

The title compound consists of two fluorophenyl groups and one butyl group equatorially oriented on a piperidine ring, which adopts a chair conformation. The dihedral angle between the mean planes of the phenyl rings is 72.1 (1)°. In the crystal, weak N—H⋯O and C—H⋯F interactions, which form  [14] motifs, link the molecules into infinite C(6) chains propgagating along [001].
[14] motifs, link the molecules into infinite C(6) chains propgagating along [001].
Keywords: piperidin-4-one, crystal structure, Hirshfeld surface
Abstract
The title compound, C21H23F2NO, consists of two fluorophenyl groups and one butyl group equatorially oriented on a piperidine ring, which adopts a chair conformation. The dihedral angle between the mean planes of the phenyl rings is 72.1 (1)°. In the crystal, N—H⋯O and weak C—H⋯F interactions, which form R 2 2[14] motifs, link the molecules into infinite C(6) chains propagating along [001]. A weak C—H⋯π interaction is also observed. A Hirshfeld surface analysis of the crystal structure indicates that the most significant contributions to the crystal packing are from H⋯H (53.3%), H⋯C/C⋯H (19.1%), H⋯F/F⋯H (15.7%) and H⋯O/O⋯H (7.7%) contacts. Density functional theory geometry-optimized calculations were compared to the experimentally determined structure in the solid state and used to determine the HOMO–LUMO energy gap and compare it to the UV–vis experimental spectrum.
Chemical context
Piperidin-4-one compounds have various biological properties and have applications as anti-viral, antitumor, and antihistaminic agents (El-Subbagh et al., 2000 ▸; Mobio et al., 1989 ▸; Katritzky & Fan, 1990 ▸; Arulraj et al., 2020 ▸). 2,6-Disubstituted piperidine-4-ones commonly adopt a chair conformation for the heterocyclic ring (see, for example, Rajkumar et al., 2018 ▸). However, on varying the substituents attached to the phenyl ring, the conformation of the ring may change (e.g. Ramachandran et al., 2007 ▸; Arulraj et al., 2020 ▸). Additionally, the attached functional group on the crystalline compound is important to determine the activity of the compound in the area of drug discovery.
As part of our studies in this area, we now describe the synthesis and structure of the title compound, C21H23F2NO, (I), in order to establish the structural effects of the butyl and fluoro groups on the conformation. DFT calculations and a Hirshfeld analysis have also been carried out.
Structural commentary
Compound (I) crystallizes in space group P21/c with one molecule in the asymmetric unit (Fig. 1 ▸). In the arbitrarily chosen asymmetric unit, the stereogenic centres have the following configurations: C1 S, C2 R and C5 R, but crystal symmetry generates a racemic mixture. The piperidine ring adopts a slightly distorted chair conformation with puckering parameters Q = 0.5864 (16) Å, θ = 6.56 (15)°, φ = 356.9 (14)°. The dihedral angles for the C1–C5/N1 (all atoms) piperidine (A), C6–C11 fluorophenyl (B) and C12–C17 fluorophenyl (C) rings are A/B = 65.50 (8), A/C = 73.87 (8) and B/C = 72.11 (8)°. The substituents on the piperidine ring adopt equatorial orientations with the keto oxygen atom being anti-clinal [O1—C3—C2—C1 = −124.44 (16)°]. The butyl group lies in a syn-periplanar orientation [O1—C3—C2—C18 = 0.7 (2)] while the fluorophenyl groups are both anti-clinal [N1—C5—C6—C7 = −148.28 (13) and N1—C1—C12—C17 = −75.42 (16)°]. The sum of the bond angles around N1 is 336.8°, which is consistent with sp 3 hybridization for this atom (Beddoes et al., 1986 ▸).
Figure 1.

A view of the molecular structure of C21H23F2NO, showing displacement ellipsoids drawn at the 30% probability level.
Supramolecular features
N1—H1⋯O1 and weak C7—H7⋯F1 interactions are observed in the crystal of (I) (Table 1 ▸, Fig. 2 ▸), which form [14] graph-set ring motifs and infinite C(6) chains (via the N—H⋯O bond) along [001]. Some longer C—H⋯O and C—H⋯F contacts are also present as well as a single weak C—H⋯π interaction (Table 1 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
Cg3 is the centroid of the C12–C17 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O1i | 1.05 | 2.06 | 3.0921 (16) | 165 |
| C7—H7⋯F1ii | 0.95 | 2.52 | 3.3291 (18) | 143 |
| C10—H10⋯O1iii | 0.95 | 2.66 | 3.470 (2) | 144 |
| C16—H16⋯F2iv | 0.95 | 2.62 | 3.3680 (18) | 136 |
| C21—H21C⋯F2ii | 0.98 | 2.58 | 3.489 (2) | 154 |
| C21—H21A⋯Cg3v | 0.98 | 2.95 | 3.793 (2) | 145 |
Symmetry codes: (i)  ; (ii)
; (ii) 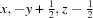 ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  .
.
