

Abstract
The Hepatitis B Virus (HBV) ribonuclease H (RNaseH) is a promising but unexploited drug target. Here, we synthesized and analyzed a library of 57 amide-containing α-hydroxytropolones (αHTs) as potential leads for HBV drug development. Fifty percent effective concentrations ranged from 0.31 to 54 μM, with selectivity indexes in cell culture of up to 80. Activity against the HBV RNaseH was confirmed in semi-quantitative enzymatic assays with recombinant HBV RNaseH. The compounds were overall poorly active against human ribonuclease H1, with 50% inhibitory concentrations of 5.1 to >1,000 μM. The αHTs had modest activity against growth of the fungal pathogen Cryptococcus neoformans, but had very limited activity against growth of the Gram - bacterium Escherichia coli and the Gram + bacterium Staphylococcus aureus, indicating substantial selectivity for HBV. A molecular model of the HBV RNaseH templated against the Ty3 RNaseH was generated. Docking the compounds to the RNaseH revealed the anticipated binding pose with the divalent cation coordinating motif on the compounds chelating the two Mn++ ions modeled into the active site. These studies reveal that that amide αHTs can be strong, specific HBV inhibitors that merit further assessment toward becoming anti-HBV drugs.
Keywords: Hepatitis B Virus, Ribonuclease H, α-Hydroxytropolones, Molecular modeling
1. Introduction
Hepatitis B virus (HBV) is an enveloped virus with a small, partially double-stranded DNA genome that replicates by reverse transcription (Seeger et al., 2013; Summers and Mason, 1982). The virus chronically infects over 250 million people worldwide and kills nearly 900,000 annually (Polaris Observatory, 2018) by inducing hepatitis, fibrosis, cirrhosis, liver failure, and hepatocellular carcinoma (Trepo et al., 2014). HBV replicates non-cytolytically in hepatocytes, and cells secrete HBV continually once an infection is established. HBV reverse transcription requires coordinate action of two virally encoded enzymes located on adjacent domains of the viral polymerase protein, the reverse transcriptase and the ribonuclease H (RNaseH).
Treatment for HBV infection employs nucleos(t)ide analogs or pegylated interferon α. Nucleos(t)ide analog therapy, usually with Entecavir or Tenofovir, suppresses viremia to undetectable levels in up to 76% of HBeAg-positive patients and up to 93% of HBeAg-negative patients after one year of therapy, and it normalizes serum alanine transferase levels that reflect ongoing liver damage in up to 78% of patients (Trepo et al., 2014). Unfortunately, treatment does not fully stop disease progression, and death rates from hepatocellular carcinoma are reduced only 2–4 fold after many years of treatment (Liaw, 2013). Furthermore, treatment is rarely curative and HBV viremia resurges in most patients following drug withdrawal (Ghany, 2017). Conditions under which nucleos(s)tide analog treatment can be stopped are not well understood, so the large majority patients undergoing treatment remain on drug indefinitely. Pegylated interferon α treatment for 24–48 weeks is used in a minority of patients. This results in durable off-therapy reduction of HBV serum titers (HBeAg seroconversion) in ~30% of patients (Trepo et al., 2014). Overall, only a few percent of patients treated with either nucleos(t)ide analogs or pegylated interferon α clear HBV DNA and HBV surface antigens in serum, the definition of a functional cure (Block et al., 2013; Levrero et al., 2016). It is believed that curative HBV therapies will require combination treatment using multiple drugs with complementary mechanisms of action that together suppress HBV replication and enhance antiviral immune responses (Fanning et al., 2019; Revill et al., 2019). Consequently, extensive efforts are ongoing to develop new HBV therapies.
RNaseHs cleave RNA when it is in an RNA:DNA heteroduplex. Catalysis is promoted by two Mg++ ions held in the active site by a “DEDD” carboxylate motif (Nowotny, 2009). The Mg++ ions activate a water molecule for nucleophilic attack on the scissile phosphodiester bond (Klumpp et al., 2003; Nowotny and Yang, 2006). Humans express two RNaseHs, RNaseH 1 and 2 (Cerritelli and Crouch, 2009). RNaseH 1 helps promote genome maintenance by suppressing accumulation of R-loops in DNA and is critical for mitochondrial DNA synthesis. RNaseH 2 is a repair enzyme that helps remove RNA bases incorporated into DNA. The HBV RNaseH is more similar to human RNaseH 1 than 2.
The biological role of the HBV RNaseH is to remove the viral pregenomic RNA (the RNA phase of the genome) after it has been copied into DNA so that the second DNA strand can be synthesized. Homology alignments and mutational analyses reveal the HBV RNaseH DEDD active site motif to be residues D702, E731, D750, and D790 (numbering for HBV strain adw2) (Gerelsaikhan et al., 1996; Tavis et al., 2013). Failure of RNaseH activity during HBV reverse transcription causes the first DNA strand (the minus-polarity strand) to truncate in about 80% of the molecules, induces accumulation of RNA:DNA heteroduplexes within viral capsids, and blocks formation of the second HBV DNA strand (the plus-polarity strand) (Gerelsaikhan et al., 1996; Hu et al., 2013). This abrogates production of functional viral genomes. No drugs exist against the HBV RNaseH despite its essential role in viral replication.
Recent technical advances have made anti-RNaseH screening feasible, and over 150 compounds have been found to inhibit HBV replication by blocking the RNaseH (Cai et al., 2014; Edwards et al., 2017; Edwards et al., 2019; Hu et al., 2013; Huber et al., 2017; Lomonosova et al., 2017a; Lu et al., 2015; Tavis et al., 2013). These inhibitors are primarily from four chemotypes, the α-hydroxytropolones (αHT), N-hydroxyisoquinolinediones, N-hydroxypyridinediones (HPD), and N-hydroxynapthyridinones. All known HBV RNaseH inhibitors appear to act by chelating the Mg++ ions in the RNaseH active site.
HBV RNaseH inhibitors suppress replication of HBV or activity of the HBV RNaseH of isolates from genotypes A, B, C, D, and H with roughly equivalent efficiency [(Hu et al., 2013; Lu et al., 2016) and unpublished]. They act synergistically with the nucleoside analog Lamivudine and additively with the capsid assembly modifier Hap12. Interestingly, RNaseH inhibitors from different chemotypes can also be synergistic with one another, with dose reduction indexes up to 9.5 (Edwards et al., 2019; Lomonosova et al., 2017b). An HPD and an αHT have been shown to suppress HBV replication in a chimeric mouse model in which the animals have humanized livers (Long et al., 2018). Interpretation of this in vivo experiment is limited due to use of unoptimized primary screening hits and sub-optimal formulation, but it revealed suppression of HBV serum titers of up to 1.4 log10 after two weeks of treatment followed by rebound in the viral titers upon compound withdrawal, as expected for short-term treatment with a replication inhibitor.
