

Abstract
Background
As regulators of gene expression, microRNAs (miRNAs) are increasingly recognized as critical biomarkers of human diseases. Till now, a series of computational methods have been proposed to predict new miRNA-disease associations based on similarity measurements. Different categories of features in miRNAs are applied in these methods for miRNA-miRNA similarity calculation. Benchmarking tests on these miRNA similarity measures are warranted to assess their effectiveness and robustness.
Results
In this study, 5 categories of features, i.e. miRNA sequences, miRNA expression profiles in cell-lines, miRNA expression profiles in tissues, gene ontology (GO) annotations of miRNA target genes and Medical Subject Heading (MeSH) terms of miRNA-associated diseases, are collected and similarity values between miRNAs are quantified based on these feature spaces, respectively. We systematically compare the 5 similarities from multi-statistical views.
Furthermore, we adopt a rule-based inference method to test their performance on miRNA-disease association predictions with the similarity measurements. Comprehensive comparison is made based on leave-one-out cross-validations and a case study. Experimental results demonstrate that the similarity measurement using MeSH terms performs best among the 5 measurements. It should be noted that the other 4 measurements can also achieve reliable prediction performance. The best-performed similarity measurement is used for new miRNA-disease association predictions and the inferred results are released for further biomedical screening.
Conclusions
Our study suggests that all the 5 features, even though some are restricted by data availability, are useful information for inferring novel miRNA-disease associations. However, biased prediction results might be produced in GO- and MeSH-based similarity measurements due to incomplete feature spaces. Similarity fusion may help produce more reliable prediction results. We expect that future studies will provide more detailed information into the 5 feature spaces and widen our understanding about disease pathogenesis.
Keywords: miRNA-disease association, Similarity measurement, Performance evaluation
Background
miRNAs are a large family of endogenous non-coding RNA molecules with approximately 22 nucleotides in length. They regulate the expression of their targeted messenger RNAs (mRNAs) through base pairing for cleavage or translational repression [1, 2]. To data, a great number of studies have identified that miRNAs are involved in various crucial biological processes, such as tissue development, cell proliferation and cell death. For example, Sabirzhanov et al. [3] found that a miRNA entitled miR-711 played a role in neuronal cell death by directly targeting the mRNA Ang-1 and decreasing its expression. Therefore, the dysfunctions of miRNAs would be associated with the pathogenesis and progression of a spectrum of human complex diseases (e.g. leukemia and cancers) [4]. In addition, as regulators of multiple genes, miRNAs harbor particular therapeutic effects [5–7] and research efforts [8–10] have demonstrated that miRNAs have the potential to become drug targets for disease treatments.
Given the importance of miRNAs in human health, several databases [4, 11, 12], which record associations between miRNAs and diseases by text-mining the published literature, have been launched as valuable resources for public use. In order to reduce the cost of biomedical experiments, computational methods [13–36] have been continuously presented to predict novel miRNA-disease associations for further experimental screening. The hypothesis behind these algorithms is that miRNAs with similar functions would be associated with diseases with similar phenotypes, and vice versa [37]. For instance, Chen et al. [13] adopted random walks on a miRNA-miRNA functional similarity network [38] to prioritize potential miRNAs for diseases of interest. Based on matched miRNA and mRNA expression profiles, Xu et al. [39] systematically identified the most promising miRNAs for cancers through inferred similarity values between miRNA target genes and known disease genes. To improve prediction accuracy, Liu et al. [22] integrated multiple data sources (e.g. miRNA-target gene associations and miRNA-lncRNA associations) for similarity calculation and implemented random walks on miRNA-disease heterogeneous networks for novel miRNA-disease association predictions. More recently, Yang et al. [40] computed similarity between miRNAs using a new GO semantic similarity metric based on miRNA target genes, and proposed a modified correlation model to infer miRNA-disease associations.
These computational approaches constitute an essential alternative to experimental assays. For these methods, it is no doubt similarity measurements are a key factor in determining their prediction accuracy. As to miRNA-miRNA similarity calculation, diverse categories of features, including miRNA sequences, expression profiles of miRNAs and GO of miRNA target genes, have been employed in these methods. However, as far as we know, there are few efforts made in comprehensively comparing the effects of miRNA similarity values, obtained from different features, on inferring novel miRNA-disease associations.