Figure 2.

Crystal packing for C21H23F2NO viewed along the a-axis direction. Dashed lines indicate N—H⋯O hydrogen bonds and weak C—H⋯F interactions forming (14) loops and infinite C(6) chains (via the N—H⋯O bond) along the c-axis direction.
Hirshfeld surface analysis
A Hirshfeld surface (HS) analysis (Spackman & Jayatilaka, 2009 ▸) was carried out using CrystalExplorer17.5 (Turner et al., 2017 ▸) to visualize the intermolecular interactions in (I). The bright-red spot near H1 indicates its role as a hydrogen-bond donor to O1 (Fig. 3 ▸) and another red region near H7 correlates with the C7—H7⋯F1 interaction. The shape-index of the HS represents a way to visualize π–π stacking by the presence of red and/or blue triangles but there are none in in the title compound (see Figure S1 in the supporting information). The curvedness of the HS can be used to divide the molecular surface into contact patches with each neighbouring molecule thereby using it to define a coordination number in the crystal (see Figure S2 in the supporting information).
Figure 3.

A view of the three-dimensional Hirshfeld surface for C21H23F2NO, plotted over d norm in the range −0.39 to 1.31 a.u.
Two-dimensional fingerprint plots show the relative contributions of the various types of contacts to the Hirshfeld surface for (I) (McKinnon et al., 2007 ▸). The overal plot is shown in Fig. 4 ▸ a. The H⋯H contacts (53.3%) are the most important interactions (Fig. 4 ▸ b), presumably because of the large hydrogen content of (I), with a pair of blue-coloured blunt spikes directing towards the bottom left, in the region 1.20 Å < (d e + d i) < 1.19 Å. The pair of wings for the H⋯C/C⋯H contacts (Fig. 4 ▸ c; 19.1% contribution to the HS) is in the region 1.04 Å < (d e + d i) < 1.58 Å and includes the weak C—H⋯π interaction. The H⋯F/F⋯H contacts (Fig. 4 ▸ d; 15.7% contribution) are seen as a pair of wings in the region 1.04 Å < (d e + d i) < 1.38 Å. The wings for the H⋯O/O⋯H contacts (Fig. 4 ▸ e; 7.7% contribution) are in the region of 0.88 Å < (d e + d i) < 1.20 Å while the blunt wings in the plot for F⋯F contacts (Fig. 4 ▸ f; 2.6%) are in the region 1.60 Å < (d e + d i) < 1.70 Å. The C⋯C contacts (Fig. 4 ▸ g) make a negligible 0.1% contribution and are viewed as a dash pattern pointing diagonally left. The O⋯O contacts (Fig. 4 ▸ h) make no contribution to the HS. The most significant of these contributions to the overall Hirshfeld surface are shown in Figure S3 in the supporting information.
Figure 4.

A view of the two-dimensional fingerprint plots for C21H23F2NO, showing (a) all interactions, and separated into (b) H⋯H, (c) H⋯C/C⋯H, (d) H⋯F/F⋯H, (e) O⋯H/H⋯O, (f) F⋯F, (g) C—C and (h) O⋯O interactions. The d i and d e values are the closest internal and external distances (in Å) from given points on the Hirshfeld surface contacts.
DFT Calculations
A density functional theory (DFT) geometry-optimized calculation for (I) was carried out using WebMo Pro (Schmidt & Polik, 2007 ▸) in the GAUSSIAN 09 program package (Frisch et al., 2009 ▸) using the 6-31+G(d) basis set (Hehre et al., 1986 ▸). The starting geometry was taken from the crystal structure and no solvent correction was applied. A comparison of bond angles and bond distances in the crystal to those from the DFT calculation are listed in supplementary Table S1, which generally shows good agreement. An overlay of the geometry-optimized calculation with the crystal structure has an r.m.s. deviation of 0.478 Å. The major difference between the experimental and calculated structures occurs in the orientation of the C12–C17 rings, which are rotated by 41.8 (6)° with respect to each other.
The calculated energies (eV) for the frontier molecular orbitals are shown in Fig. 5 ▸ and key parameters are listed in supplementary Table S2. Both the HOMO and HOMO−1 are localized largely on the piperidine ring. For the LUMO, LUMO+1 and LUMO+2, the orbitals are delocalized over the piperidine ring as well as both phenyl rings. The observed UV/vis absorption spectrum (Fig. 6 ▸) shows two band envelopes with λmax values located at ca 256 and 216 nm (∼4.84 and 5.74 eV). The molar extinction coefficients, ∊, are 1.12 and 2.50 l mol−1 cm−1, respectively. We tentatively assign the first absorption band envelope at 256 nm to overlapping contributions from HOMO → LUMO (energy gap 5.71 eV), HOMO → LUMO+1 (5.83 eV) and HOMO−1 → LUMO (5.82 eV). The band at 216 nm is assigned to overlapping contributions from HOMO → LUMO+2 (5.89 eV), HOMO−1 → LUMO+1 (5.95 eV) and HOMO−1 → LUMO+2 (6.01 eV).
