

In the title molecule, the imidazolopyridine and phenyl rings are nearly co-planar, and the 12-bromododecyl side chain is directed so as to give the complete molecule a V-shape.
Keywords: crystal structure, imidazole, pyridine, π-stacking, hydrogen bond, disorder
Abstract
The title compound, C24H30Br2N4O2, consists of a 2-(4-nitrophenyl)-4H-imidazo[4,5-b]pyridine entity with a 12-bromododecyl substituent attached to the pyridine N atom. The middle eight-carbon portion of the side chain is planar to within 0.09 (1) Å and makes a dihedral angle of 21.9 (8)° with the mean plane of the imidazolopyridine moiety, giving the molecule a V-shape. In the crystal, the imidazolopyridine units are associated through slipped π–π stacking interactions together with weak C—HPyr⋯ONtr and C—HBrmdcyl⋯ONtr (Pyr = pyridine, Ntr = nitro and Brmdcyl = bromododecyl) hydrogen bonds. The 12-bromododecyl chains overlap with each other between the stacks. The terminal –CH2Br group of the side chain shows disorder over two resolved sites in a 0.902 (3):0.098 (3) ratio. Hirshfeld surface analysis indicates that the most important contributions for the crystal packing are from H⋯H (48.1%), H⋯Br/Br⋯H (15.0%) and H⋯O/O⋯H (12.8%) interactions. The optimized molecular structure, using density functional theory at the B3LYP/ 6–311 G(d,p) level, is compared with the experimentally determined structure in the solid state. The HOMO–LUMO behaviour was elucidated to determine the energy gap.
Chemical context
Imidazole derivatives are a class of heterocyclic compounds exhibiting pharmacological activities across a wide range of therapeutic targets (El Kazzouli et al., 2011 ▸; Martínez-Urbina et al., 2010 ▸). Imidazopyridine derivatives are used in medicinal chemistry because of their biological and pharmacological properties, in particular as anti-inflammatory, anti-cancer, antiviral, anti-osteoporotic, anti-parasitic and antihypertensive agents. Certain compounds with an imidazopyridine skeleton are used to treat psychiatric and autoimmune disorders (Dymínska, 2015 ▸; Bababdani & Mousavi, 2013 ▸). They have also been approved as effective antifungal agents and exhibit antimicrobial activities (Devi et al., 2018 ▸), some of which can be used for the treatment of disorders characterized by activation of the Wnt signaling pathway (cancer, abnormal cell proliferation, angiogenesis, fibrous disorders, bone or cartilage and arthritis), as well as genetic and neurological diseases. On the other hand, imidazopyridine compounds, which have a specificity to GPR4 as negative allosteric modulators (Tobo et al., 2015 ▸), can also be used for the treatment of gastric and/or duodenal ulcers. Several imidazo[4,5-b]pyridine derivatives have also been reported as corrosion inhibitors of steel in an acidic environment (Bouayad et al., 2018 ▸; Sikine et al., 2016 ▸; Yadav et al., 2014 ▸), and also as inhibitors of a nanomolar rhodesaine (Ehmke et al., 2013 ▸) or the Et-PKG enzyme (Cheng et al., 2010 ▸).
Following our research work directed at obtaining new heterocyclic compounds having an imidazo[4,5-b]pyridine moiety, we were interested in the condensation of imidazo[4,5-b]pyridine derivatives with di-halogenated chains under phase-transfer catalysis (PTC) conditions. We report herein the synthesis and the molecular and crystal structures of the title compound (I) together with Hirshfeld surface analysis and DFT calculations for comparison with the experimentally determined molecular structure in the solid state.
Structural commentary
The molecule of (I) consists of an imidazolopyridine moiety to which a nitrophenyl group is attached to the C atom (C6) of the five-membered ring. The 12-bromododecyl side chain is attached to one of the two N atoms (N1). The imidazolopyridine moiety is planar to within 0.0208 (15) Å (r.m.s. deviation = 0.0122 Å) with atom C3 being the most distant from the least-squares plane. The phenyl ring (C7–C12) is inclined to the above plane by only 1.6 (1)°, making the two ring systems essentially planar. The C4—N1—C13—C14 torsion angle (C13 and C14 are the first two atoms of the 12-bromododecyl chain) is 95.7 (2)°. The C15–C22 portion of the 12-bromododecyl chain is approximately planar [maximum deviation from the least-squares plane running through the eight C atoms is 0.09 (1) Å for C20], and this plane is inclined to that of the imidazolopyridine moiety by 21.9 (8)°, giving the molecule an overall V-shape (Fig. 1 ▸). The terminal C24—Br2 portion of the 12-bromododecyl chain is disordered over two resolved sites in a refined ratio of 0.902 (3):0.098 (3).
Figure 1.

The asymmetric unit of (I) with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. Only the major part of the disordered –CH2Br group is shown.
Supramolecular features
In the crystal, sloped stacks of molecules extending along the a-axis direction are formed by slipped π–π stacking interactions between the N1/C1–C5 and C7–C12 rings, with a centroid-to-centroid distance of 3.5308 (13) Å, a dihedral angle of 1.83 (9)° and a slippage of 1.192 Å. The angle between the plane defined by the relevant centroids in the stack and (001) is 19.210 (11)°. The stacks are connected by weak C—HPyr⋯ONtr (Pyr = pyridine and Ntr = nitro) hydrogen bonds (Table 1 ▸, Fig. 2 ▸). Additional linkage is accomplished by C—HBrmdcyl⋯ONtr (Brmdcyl = bromododecyl) hydrogen bonds that influence the arrangement of the 12-bromododecyl chains between adjacent stacks (Table 1 ▸, Fig. 3 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C4—H4⋯O1i | 0.95 | 2.43 | 3.171 (3) | 135 |
| C24—H24A⋯O2ii | 0.99 | 2.53 | 3.372 (3) | 142 |
Symmetry codes: (i) 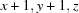 ; (ii)
; (ii)  .
.
Figure 2.