αHTs (Meck et al., 2014) contain a seven-carbon non-benzenoid aromatic ring with three adjacent oxygen appendages on the ring (Fig. 1A). αHTs presumably bind directly to the HBV RNaseH active site, in part through coordination of the two Mg++ ions, as demonstrated by the necessity of the oxygen trident for their inhibition of HBV RNaseH in biochemical assays [(Cai et al., 2014; Lu et al., 2015) and unpublished]. This conclusion is strengthened by crystal studies showing binding of an αHT into the active site of the HIV RNaseH promoted by coordination of the active site cations (Himmel et al., 2009). αHTs as a chemotype have broad anti-microbial activities, with efficacy against pathogens in addition to HBV and HIV that include Cryptococcus neoformans (Donlin et al., 2017), Herpes simplex virus (Ireland et al., 2016; Tavis et al., 2014), Escherichia coli, Staphylococcus aureus, and Acintobacter baumanniij (Cao et al., 2018), but individual molecules usually have good specificity profiles among the pathogens tested (Agyemang et al., 2019; Ireland et al., 2016).
Fig. 1. Structure and synthesis of α-hydroxytropolones (αHTs).

A. Representative examples of natural product and synthetic αHTs with antiviral activity against HBV, along with their 50% effective concentration values for viral replication inhibition and selectivity indexes (SI) based on toxicity in HepG2-DES19 cells. B. Final-step amidation sequence used to create a library of amide-containing αHTs.
Previous studies by our groups have explored the ability of αHTs to inhibit the HBV RNaseH in biochemical assays and HBV replication in cells (Agyemang et al., 2019; Hu et al., 2013; Lomonosova et al., 2017a; Lu et al., 2015). Here, we expanded these studies to assess a library of novel amide-containing αHTs as potential leads for HBV drug development. We also evaluated efficacy of these compounds against C. neoformans, E. coli, and S. aureus to assess their selectivity for HBV.
2. Methods
2.1. Compound synthesis
All compounds were synthesized using a recently described final-step amidation strategy (Fig. 1B) (Berkowitz et al., 2018), which reported synthesis of compounds 384, 388–391, 539, and 873. 712 was characterized in (Hirsch et al., 2019). Details of synthesis for the remaining componds, including 1H NMR spectra, are in the Supplemental Data. All compounds were ≥95% pure, dissolved in 100% DMSO at 10 mM, and stored at −25°C in opaque tubes.
2.2. HBV replication inhibition assays
HepDES19 cells are HepG2 human hepatoblastoma cells stably transfected with an HBV genotype D genome under the control of a tetracycline-repressible promoter (Guo et al., 2007). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) with 1 μg/mL tetracycline.
HepDES19 cells were seeded in 96-well plates at 4 × 104 cells per well in the absence of tetracycline. Compounds in a final DMSO concentration of 1% were added 2 days later and cells were incubated with compounds for 3 days. Cells were washed in 200 μL of phosphate buffered saline (PBS) and lysed in 150 μL of core lysis buffer (10 mM Tris pH 7.4, 1% Tween20, 150 mM NaCl). Cells were incubated at room temperate on an orbital shaker at 350 rpm for 40 minutes. Cell lysate was transferred to a 96 well polymerase chain reaction (PCR) plate and centrifuged at 3300 x g for 5 minutes. The supernatant (50 μL) was transferred to a 96-well PCR plate, brought to 5 mM CaCl2, and mixed with 20 units of micrococcal nuclease. The lysate was incubated for 1 hour at 37°C, and then the nuclease was inactivated at 70°C for ten minutes. Qiagen protease (0.005 Anson units) was added to the lysate, the mixture was incubated overnight, and then the protease was then inactivated at 95°C for ten minutes.
The lysate was used as the template for strand-preferential quantitative polymerase chain reaction (q-PCR) analysis. Quantitative PCR was performed with 40 cycles of 95 °C for 15s and 60 °C for 1 minute employing the Kappa Probe Force universal PCR master mix. The primers and probe (IDT Inc.) for the plus-polarity DNA strand were 5’CATGAACAAGAGATGATTAGGCAGAG3’, 5’GGAGGCTGTAGGCATAAATTGG3’, and 5’/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. The primers and probe for the minus-polarity DNA strand were 5’GCAGATGAGAAGGCACAGA3’, 5’CTTCTCCGTCTGCCGTT3’, and 5’/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ. Fifty percent effective concentrations (EC50) were calculated from the plus-polarity DNA data with GraphPad Prism using the four-parameter variable slope algorithm.
2.3. Cytotoxicity assays
Cell viability was measured in the presence of the compounds in HepDES19 cells was using the CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) (MTS) as done previously (Edwards et al., 2017). Cells were seeded in 96 well plates at 1 × 104 cells/well in the absence of tetracycline. Compounds were added 2 days later, cells were incubated for 72 hours in the presence of compound, and then MTS solution was added to the cells as directed by the manufacturer. Cells were incubated at 37°C for 1 hour and absorbance was measured at 490 nm. Fifty percent cytotoxic concentration (CC50) values were calculated with GraphPad Prism using the three-parameter variable-response log(inhibitor)-versus-response algorithm with the bottom value set to zero.
2.4. Ribonuclease H assays
Recombinant HBV and human RNaseH1 enzymes were purified by nickel-affinity chromatography from E. coli cells as previously described (Villa et al., 2016). The HBV RNaseH version used was MBP-HRHgtCΔ5, a genotype C isolate which lacks the C-terminal 35 amino acids of the HBV RNaseH domain that are predicted to be unstructured. Quantitative inhibition of the human RNaseH1 in biochemical assays was assessed using a FRET-based RNaseH assay under conditions previously reported for an RNaseH molecular beacon assay (Edwards et al., 2017). The heteroduplex substrate was formed by annealing oligonucleotide RHSF5 (5’-rGrArUrCrUrGrArGrCrCrUrGrGrGrArGrCrU/6FAM) to DQ9 (5’-IABkFQ/AGCTCCCAGGCTCAGATC) (IDT, Inc.) and used at 12.5 nM. Cleavage of the RNA strand released the fluorescein from quenching, increasing fluorescence. Reactions were monitored for 90 min at 37°C in a plate reader and initial reaction rates were calculated. 50% inhibitory concentrations (IC50) were calculated for suppression of the reaction rate by non-linear curve fitting using the four-parameter variable slope algorithm in GraphPad Prism.
2.5. Cryptococcus neoformans replication inhibition assays
Fungal growth inhibition assays were performed with C. neoformans var. grubii, KN99 (Donlin et al., 2017). Cells were passaged on YPD (1% yeast extract, 2% yeast peptone, 2% dextrose) agar plates and grown overnight at 30°C in YPD liquid medium prior to diluting for determination of minimal 80% inhibitory concentrations (MIC80). Cells were diluted to OD650 0.001 in YNB-02 02 (0.67% yeast nitrogen base, 0.2% dextrose, pH 7.0 with 50 mM MOPs) + 1% DMSO, incubated with compounds for 2 days, and OD650 was read. MIC80 values were defined as the concentration where cell density was ≤80% relative to the DMSO vehicle-treated control culture.