Figure 5.

Schematic MO diagram.
Figure 6.

UV–vis spectrum of C21H23F2NO
Database survey
A search in the Cambridge Crystallographic Database (CSD version 2.0.4 of December 2019; Groom et al.. 2016 ▸) for the 2,6-diphenylpiperidin-4-one skeleton resulted in 240 hits, which was refined to 44 matches by removing those structures in which the title skeleton substructure was combined with larger molecules. The four most closely related remaining structures based on the pendant arms of the 2,6 diphenylpiperidine-4-one central substructure are 2,6-diphenyl-3-isopropylpiperidin-4-one (ACEZUD; Nilofar Nissa et al., 2001 ▸), t(3)-pentyl-r(2),c6)-diphenylpiperidin-4-one (RUGLOV; Gayathri et al., 2009 ▸), 3-(2-chloroethyl)-r(2),c(6)-diphenylpiperidin-4-one (PEXDII; Rajkumar et al., 2018 ▸) and 3-(2-chloroethyl)-r(2),c(6)-bis(4-fluorophenyl)piperidin-4-one (PEXDOO; Rajkumar et al., 2018 ▸). The piperidine ring in the title compound is in a slightly distorted chair conformation, similar to that observed in ACEZUD and PEXDOO but different from the chair conformation seen in RUGLOV and PEXDII. The dihedral angle between the mean planes of pendant phenyl rings is 72.(1)° in the title compound compared to 76.1 (1)° in PEXDOO, whereas it is 59.90 (5), 59.1 (1) and 63.4 (1)° in RUGLOV, PEXDII and ACEZUD, respectively. In all five compounds, various N—H⋯O and weak C—H⋯O, C—H⋯π or C—H⋯F interactions occur in the crystal.
Synthesis and crystallization
A mixture of ammonium acetate (0.100 mol, 7.71 g), 4-fluorobenzaldehyde (0.200 mol, 22.0 ml) and 2-heptanone (0.100 mol, 14.2 ml) in distilled ethanol was heated first to boiling. After cooling, the viscous liquid obtained was dissolved in ether (200 ml) and shaken with 100 ml concentrated hydrochloric acid. The precipitated hydrochloride of 3-butyl-2,6-bis(4-fluorophenyl)piperidin-4-one was removed by filtration and washed first with a 50 ml mixture of ethanol and ether (1:1) and then with ether to remove most of the coloured impurities. The resulting yellowish base was liberated from an alcoholic solution by adding aqueous ammonia (15 ml) and then diluted with water (200 ml). Then, 1.0 g of the crude sample was dissolved in 100 ml of absolute alcohol, warmed until the sample dissolved, and 2.0 g of animal charcoal added in the resulting solution. The hot solution was filtered and the procedure repeated again. The filtered solution was left for 48 h and colourless prisms of (I) were collected in 75% yield. Analysis for C21H23F2NO (%): found C 74.24, H 6.16, N 4.03; calculated C 73.45, H 6.75, N 4.08; melting point 381.5 K.
FT–IR (cm−1) (KBr): 3287 (νN—H), 3134, 2929, 2866 (νC—H), 1702 (νC=O), 1605, 1508 (νC=C), 793 (νC—Cl); 1H NMR (400 MHz, CDCl3): δ 7.01–7.45 (m, aromatic protons), 4.04 (d, H6 proton), 3.68 (s, H2 proton), 2.67 (t, H5a proton), 2.56 (dd, H5e proton), 2.0 (NH proton), 0.95–1.0 CH2(3), 1.09–1.15 CH2(2), 1.59–1.63 CH2(1), 0.74, (t, CH3 alkyl proton); 13C NMR (400 MHz, CDCl3): δ 129.16, 129.38, 128.18, 128.10, 115.64, 115.56, 115.43, 115.35 (aromatic carbon atoms), 138.52 and 137.64 (aromatic ipso carbon atoms), 66.33 (C2), 57.50 (C3), 208.7 (C4), 51.63 (C5), 61.08 (C6), 24.30 C18H2, 29.71 C19H2, 22.75 C20H2, 13.81 C21H3.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The C-bound H atoms were geometrically placed (C—H = 0.93–0.98 Å) and refined as riding atoms. The N-bound H atom was located in a difference map and its position was fixed. The methyl group was allowed to rotate, but not to tip, to best fit the electron density. The constraint U iso(H) = 1.2U eq(carrier) or 1.5U eq(methyl carrier) was applied in all cases.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C21H23F2NO |
| M r | 343.40 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 173 |
| a, b, c (Å) | 5.4945 (3), 25.0707 (13), 12.9811 (9) |
| β (°) | 93.497 (6) |
| V (Å3) | 1784.83 (18) |
| Z | 4 |
| Radiation type | Cu Kα |
| μ (mm−1) | 0.76 |
| Crystal size (mm) | 0.42 × 0.36 × 0.35 |
| Data collection | |
| Diffractometer | Rigaku Oxford Diffraction Gemini Eos |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2019 ▸) |
| T min, T max | 0.803, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 6900, 3404, 3045 |
| R int | 0.027 |
| (sin θ/λ)max (Å−1) | 0.614 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.045, 0.126, 1.04 |
| No. of reflections | 3404 |
| No. of parameters | 228 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.24 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989020004636/hb7882sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020004636/hb7882Isup2.hkl
Theoretical chemistry data and Hirshfeld figures. DOI: 10.1107/S2056989020004636/hb7882sup3.docx
CCDC reference: 1994539
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors would like to acknowledge Vellore Institute of Technology, Tamilnadu, India for recording the NMR spectra, the Indian Institute of Technology (IIT), Chennai, Tamilnadu, India for recording the FT–IR and UV–Visible spectra and extend their thanks to the Principal, Dr V. Ramnath, Chairman, Mr R. Sattanathan, and Treasurer, Mr T. Ramalingam, of Thiruvalluvar Arts and Science College for giving permission to carry out research work in the Chemistry Laboratory.