In this study, we first download 5 types of features from miRNAs and calculate their pairwise similarity values based on these feature spaces. Statistical tests are made on the datasets to compare properties of the similarity measurements. Then, we apply the similarity measurements for miRNA-disease association predictions using a popular rule-based inference method. Leave-one-out cross-validations and a case study are implemented for performance assessment and comparison. The best-performed similarity dataset is further used for new miRNA-disease association predictions. Finally, we comprehensively discuss the impacts of the 5 features on similarity calculation and miRNA-disease association predictions from multiple viewpoints, which we expect would provide a reference for biologists when investigating the functions of miRNAs.
Results
Overview of the 5 types of similarity measurements
In this study, we collect 5 types of features in miRNAs for pairwise similarity measurements (see Methods). For fair comparison, we use the latest information in each type for similarity calculation.
Table 1 provides a whole view of the information in the 5 datasets. Because of difference in feature availability, the numbers of miRNAs in the 5 datasets vary much with the highest number 2656 in seqSim and the lowest 1044 in MeSHSim, of which 205 miRNAs are commonly-owned. The distributions of pairwise similarity values in the 5 datasets can be seen in Fig. 1. We further use a boxplot (Fig. 2) to represent the similarity values in the 5 datasets. Four types of statistical results (mean value, standard deviation, skewness and kurtosis) of similarity values in the 5 datasets are calculated and we list them in Table 2.
Table 1.
An overview of the 5 types of similarity measurements for miRNAs
| Name of similarity matrix | No. of miRNAs | Feature for similarity calculation | Year of data published |
|---|---|---|---|
| seqSim | 2656 | miRNA sequences | 2018 |
| celllineSim | 2295 | expression profiles in cell lines | 2017 |
| tissueSim | 2300 | expression profiles in tissues | 2017 |
| GOSim | 2588 | GO of miRNA target genes | 2018 |
| MeSHSim | 1044 | MeSH terms of miRNA-associated diseases | 2019 |
Fig. 1.

The distributions of pairwise similarity values of miRNAs in the 5 datasets
Fig. 2.

Boxplot of similarity values of miRNAs in the 5 datasets
Table 2.
Four types of statistical results of similarity values in the 5 datasets
| mean value | standard deviation | skewness | kurtosis | |
|---|---|---|---|---|
| seqSim | 0.1682 | 0.0783 | 0.8617 | 5.5952 |
| celllineSim | 0.0860 | 0.1436 | 3.4613 | 16.1542 |
| tissueSim | 0.1230 | 0.2063 | 2.3258 | 7.7156 |
| GOSim | 0.7925 | 0.1101 | −1.5978 | 6.0010 |
| MeSHSim | 0.1301 | 0.1690 | 1.3665 | 4.8867 |
Similarly, we show the distributions of similarity values for the 205 common miRNAs in the 5 datasets in Additional file 1. We also apply a boxplot (Additional file 2) to illustrate similarity values for the 205 common miRNAs. Mean values, standard deviation, skewness and kurtosis for the 205 miRNAs are available at Additional file 3. We discover from the statistical analyses that for each dataset the distributions of similarity values of the whole miRNAs can be well represented by those of the 205 common miRNAs.
Prediction performance evaluation of the whole miRNAs in each of the 5 datasets
To compare the prediction performance, we first conduct leave-one-out cross-validations for the whole miRNAs in each of the 5 similarity measurements. As shown in Table 3, MeSHSim receives the highest average values of ROC-AUC and PR-AUC and performs best in the 5 datasets. The average ROC-AUC value for MeSHSim is 0.0389, 0.0394, 0.0406 and 0.0430 higher than these for the other 4 datasets, respectively. Meanwhile, the average PR-AUC value for MeSHSim increases by 0.0204, 0.0123, 0.0114 and 0.0265 compared with these for the other 4 datasets, respectively. Note that the other 4 similarity measurements also receive reliable prediction performance.
Table 3.