Detail of the intermolecular interactions viewed along the c-axis direction. The weak C—HPyr⋯ONtr and C—HBrmdcyl⋯ONtr (Pyr = pyridine, Ntr = nitro and Brmdcyl = bromododecyl) hydrogen bonds and π–π stacking interactions are depicted, respectively, by black and orange dashed lines.
Figure 3.

A partial packing diagram projected onto (581) with intermolecular interactions depicted as in Fig. 2 ▸.
Hirshfeld surface analysis
In order to quantify and visualize the intermolecular interactions in the crystal of (I), a Hirshfeld surface (HS) analysis (Hirshfeld, 1977 ▸; Spackman & Jayatilaka, 2009 ▸) was carried out by using Crystal Explorer 17.5 (Turner et al., 2017 ▸). In the HS plotted over d norm (Fig. 4 ▸), the white surface indicates contacts with distances equal to the sum of van der Waals radii, and the red and blue colours indicate distances shorter (in close contact) or longer (distant contact) than the van der Waals radii, respectively (Venkatesan et al., 2016 ▸). The overall two-dimensional fingerprint plot, Fig. 5 ▸ a, and those delineated into H⋯H, H⋯Br/Br⋯H, H⋯O/O⋯H, H⋯C/C⋯H, H⋯N/N⋯H, C⋯C, C⋯Br/Br⋯C and C⋯N/N⋯C contacts (McKinnon et al., 2007 ▸) are illustrated in Fig. 5 ▸ b–i, respectively, together with their relative contributions to the HS. The large number of these contacts suggests that van der Waals interactions and hydrogen bonding play the major roles in the crystal packing (Hathwar et al., 2015 ▸). The most important interaction is H⋯H contributing 48.1% to the overall crystal packing, Fig. 5 ▸ b, followed by H⋯Br/Br⋯H, Fig. 5 ▸ c, contacts at 15.0%, H⋯O/O⋯H, Fig. 5 ▸ d, contacts at 12.8%, H⋯C/C⋯H, Fig. 5 ▸ e, contacts at 6.0%, H⋯/N⋯H, Fig. 5 ▸ f, contacts at 5.8%, C⋯C, Fig. 5 ▸ g contacts at 3.7%, C⋯Br/Br⋯C, Fig. 5 ▸ h, contacts at 3.5%, and C⋯N/N⋯C, Fig. 5 ▸ i, contacts at 1.6%.
Figure 4.

View of the three-dimensional Hirshfeld surface of the title compound plotted over d norm in the range −0.1956 to 1.3971 a.u..
Figure 5.

Two-dimensional fingerprint plots for (I), showing (a) all interactions, and delineated into (b) H⋯H, (c) H⋯Br/Br⋯H, (d) H⋯O/O⋯H, (e) H⋯C/C⋯H, (f) H ⋯N/N⋯H, (g) C⋯C, (h) C⋯Br/Br⋯C and (i) C⋯N/N⋯C interactions. The d i and d e values are the closest internal and external distances (in Å) from given points on the Hirshfeld surface contacts.
DFT calculations
The optimized structure of the molecule in the gas phase was calculated via density functional theory (DFT) using the standard B3LYP functional and 6–311G(d,p) basis-set calculations (Becke, 1993 ▸) as implemented in GAUSSIAN 09 (Frisch et al., 2009 ▸). The theoretical and experimental bond lengths and angles are in good agreement (Table 2 ▸). Results for E HOMO and E LUMO energies, electronegativity (χ), hardness (η), potential (μ), electrophilicity (ω) and softness (σ) are collated in Table 3 ▸. The electron transition from the HOMO to the LUMO energy level is shown in Fig. 6 ▸. The energy band gap [ΔE = E LUMO – E HOMO] of the molecule is 1.6731 eV, and the calculated frontier molecular orbital energies, E HOMO and E LUMO, are −4.0238 and −2.3507 eV, respectively.
Table 2. Comparison of selected (X-ray and DFT) geometric data (Å, °) for (I).
| Bonds/angles | X-ray | B3LYP/6–311G(d,p) |
|---|---|---|
| Br1—C1 | 1.8954 (19) | 1.94573 |
| Br2—C24 | 1.953 (3) | 2.02642 |
| O1—N4 | 1.224 (2) | 1.28476 |
| O2—N4 | 1.233 (2) | 1.28545 |
| N1—C5 | 1.357 (2) | 1.36426 |
| N1—C4 | 1.361 (2) | 1.42977 |
| N1—C13 | 1.473 (3) | 1.47563 |
| N2—C5 | 1.336 (2) | 1.35689 |
| N2—C6 | 1.372 (3) | 1.40945 |
| N3—C6 | 1.346 (2) | 1.40939 |
| N3—C1 | 1.372 (2) | 1.38222 |
| N4—C10 | 1.465 (2) | 1.42118 |
| C5—N1—C4 | 118.57 (17) | 117.37 |
| C5—N1—C13 | 119.67 (16) | 119.58 |
| C4—N1—C13 | 121.75 (17) | 120.96 |
| C5—N2—C6 | 100.79 (16) | 101.37 |
| C6—N3—C1 | 102.17 (16) | 101.96 |
| O1—N4—O2 | 123.30 (19) | 122.47 |
| O1—N4—C10 | 118.48 (18) | 118.78 |
| O2—N4—C10 | 118.22 (18) | 118.74 |
| N3—C1—C2 | 132.04 (18) | 133.14 |
| N3—C1—C5 | 107.59 (16) | 105.38 |
| C2—C1—C5 | 120.37 (18) | 121.46 |
Table 3. Calculated molecular energies for (I).
| Total Energy, TE (eV) | −175539.349 |
| E HOMO (eV) | −4.0238 |
| E LUMO (eV) | −2.3507 |
| Gap, ΔE (eV) | 1.6731 |
| Dipole moment, μ (Debye) | 15.7142 |
| Ionization potential, I (eV) | 4.0238 |
| Electron affinity, A | 2.3507 |
| Electronegativity, χ | 3.1872 |
| Hardness, η | 0.8366 |
| Electrophilicity index, ω | 3.0715 |
| Softness, σ | 1.1954 |
| Fraction of electron transferred, ΔN | 2.2788 |
Figure 6.