2.6. Bacterial replication inhibition assays
MIC80s were determined by the broth microdilution method recommended by the Clinical and Laboratory Standards Institute (Clinical and Laboratory Standards Institute, 2015). Overnight bacterial cultures were diluted to 5 × 105 colony forming units/mL in cation-adjusted Mueller-Hinton broth, compounds were added, and OD630 was read after 16−24 hours incubation at 35°C. MIC80 values were defined as the concentration of compound that inhibited bacteria growth ≥80% compared to an untreated control cultures.
2.7. Molecular modeling and docking
The structure of the HBV RNaseH domain was predicted by homology modeling using a HBV RNaseH genotype C sequence using the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2). The crystal structures of the RNaseH domains of Escherichia coli (strain K12) (PDB 1RNH), Shewanella oneidensis (strain MR-1) (2E4L), Human immunodeficiency virus type 1, group M, subtype B (isolate BH10) (HIV-1) (5J1E), Thermus thermophilus (strain HB8 / ATCC 27634 / DSM 579) (1RIL), and Ty3 reverse transcriptase (4O8L) were used as homology structural templates. The structure used in the molecular docking studies was based on the Ty3 threading model. The Mn++ ions and a reference αHT inhibitor were placed by superimposing the DEDD binding motif of the HBV homology model to that of HIV 1 RNaseH domain bound to β-thujaplicinol (3K2P). The structure of the complex was refined using the Protein Preparation facility of the Maestro program (Schrodinger, LLC).
Compounds were built in Maestro and prepared using the LigPrep facility in their singly and doubly ionized protonation states to facilitate metal binding. Molecular docking of the compounds into the HBV RNaseH homology model was performed using Glide SP (Schrodinger, LLC) with default settings using the position of bound inhibitor as the center of the docking grid and using metal constraints to place the ionized oxygen atoms of the αHT compounds in proximity to the Mn++ ions. Docking was also done without the constraint that the compound contact the Mn++ ions. Three excluded volume spherical regions were placed to mimic a bound nucleic acid substrate to filter out binding poses in which the inhibitors would clash with the fully-bound substrate (Zhang et al., 2016).
3. Results
3.1. Anti-HBV activity of amide-containing α-hydroxytropolones
57 novel amide-containing αHTs were synthesized employing a final-step amidation strategy (Fig. 1B) (Berkowitz et al., 2018). These molecules can be divided into 20 piperidine- or piperizine-based analogs (Table 1), 7 benzylamine-based compounds (Table 2), 8 aniline-based derivatives (Table 3), 9 amino acid-derived analogs (Table 4), and 13 molecules falling outside those classifications (Table 5).
Table 1.
Piperidine and piperizine derivatives
| HBV replication inhibition | HBV RNaseH inhibition | |||||
|---|---|---|---|---|---|---|
| Entry | Compound number | NR1R2 | EC50 (μM) | CC50 (μM) | SI | IC50 (μM) |
| 1 | 390 | 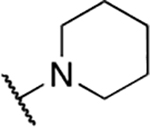 |
0.31 ± 0.13 | 24.9 ± 11.6 | 80.3 | 23.3 ± 9.1 |
| 2 | 404 | 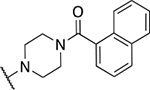 |
0.33 ± 0.13 | 4.5 ± 1.4 | 13.7 | 27.8 ± 11.6 |
| 3 | 391 |  |
0.46 ± 0.22 | 37.5 ± 4.8 | 81.5 | 47.6 ± 0 |
| 4 | 800 | 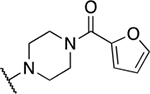 |
0.52 ± 0.18 | 16.5 ± 11.4 | 31.7 | 45.6 ± 2.2 |
| 5 | 876 | 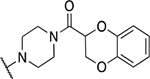 |
0.70 ± 0.36 | 9.2 ± 3.2 | 13.1 | 18.5 ± 5.6 |
| 6 | 872 | 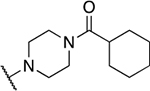 |
0.84 ± 0.37 | 14.4 ± 3.7 | 17.2 | 42.5 ± 31.2 |
| 7 | 1016 | 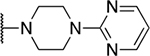 |
0.90 ± 0.00 | 24.6 ± 40.0 | 27.3 | 74.1 ± 73.9 |
| 8 | 877 | 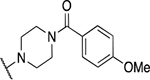 |
0.98 ± 0.45 | 16.5 ± 1.4 | 16.8 | 50.2 ± 21.7 |
| 9 | 920 | 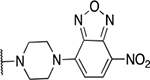 |
1.03 ± 0.3 | 19.3 ± 14.6 | 18.7 | 4.1 ± 2.2 |
| 10 | 919 | 1.08 ± 0.29 | 5.5 ± 1.9 | 5.1 | 1000 ± 0 | |
| 11 | 871 | 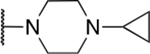 |
1.5 ± 0.36 | 36.6 ± 3.6 | 24.4 | 166 ± 99 |
| 12 | 802 | 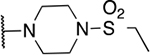 |
1.19 ± 1.41 | 27.1 ± 31.2 | 22.7 | 85.3 ± 29.9 |
| 13 | 803 | 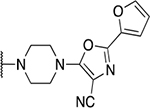 |
1.30 ± 0.00 | 23.7 ± 22.9 | 18.2 | 158 ± 192 |
| 14 | 873 | 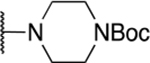 |
1.43 ±0.67 | 19.5 ± 8.7 | 13.7 | 46.5 ± 39.5 |
| 15 | 918 | 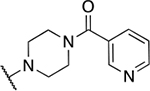 |
1.45 ± 0.40 | 43.8 ± 10.7 | 30.2 | 82.2 ± 25.1 |
| 16 | 875 | 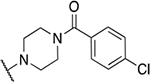 |
1.47 ±1.48 | 4.7 ± 1.8 | 3.2 | 9.7 ± 4.6 |
| 17 | 917 | 1.83 ± 1.3 | 8.9 ± 4.1 | 4.9 | 9.7 ± 3.8 | |
| 18 | 1020 |  |
2.65 ± 0.35 | 7.8 ± 3.7 | 2.9 | 8.9 ± 0.4 |
| 19 | 1021 | 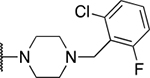 |
3.05 ± 0.64 | 10.4 ± 7.3 | 3.3 | 17.9 ± 8.8 |
| 20 | 1017 | 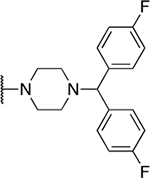 |
5.60 ± 0.00 | 8.7 ± 2.8 | 1.6 | 122 ± 1.2 |
Table 2.