Comparison of average values of ROC-AUC and PR-AUC received based on HMDD V3.2 for the whole miRNAs in each of the 5 similarity datasets by leave-one-out cross-validations
| Average ROC-AUC value | Average PR-AUC value | |
|---|---|---|
| seqSim | 0.8880 | 0.2291 |
| celllineSim | 0.8875 | 0.2372 |
| tissueSim | 0.8863 | 0.2381 |
| GOSim | 0.8839 | 0.2230 |
| MeSHSim | 0.9269 | 0.2495 |
Note:The bold value indicated the highest one in each column
In addition, we implement paired t-tests to measure whether the ROC-AUC values and PR-AUC values obtained by MeSHSim across the whole miRNAs are significantly higher than these in the other 4 datasets. The calculated p-values are available at Table 4. The statistical results demonstrate MeSHSim can mostly achieve significantly better performance than all the other 4 measurements at the significance level 0.05.
Table 4.
Pairwise comparison with paired t-tests on the performance results obtained by MeSHSim and the other 4 measurements
| seqSim | celllineSim | tissueSim | GOSim | |
|---|---|---|---|---|
| p-value between MeSHSim and another similarity measurement based on ROC-AUC values | 1.90783E-20 | 6.63463E-19 | 4.96762E-19 | 4.63222E-23 |
| p-value between MeSHSim and another similarity measurement based on PR-AUC values | 0.023617 | 0.185257 | 0.221117 | 0.00308 |
Higher precision and recall values within the top k ranking list indicate more positive testing samples (real miRNA-disease associations in our study) are successfully predicted. The average precision and recall values across the whole miRNAs in the 5 datasets within the top k candidates are illustrated in Fig. 3 and Fig. 4, respectively. The two figures demonstrate that MeSHSim consistently outperforms the other 4 measurements at different k cutoffs.
Fig. 3.

Comparison of average PRE values in the top-k predictions for the whole miRNAs in each of the 5 datasets by leave-one-out cross-validations based on HMDD V3.2
Fig. 4.

Comparison of average REC values in the top-k predictions for the whole miRNAs in each of the 5 datasets by leave-one-out cross-validations based on HMDD V3.2
Prediction performance evaluation of the 205 common miRNAs in each of the 5 datasets
Considering the numbers of miRNAs in each of the 5 similarity datasets are different, we further choose the 205 common miRNAs in the 5 datasets to carry out leave-one-out cross-validation experiments to test their prediction performance.
As shown in Table 5, MeSHSim receives the highest average values of ROC-AUC and PR-AUC and performs best in the 5 datasets. The average ROC-AUC value for MeSHSim is 0.0267, 0.0363, 0.0372 and 0.0296 higher than these for the other 4 datasets, respectively. Meanwhile, the average PR-AUC value for MeSHSim increases by 0.0536, 0.0729, 0.0714 and 0.0606 compared with these for the other 4 datasets, respectively. Table 5 also suggests that the other 4 similarity measurements are able to achieve reliable prediction performance.
Table 5.
Comparison of average values of ROC-AUC and PR-AUC received based on HMDD V3.2 for the 205 common miRNAs in the 5 similarity datasets by leave-one-out cross-validations
| Average ROC-AUC value | Average PR-AUC value | |
|---|---|---|
| seqSim | 0.9114 | 0.1366 |
| celllineSim | 0.9018 | 0.1173 |
| tissueSim | 0.9009 | 0.1188 |
| GOSim | 0.9085 | 0.1296 |
| MeSHSim | 0.9381 | 0.1902 |
Note:The bold value indicated the highest one in each column
Paired t-tests are implemented to measure whether the ROC-AUC values and PR-AUC values obtained by MeSHSim across the 205 common miRNAs are significantly higher than these in the other 4 datasets. The calculated p-values are available at Table 6, and statistical results demonstrate MeSHSim achieves significantly better performance than all the other 4 measurements at the significance level 0.05.
Table 6.
Pairwise comparison with paired t-tests on the performance results obtained by MeSHSim and the other 4 measurements across the 205 common miRNAs
| seqSim | celllineSim | tissueSim | GOSim | |
|---|---|---|---|---|
| p-value between MeSHSim and another similarity measurement based on ROC-AUC values | 0.002349188 | 6.66612E-05 | 4.42713E-05 | 0.000785147 |
| p-value between MeSHSim and another similarity measurement based on PR-AUC values | 0.000938522 | 3.71868E-06 | 6.19924E-06 | 0.000176833 |
The average precision and recall values across the 205 common miRNAs in the 5 datasets within the top k candidates are illustrated in Fig. 5 and Fig. 6, respectively. We can conclude from the 2 figures that MeSHSim consistently outperforms the other 4 measurements at various k cutoffs.