Shapes of HOMO and LUMO of (I) and the energy band gap between them.
Database survey
The importance of benzimidazole derivatives is highlighted by a search of the Cambridge Structural Database (CSD, updated to November 2019; Groom et al., 2016 ▸) using a fragment allowing for substituents at the 6-position and having a single carbon atom at the 3- and 4-positions, which resulted in over 1900 hits. Restricting the search to exclude metal complexes and using the fragment (II) (Fig. 7 ▸) yielded eight hits most comparable to (I). These are of the general type (III) (Fig. 7 ▸) with R′′ = H, R = 4-[Ph2C=C(Ph)]C6H4, R′ = n-Bu (VEDKEX; Zhang et al., 2018 ▸), n-hexyl (VEDHUK; Zhang et al., 2018 ▸) and R′ = 6-(9H-carbazol-9-yl)hexyl, R = Ph (DUKJAV; Zhao et al., 2009 ▸). Additional ones have R′′ = COOMe, R′ = n-Bu, R = 3,4-Cl2C6H3 (ABEJAT; Arslan et al., 2004 ▸), 2-[2-(cinnamylthio)benzo[d]oxazol-5-yl (TAPVIR; Chanda et al., 2012 ▸), and the set is completed by those with R′ = n-Bu, R′′ = CN, R = 3-ClC6H4 (WEWVIE; Kazak et al., 2006 ▸) or R= 3,4-(MeO)2C6H3 (WEWVOK; Kazak et al., 2006 ▸) and R′′ = NO2, R′ = n-Bu, R = 2-(pTosNH)C6H4 (BUXDUV; Burlov et al., 2016 ▸). In all of the matches where R′ is an alkyl chain, the base of the chain is approximately perpendicular to the benzimidazole plane but not all of them have the remainder of the chain in a fully extended conformation and in no instances are structures seen in which the alkyl chains overlap. Part of the reason is that the butyl group is not long enough to counteract the packing interactions involving the benzimidazole moiety and the substituents in the 2-position. The possible exception is in DUKJAV where π–π stacking interactions appear to occur between the benzimidazole units and between the carbazole units. Also, all of the related molecules identified have a dihedral angle between the plane of the benzimidazole unit and the plane of the aromatic ring in the 2-position of 28–48° while in (I) the two are nearly coplanar [1.6 (1)°]. This is likely due to packing considerations.
Figure 7.

Lewis structures of fragments (II) and (III) used in the database search.
Synthesis and crystallization
To a solution of 6-bromo-2-(4′-nitrophenyl)-3H-imidazo[4,5-b]pyridine (0.4 g, 1.25 mmol), potassium carbonate (2.2 equivalents; 0.38 g, 2.75 mmol) and tetra-n-butylammonium bromide (0.2 equivalents; 0.061 g, 0.187 mmo)l in 40 ml of DMF were added in small portions to 1.5 equivalents of 1,12-dibromododecane. The resulting mixture was stirred magnetically at room temperature for 48 h. After removal of the salts and evaporation of DMF under reduced pressure, the product was separated by chromatography on a column of silica gel using a mixture of hexane/dichloromethane: 1/4 (v/v) as the mobile phase. Orange single crystals suitable for X-ray diffraction were obtained by slow evaporation of the eluant.
Refinement
Crystal, data collection and refinement details are presented in Table 4 ▸. Hydrogen atoms were included as riding contributions in idealized positions with isotropic displacement parameters tied to those of the attached atoms. The terminal C24—Br2 portion of the 12-bromododecyl chain is disordered over two resolved sites in a 0.902 (3)/0.098 (3) ratio. The two components of the disorder were refined with restraints so that their bond lengths and angles are comparable.
Table 4. Experimental details.
| Crystal data | |
| Chemical formula | C24H30Br2N4O2 |
| M r | 566.34 |
| Crystal system, space group | Triclinic, P

|
| Temperature (K) | 150 |
| a, b, c (Å) | 6.3291 (11), 9.8911 (17), 21.133 (4) |
| α, β, γ (°) | 76.480 (2), 84.965 (3), 73.891 (2) |
| V (Å3) | 1235.4 (4) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 3.31 |
| Crystal size (mm) | 0.48 × 0.23 × 0.20 |
| Data collection | |
| Diffractometer | Bruker SMART APEX CCD |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.23, 0.56 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 24139, 6598, 5337 |
| R int | 0.040 |