Benzyl amine derivatives
| HBV replication inhibition | HBV RNaseH inhibiton | |||||
|---|---|---|---|---|---|---|
| Entry | Compound number | NR1R2 | EC50 (μM) | CC50 (μM) | SI | IC50 (μM) |
| 1 | 388 | 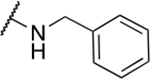 |
1.22 ± 1.29 | 19.8 ± 3.0 | 16.2 | 77.6 ± 66.2 |
| 2 | 807 | 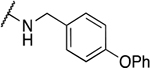 |
1.7 ± 1.01 | 6.0 ± 6.5 | 3.5 | 35.6 ± 38.0 |
| 3 | 809 | 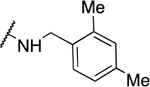 |
1.70 ± 0.46 | 5.8 ± 6.5 | 3.4 | 147 ± 94.1 |
| 4 | 806 | 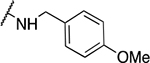 |
1.8 ± 0.1 | 15.9 ± 6.9 | 8.8 | 122 ± 53.8 |
| 5 | 539 | 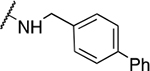 |
3.17 ± 1.91 | 3.6 ± 1.8 | 1.1 | 98.3 ± 41.9 |
| 6 | 804 | 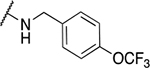 |
4.5 ± 0.15 | 30.1 ± 18.0 | 6.6 | 284 ± 226 |
| 7 | 1019 | 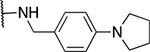 |
5.0 ± 0.42 | 10.7 ± 6.0 | 2.1 | 129 ± 43 |
Table 3.
Aniline derivatives
| HBV replication inhibition | HBV RNaseH inhibiton | |||||
|---|---|---|---|---|---|---|
| Entry | Compound number | NR1R1 | EC50 (μM) | CC50 (μM) | SI | IC50 (μM) |
| 1 | 389 | 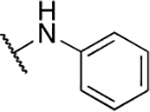 |
0.43 ± 0.23 | 21.2 ± 9.5 | 49.3 | 200 ± 190 |
| 2 | 711 | 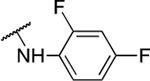 |
0.75 ± 0.35 | 4.6 ± 0.8 | 6.1 | NA |
| 3 | 836 | 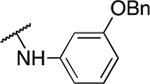 |
3.32 ± 0.67 | 5.4 ± 3.5 | 1.7 | 78.9 ± 49 |
| 4 | 799 | 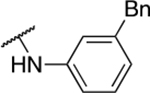 |
3.25 ± 0.78 | 5.3 ± 3.1 | 1.6 | 78.5 ± 60 |
| 5 | 798 | 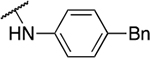 |
3.47 ± 0.49 | 3.3 ± 1.8 | 0.9 | 58.3 ± 49 |
| 6 | 835 | 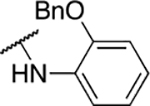 |
5.07 ± 2.45 | 10.3 ± 5.2 | 2 | 86.0 ± 42 |
| 7 | 834 |  |
5.30 ± 0.10 | 16.4 ± 11.2 | 3.1 | 83.5 ± 69 |
| 8 | 837 | 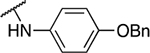 |
6.0 ± 1.98 | 8.7 ± 3.4 | 1.5 | 119 ± 89 |
Table 4.
Amino acid derivatives
| HBV replication inhibition | HBV RNaseH inhibiton | |||||
|---|---|---|---|---|---|---|
| Entry | Compound number | R1 | EC50 (μM) | CC50 (μM) | SI | IC50 (μM) |
| 1 | 797 | 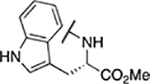 |
0.91 ± 0.27 | 13.9 ± 13.3 | 15.3 | 52.3 ± 8.2 |
| 2 | 793 | 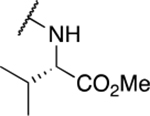 |
1.05 ± 0.38 | 18.3 ± 9.8 | 17.4 | 110 ± 10.5 |
| 3 | 796 | 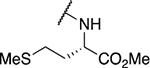 |
1.10 ± 0.15 | 36.0 ± 24.1 | 32.7 | 25.4 ± 26.3 |
| 4 | 870 | 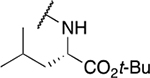 |
4.2 ± 0.71 | 9.1 ± 2.7 | 2.2 | 76.7 ± 94.4 |
| 5 | 867 | 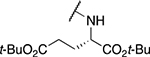 |
5.25 ± 0.78 | 5.2 ± 1.3 | 1 | 567 ± 474 |
| 6 | 794 | 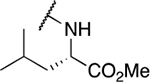 |
7.02 ± 4.74 | 14.7 ± 9.6 | 2.1 | 36.7 ± 14.6 |
| 7 | 869 | 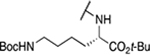 |
13.6 ± 0.57 | 14.6 ± 3.4 | 1.1 | 769 ± 462 |
| 8 | 795 | 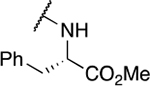 |
41.2 ± 39.7 | 12.7 ± 5.5 | 0.3 | 31.3 ± 1.4 |
| 9 | 868 | 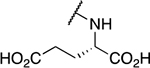 |
54.5 ± 4.95 | 100 ± 0 | 1.8 | 40.7 ± 25.4 |
Table 5.
Miscellaneous amide derivatives
| HBV replication inhibition | HBV RNaseH inhibiton | |||||
|---|---|---|---|---|---|---|
| Entry | Compound number | NR1R2 | EC50 (μM) | CC50 (μM) | SI | IC50 (μM) |
| 1 | 808 | 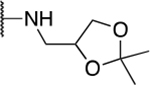 |
0.48 ± 0.17 | 14.9 ± 8.6 | 31.2 | 24.9 ± 5.5 |
| 2 | 707 | 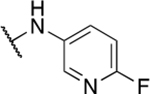 |
0.54 ± 0.01 | 19.4 ± 8.4 | 35.9 | NA |
| 3 | 384 | 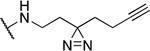 |
0.66 ± 0.67 | 17.6 ± 8.9 | 26.6 | 48.0 ± 31.8 |
| 4 | 712 | 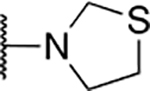 |
0.77 ± 0.49 | 6.4 ± 0.8 | 8.3 | 47.5 ± 7.9 |
| 5 | 810 | 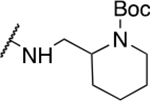 |
0.99 ± 0.52 | 10.9 ± 11.2 | 11 | 41.6 ± 9.3 |
| 6 | 1018 | 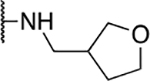 |
2.3 ± 0.28 | 41.2 ± 33.4 | 17.9 | 71.0 ± 21.6 |
| 7 | 805 | 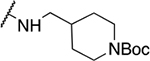 |
2.45 ± 0.59 | 19.5 ± 10.8 | 8 | 78.7 ± 46.0 |
| 8 | 1039 | 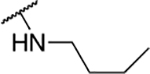 |
3.35 ± 2.05 | 8.7 ± 2.8 | 2.6 | 33.9 ± 2.8 |
| 9 | 710 | 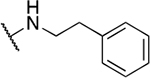 |
4.17 ± 4.04 | 4.5 ± 3.0 | 1.1 | 65.7 ± 18.1 |
| 10 | 709 | 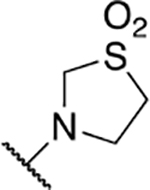 |
4.23 ± 6.52 | 19.5 ± 6.7 | 4.6 | 81.0 ± 28.7 |
| 11 | 801 | 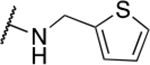 |
4.9 ± 1.67 | 61.1 ± 22.8 | 12.5 | 603 ± 354 |
| 12 | 874 | 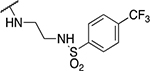 |
5.75 ± 1.20 | 17.7 ± 6.7 | 3.1 | 597 ± 124 |
| 13 | 708 | 6.5 ± 5.24 | 22.6 ± 12.8 | 3.4 | 58.7 ± 15.5 | |
Efficacy against HBV replication in culture was measured in HepDES19 cells (Guo et al., 2007), a HepG2 hepatoblastoma derivative carrying an integrated, tetracycline-repressible copy of the HBV genome. HBV reverse transcription proceeds normally in these cells and produces the normal distribution of capsid-associated HBV nucleic acids and the nuclear cccDNA, making it an efficient system for screening of compounds that directly target reverse transcription. Cells were plated in the absence of tetracycline to induce HBV reverse transcription in the cells. Two days later compounds were added to the medium in a final concentration of 1% DMSO. Three days later, cells were lysed, non-encapsidated cellular DNA (including the integrated transgene sequences) was digested, and capsids were lysed. The positive- and negative-polarity strands of the encapsidated HBV DNA were independently measured in these lysates using a strand-preferential qPCR assay (Edwards et al., 2019) that has about a 50-fold discrimination in its ability to distinguish the two strands. Fifty percent effective (EC50) values were calculated from the decline in the plus-polarity DNA because the plus-polarity DNA strand cannot be made in the absence of RNaseH activity, whereas substantial amounts of truncated minus-polarity DNAs accumulate. Fig. 2 shows example strand-preferential qPCR assays. Cytotoxicity was measured in HepDES19 cells following the same compound addition pattern to discriminate reductions in progeny DNA due to antiviral efficacy from reactions due to cell death. CC50 values were measured using an MTS assay that measures mitochondrial function because some tropolones have been reported to interfere with mitochondrial metabolism (Nakagawa and Tayama, 1998), and hence the MTS assay is likely to be sensitive to early adverse effects of the compounds on the cells. Fig. 2 also shows example CC50 experiments.