Fig. 5.

Comparison of average PRE values in the top-k predictions for the 205 common miRNAs in the 5 datasets by leave-one-out cross-validations based on HMDD V3.2
Fig. 6.

Comparison of average REC values in the top-k predictions for the 205 common miRNAs in the 5 datasets by leave-one-out cross-validations based on HMDD V3.2
A case study
To further compare their abilities to predict potential disease candidates for miRNAs in the 5 datasets, we conduct a case study on hsa-mir-2861. The whole 894 disease candidates in the benchmarking dataset are ranked according to our method. We choose the top k (k = 10, 20, 40, 60, 80 and 100) predicted results for confirmation. We list the numbers of verified results in Table 7, which indicates the superiority of MeSHSim in screening the most predicted miRNA-disease associations.
Table 7.
Confirmed numbers of the top-k predicted results of hsa-mir-2861 in the 5 datasets
| Top 10 | Top 20 | Top 40 | Top 60 | Top 80 | Top 100 | |
|---|---|---|---|---|---|---|
| number of confirmed predictions (seqSim) | 0 | 0 | 1 | 3 | 4 | 4 |
| number of confirmed predictions (celllineSim) | 0 | 1 | 1 | 2 | 4 | 4 |
| number of confirmed predictions (tissueSim) | 0 | 0 | 1 | 2 | 4 | 4 |
| number of confirmed predictions (GOSim) | 0 | 1 | 1 | 2 | 4 | 4 |
| number of confirmed predictions (MeSHSim) | 0 | 1 | 3 | 3 | 5 | 5 |
Predictions of new miRNA-disease associations
After extensive comparison, we choose the best-performed similarity measurement MeSHSim to conduct comprehensive predictions of unknown associations between miRNAs and diseases. Experimentally verified miRNA-disease associations are downloaded from HMDD V3.2. In this inference proceeding, we train the method MBSI (see Method) with all known associations. We rank the non-interacting pairs according to their scores derived from Eq. (1) and extract the top 10 predicted results for each miRNA. The list of predicted associations can be seen in Additional file 4.
Discussion
In this study, 5 types of features are applied for miRNA similarity calculation. From the viewpoint of data sources, miRNA sequences are the most available, which is confirmed by the numbers of miRNAs in Table 1. As to miRNA expressions, accumulating data are available thanks to biomedical advance. However, it is known that quantitative values of miRNA expressions are affected by factors like library preparation protocols and adapter trimming steps. Therefore, robust pipelines to measure the expression values are well needed. Regarding GOSim, functional annotations for miRNAs are scarce in public databases and predicted miRNA target genes are integrated in Reference [40] for similarity calculation. False positive rate of predicted target genes would affect the similarity results and final prediction performance. For MeSHSim, it quantifies miRNA functional similarity based on MeSH terms derived from existing miRNA-associated diseases. The number of miRNAs in this dataset would therefore be greatly constrained. Because of incomplete data of experimentally supported miRNA-disease associations, the calculated similarity values in MeSHSim may be biased.
Experimental results demonstrate that MeSHSim performs best and the other 4 similarity measurements can also achieve stable and reliable prediction abilities. This can be explained with two biological facts, i.e. miRNAs target mRNAs through base pairing and a change in the expression level of a particular miRNA would lead to severe pathological conditions. Therefore, we expect that seamless integration of the 5 kinds of features for similarity measurements would produce more reliable prediction results.
For algorithms to infer miRNA-disease associations, the cold-start problem, in which associated diseases need to be predicted for a totally new miRNA, is a challenge that needs to be properly addressed. Strictly speaking, the similarity values in MeSHSim should be re-calculated before each round of cross validation is implemented in our study. As these values are computed based on known miRNA-disease associations, algorithms using MeSHSim for predictions suffer from the cold-start problem. Compared with MeSHSim, the other 4 similarity measurements do not encounter such challenge.