supplementary crystallographic information
Crystal data
| C21H23F2NO | F(000) = 728 |
| Mr = 343.40 | Dx = 1.278 Mg m−3 |
| Monoclinic, P21/c | Cu Kα radiation, λ = 1.54184 Å |
| a = 5.4945 (3) Å | Cell parameters from 3131 reflections |
| b = 25.0707 (13) Å | θ = 0.8–1.0° |
| c = 12.9811 (9) Å | µ = 0.76 mm−1 |
| β = 93.497 (6)° | T = 173 K |
| V = 1784.83 (18) Å3 | Prism, colourless |
| Z = 4 | 0.42 × 0.36 × 0.35 mm |
Data collection
| Rigaku Oxford Diffraction Gemini Eos diffractometer | 3404 independent reflections |
| Radiation source: fine-focus sealed X-ray tube | 3045 reflections with I > 2σ(I) |
| Detector resolution: 16.0416 pixels mm-1 | Rint = 0.027 |
| ω scans | θmax = 71.3°, θmin = 3.5° |
| Absorption correction: multi-scan (CrysAlisPro; Rigaku OD, 2019) | h = −6→6 |
| Tmin = 0.803, Tmax = 1.000 | k = −30→26 |
| 6900 measured reflections | l = −9→15 |
Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.045 | w = 1/[σ2(Fo2) + (0.0664P)2 + 0.4364P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.126 | (Δ/σ)max < 0.001 |
| S = 1.04 | Δρmax = 0.26 e Å−3 |
| 3404 reflections | Δρmin = −0.24 e Å−3 |
| 228 parameters | Extinction correction: SHELXL (Sheldrick 2015b), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.0035 (5) |
| Primary atom site location: dual |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 1.1088 (2) | 0.45264 (4) | 0.91794 (7) | 0.0458 (3) | |
| F2 | 0.2626 (2) | 0.04577 (4) | 0.65950 (9) | 0.0493 (3) | |
| O1 | 0.9789 (3) | 0.28871 (5) | 0.32525 (9) | 0.0439 (3) | |
| N1 | 0.8301 (2) | 0.26447 (5) | 0.61588 (9) | 0.0265 (3) | |
| H1 | 0.859899 | 0.250816 | 0.692480 | 0.032* | |
| C1 | 1.0000 (3) | 0.30675 (6) | 0.58925 (11) | 0.0267 (3) | |
| H1A | 1.163280 | 0.290264 | 0.580635 | 0.032* | |
| C2 | 0.9099 (3) | 0.33248 (6) | 0.48525 (11) | 0.0278 (3) | |
| H2 | 0.745757 | 0.348399 | 0.494336 | 0.033* | |
| C3 | 0.8784 (3) | 0.28808 (6) | 0.40558 (11) | 0.0312 (3) | |
| C4 | 0.7247 (3) | 0.24148 (6) | 0.43663 (12) | 0.0341 (4) | |
| H4A | 0.729221 | 0.212850 | 0.384262 | 0.041* | |
| H4B | 0.553231 | 0.252968 | 0.440962 | 0.041* | |
| C5 | 0.8253 (3) | 0.22036 (6) | 0.54247 (11) | 0.0282 (3) | |
| H5 | 0.996468 | 0.207883 | 0.535604 | 0.034* | |
| C6 | 0.6767 (3) | 0.17398 (6) | 0.57869 (11) | 0.0261 (3) | |
| C7 | 0.7422 (3) | 0.12243 (6) | 0.55255 (12) | 0.0303 (3) | |
| H7 | 0.883737 | 0.116949 | 0.515379 | 0.036* | |
| C8 | 0.6046 (3) | 0.07877 (6) | 0.57975 (13) | 0.0344 (4) | |
| H8 | 0.649053 | 0.043603 | 0.561167 | 0.041* | |
| C9 | 0.4023 (3) | 0.08793 (6) | 0.63432 (12) | 0.0340 (4) | |
| C10 | 0.3355 (3) | 0.13790 (7) | 0.66510 (13) | 0.0351 (4) | |
| H10 | 0.198118 | 0.142774 | 0.705110 | 0.042* | |
| C11 | 0.4740 (3) | 0.18125 (6) | 0.63626 (12) | 0.0314 (3) | |
| H11 | 0.429534 | 0.216223 | 0.656111 | 0.038* | |
| C12 | 1.0247 (3) | 0.34625 (6) | 0.67737 (11) | 0.0265 (3) | |