Fig 2. Example HBV replication inhibition and CC50 experiments.

Left, Replication inhibition. Representative assays are shown. Values are the average ± standard deviation from multiple assays. Black, plus-polarity DNA; gray, minus-polarity DNA. Right, Cytotoxicity. Representative MTS assays are shown. Values are the average ± standard deviation from multiple assays. Representative assays with the nucleoside analog Lamivudine are shown for context.
EC50 values for the amides against HBV replication ranged from 0.31 μM (390) to 54 μM (868). CC50s for the amides ranged from 3.3 μM (798) to >100 μM (868), indicating that almost all compounds had some detectable cytotoxicity, although it was moderate(20 – 50 μM) to functionally negligible (>50 μM) for many compounds. Selectivity index values (SI, CC50/EC50) for HBV varied from 80 (390) to 0.3 (795). TI values ≤1 indicate that the apparent efficacy against HBV replication is due primarily to cytotoxicity, and values from ~1 – 3 indicate that much of the apparent efficacy is likely due to cytotoxicity. There were 40 compounds with TI values >3.0, indicating that 74% of the 57 amides were HBV replication inhibitors.
3.2. Inhibition of the HBV RNaseH
αHTs tested to date inhibit recombinant HBV RNaseH, cause preferential suppression of the HBV plus-polarity DNA strand that is reflective of RNaseH inhibition (Fig. 2) (Hu et al., 2013; Lomonosova et al., 2017a; Lu et al., 2015; Lu et al., 2016), and the initial αHT hit against HBV (β-thujaplicinol, 46 in our numbering system) induces dose-dependent accumulation of RNA:DNA heteroduplexes within HBV capsids (Hu et al., 2013).
To extend these results, we measured 50% inhibitory concentration (IC50) values for 55 compounds examined in this study. The HBV RNaseH inhibition assay can detect RNaseH inhibitors, but it is insufficiently quantitative compared to inhibition of viral replication in culture, and at least for the N-hydroxyisoquinolinedione chemotype, is prone to false-negative results (Edwards et al., 2017; Edwards et al., 2019). Therefore, the assay is only semi-quantitative, presumably due to limitations to the recombinant enzyme that carries just the RNaseH domain of the four-domain HBV polymerase protein. Eight compounds inhibited recombinant HBV RNaseH with low IC50s (apparent IC50 = ≤25 μM), 32 were intermediate inhibitors (>25 to ≤100 μM), and 15 were poor inhibitors or lacked detectable inhibition (>100 μM) (Tables 1–5 and S1).
3.3. Selectivity against human RNase H1
Inhibition of human RNase H1 (huRH1) was measured because it is a likely off-target enzyme. huRH1 assays were performed with purified recombinant huRH1 under the same conditions as were used for the HBV RNaseH. In contrast to the HBV RNaseH, huRH1 data can be interpreted quantitatively (Edwards et al., 2019), so IC50 values were determined by titrating compounds in the RNaseH assays and observing their effects on the initial rates of the reactions. IC50 values ranged from 5.1 μM (1016) to >1,000 μM (919) (Table S1). There was no correlation between huRH1 IC50 values and CC50s, indicating that inhibition of huRH1 is not the primary driver of cytotoxicity during these three day assays.
3.4. Selectivity vs. Cryptococcus neoformans growth
MIC80 values against the fungal pathogen C. neoformans were measured by growing the cells in the presence of the compound for 48 hours and comparing growth of compound-treated cells to growth of the vehicle-treated cells. MIC80 values for the amide α-hydroxytropolones against C. neoformans ranged from 1 to 50 μM (Table S1).
3.5. Selectivity bacteria
MIC80 values against the bacterial pathogens E. coli (Gram -) and Staphylococcus aureus (Gram +) were determined by growing the cells in the presence of the compound for 16–24 hours and comparing growth of compound-treated cells to growth of the mock-treated cells. Only 8 compounds had measurable MIC80 values against the bacteria (Table S1), with the lowest MIC80 being 30 μM.
3.6. Modeling the HBV RNaseH structure and compound docking studies
The structure of the HBV RNaseH was predicted using five evolutionarily divergent RNaseHs for which structures are available as templates (from E. coli, Shewanella oneidensis, HIV-1, Thermus thermophilus, and Saccharomyces cerevisiae retrotransposon Ty3). The folds for the N-terminal ~60 amino acids in all five structures were readily superimposable, but only the Ty3 RNaseH fold extended past the C-terminal DEDD residue of the HBV RNaseH active site. The predicted fold based on the Ty3 structure also had the highest confidence score (89%), despite the amino acid identity in the sequence alignment being only 20%. Two Mn++ ions were inserted into positions analogous to their locations in the HIV RNaseH, and the model was refined using the Protein Preparation facility of the Maestro program in the Schrodinger suite. The final model (Fig. 3A) included 97 amino acids of the HBV RNaseH domain corresponding to residues 698–794 of the polymerase protein from the reference HBV ADW2 sequence (genotype A). This model comprised all but the N-terminal five and C-terminal 16 residues of the MBP-HRHgtCΔ5 protein used for the biochemical studies.