We focus only on the impact of miRNA similarity on miRNA-disease association predictions in this study. It is worthy pointing out that disease similarity is also vital for these similarity-based methods to improve their prediction performance, which is a further research topic.
Conclusions
Pairwise miRNA similarity measurement is an important step for miRNA-disease association predictions. In this study, we collect 5 feature spaces in miRNAs for similarity calculation and apply the similarity values to miRNA-disease association predictions. We comprehensively compare the statistical properties of the similarity values and systematically evaluate their inference performance on one independent benchmarking dataset. Although satisfied experimental results are received in all the 5 datasets, researchers should be cautious of the potential bias caused by some similarity measurements. Approaches allowing similarity fusion are in need for achieving more reliable prediction results.
Methods
Data preparation
We exploit 5 widely-used features for miRNA-miRNA similarity measurements. All similarity measures are symmetrically normalized to be in the range of (0, 1). The miRNA-miRNA similarity measures are as follows.
Sequence-based similarity between miRNAs: We download nucleotide sequences of miRNAs from the latest version of miRBase (http://www.mirbase.org/) [41]. The fasta format sequences of 2656 mature miRNAs in Homo sapiens in the database are kept and the sequences of miRNAs in other species are removed. The sequence similarity between two miRNAs is computed using needleall (http://www.bioinformatics.nl/cgi-bin/emboss/needleall). The parameters for this tool are set according to default values (Matrix file = EDNAfull, Gap opening penalty = 10, Gap extension penalty = 0.5). We refer to the 2656 × 2656 sequence similarity matrix as seqSim.
Expression-profile-in-cell-line-based similarity between miRNAs: We download expression profiles of miRNAs in 24 different types of cell-lines from miRmine (http://guanlab.ccmb.med.umich.edu/mirmine/) [42]. After merging miRNAs with the same name and deleting miRNAs with whole expression values of 0, we obtain 2295 mature miRNAs. Absolute values of Pearson correlation coefficient (PCC) between the expression profiles are computed as the measurement of similarity for the miRNAs. We refer to the 2295 × 2295 expression similarity matrix as celllineSim.
Expression-profile-in-tissue-based similarity between miRNAs: We download expression profiles of miRNAs in 16 different types of human tissues and bio fluids from miRmine (http://guanlab.ccmb.med.umich.edu/mirmine/) [42]. We take the same data processing steps as these in celllineSim and obtain 2300 mature miRNAs. We refer to the 2300 × 2300 expression similarity matrix as tissueSim.
GO-of-target-gene-based similarity between miRNAs: Recently, Yang et al. [40] developed a method entitled MiRGOFS to measure the functional similarity for 2588 miRNAs based on GO annotations of their target genes. We download the similarity results from their study. To normalize the raw data, we divide the value of each element before the diagonal one in each row (and column) by the value of the diagonal element and obtain a symmetric similarity matrix. Note that the normalized similarity matrix in Reference [40] was unsymmetric. We refer to the 2588 × 2588 similarity matrix as GOSim.
MeSH-term-of-disease-based similarity between miRNAs: In 2010, Wang et al. [38] presented a method MISIM to infer pairwise functional similarity for miRNAs based on MeSH terms of miRNA-associated diseases. More recently, an improved and updated version of MISIM (MISIM V2.0 [43]) was released. We download the similarity values of 1044 miRNAs from MISIM V2.0 (http://www.lirmed.com/misim/) and refer to the 1044 × 1044 similarity matrix as MeSHSim.
miRNA-disease association discovering
We adopt one popular rule-based inference method, miRNA-based similarity inference (MBSI) [15], to discover miRNA-disease associations with the similarities obtained from the above section.
We postulate in MBSI if a miRNA is implicated in a disease, similar miRNAs might also be associated with the disease (see Fig. 7). For a pair of miRNA-disease association (mi, dj), the inference score of the pair is calculated as,
| 1 |
where mi and dj denote miRNA i and disease j, Sim(mi, ml) is the similarity value between mi and ml, and alj =1if there is an existing association between ml and dj, otherwise alj =0. A higher score received from Eq. (1) indicates more confidence in a predicted association.
Fig. 7.