| (sin θ/λ)max (Å−1) | 0.688 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.035, 0.095, 1.03 |
| No. of reflections | 6598 |
| No. of parameters | 293 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.72, −0.80 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989020005228/wm5551sup1.cif
Supporting information file. DOI: 10.1107/S2056989020005228/wm5551Isup3.cdx
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020005228/wm5551Isup4.hkl
Supporting information file. DOI: 10.1107/S2056989020005228/wm5551Isup4.cml
CCDC reference: 1996788
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C24H30Br2N4O2 | Z = 2 |
| Mr = 566.34 | F(000) = 576 |
| Triclinic, P1 | Dx = 1.522 Mg m−3 |
| a = 6.3291 (11) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 9.8911 (17) Å | Cell parameters from 9900 reflections |
| c = 21.133 (4) Å | θ = 2.2–29.0° |
| α = 76.480 (2)° | µ = 3.31 mm−1 |
| β = 84.965 (3)° | T = 150 K |
| γ = 73.891 (2)° | Column, light orange |
| V = 1235.4 (4) Å3 | 0.48 × 0.23 × 0.20 mm |
Data collection
| Bruker SMART APEX CCD diffractometer | 6598 independent reflections |
| Radiation source: fine-focus sealed tube | 5337 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.040 |
| Detector resolution: 8.3333 pixels mm-1 | θmax = 29.3°, θmin = 2.0° |
| φ and ω scans | h = −8→8 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | k = −13→13 |
| Tmin = 0.23, Tmax = 0.56 | l = −29→29 |
| 24139 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.095 | H-atom parameters constrained |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0494P)2 + 0.370P] where P = (Fo2 + 2Fc2)/3 |
| 6598 reflections | (Δ/σ)max = 0.002 |
| 293 parameters | Δρmax = 0.72 e Å−3 |
| 0 restraints | Δρmin = −0.80 e Å−3 |
Special details
| Experimental. The diffraction data were obtained from 3 sets of 400 frames, each of width 0.5° in ω, colllected at φ = 0.00, 90.00 and 180.00° and 2 sets of 800 frames, each of width 0.45° in φ, collected at ω = -30.00 and 210.00°. The scan time was 10 sec/frame. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. H-atoms attached to carbon were placed in calculated positions (C—H = 0.95 - 0.99 Å). All were included as riding contributions with isotropic displacement parameters 1.2 - 1.5 times those of the attached atoms. Br2 is disordered over two sites in a 0.902 (3)/0.098 (32) ratio with the two components refined with restraints that they have comparable geometries. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Br1 | 1.82823 (3) | 0.88087 (2) | 0.00828 (2) | 0.02928 (7) | |
| Br2 | −0.34993 (9) | 0.51084 (14) | 0.63610 (3) | 0.05439 (19) | 0.902 (3) |
| Br2A | −0.3773 (9) | 0.5733 (11) | 0.6273 (3) | 0.05439 (19) | 0.098 (3) |
| O1 | 0.4691 (3) | 0.16270 (19) | 0.13274 (10) | 0.0470 (4) | |
| O2 | 0.3734 (3) | 0.2314 (2) | 0.22339 (9) | 0.0468 (4) | |
| N1 | 1.3755 (3) | 0.79656 (18) | 0.15128 (8) | 0.0244 (3) | |
| N2 | 1.1281 (3) | 0.64593 (18) | 0.16156 (8) | 0.0239 (3) | |
| N3 | 1.2508 (3) | 0.55633 (18) | 0.06847 (8) | 0.0234 (3) | |
| N4 | 0.4845 (3) | 0.23168 (19) | 0.17246 (10) | 0.0317 (4) | |
| C1 | 1.3664 (3) | 0.6514 (2) | 0.07409 (9) | 0.0222 (4) | |
| C2 | 1.5326 (3) | 0.6991 (2) | 0.03550 (10) | 0.0241 (4) | |
| H2 | 1.588946 | 0.665956 | −0.002911 | 0.029* | |
| C3 | 1.6108 (3) | 0.7989 (2) | 0.05705 (10) | 0.0241 (4) | |
| C4 | 1.5370 (3) | 0.8446 (2) | 0.11371 (10) | 0.0250 (4) | |
| H4 | 1.599544 | 0.910484 | 0.126734 | 0.030* | |
| C5 | 1.2880 (3) | 0.7032 (2) | 0.13151 (9) | 0.0223 (4) | |
| C6 | 1.1134 (3) | 0.5596 (2) | 0.12076 (9) | 0.0225 (4) | |
| C7 | 0.9500 (3) | 0.4757 (2) | 0.13404 (9) | 0.0223 (4) | |
| C8 | 0.9326 (3) | 0.3904 (2) | 0.09185 (10) | 0.0256 (4) | |
| H8 | 1.027479 | 0.386290 | 0.054538 | 0.031* | |
| C9 | 0.7781 (3) | 0.3117 (2) | 0.10385 (10) | 0.0266 (4) | |
| H9 | 0.764906 | 0.253965 | 0.074998 | 0.032* | |
| C10 | 0.6431 (3) | 0.3189 (2) | 0.15897 (10) | 0.0249 (4) | |
| C11 | 0.6535 (3) | 0.4038 (2) | 0.20136 (10) | 0.0272 (4) | |