Fig. 3. HBV RNaseH homology model and in silico docking of αHTs to the HBV RNaseH active site.

A. Homology model for the HBV RNaseH. Purple spheres represent Mn++ ions modeled into the active site. B. 390 (green) and 404 (magenta) docked into the HBV RNaseH active site. C. 390 (green) and 120 (red) docked into the active site. D. Alternative binding pose of 920. The active site Mn++ ions used during docking are shown as blue spheres near the upper left of panels B-D.
This predicted model was used to dock the αHTs into the HBV RNaseH active site using Glide SP within the Schrodinger suite. Based on the earlier HIV RNase H modeling (Zhang et al., 2016), the docking calculation was set to yield those poses that are less likely to clash with partially bound, but displaced, DNA/RNA duplex substrate in the event that it remained bound to the RT in the enzymatic assay. Compound 120 and all but one of the 57 amide αHTs were successfully docked into the RNaseH active site at a pH range of 6–8 (Figs. 3B–D, docking scores in Table S2). Multiple binding poses were often observed for a given molecule, although with only a few exceptions, at least one of the poses had the troponoid oxygen triad chelated to both metals. This was the expected pose based on binding of the αHT β-thujaplicinol to the HIV RNaseH active site (Himmel et al., 2009). In this pose, the appendages are generally directed towards His728, which sits between N751 on one face creating a pocket (s1), and P705 and T706 residues on the other face creating a second pocket (s2). Piperazine-based analogs with distal aromatic appendages (i.e. 404, Fig. 3B) tend to engage pocket s2, facilitating pi-pi stacking with H728. The piperazine linker is similarly positioned to that of the piperidine appendage (390), suggesting that this engagement with H728 happens without distorting the natural conformational preference of the piperazine. The cyclohexyl group of 120 also engages in a similar configuration (Fig. 3C), but the tetrahedral nature of the ketone relative to the planar nature of the amide leads to steric interactions with P705.
Intriguingly, an inverted binding pose was suggested for 920, in which the nitrobenzoxadiazol group chelates both metal ions in the metalloenzyme core through a similar binding pose and the tropolone OH participates in hydrogen bonding with T706 (Fig. 3D). Significance of this binding pose was tested by purchasing six non-tropolone analogs of 920 and measuring their activity against the HBV RNaseH in biochemical assays. All of the compounds were either inactive or markedly less active than 920 (Fig. 4)..
Fig. 4. Nitrobenzoxadiazol derivatives used to test the alternative binding pose of 390.

IC50 values are against the HBV RNaseH.
4. Discussion
Our prior studies with the αHTs vs. HBV replication identified the methyl ketone 110 (EC50 = 0.25 μM, TI = 128) as the best αHT by both EC50 and TI value [(Lu et al., 2015) and unpublished]. Despite assessment of 17 additional ketones, we were unable to improve efficacy over 110. Similarly, attempts to improve efficacy with R3 sulfone and sulfide derivatives led to a largely flat SAR against HBV replication, but with reduced TI values due to elevated cytotoxicity (Agyemang et al., 2019). We recently described a final-step amidation approach to αHTs that allowed for the incorporation of a dynamic range of functional groups that were incompatible with prior αHT synthetic methods (Berkowitz et al., 2018). Leveraging this method, a diverse range of 52 new amide-functionalized αHTs were synthesized, and along with five additional molecules previously described, provided 57 amide-containing αHTs (Tables 1–5).
EC50 values for this library ranged from 0.31 – 54 μM, with 16 of the 57 novel amide αHTs (28%) having sub-micromolar EC50s, seven of which had values below 0.6 μM. Cytotoxicity was also high for certain members of the library, and CC50s ranged from 3.3 – 100 μM. Furthermore, while a substantial percentage of molecules had CC50 values below 20 μM, this cytotoxicity did not correlate closely with antiviral activity, and some of the more potent compounds (390, 391, and 389) were among the least cytotoxic (CC50 >20 μM), leading to SI values up to 80.
The high hit-rate of the amide library compared to previously tested molecules may imply a preference for the amide moiety. Indeed, a dramatic loss in activity is observed by comparing potent piperidine amide 390 (EC50 = 0.31 μM) with structurally analogous cyclohexyl ketone 120 (EC50 = 3.3 μM) (Fig. 1A) (Lomonosova et al., 2017a). This difference could be the result of amide aryl C-(O) and C-N bonds being more rigid than those of ketones, and thus the entropic penalties for binding may not be as severe. Alternatively, the stronger Lewis basicity of the amide carbonyl or structural differences due to the hybridization of the amide could also be beneficial.
Perhaps due to a similar preference for rigidity, piperazine-based analogs that are close structural homologs to 390 appear to have additional structural advantages. These molecules constitute 35% of the molecules (20 molecules), but over half of the ones with EC50 values < 0.6 μM (4 of 7 amide αHTs), and 8 of the 16 molecules with sub-micromolar EC50s. Furthermore, only three of the piperazine analogs had EC50 values >2.0 μM (5%) as compared to 44% (25/57) of the library as a whole. These less-potent molecules included two diaryl analogs (1020 and 1017) with significant steric bulk that could impede target binding, and one with a methylene appendage (1021) whose flexibility might lead to greater entropic penalties. Similarly, amino-acid and benzyl amide-derived αHTs are also flexible classes, and they are mostly less active, with only one of the 16 molecules having a sub-micromolar EC50 (797, EC50 = 0.91 μM) and over half (9 of 16, 56%) having EC50 values >3 μM. In contrast, aniline is rigid, and 389 and 711 are both sub-micromolar inhibitors. The remaining aniline class is comprised of molecules with additional flexible side-chains, and they are all weaker inhibitors (EC50 = 3.3 – 6.0 μM).
The amides were also tested for the ability to inhibit the HBV RNaseH in biochemical assays, with IC50 values ranging from 4.0 – 1000 μM. Activity against the RNaseH does not correlate well with replication inhibition, presumably in part due to the need to express the active recombinant enzyme as a fragment of the larger HBV polymerase protein that carries the RNaseH activity (Edwards et al., 2019). Consequently the HBV RNaseH IC50 values must be viewed only semi-quantitatively. However, the data do support some broader conclusions. First, only 1 of the 17 molecules that had IC50 values > 100 μM had an EC50 against viral replication <1 μM. Second, the piperidine amides were among the best compounds in both the enzymatic assay and in the cell-based replication inhibition assays. For example, 13 of 20 (65%) piperidine amides had IC50 values <50 μM, as compared to 28% of the remaining library, and all 5 molecules with IC50 values under 20 μM were piperidine amides. However, within the piperazine-based amide series, there was no correlation between IC50 and EC50 values.