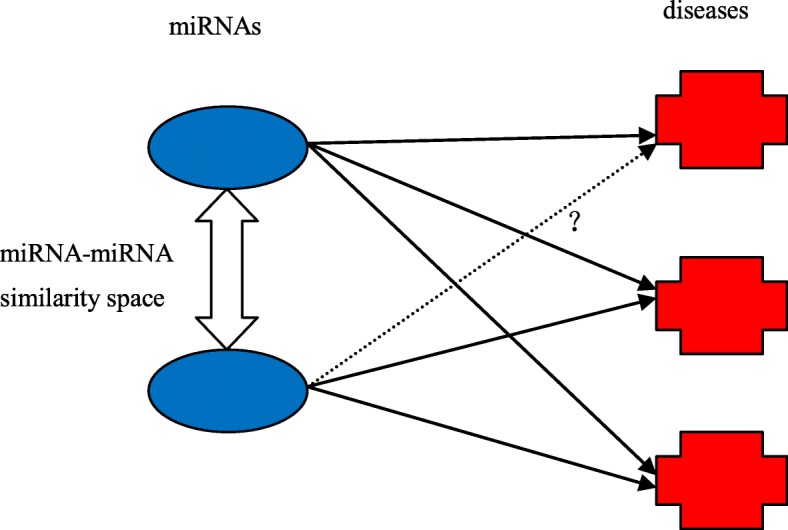
The principle behind new miRNA-disease association predictions. If a miRNA with unknown interaction profile shares a similar property with another miRNA with known interaction profile property, the former may also share the same interaction profile with the latter
Validation and evaluation metrics
We obtain a benchmarking dataset from HMDD V3.2 which contained experimentally supported miRNA-disease associations. This gold-standard dataset is regarded as true positive samples and is used for performance test.
We implement leave-one-out cross-validations to evaluate the prediction performance. Specifically, each miRNA is taken out once for testing and the remaining miRNAs for training. For each testing miRNA, all its association information is removed and the predicted scores for its associations with diseases are derived from Eq. (1). We rank the entire disease set for the testing miRNA according to the scores.
For each testing miRNA, we take the known miRNA-disease associations as positive instances. For each specific ranking threshold, if the score of a predicted miRNA-disease association is above the threshold, it is considered as a true positive. Otherwise, it is taken as a false positive. True positive rate (TPR), false positive rate (FPR), precision (PRE) and recall (REC) are calculated as follows by varying thresholds to plot ROC and PR curves,
| 2 |
| 3 |
| 4 |
| 5 |
where TP and TN are the numbers of correctly predicted positive and negative samples. FP and FN are the numbers of misidentified positive and negative samples. We use values of area under the ROC and PR curves (AUC) to demonstrate the prediction ability. We also measure the PRE and REC within the top 5, top 10 and top 20 candidates in the ranking list, because biologists are more interested in the top predictions.
Supplementary information
Additional file 1. The distributions of pairwise similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 2. Boxplot of similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 3. Four types of statistical results of similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 4. The top 10 predicted results for miRNAs in MeSHSim.
Acknowledgements
We are grateful to Dr. Wei Lan at Guangxi University for useful discussions. We thank Prof. Qinghua Cui at School of Basic Medical Sciences of Peking University for his help. We thank Prof. FangXiang Wu at University of Saskatchewan, Canada, for polishing the manuscript.
Abbreviations
- miRNAs
microRNAs
- mRNAs
Messenger RNAs
- GO
Gene ontology
- PCC
Pearson correlation coefficient
- MeSH
Medical Subject Headings
- MBSI
miRNA-based similarity inference
Authors’ contributions
HC and RG collected the data. HC, RG, GL, WZ and ZZ conceived and designed the experiments. HC and RG implemented the experiments. HC, GL and WZ analyzed the results. HC and RG wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China under Grant 61862026 and by the Natural Science Foundation of Jiangxi, China, under Grant 20181BAB202008. The funding bodies did not play any roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hailin Chen, Email: chl_mail@csu.edu.cn.
Ruiyu Guo, Email: 772123112@qq.com.
Guanghui Li, Email: ghli16@163.com.
Wei Zhang, Email: wzhang_math@whu.edu.cn.
Zuping Zhang, Email: zpzhang@mail.csu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-020-3515-9.