| H11 | 0.557276 | 0.407835 | 0.238384 | 0.033* | |
| C12 | 0.8083 (3) | 0.4831 (2) | 0.18856 (10) | 0.0254 (4) | |
| H12 | 0.817963 | 0.542629 | 0.216954 | 0.031* | |
| C13 | 1.2921 (3) | 0.8451 (2) | 0.21174 (10) | 0.0277 (4) | |
| H13A | 1.407583 | 0.875235 | 0.229259 | 0.033* | |
| H13B | 1.259112 | 0.763605 | 0.244503 | 0.033* | |
| C14 | 1.0856 (4) | 0.9702 (2) | 0.20091 (10) | 0.0303 (4) | |
| H14A | 0.979474 | 0.946902 | 0.176234 | 0.036* | |
| H14B | 1.124005 | 1.057448 | 0.174668 | 0.036* | |
| C15 | 0.9786 (4) | 1.0010 (2) | 0.26569 (11) | 0.0354 (5) | |
| H15A | 1.093787 | 1.003377 | 0.293931 | 0.042* | |
| H15B | 0.872569 | 1.097574 | 0.257308 | 0.042* | |
| C16 | 0.8594 (4) | 0.8905 (3) | 0.30167 (12) | 0.0422 (6) | |
| H16A | 0.766800 | 0.873698 | 0.270434 | 0.051* | |
| H16B | 0.970288 | 0.798211 | 0.317524 | 0.051* | |
| C17 | 0.7154 (5) | 0.9319 (3) | 0.35888 (13) | 0.0481 (7) | |
| H17A | 0.608784 | 1.026430 | 0.343710 | 0.058* | |
| H17B | 0.808726 | 0.943166 | 0.391508 | 0.058* | |
| C18 | 0.5895 (5) | 0.8219 (3) | 0.39151 (13) | 0.0498 (7) | |
| H18A | 0.499382 | 0.809373 | 0.358399 | 0.060* | |
| H18B | 0.697061 | 0.727992 | 0.407038 | 0.060* | |
| C19 | 0.4406 (5) | 0.8604 (3) | 0.44815 (14) | 0.0551 (8) | |
| H19A | 0.334976 | 0.955515 | 0.433229 | 0.066* | |
| H19B | 0.530537 | 0.869341 | 0.482227 | 0.066* | |
| C20 | 0.3130 (5) | 0.7502 (3) | 0.47773 (13) | 0.0511 (7) | |
| H20A | 0.418487 | 0.653915 | 0.489418 | 0.061* | |
| H20B | 0.215232 | 0.746721 | 0.444409 | 0.061* | |
| C21 | 0.1746 (5) | 0.7810 (3) | 0.53790 (14) | 0.0504 (7) | |
| H21A | 0.268887 | 0.794690 | 0.569570 | 0.060* | |
| H21B | 0.057909 | 0.872000 | 0.525371 | 0.060* | |
| C22 | 0.0687 (5) | 0.6614 (3) | 0.57043 (12) | 0.0434 (6) | |
| H22A | 0.182702 | 0.568038 | 0.577361 | 0.052* | |
| H22B | −0.041627 | 0.656911 | 0.541078 | 0.052* | |
| C23 | −0.0432 (5) | 0.6828 (3) | 0.63564 (12) | 0.0434 (6) | |
| H23A | 0.059462 | 0.705909 | 0.661635 | 0.052* | |
| H23B | −0.173900 | 0.766726 | 0.627551 | 0.052* | |
| C24 | −0.1145 (4) | 0.5542 (3) | 0.67503 (11) | 0.0404 (5) | 0.902 (3) |
| H24A | −0.164784 | 0.571989 | 0.718783 | 0.049* | 0.902 (3) |
| H24B | 0.014190 | 0.468801 | 0.680713 | 0.049* | 0.902 (3) |
| C24A | −0.1145 (4) | 0.5542 (3) | 0.67503 (11) | 0.0404 (5) | 0.098 (3) |
| H24C | −0.150774 | 0.561926 | 0.720811 | 0.049* | 0.098 (3) |
| H24D | −0.001810 | 0.462587 | 0.673847 | 0.049* | 0.098 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.02318 (11) | 0.02607 (11) | 0.04000 (13) | −0.01178 (8) | 0.00655 (8) | −0.00638 (8) |
| Br2 | 0.0548 (2) | 0.0723 (6) | 0.04320 (19) | −0.0293 (3) | −0.00326 (15) | −0.0109 (3) |
| Br2A | 0.0548 (2) | 0.0723 (6) | 0.04320 (19) | −0.0293 (3) | −0.00326 (15) | −0.0109 (3) |
| O1 | 0.0496 (11) | 0.0440 (10) | 0.0645 (12) | −0.0309 (8) | 0.0154 (9) | −0.0294 (9) |
| O2 | 0.0465 (10) | 0.0549 (11) | 0.0518 (11) | −0.0357 (9) | 0.0175 (8) | −0.0171 (9) |
| N1 | 0.0216 (8) | 0.0245 (8) | 0.0304 (8) | −0.0099 (6) | 0.0014 (7) | −0.0087 (7) |
| N2 | 0.0224 (8) | 0.0255 (8) | 0.0276 (8) | −0.0119 (7) | 0.0019 (6) | −0.0073 (7) |
| N3 | 0.0196 (8) | 0.0236 (8) | 0.0301 (8) | −0.0089 (6) | 0.0003 (6) | −0.0082 (7) |
| N4 | 0.0279 (9) | 0.0267 (9) | 0.0437 (11) | −0.0140 (7) | 0.0035 (8) | −0.0074 (8) |
| C1 | 0.0182 (9) | 0.0219 (9) | 0.0274 (9) | −0.0062 (7) | −0.0007 (7) | −0.0063 (7) |
| C2 | 0.0187 (9) | 0.0224 (9) | 0.0306 (10) | −0.0058 (7) | 0.0013 (7) | −0.0053 (8) |
| C3 | 0.0170 (9) | 0.0210 (9) | 0.0334 (10) | −0.0066 (7) | 0.0015 (7) | −0.0033 (8) |
| C4 | 0.0216 (9) | 0.0211 (9) | 0.0344 (10) | −0.0091 (7) | −0.0002 (8) | −0.0066 (8) |
| C5 | 0.0202 (9) | 0.0219 (9) | 0.0266 (9) | −0.0080 (7) | −0.0005 (7) | −0.0059 (7) |
| C6 | 0.0194 (9) | 0.0228 (9) | 0.0263 (9) | −0.0072 (7) | −0.0020 (7) | −0.0049 (7) |
| C7 | 0.0188 (9) | 0.0211 (9) | 0.0274 (9) | −0.0065 (7) | −0.0014 (7) | −0.0044 (7) |
| C8 | 0.0218 (9) | 0.0252 (9) | 0.0322 (10) | −0.0091 (8) | 0.0033 (8) | −0.0088 (8) |