| C13 | 1.2301 (3) | 0.34492 (6) | 0.74451 (13) | 0.0350 (4) | |
| H13 | 1.353733 | 0.319324 | 0.734112 | 0.042* | |
| C14 | 1.2594 (3) | 0.38022 (7) | 0.82671 (13) | 0.0389 (4) | |
| H14 | 1.400409 | 0.378901 | 0.872784 | 0.047* | |
| C15 | 1.0794 (3) | 0.41700 (6) | 0.83961 (12) | 0.0327 (4) | |
| C16 | 0.8701 (3) | 0.41935 (6) | 0.77634 (12) | 0.0347 (4) | |
| H16 | 0.747117 | 0.444956 | 0.787708 | 0.042* | |
| C17 | 0.8432 (3) | 0.38332 (6) | 0.69539 (12) | 0.0312 (3) | |
| H17 | 0.698502 | 0.383920 | 0.651444 | 0.037* | |
| C18 | 1.0774 (3) | 0.37699 (6) | 0.45103 (12) | 0.0300 (3) | |
| H18A | 1.214626 | 0.360763 | 0.416194 | 0.036* | |
| H18B | 1.146589 | 0.395904 | 0.513051 | 0.036* | |
| C19 | 0.9510 (3) | 0.41772 (6) | 0.37807 (12) | 0.0347 (4) | |
| H19A | 1.075666 | 0.442006 | 0.352266 | 0.042* | |
| H19B | 0.873544 | 0.398610 | 0.317901 | 0.042* | |
| C20 | 0.7587 (3) | 0.45074 (7) | 0.42774 (14) | 0.0409 (4) | |
| H20A | 0.828276 | 0.464904 | 0.494378 | 0.049* | |
| H20B | 0.619586 | 0.427418 | 0.442398 | 0.049* | |
| C21 | 0.6662 (4) | 0.49699 (7) | 0.36054 (15) | 0.0453 (4) | |
| H21A | 0.800002 | 0.522000 | 0.351000 | 0.068* | |
| H21B | 0.534931 | 0.515376 | 0.394092 | 0.068* | |
| H21C | 0.603886 | 0.483414 | 0.293211 | 0.068* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0703 (7) | 0.0351 (5) | 0.0321 (5) | −0.0142 (5) | 0.0041 (5) | −0.0113 (4) |
| F2 | 0.0580 (6) | 0.0342 (5) | 0.0559 (7) | −0.0176 (5) | 0.0041 (5) | 0.0125 (5) |
| O1 | 0.0700 (8) | 0.0380 (6) | 0.0248 (6) | −0.0140 (6) | 0.0122 (5) | −0.0041 (5) |
| N1 | 0.0356 (6) | 0.0226 (6) | 0.0212 (6) | −0.0038 (5) | 0.0004 (5) | 0.0010 (4) |
| C1 | 0.0301 (7) | 0.0244 (7) | 0.0255 (7) | −0.0014 (5) | 0.0015 (5) | 0.0005 (6) |
| C2 | 0.0342 (7) | 0.0259 (7) | 0.0233 (7) | −0.0032 (6) | 0.0018 (6) | 0.0009 (6) |
| C3 | 0.0411 (8) | 0.0301 (7) | 0.0219 (7) | −0.0048 (6) | −0.0009 (6) | 0.0028 (6) |
| C4 | 0.0449 (9) | 0.0313 (8) | 0.0258 (8) | −0.0093 (6) | −0.0010 (6) | −0.0008 (6) |
| C5 | 0.0340 (7) | 0.0244 (7) | 0.0262 (7) | −0.0035 (6) | 0.0026 (6) | 0.0002 (6) |
| C6 | 0.0309 (7) | 0.0227 (7) | 0.0244 (7) | −0.0013 (5) | −0.0010 (5) | 0.0010 (5) |
| C7 | 0.0353 (7) | 0.0276 (7) | 0.0281 (7) | 0.0002 (6) | 0.0027 (6) | −0.0029 (6) |
| C8 | 0.0467 (9) | 0.0216 (7) | 0.0344 (8) | 0.0011 (6) | −0.0031 (7) | −0.0008 (6) |
| C9 | 0.0391 (8) | 0.0284 (8) | 0.0338 (8) | −0.0087 (6) | −0.0040 (6) | 0.0093 (6) |
| C10 | 0.0335 (7) | 0.0358 (8) | 0.0365 (9) | −0.0006 (6) | 0.0063 (6) | 0.0047 (6) |
| C11 | 0.0343 (7) | 0.0243 (7) | 0.0356 (8) | 0.0029 (6) | 0.0033 (6) | 0.0009 (6) |
| C12 | 0.0335 (7) | 0.0229 (7) | 0.0231 (7) | −0.0047 (5) | 0.0029 (5) | 0.0025 (5) |
| C13 | 0.0371 (8) | 0.0334 (8) | 0.0338 (8) | 0.0041 (6) | −0.0029 (6) | −0.0032 (6) |
| C14 | 0.0398 (8) | 0.0430 (9) | 0.0327 (9) | −0.0049 (7) | −0.0075 (7) | −0.0048 (7) |