References
- 1.Ambros V. The functions of animal microRNAs. NATURE. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. CELL. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Sabirzhanov B, Faden AI, Aubrecht T, Henry R, Glaser E, Stoica BA. MicroRNA-711–induced Downregulation of Angiopoietin-1 mediates neuronal cell death. J Neurotraum. 2018;35(20):2462–2481. doi: 10.1089/neu.2017.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Z, Shi J, Gao Y, Cui C, Zhang S, Li J, Zhou Y, Cui Q. HMDD v3.0: a database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2019;47(D1):D1013–D1017. doi: 10.1093/nar/gky1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews. Drug discovery. 2013;12(11):847–865. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16(3):167–179. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov. 2018;17(8):547–558. doi: 10.1038/nrd.2018.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Zhang Z. A miRNA-driven inference model to construct potential drug-disease associations for drug repositioning. Biomed Res Int. 2015;2015:1–9. doi: 10.1155/2015/406463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Zhang Z. Peng W. miRDDCR: a miRNA-based method to comprehensively infer drug-disease causal relationships. Sci Rep. 2017;7(1):15921. doi: 10.1038/s41598-017-15716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Zhang Z. Prediction of drug-disease associations for drug repositioning through drug-miRNA-disease heterogeneous network. IEEE Access. 2018;6:45281–45287. doi: 10.1109/ACCESS.2018.2860632. [DOI] [Google Scholar]
- 11.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y: miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database):D98-104. [DOI] [PMC free article] [PubMed]
- 12.Yang Z, Wu L, Wang A, Tang W, Zhao Y, Zhao H, Teschendorff AE. dbDEMC 2.0: updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017;45(D1):D812–D818. doi: 10.1093/nar/gkw1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Liu M, Yan G. RWRMDA: predicting novel human microRNA–disease associations. Mol BioSyst. 2012;8(10):2792. doi: 10.1039/c2mb25180a. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Zhang Z. Prediction of associations between OMIM diseases and MicroRNAs by random walk on OMIM disease similarity network. Sci World J. 2013;2013:1–6. doi: 10.1155/2013/204658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Zhang Z. Similarity-based methods for potential human microRNA-disease association prediction. BMC Med Genet. 2013;6(1):12. doi: 10.1186/1755-8794-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Yan G. Semi-supervised learning for potential human microRNA-disease associations inference. SCI REP-UK. 2014;4(1). [DOI] [PMC free article] [PubMed]
- 17.Zou Q, Li J, Hong Q, Lin Z, Wu Y, Shi H, Ju Y. Prediction of MicroRNA-disease associations based on social network analysis methods. Biomed Res Int. 2015;2015:1–9. doi: 10.1155/2015/810514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao B, Ding S, Chen H, Li Z, Cai L. Identifying human microRNA–disease associations by a new diffusion-based method. J Bioinf Comput Biol. 2015;13(04):1550014. doi: 10.1142/S0219720015500146. [DOI] [PubMed] [Google Scholar]
- 19.Lan W, Wang J, Li M, Liu J, Pan Y. Predicting microRNA-disease associations by integrating multiple biological information. In: International Conference on Bioinformatics and Biomedicine (BIBM).: IEEE; 2015:183–188.