| C9 | 0.0232 (10) | 0.0243 (9) | 0.0366 (11) | −0.0095 (8) | 0.0006 (8) | −0.0114 (8) |
| C10 | 0.0207 (9) | 0.0214 (9) | 0.0340 (10) | −0.0098 (7) | 0.0004 (8) | −0.0039 (8) |
| C11 | 0.0231 (10) | 0.0302 (10) | 0.0292 (10) | −0.0108 (8) | 0.0020 (8) | −0.0050 (8) |
| C12 | 0.0245 (10) | 0.0275 (10) | 0.0274 (10) | −0.0109 (8) | −0.0010 (8) | −0.0070 (8) |
| C13 | 0.0288 (11) | 0.0302 (10) | 0.0292 (10) | −0.0116 (8) | 0.0019 (8) | −0.0129 (8) |
| C14 | 0.0322 (11) | 0.0288 (10) | 0.0312 (10) | −0.0111 (9) | 0.0041 (8) | −0.0073 (9) |
| C15 | 0.0383 (13) | 0.0298 (11) | 0.0386 (12) | −0.0096 (9) | 0.0092 (10) | −0.0117 (9) |
| C16 | 0.0457 (14) | 0.0465 (14) | 0.0412 (13) | −0.0219 (12) | 0.0138 (11) | −0.0168 (11) |
| C17 | 0.0528 (16) | 0.0397 (14) | 0.0486 (15) | −0.0129 (12) | 0.0184 (12) | −0.0102 (11) |
| C18 | 0.0584 (17) | 0.0536 (16) | 0.0413 (14) | −0.0229 (14) | 0.0172 (12) | −0.0148 (12) |
| C19 | 0.0665 (19) | 0.0431 (15) | 0.0488 (15) | −0.0142 (13) | 0.0258 (14) | −0.0070 (12) |
| C20 | 0.0589 (18) | 0.0568 (17) | 0.0365 (13) | −0.0198 (14) | 0.0158 (12) | −0.0089 (12) |
| C21 | 0.0590 (17) | 0.0378 (14) | 0.0466 (15) | −0.0096 (12) | 0.0208 (13) | −0.0058 (11) |
| C22 | 0.0490 (15) | 0.0473 (14) | 0.0335 (12) | −0.0146 (12) | 0.0101 (11) | −0.0101 (11) |
| C23 | 0.0486 (15) | 0.0449 (14) | 0.0370 (12) | −0.0135 (12) | 0.0122 (11) | −0.0129 (11) |
| C24 | 0.0434 (14) | 0.0483 (14) | 0.0307 (11) | −0.0158 (11) | 0.0009 (10) | −0.0069 (10) |
| C24A | 0.0434 (14) | 0.0483 (14) | 0.0307 (11) | −0.0158 (11) | 0.0009 (10) | −0.0069 (10) |
Geometric parameters (Å, º)
| Br1—C3 | 1.8954 (19) | C14—H14A | 0.9900 |
| Br2—C24 | 1.953 (3) | C14—H14B | 0.9900 |
| Br2A—C24A | 1.962 (5) | C15—C16 | 1.520 (3) |
| O1—N4 | 1.224 (2) | C15—H15A | 0.9900 |
| O2—N4 | 1.233 (2) | C15—H15B | 0.9900 |
| N1—C5 | 1.357 (2) | C16—C17 | 1.514 (3) |
| N1—C4 | 1.361 (2) | C16—H16A | 0.9900 |
| N1—C13 | 1.473 (3) | C16—H16B | 0.9900 |
| N2—C5 | 1.336 (2) | C17—C18 | 1.526 (4) |
| N2—C6 | 1.372 (3) | C17—H17A | 0.9900 |
| N3—C6 | 1.346 (2) | C17—H17B | 0.9900 |
| N3—C1 | 1.372 (2) | C18—C19 | 1.511 (4) |
| N4—C10 | 1.465 (2) | C18—H18A | 0.9900 |
| C1—C2 | 1.392 (3) | C18—H18B | 0.9900 |
| C1—C5 | 1.423 (3) | C19—C20 | 1.520 (4) |
| C2—C3 | 1.396 (3) | C19—H19A | 0.9900 |
| C2—H2 | 0.9500 | C19—H19B | 0.9900 |
| C3—C4 | 1.376 (3) | C20—C21 | 1.521 (4) |
| C4—H4 | 0.9500 | C20—H20A | 0.9900 |
| C6—C7 | 1.467 (3) | C20—H20B | 0.9900 |
| C7—C8 | 1.393 (3) | C21—C22 | 1.517 (4) |
| C7—C12 | 1.400 (3) | C21—H21A | 0.9900 |
| C8—C9 | 1.383 (3) | C21—H21B | 0.9900 |
| C8—H8 | 0.9500 | C22—C23 | 1.526 (3) |
| C9—C10 | 1.387 (3) | C22—H22A | 0.9900 |
| C9—H9 | 0.9500 | C22—H22B | 0.9900 |
| C10—C11 | 1.380 (3) | C23—C24A | 1.507 (3) |
| C11—C12 | 1.389 (3) | C23—C24 | 1.507 (3) |
| C11—H11 | 0.9500 | C23—H23A | 0.9900 |
| C12—H12 | 0.9500 | C23—H23B | 0.9900 |
| C13—C14 | 1.521 (3) | C24—H24A | 0.9900 |
| C13—H13A | 0.9900 | C24—H24B | 0.9900 |
| C13—H13B | 0.9900 | C24A—H24C | 0.9900 |
| C14—C15 | 1.531 (3) | C24A—H24D | 0.9900 |
| C5—N1—C4 | 118.57 (17) | C17—C16—C15 | 115.0 (2) |
| C5—N1—C13 | 119.67 (16) | C17—C16—H16A | 108.5 |
| C4—N1—C13 | 121.75 (17) | C15—C16—H16A | 108.5 |
| C5—N2—C6 | 100.79 (16) | C17—C16—H16B | 108.5 |
| C6—N3—C1 | 102.17 (16) | C15—C16—H16B | 108.5 |
| O1—N4—O2 | 123.30 (19) | H16A—C16—H16B | 107.5 |
| O1—N4—C10 | 118.48 (18) | C16—C17—C18 | 113.4 (2) |
| O2—N4—C10 | 118.22 (18) | C16—C17—H17A | 108.9 |
| N3—C1—C2 | 132.04 (18) | C18—C17—H17A | 108.9 |
| N3—C1—C5 | 107.59 (16) | C16—C17—H17B | 108.9 |
| C2—C1—C5 | 120.37 (18) | C18—C17—H17B | 108.9 |
| C1—C2—C3 | 115.51 (18) | H17A—C17—H17B | 107.7 |
| C1—C2—H2 | 122.2 | C19—C18—C17 | 115.1 (2) |
| C3—C2—H2 | 122.2 | C19—C18—H18A | 108.5 |
| C4—C3—C2 | 123.24 (18) | C17—C18—H18A | 108.5 |
| C4—C3—Br1 | 115.85 (15) | C19—C18—H18B | 108.5 |
| C2—C3—Br1 | 120.92 (15) | C17—C18—H18B | 108.5 |
| N1—C4—C3 | 120.73 (18) | H18A—C18—H18B | 107.5 |