A homology model of HBV RNaseH was developed by threading against the Ty3 RNaseH structure (Fig. 3). Two Mn++ ions were readily accommodated in the model in positions analogous to their binding to the HIV RNaseH, and the active site was shallow, again in agreement with the structure of the HIV enzyme (Himmel et al., 2009). Docking of the amide αHTs into the model often revealed several binding poses per each molecule, although in virtually all cases, one of these poses had the troponoid oxygen triad chelated to both metals. This is consistent with crystal structures of αHTs bound to related dinuclear metalloenzymes such as the HIV RT RNaseH (Himmel et al., 2009), and our observation that ablating the oxygen trident on the αHT eliminates inhibition of HBV replication [(Cai et al., 2014; Lu et al., 2015) and unpublished]. A possible inverted binding pose was revealed for 920 in which the nitrobenzoxadiazol moiety chelates the cations. Five of the six non-troponoid 920 analogs detectably inhibited the RNaseH (IC50 <500 μM), and 1131 that was selected based on potential interactions with T706 was the only analog with an IC50 <100 μM. Therefore, this inverted pose is appears likely to be real but to contribute only modestly to efficacy of 920. (Fig. 4). However, there was only a very weak correlation for the amide compounds between docking score and HBV RNaseH IC50s, and almost no correlation with efficacy against viral replication. Therefore, the model appears to reveal general aspects of the compounds’ binding poses, but it cannot yet identify details of their interactions with the enzyme.
This metal-chelating mechanism is very similar to that used by the HIV integrase inhibitors Bictegravir, Dolutegravir, Elvitegravir, and Raltegravir. In addition, the Influenza Virus PA cap-snatching endonuclease inhibitor Baloxavir marboxil was recently approved by the FDA (Yang, 2019). Baloxavir inhibits the PA endonuclease using the same metal chelating mechanism that the αHTs presumably use against the HBV RNaseH (Omoto et al., 2018). Furthermore, the US Food and Drug Administration had approved 62 drugs that act by coordinating active-site cations in metalloenzymes as of 2017 (Chen et al., 2019). Therefore, the metal chelation mechanism employed by HBV RNaseH inhibitors is a well-established drug mechanism, and the challenges to HBV drug development are the normal ones of achieving adequate efficacy and selectivity while maintaining suitable pharmacological parameters.
Selectivity will be important to establish for compounds that act by metal chelation due to the large number of enzymes in cells with divalent cations in their active sites. Unfortunately, the semi-quantitative nature of the HBV RNaseH assay prevents rigorous calculation of biochemical selectivity indexes. The limited observations possible reveal that the HBV RNaseH IC50 values correlate only weakly with those of huRH1 (R2 = 0.0.34), and that the semi-quantitative selectivity indexes for human vs. HBV RNaseH IC50s range from 14-fold more potent for huRH1 (867), to 22-fold more potent for the HBV RNaseH (808). The best available quantitative assessment of selectivity is an apparent selectivity index that compares EC50s for HBV replication inhibition to IC50s against HBV huRH1 (SIapparent = IC50 huRH1 / EC50 HBV replication). SIapparent values averaged 105 and ranged from 1 to >900, with 43 compounds having values ≥20. Therefore, biochemical selectivity against huRH1 is present, although it cannot be rigorously calculated.
Additional perspective on selectivity was obtained by evaluating the activity of the amide αHTs against growth of the fungus C. neoformans and two bacteria, E. coli and S. aureus, and by considering the SIs in human cells (Table S1). Forty-nine of the 57 αHTs detectably inhibited C. neoformans (MIC80 <50 μM), with an average MIC80 value of 17 μM, and over half of the molecules (33) had values <10 μM. However, the C. neoformans SAR was distinct from that against HBV replication. Activity against C. neoformans heavily favored the benzyl amide-derivative series, with all but a single molecule having an MIC80 less than 6 μM. On the other hand, the piperazine-derived analogs that seemed privileged for activity against HBV replication were considerably less potent against C. neoformans, with only one from this series having an MIC80 < 6 μM. The library was largely inactive against the bacteria, with only 8 of the 57 amide αHTs having measurable MIC80s against either E. coli or S. aureus and the lowest MIC80 being 30 μM for 710 vs. E. coli. The failure of the amide αHTs to inhibit bacterial growth is not a general feature of the αHTs because other troponoid subclasses can enter cells and inhibit bacterial growth with MIC80 values as low as 9 μM against E. coli (Cao et al., 2018). Finally, the SI values for HBV replication vs. cytotoxicity in cell culture are affected by the sum of the off-target effects on cells. SIs for the amide αHTs average 18 (range 0.3 – 80). Although some of the amide αHTs were clearly cytopathic, 26 of 57 (46%) compounds had SIs >10. Therefore, selectivity for HBV over cellular and microbial targets exists but will need to be improved as the compound series advances.
5. Conclusions
These data indicate that amide αHTs can be strong, specific HBV inhibitors merit further development toward becoming anti-HBV drugs for use in combination therapies to improve treatment for chronically infected patients. Key issues to be addressed include mitigating the moderate cytotoxicity seen with this compound series and continuing to improve efficacy to single-digit or sub-nanomolar levels while maintaining selectivity for the HBV enzyme. Finally, the amide αHTs are now advanced enough to merit optimization of critical pharmacological parameters such as cellular permeability, stability in body fluids, and pharmacokinetics.
Supplementary Material
Supplemental Data. Validation of compounds synthesized for this study.
Table S1. All screening data for compounds in this study.
Table S2. Docking scores for RNaseH inhibitors to the HBV RNaseH homology model.
Highlights.