- 20.Peng W, Lan W, Yu Z, Wang J, Pan Y. A framework for integrating multiple biological networks to predict MicroRNA-disease associations. IEEE T Nanobiosci. 2017;16(2):100–107. doi: 10.1109/TNB.2016.2633276. [DOI] [PubMed] [Google Scholar]
- 21.Chen X, Huang L. LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput Biol. 2017;13(12):e1005912. doi: 10.1371/journal.pcbi.1005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Zeng X, He Z, Zou Q. Inferring MicroRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2017;14(4):905–915. doi: 10.1109/TCBB.2016.2550432. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Ding P, Liang C, Cao B, Chen X. Collective prediction of disease-associated miRNAs based on transduction learning. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2017;14(6):1468–1475. doi: 10.1109/TCBB.2016.2599866. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Wang L, Qu J, Guan N, Li J. Predicting miRNA-disease association based on inductive matrix completion. BIOINFORMATICS. 2018;34(24):4256–4265. doi: 10.1093/bioinformatics/bty503. [DOI] [PubMed] [Google Scholar]
- 25.Zeng X, Liu L, Lü L, Zou Q. Prediction of potential disease-associated microRNAs using structural perturbation method. BIOINFORMATICS. 2018;34(14):2425–2432. doi: 10.1093/bioinformatics/bty112. [DOI] [PubMed] [Google Scholar]
- 26.Zhong Y, Xuan P, Wang X, Zhang T, Li J, Liu Y, Zhang W. A non-negative matrix factorization based method for predicting disease-associated miRNAs in miRNA-disease bilayer network. Bioinformatics. 2018;34(2):267–277. doi: 10.1093/bioinformatics/btx546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao Q, Luo J, Liang C, Cai J, Ding P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics. 2018;34(2):239–248. doi: 10.1093/bioinformatics/btx545. [DOI] [PubMed] [Google Scholar]
- 28.Li G, Luo J, Xiao Q, Liang C, Ding P. Predicting microRNA-disease associations using label propagation based on linear neighborhood similarity. J Biomed Inform. 2018;82:169–177. doi: 10.1016/j.jbi.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Chen X, Gong Y, Zhang DH, You ZH, Li ZW. DRMDA: deep representations-based miRNA-disease association prediction. J Cell Mol Med. 2018;22(1):472–485. doi: 10.1111/jcmm.13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan W, Wang J, Li M, Liu J, Wu F, Pan Y. Predicting MicroRNA-disease associations based on improved MicroRNA and disease similarities. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2018;15(6):1774–1782. doi: 10.1109/TCBB.2016.2586190. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Yin J, Qu J, Huang L. MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput Biol. 2018;14(8):e1006418. doi: 10.1371/journal.pcbi.1006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Y, Zhang H, Liang C, Dong X. KATZMDA: prediction of miRNA-disease associations based on KATZ model. IEEE Access. 2018;6:3943–3950. doi: 10.1109/ACCESS.2017.2754409. [DOI] [Google Scholar]
- 33.Chen X, Zhu C, Yin J. Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput Biol. 2019;15(7):e1007209. doi: 10.1371/journal.pcbi.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang C, Yu S, Luo J. Adaptive multi-view multi-label learning for identifying disease-associated candidate miRNAs. PLoS Comput Biol. 2019;15(4):e1006931. doi: 10.1371/journal.pcbi.1006931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Zhang Z, Feng D. Prediction and interpretation of miRNA-disease associations based on miRNA target genes using canonical correlation analysis. BMC Bioinformatics. 2019;20(1):404. doi: 10.1186/s12859-019-2998-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Li Z, Guo W, Yang W, Huang F. A fast linear neighborhood similarity-based network link inference method to predict microRNA-disease associations. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2019;1. [DOI] [PubMed]
- 37.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008;3(10):e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26(13):1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 39.Xu C, Ping Y, Li X, Zhao H, Wang L, Fan H, Xiao Y, Li X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol BioSyst. 2014;10(11):2800–2809. doi: 10.1039/C4MB00353E. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Fu X, Qu W, Xiao Y, Shen H. MiRGOFS: a GO-based functional similarity measurement for miRNAs, with applications to the prediction of miRNA subcellular localization and miRNA-disease association. Bioinformatics. 2018;34(20):3547–3556. doi: 10.1093/bioinformatics/bty343. [DOI] [PubMed] [Google Scholar]
- 41.Kozomara A, Birgaoanu M. Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panwar B, Omenn GS. Guan Y. miRmine: a database of human miRNA expression profiles. Bioinformatics. 2017;33(10):1554–1560. doi: 10.1093/bioinformatics/btx019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Zhang S, Wan Y, Zhao Y, Shi J, Zhou Y, Cui Q. MISIM v2.0: a web server for inferring microRNA functional similarity based on microRNA-disease associations. Nucleic Acids Res. 2019;47(W1):W536–W541. doi: 10.1093/nar/gkz328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. The distributions of pairwise similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 2. Boxplot of similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 3. Four types of statistical results of similarity values of the 205 common miRNAs in the 5 datasets.
Additional file 4. The top 10 predicted results for miRNAs in MeSHSim.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.