : 91–96, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev : 1577–1590, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal : 2111–2124, 2006. [DOI] [PubMed] [Google Scholar]
- 61.Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun : 8701, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension : 241–248, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc MG, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J : 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 64.Toba M, Alzoubi A, O'Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol : H243–H250, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell : 662–673, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A : 18255–18260, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation : 1883–1891, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Wang HW, Huang TS, Lo HH, Huang PH, Lin CC, Chang SJ, Liao KH, Tsai CH, Chan CH, Tsai CF, Cheng YC, Chiu YL, Tsai TN, Cheng CC, Cheng SM. Deficiency of the microRNA-31-microRNA-720 pathway in the plasma and endothelial progenitor cells from patients with coronary artery disease. Arterioscler Thromb Vasc Biol : 857–869, 2014. [DOI] [PubMed] [Google Scholar]
- 69.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest : 1497–1504, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan H, Wang S, Yu H, Zhu J, Chen C. Molecular pathways and functional analysis of miRNA expression associated with paclitaxel-induced apoptosis in hepatocellular carcinoma cells. Pharmacology : 167–174, 2013. [DOI] [PubMed] [Google Scholar]
- 71.Yu B, Gong M, Wang Y, Millard RW, Pasha Z, Yang Y, Ashraf M, Xu M. Cardiomyocyte protection by GATA-4 gene engineered mesenchymal stem cells is partially mediated by translocation of miR-221 in microvesicles. PLoS One : e73304, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, Chang JJ, Xu F, Ma XJ, Wu Y, Li WC, Wang HJ, Huang GY, Ma D. MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of fallot. Can J Cardiol : 1695–1703, 2013. [DOI] [PubMed] [Google Scholar]
- 73.Zhang WG, Chen L, Dong Q, He J, Zhao HD, Li FL, Li H. Mmu-miR-702 functions as an anti-apoptotic mirtron by mediating ATF6 inhibition in mice. Gene : 235–242, 2013. [DOI] [PubMed] [Google Scholar]
- 74.Zhao J, Lei T, Xu C, Li H, Ma W, Yang Y, Fan S, Liu Y. MicroRNA-187, down-regulated in clear cell renal cell carcinoma and associated with lower survival, inhibits cell growth and migration though targeting B7–H3. Biochem Biophys Res Commun : 439–444, 2013. [DOI] [PubMed] [Google Scholar]
- 75.Zhou Y, Pan P, Yao L, Su M, He P, Niu N, McNutt MA, Gu J. CD117-positive cells of the heart: progenitor cells or mast cells? J Histochem Cytochem : 309–316, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]