| N1—C4—H4 | 119.6 | C18—C19—C20 | 113.3 (3) |
| C3—C4—H4 | 119.6 | C18—C19—H19A | 108.9 |
| N2—C5—N1 | 126.94 (18) | C20—C19—H19A | 108.9 |
| N2—C5—C1 | 111.52 (17) | C18—C19—H19B | 108.9 |
| N1—C5—C1 | 121.53 (17) | C20—C19—H19B | 108.9 |
| N3—C6—N2 | 117.92 (17) | H19A—C19—H19B | 107.7 |
| N3—C6—C7 | 121.76 (17) | C19—C20—C21 | 114.2 (3) |
| N2—C6—C7 | 120.31 (17) | C19—C20—H20A | 108.7 |
| C8—C7—C12 | 119.67 (18) | C21—C20—H20A | 108.7 |
| C8—C7—C6 | 120.18 (18) | C19—C20—H20B | 108.7 |
| C12—C7—C6 | 120.14 (18) | C21—C20—H20B | 108.7 |
| C9—C8—C7 | 120.52 (19) | H20A—C20—H20B | 107.6 |
| C9—C8—H8 | 119.7 | C22—C21—C20 | 113.0 (2) |
| C7—C8—H8 | 119.7 | C22—C21—H21A | 109.0 |
| C8—C9—C10 | 118.46 (19) | C20—C21—H21A | 109.0 |
| C8—C9—H9 | 120.8 | C22—C21—H21B | 109.0 |
| C10—C9—H9 | 120.8 | C20—C21—H21B | 109.0 |
| C11—C10—C9 | 122.63 (18) | H21A—C21—H21B | 107.8 |
| C11—C10—N4 | 119.21 (18) | C21—C22—C23 | 112.7 (2) |
| C9—C10—N4 | 118.16 (18) | C21—C22—H22A | 109.0 |
| C10—C11—C12 | 118.37 (19) | C23—C22—H22A | 109.0 |
| C10—C11—H11 | 120.8 | C21—C22—H22B | 109.0 |
| C12—C11—H11 | 120.8 | C23—C22—H22B | 109.0 |
| C11—C12—C7 | 120.34 (19) | H22A—C22—H22B | 107.8 |
| C11—C12—H12 | 119.8 | C24A—C23—C22 | 114.3 (2) |
| C7—C12—H12 | 119.8 | C24—C23—C22 | 114.3 (2) |
| N1—C13—C14 | 112.14 (17) | C24—C23—H23A | 108.7 |
| N1—C13—H13A | 109.2 | C22—C23—H23A | 108.7 |
| C14—C13—H13A | 109.2 | C24—C23—H23B | 108.7 |
| N1—C13—H13B | 109.2 | C22—C23—H23B | 108.7 |
| C14—C13—H13B | 109.2 | H23A—C23—H23B | 107.6 |
| H13A—C13—H13B | 107.9 | C23—C24—Br2 | 114.01 (18) |
| C13—C14—C15 | 111.20 (18) | C23—C24—H24A | 108.8 |
| C13—C14—H14A | 109.4 | Br2—C24—H24A | 108.8 |
| C15—C14—H14A | 109.4 | C23—C24—H24B | 108.8 |
| C13—C14—H14B | 109.4 | Br2—C24—H24B | 108.8 |
| C15—C14—H14B | 109.4 | H24A—C24—H24B | 107.6 |
| H14A—C14—H14B | 108.0 | C23—C24A—Br2A | 99.5 (3) |
| C16—C15—C14 | 113.51 (19) | C23—C24A—H24C | 111.9 |
| C16—C15—H15A | 108.9 | Br2A—C24A—H24C | 111.9 |
| C14—C15—H15A | 108.9 | C23—C24A—H24D | 111.9 |
| C16—C15—H15B | 108.9 | Br2A—C24A—H24D | 111.9 |
| C14—C15—H15B | 108.9 | H24C—C24A—H24D | 109.6 |
| H15A—C15—H15B | 107.7 | ||
| C6—N3—C1—C2 | −179.2 (2) | C6—C7—C8—C9 | −179.63 (18) |
| C6—N3—C1—C5 | 0.9 (2) | C7—C8—C9—C10 | −0.5 (3) |
| N3—C1—C2—C3 | 179.6 (2) | C8—C9—C10—C11 | 1.5 (3) |
| C5—C1—C2—C3 | −0.4 (3) | C8—C9—C10—N4 | −178.13 (18) |
| C1—C2—C3—C4 | 2.3 (3) | O1—N4—C10—C11 | 176.3 (2) |
| C1—C2—C3—Br1 | −177.37 (13) | O2—N4—C10—C11 | −3.8 (3) |
| C5—N1—C4—C3 | −0.3 (3) | O1—N4—C10—C9 | −4.1 (3) |
| C13—N1—C4—C3 | −179.31 (18) | O2—N4—C10—C9 | 175.81 (19) |
| C2—C3—C4—N1 | −2.0 (3) | C9—C10—C11—C12 | −1.0 (3) |
| Br1—C3—C4—N1 | 177.68 (14) | N4—C10—C11—C12 | 178.54 (18) |
| C6—N2—C5—N1 | −178.66 (19) | C10—C11—C12—C7 | −0.3 (3) |
| C6—N2—C5—C1 | −0.1 (2) | C8—C7—C12—C11 | 1.2 (3) |
| C4—N1—C5—N2 | −179.38 (18) | C6—C7—C12—C11 | −179.94 (18) |
| C13—N1—C5—N2 | −0.4 (3) | C5—N1—C13—C14 | −83.3 (2) |
| C4—N1—C5—C1 | 2.2 (3) | C4—N1—C13—C14 | 95.7 (2) |
| C13—N1—C5—C1 | −178.80 (18) | N1—C13—C14—C15 | 169.76 (17) |
| N3—C1—C5—N2 | −0.5 (2) | C13—C14—C15—C16 | −74.4 (3) |
| C2—C1—C5—N2 | 179.53 (17) | C14—C15—C16—C17 | −168.7 (2) |
| N3—C1—C5—N1 | 178.14 (17) | C15—C16—C17—C18 | 177.0 (2) |
| C2—C1—C5—N1 | −1.8 (3) | C16—C17—C18—C19 | −179.0 (3) |
| C1—N3—C6—N2 | −1.0 (2) | C17—C18—C19—C20 | 178.2 (3) |
| C1—N3—C6—C7 | 178.18 (17) | C18—C19—C20—C21 | 175.8 (3) |
| C5—N2—C6—N3 | 0.7 (2) | C19—C20—C21—C22 | −173.9 (3) |
| C5—N2—C6—C7 | −178.50 (17) | C20—C21—C22—C23 | 172.0 (3) |
| N3—C6—C7—C8 | −0.5 (3) | C21—C22—C23—C24A | −169.6 (2) |
| N2—C6—C7—C8 | 178.74 (18) | C21—C22—C23—C24 | −169.6 (2) |
| N3—C6—C7—C12 | −179.32 (18) | C22—C23—C24—Br2 | −66.7 (3) |
| N2—C6—C7—C12 | −0.1 (3) | C22—C23—C24A—Br2A | −76.3 (3) |
| C12—C7—C8—C9 | −0.8 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C4—H4···O1i | 0.95 | 2.43 | 3.171 (3) | 135 |
| C24—H24A···O2ii | 0.99 | 2.53 | 3.372 (3) | 142 |
Symmetry codes: (i) x+1, y+1, z; (ii) −x, −y+1, −z+1.