Amide-containing α-hydroxytropolones can be strong, specific HBV inhibitors
Molecular modeling reveals a probable cation-chelating binding pose
Selectivity versus human ribonuclease H1 and microbial pathogens is present
Acknowledgements
This work was supported by NIH grants R01 AI122669, SC1 GM111158, and an NSF CAREER grant to EG (1750511). The funders had no role in study design, data interpretation, or the publication process. JET and RPM are inventors on patent applications covering use of these compounds against HBV. No other authors have competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Agyemang NB, Kukla CR, Edwards TC, Li Q, Langen MK, Schaal A, Franson AD, Casals AG, Donald KA, Yu AJ, Donlin MJ, Morrison LA, Tavis JE, Murelli RP, 2019. Divergent synthesis of a thiolate-based α-hydroxytropolone library with a dynamic bioactivity profile. RSC Advances 9, 34227–34234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz AJ, Abdelmessih RG, Murelli RP, 2018. Amidation Strategy for Final-Step alpha-Hydroxytropolone Diversification. Tetrahedron Lett 59, 3026–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT, 2013. Chronic hepatitis B: What should be the goal for new therapies? Antiviral Res 98, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE, 2014. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Orth C, Donlin MJ, Adegboyega P, Meyers MJ, Murelli RP, Elagawany M, Elgendy B, Tavis JE, 2018. Synthesis and Evaluation of Troponoids as a New Class of Antibiotics. ACS Omega 3, 15125–15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ, 2009. Ribonuclease H: the enzymes in eukaryotes. FEBS J 276, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Adamek RN, Dick BL, Credille CV, Morrison CN, Cohen SM, 2019. Targeting Metalloenzymes for Therapeutic Intervention. Chem Rev 119, 1323–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical and Laboratory Institute, 2015. Methods for dilution antimicrobial susceptibilty tests for bacterian that grow aerobically; Approved Standard -- Tenth Edition. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- Donlin MJ, Zunica A, Lipnicky A, Garimallaprabhakaran AK, Berkowitz AJ, Grigoryan A, Meyers MJ, Tavis JE, Murelli RP, 2017. Troponoids can inhibit growth of the human fungal pathogen Cryptococcus neoformans. Antimicrob Agents Chemother 61, e02574–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK, Morrison LA, Bailly F, Cotelle P, Giannakopoulou E, Zoidis G, Tavis JE, 2017. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antiviral Res 143, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TC, Mani N, Dorsey B, Kakarla R, Rijnbrand R, Sofia MJ, Tavis JE, 2019. Inhibition of HBV replication by N-hydroxyisoquinolinedione and N-hydroxypyridinedione ribonuclease H inhibitors. Antiviral Res 164, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning GC, Zoulim F, Hou J, Bertoletti A, 2019. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov. 18:827–844. [DOI] [PubMed] [Google Scholar]
- Gerelsaikhan T, Tavis JE, Bruss V, 1996. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J.Virol. 70, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghany MG, 2017. Current treatment guidelines of chronic hepatitis B: The role of nucleos(t)ide analogues and peginterferon. Best practice & research. Clinical gastroenterology 31, 299–309. [DOI] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT, 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81, 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD Jr., Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E, 2009. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 17, 1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch DR, Metrano AJ, Stone EA, Storch G, Miller SJ, Murelli RP, 2019. Troponoid Atropisomerism: Studies on the Configurational Stability of Tropone-Amide Chiral Axes. Organic letters 21, 2412–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Cheng X, Cao F, Huang A, Tavis JE, 2013. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99, 221–229. [DOI] [PubMed] [Google Scholar]
- Huber AD, Michailidis E, Tang J, Puray-Chavez MN, Boftsi M, Wolf JJ, Boschert KN, Sheridan MA, Leslie MD, Kirby KA, Singh K, Mitsuya H, Parniak MA, Wang Z, Sarafianos SG, 2017. 3-Hydroxypyrimidine-2,4-Diones as Novel Hepatitis B Virus Antivirals Targeting the Viral Ribonuclease H. Antimicrob Agents Chemother 61, e00245–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland PJ, Tavis JE, D’Erasmo MP, Hirsch DR, Murelli RP, Cadiz MM, Patel BS, Gupta AK, Edwards TC, Korom M, Moran EA, Morrison LA, 2016. Synthetic alpha-Hydroxytropolones Inhibit Replication of Wild-Type and Acyclovir-Resistant Herpes Simplex Viruses. Antimicrob Agents Chemother 60, 2140–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong KI, Overton H, Parkes KE, Cammack N, Martin JA, 2003. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 31, 6852–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero M, Testoni B, Zoulim F, 2016. HBV cure: why, how, when? Curr Opin Virol 18, 135–143. [DOI] [PubMed] [Google Scholar]
- Liaw YF, 2013. Impact of therapy on the outcome of chronic hepatitis B. Liver international : official journal of the International Association for the Study of the Liver 33 Suppl 1, 111–115. [DOI] [PubMed] [Google Scholar]
- Lomonosova E, Daw J, Garimallaprabhakaran AK, Agyemang NB, Ashani Y, Murelli RP, Tavis JE, 2017a. Efficacy and cytotoxicity in cell culture of novel alpha-hydroxytropolone inhibitors of hepatitis B virus ribonuclease H. Antiviral Res 144, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosova E, Zlotnick A, Tavis JE, 2017b. Synergistic Interactions between Hepatitis B Virus RNase H Antagonists and Other Inhibitors. Antimicrob Agents Chemother 61, e02441–02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KR, Lomonosova E, Li Q, Ponzar NL, Villa JA, Touchette E, Rapp S, Liley RM, Murelli RP, Grigoryan A, Buller RM, Wilson L, Bial J, Sagartz JE, Tavis JE, 2018. Efficacy of hepatitis B virus ribonuclease H inhibitors, a new class of replication antagonists, in FRG human liver chimeric mice. Antiviral Res 149, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D’Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE, 2015. Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antimicrob Agents Chemother 59, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Villa JA, Donlin M, Edwards TC, Cheng X, Heier RF, Meyers MJ, Tavis JE, 2016. Hepatitis B virus genetic diversity has minimal impact on sensitivity of the viral ribonuclease H to inhibitors. Antiviral Res 135, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meck C, D’Erasmo MP, Hirsch DR, Murelli RP, 2014. The biology and synthesis of alpha-hydroxytropolones. MedChemComm 5, 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, Tayama K, 1998. Mechanism of mitochondrial dysfunction and cytotoxicity induced by tropolones in isolated rat hepatocytes. Chemico-biological interactions 116, 45–60. [DOI] [PubMed] [Google Scholar]
- Nowotny M, 2009. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 10, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Yang W, 2006. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 25, 1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto S, Speranzini V, Hashimoto T, Noshi T, Yamaguchi H, Kawai M, Kawaguchi K, Uehara T, Shishido T, Naito A, Cusack S, 2018. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci Rep 8, 9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polaris Observatory C, 2018. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol 3, 383–403. [DOI] [PubMed] [Google Scholar]
- Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, Levrero M, Li W, Liang TJ, Lim SG, Lu F, Penicaud MC, Tavis JE, Thimme R, Members of the ICEHBVWG, Chairs I-HSG, Advisors I-HS, Zoulim F, 2019. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol. 4, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Zoulim F, Mason WS, 2013. Hepadnaviruses, in: Knipe DM, Howley PM (Eds.), Fields Virology, 6 ed. Lippincott Williams & Wilkins, Philadelphia PA, pp. 2185–2221. [Google Scholar]
- Summers J, Mason WS, 1982. Replication of the Genome of a Hepatitis B-Like Virus by Reverse Transcription of an RNA Intermediate. Cell 29, 403–415. [DOI] [PubMed] [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG, 2013. The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS pathogens 9, e1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA, 2014. Inhibitors of nucleotidyl transferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother 58, 7451–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepo C, Chan HL, Lok A, 2014. Hepatitis B virus infection. Lancet 384, 2053–2063. [DOI] [PubMed] [Google Scholar]
- Villa JA, Pike DP, Patel KB, Lomonosova E, Lu G, Abdulqader R, Tavis JE, 2016. Purification and enzymatic characterization of the hepatitis B virus ribonuclease H, a new target for antiviral inhibitors. Antiviral Res 132, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, 2019. Baloxavir Marboxil: The First Cap-Dependent Endonuclease Inhibitor for the Treatment of Influenza. Ann Pharmacother 53, 754–759. [DOI] [PubMed] [Google Scholar]
- Zhang B, D’Erasmo MP, Murelli RP, Gallicchio E, 2016. Free Energy-Based Virtual Screening and Optimization of RNase H Inhibitors of HIV-1 Reverse Transcriptase. ACS Omega 1, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data. Validation of compounds synthesized for this study.
Table S1. All screening data for compounds in this study.
Table S2. Docking scores for RNaseH inhibitors to the HBV RNaseH homology model.