| C15 | 0.0496 (9) | 0.0256 (7) | 0.0235 (7) | −0.0119 (6) | 0.0064 (6) | −0.0038 (6) |
| C16 | 0.0430 (8) | 0.0301 (8) | 0.0316 (8) | 0.0033 (6) | 0.0074 (6) | −0.0015 (6) |
| C17 | 0.0321 (7) | 0.0344 (8) | 0.0270 (7) | 0.0004 (6) | 0.0006 (6) | −0.0012 (6) |
| C18 | 0.0348 (7) | 0.0290 (7) | 0.0266 (7) | −0.0063 (6) | 0.0045 (6) | 0.0004 (6) |
| C19 | 0.0460 (9) | 0.0294 (8) | 0.0293 (8) | −0.0052 (6) | 0.0073 (6) | 0.0038 (6) |
| C20 | 0.0513 (10) | 0.0329 (8) | 0.0396 (9) | −0.0004 (7) | 0.0114 (8) | 0.0073 (7) |
| C21 | 0.0581 (11) | 0.0334 (9) | 0.0442 (10) | 0.0035 (8) | 0.0022 (8) | 0.0044 (7) |
Geometric parameters (Å, º)
| F1—C15 | 1.3559 (17) | C10—H10 | 0.9500 |
| F2—C9 | 1.3581 (17) | C10—C11 | 1.391 (2) |
| O1—C3 | 1.2096 (19) | C11—H11 | 0.9500 |
| N1—H1 | 1.0549 | C12—C13 | 1.384 (2) |
| N1—C1 | 1.4678 (17) | C12—C17 | 1.393 (2) |
| N1—C5 | 1.4590 (18) | C13—H13 | 0.9500 |
| C1—H1A | 1.0000 | C13—C14 | 1.388 (2) |
| C1—C2 | 1.5498 (19) | C14—H14 | 0.9500 |
| C1—C12 | 1.5128 (19) | C14—C15 | 1.370 (2) |
| C2—H2 | 1.0000 | C15—C16 | 1.373 (2) |
| C2—C3 | 1.522 (2) | C16—H16 | 0.9500 |
| C2—C18 | 1.5296 (19) | C16—C17 | 1.387 (2) |
| C3—C4 | 1.511 (2) | C17—H17 | 0.9500 |
| C4—H4A | 0.9900 | C18—H18A | 0.9900 |
| C4—H4B | 0.9900 | C18—H18B | 0.9900 |
| C4—C5 | 1.543 (2) | C18—C19 | 1.530 (2) |
| C5—H5 | 1.0000 | C19—H19A | 0.9900 |
| C5—C6 | 1.5121 (19) | C19—H19B | 0.9900 |
| C6—C7 | 1.389 (2) | C19—C20 | 1.517 (2) |
| C6—C11 | 1.391 (2) | C20—H20A | 0.9900 |
| C7—H7 | 0.9500 | C20—H20B | 0.9900 |
| C7—C8 | 1.388 (2) | C20—C21 | 1.520 (2) |
| C8—H8 | 0.9500 | C21—H21A | 0.9800 |
| C8—C9 | 1.373 (2) | C21—H21B | 0.9800 |
| C9—C10 | 1.372 (2) | C21—H21C | 0.9800 |
| C1—N1—H1 | 113.1 | C6—C11—C10 | 120.76 (14) |
| C5—N1—H1 | 111.4 | C6—C11—H11 | 119.6 |
| C5—N1—C1 | 112.32 (11) | C10—C11—H11 | 119.6 |
| N1—C1—H1A | 108.4 | C13—C12—C1 | 119.50 (13) |
| N1—C1—C2 | 109.37 (11) | C13—C12—C17 | 118.36 (14) |
| N1—C1—C12 | 108.87 (11) | C17—C12—C1 | 122.12 (13) |
| C2—C1—H1A | 108.4 | C12—C13—H13 | 119.3 |
| C12—C1—H1A | 108.4 | C12—C13—C14 | 121.38 (15) |
| C12—C1—C2 | 113.29 (11) | C14—C13—H13 | 119.3 |
| C1—C2—H2 | 107.8 | C13—C14—H14 | 120.9 |
| C3—C2—C1 | 107.73 (11) | C15—C14—C13 | 118.19 (15) |
| C3—C2—H2 | 107.8 | C15—C14—H14 | 120.9 |
| C3—C2—C18 | 112.44 (12) | F1—C15—C14 | 118.78 (15) |
| C18—C2—C1 | 113.01 (12) | F1—C15—C16 | 118.50 (15) |
| C18—C2—H2 | 107.8 | C14—C15—C16 | 122.72 (14) |
| O1—C3—C2 | 122.51 (14) | C15—C16—H16 | 120.9 |
| O1—C3—C4 | 122.15 (14) | C15—C16—C17 | 118.15 (15) |
| C4—C3—C2 | 115.25 (12) | C17—C16—H16 | 120.9 |
| C3—C4—H4A | 109.9 | C12—C17—H17 | 119.4 |
| C3—C4—H4B | 109.9 | C16—C17—C12 | 121.17 (14) |
| C3—C4—C5 | 109.09 (12) | C16—C17—H17 | 119.4 |
| H4A—C4—H4B | 108.3 | C2—C18—H18A | 108.7 |
| C5—C4—H4A | 109.9 | C2—C18—H18B | 108.7 |