Funding Statement
This work was funded by Tulane University grant . Hacettepe University Scientific Research Project Unit grant 013 D04 602 004 to T. Hokelek.
References
- Arslan, B., Kazak, C., Karataş, H. & Özden, S. (2004). Acta Cryst. E60, o1535–o1537.
- Bababdani, B. M. & Mousavi, M. (2013). Chemometrics & Intelligent Lab. Syst. 122, 1–11.
- Becke, A. D. (1993). J. Chem. Phys. 98, 5648–5652.
- Bouayad, K., Kandri Rodi, Y., Elmsellem, H., El Ghadraoui, E. H., Ouzidan, Y., Abdel-Rahman, I., Kusuma, H. S., Warad, I., Mague, J. T., Essassi, E. M., Hammouti, B. & Chetouani, A. (2018). Mor. J. Chem. 6, 22–34.
- Brandenburg, K. & Putz, H. (2012). DIAMOND, Crystal Impact GbR, Bonn, Germany.
- Bruker (2016). APEX3 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Burlov, A. S., Koshchienko, Y. V., Kiskin, M. A., Nikolaevskii, S. A., Garnovskii, D. A., Lermontov, A. S., Makarova, N. I., Metelitsa, A. V. & Eremenko, I. L. (2016). J. Mol. Struct. 1104, 7–13.
- Chanda, K., Maiti, B., Tseng, C.-C. & Sun, C.-M. (2012). ACS Comb. Sci. 14, 115–123. [DOI] [PubMed]
- Cheng, Z., Zhang, Y. & Zhang, W. (2010). Med. Chem. Res. 19, 1307–1325.
- Devi, N., Jana, A. K. & Singh, V. (2018). Karbala Int. J. Modern Sci. 1–7.
- Dymińska, L. (2015). Bioorg. Med. Chem. 23, 6087–6099. [DOI] [PubMed]
- Ehmke, V., Winkler, E. W., Banner, D., Haap, W., Schweizer, W. B., Rottmann, M., Kaiser, M., Freymond, C., Schirmeister, T. & Diederich, F. (2013). ChemMedChem, 8, 967–975. [DOI] [PubMed]
- El Kazzouli, S., Griffon du Bellay, A., Berteina-Raboin, S., Delagrange, P., Caignard, D. H. & Guillaumet, G. (2011). Eur. J. Med. Chem. 46, 4252–4257. [DOI] [PubMed]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Jr., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. (2009). GAUSSIAN09. Gaussian Inc., Wallingford, CT, US.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hathwar, V. R., Sist, M., Jørgensen, M. R. V., Mamakhel, A. H., Wang, X., Hoffmann, C. M., Sugimoto, K., Overgaard, J. & Iversen, B. B. (2015). IUCrJ, 2, 563–574. [DOI] [PMC free article] [PubMed]
- Hirshfeld, H. L. (1977). Theor. Chim. Acta, 44, 129–138.
- Kazak, C., Yilmaz, V. T., Goker, H. & Kus, C. (2006). Cryst. Res. Technol. 41, 528–532.
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Martínez-Urbina, M. A., Zentella, A., Vilchis-Reyes, M. A., Guzmán, A., Vargas, O., Ramírez Apan, M. T., Ventura Gallegos, J. L. & Díaz, E. (2010). Eur. J. Med. Chem. 45, 1211–1219. [DOI] [PubMed]
- McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. (2007). Chem. Commun. pp. 3814–3816. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Sikine, M., Elmsellem, H., Kandri Rodi, Y., Steli, H., Aouniti, A., Hammouti, B., Ouzidan, Y., Ouazzani Chahdi, F., Bourass, M. & Essassi, E. M. (2016). J. Mater. Environ. Sci. 7, 4620–4632.
- Spackman, M. A. & Jayatilaka, D. (2009). CrystEngComm, 11, 19–32.
- Tobo, A., Tobo, M., Nakakura, T., Ebara, M., Tomura, H., Mogi, C., Im, D., Murata, N., Kuwabara, A., Ito, S., Fukuda, H., Arisawa, M., Shuto, S., Nakaya, M., Kurose, H., Sato, K. & Okajima, F. (2015). PLoS One, 10, e0129334. [DOI] [PMC free article] [PubMed]
- Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). CrystalExplorer17. The University of Western Australia.
- Venkatesan, P., Thamotharan, S., Ilangovan, A., Liang, H. & Sundius, T. (2016). Spectrochim. Acta A, 153, 625–636. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yadav, M., Behera, D. & Kumar, S. (2014). Surf. Interface Anal. 46, 640–652.
- Zhang, T., Zhang, R., Zhao, Y. & Ni, Z. (2018). Dyes Pigments, 148, 276–285.
- Zhao, Y.-L., Yu, T.-Z. & Meng, J. (2009). Acta Cryst. E65, o3076. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989020005228/wm5551sup1.cif
Supporting information file. DOI: 10.1107/S2056989020005228/wm5551Isup3.cdx
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020005228/wm5551Isup4.hkl
Supporting information file. DOI: 10.1107/S2056989020005228/wm5551Isup4.cml
CCDC reference: 1996788
Additional supporting information: crystallographic information; 3D view; checkCIF report