| C5—C4—H4B | 109.9 | C2—C18—C19 | 114.09 (12) |
| N1—C5—C4 | 108.21 (12) | H18A—C18—H18B | 107.6 |
| N1—C5—H5 | 108.4 | C19—C18—H18A | 108.7 |
| N1—C5—C6 | 111.60 (12) | C19—C18—H18B | 108.7 |
| C4—C5—H5 | 108.4 | C18—C19—H19A | 108.8 |
| C6—C5—C4 | 111.70 (12) | C18—C19—H19B | 108.8 |
| C6—C5—H5 | 108.4 | H19A—C19—H19B | 107.7 |
| C7—C6—C5 | 119.12 (13) | C20—C19—C18 | 113.74 (13) |
| C7—C6—C11 | 118.74 (14) | C20—C19—H19A | 108.8 |
| C11—C6—C5 | 122.13 (13) | C20—C19—H19B | 108.8 |
| C6—C7—H7 | 119.4 | C19—C20—H20A | 109.0 |
| C8—C7—C6 | 121.23 (14) | C19—C20—H20B | 109.0 |
| C8—C7—H7 | 119.4 | C19—C20—C21 | 112.94 (14) |
| C7—C8—H8 | 121.0 | H20A—C20—H20B | 107.8 |
| C9—C8—C7 | 118.03 (14) | C21—C20—H20A | 109.0 |
| C9—C8—H8 | 121.0 | C21—C20—H20B | 109.0 |
| F2—C9—C8 | 118.78 (14) | C20—C21—H21A | 109.5 |
| F2—C9—C10 | 118.38 (15) | C20—C21—H21B | 109.5 |
| C10—C9—C8 | 122.84 (14) | C20—C21—H21C | 109.5 |
| C9—C10—H10 | 120.8 | H21A—C21—H21B | 109.5 |
| C9—C10—C11 | 118.34 (15) | H21A—C21—H21C | 109.5 |
| C11—C10—H10 | 120.8 | H21B—C21—H21C | 109.5 |
| F1—C15—C16—C17 | 178.97 (13) | C4—C5—C6—C11 | −88.66 (17) |
| F2—C9—C10—C11 | −177.84 (14) | C5—N1—C1—C2 | 64.80 (15) |
| O1—C3—C4—C5 | 123.39 (17) | C5—N1—C1—C12 | −170.93 (11) |
| N1—C1—C2—C3 | −54.89 (15) | C5—C6—C7—C8 | −176.87 (13) |
| N1—C1—C2—C18 | −179.73 (12) | C5—C6—C11—C10 | 177.54 (14) |
| N1—C1—C12—C13 | 102.72 (15) | C6—C7—C8—C9 | −0.6 (2) |
| N1—C1—C12—C17 | −75.42 (16) | C7—C6—C11—C10 | −1.5 (2) |
| N1—C5—C6—C7 | −148.28 (13) | C7—C8—C9—F2 | 178.51 (13) |
| N1—C5—C6—C11 | 32.63 (19) | C7—C8—C9—C10 | −1.8 (2) |
| C1—N1—C5—C4 | −64.54 (15) | C8—C9—C10—C11 | 2.5 (2) |
| C1—N1—C5—C6 | 172.17 (11) | C9—C10—C11—C6 | −0.7 (2) |
| C1—C2—C3—O1 | −124.44 (16) | C11—C6—C7—C8 | 2.2 (2) |
| C1—C2—C3—C4 | 52.03 (17) | C12—C1—C2—C3 | −176.53 (12) |
| C1—C2—C18—C19 | −155.74 (13) | C12—C1—C2—C18 | 58.63 (16) |
| C1—C12—C13—C14 | −179.62 (14) | C12—C13—C14—C15 | −0.5 (3) |
| C1—C12—C17—C16 | −179.67 (14) | C13—C12—C17—C16 | 2.2 (2) |
| C2—C1—C12—C13 | −135.36 (14) | C13—C14—C15—F1 | −178.24 (14) |
| C2—C1—C12—C17 | 46.50 (18) | C13—C14—C15—C16 | 1.7 (2) |
| C2—C3—C4—C5 | −53.09 (18) | C14—C15—C16—C17 | −1.0 (2) |
| C2—C18—C19—C20 | 66.24 (18) | C15—C16—C17—C12 | −1.0 (2) |
| C3—C2—C18—C19 | 82.03 (16) | C17—C12—C13—C14 | −1.4 (2) |
| C3—C4—C5—N1 | 55.98 (16) | C18—C2—C3—O1 | 0.7 (2) |
| C3—C4—C5—C6 | 179.22 (13) | C18—C2—C3—C4 | 177.20 (13) |
| C4—C5—C6—C7 | 90.43 (16) | C18—C19—C20—C21 | 170.21 (14) |
Hydrogen-bond geometry (Å, º)
Cg3 is the centroid of the C12–C17 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O1i | 1.05 | 2.06 | 3.0921 (16) | 165 |
| C7—H7···F1ii | 0.95 | 2.52 | 3.3291 (18) | 143 |
| C10—H10···O1iii | 0.95 | 2.66 | 3.470 (2) | 144 |
| C16—H16···F2iv | 0.95 | 2.62 | 3.3680 (18) | 136 |
| C21—H21C···F2ii | 0.98 | 2.58 | 3.489 (2) | 154 |
| C21—H21A···Cg3v | 0.98 | 2.95 | 3.793 (2) | 145 |
Symmetry codes: (i) x, −y+1/2, z+1/2; (ii) x, −y+1/2, z−1/2; (iii) x−1, −y+1/2, z+1/2; (iv) −x+1, y+1/2, −z+3/2; (v) −x+2, −y+1, −z+1.
Funding Statement
This work was funded by National Science Foundation grant CHE1039027.
References
- Arulraj, R., Sivakumar, S., Rajkumar, K., Jasinski, J. P., Kaur, M. & Thiruvalluvar, A. (2019). J. Chem. Cryst. 50, 41–51.
- Arulraj, R., Sivakumar, S., Suresh, S. & Anitha, K. (2020). Spectrochim. Acta Part A, 232, 118166. [DOI] [PubMed]
- Beddoes, R. L., Dalton, L., Joule, T. A., Mills, O. S., Street, J. D. & Watt, C. I. F. (1986). J. Chem. Soc. Perkin Trans. 2, pp. 787.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- El-Subbagh, H. I., Abu-Zaid, S. M., Mahran, M. A., Badria, F. A. & Al-Obaid, A. M. (2000). J. Med. Chem. 43, 2915–2921. [DOI] [PubMed]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Jr., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. (2009). GAUSSIAN09. Gaussian Inc., Wallingford, CT, USA.
- Gayathri, P., Jayabharathi, J., Rajarajan, G., Thiruvalluvar, A. & Butcher, R. J. (2009). Acta Cryst. E65, o3083. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hehre, W. J., Random, L., Schleyer, P. R. & Pople, J. A. (1986). Abinitio Molecular Orbital Theory. New York: Wiley.
- Katritzky, A. R. & Fan, W. J. (1990). J. Org. Chem. 55, 3205–3209.
- McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. (2007). Chem. Commun. pp. 3814–3816. [DOI] [PubMed]
- Mobio, I. G., Soldatenkov, A. T., Federov, V. O., Ageev, E. A., Sergeeva, N. D., Lin, S., Stashenku, E. E., Prostakov, N. S. & Andreeva, E. L. (1989). Khim. Farm. Zh. 23, 421–427.
- Nilofar Nissa, M., Velmurugan, D., Narasimhan, S., Rajagopal, V. & Kim, M.-J. (2001). Acta Cryst. E57, o996–o998.
- Rajkumar, K., Sivakumar, S., Arulraj, R., Kaur, M., Jasinski, J. P., Manimekalai, A. & Thiruvalluvar, A. (2018). Acta Cryst. E74, 483–486. [DOI] [PMC free article] [PubMed]
- Ramachandran, R., Parthiban, P., Doddi, A., Ramkumar, V. & Kabilan, S. (2007). Acta Cryst. E63, o4559.
- Rigaku OD (2019). CrysAlis PRO. Rigaku Americas, The Woodlands, Texas, USA.
- Schmidt, J. R. & Polik, W. F. (2007). WebMO Pro. WebMO, LLc: Holland. http://www.webmo.net
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spackman, M. A. & Jayatilaka, D. (2009). CrystEngComm, 11, 19–32.
- Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). CrystalExplorer17. The University of Western Australia.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989020004636/hb7882sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020004636/hb7882Isup2.hkl
Theoretical chemistry data and Hirshfeld figures. DOI: 10.1107/S2056989020004636/hb7882sup3.docx
CCDC reference: 1994539
Additional supporting information: crystallographic information; 3D view; checkCIF report