

Abstract
Pd-catalyzed sequences involving oxidative addition, cyclization, and termination through intermolecular nucleophile capture have tremendous utility. Indeed, they can generate a plethora of different polycyclic structures possessing a diverse range of functionality. However, one area of deficiency for Pd0/PdII variants is the ability to conclude them with oxygen-based species. Inspired by the recent discovery of one such reaction in the course of a total synthesis program, we delineate herein that it has significant strength, both in terms of substrate scope as well as the terminating oxygen nucleophile. As a result, the reaction proved critical in achieving total syntheses of two oxygenated natural products, one of which was prone to over-oxidation. Finally, a mechanistic proposal that accounts for its success is provided.
Keywords: catalysis, oxygenation, Pd, total synthesis, triquinanes
Among terpene-based natural products, there are several triquinanes and related congeners[1] including (–)-presilphiperfolan-8-ol (1),[2] which possess a 1,3-trans stereochemical arrangement of substituents at the fusions of their varied ring systems (Scheme 1, colored in blue). Given that structural homology, our group has been interested in developing a cohesive Pd-catalyzed cyclization approach to fashion the diverse targets possessing such patterning. Our previous work on the total synthesis of presilphiperfolan-8-ol (1),[3] and other literature precedents,[4] have demonstrated that the desired 1,3-trans selectivity could be achieved through a Pd-mediated migratory insertion step (from 4 to 5). However, further extensions of the process,[5] including an intermolecular oxygen nucleophile terminating functionalization (5 to 6), are needed to access molecules such as 2[6] and 3[7] that possess an oxygen adjacent to the quaternary center.
Scheme 1.

Development of a unified approach to varied terpenes of different oxidation states predicated on the development of a unique functionalization of unactivated C(sp3)─PdII intermediates of type 5.
While seemingly plausible, reports of C─O bond formation from unactivated C(sp3)─PdII intermediates are rare. Normally, such processes occur via PdIV intermediates (7→9),[8] typically requiring the presence of an additional oxidant traditionally incompatible with Pd0/PdII catalytic systems. Nevertheless, our hope was that the α-quaternary center in the alkyl─PdII intermediate 5 might enable its interception by an oxygen nucleophile through intermolecular capture by precluding any intramolecular reactions such as β─H elimination; such a strategy has proven successful in promoting other types of challenging reductive eliminations.[5e-n] In fact, we recently achieved one example of such a functionalization as part of a total synthesis of conidiogenol (16)[9] and related family members in which 14 was obtained directly from 13 following an initial C─C bond construction through migratory insertion, leading to a similarly situated quaternary center.[10] Herein, we delineate the development and scope of this overall reaction process, one that is capable of affording a range of architectures with a variety of terminating oxygen-based nucleophiles with complete diastereocontrol. Critically, this method also provides general access to the core carbon skeleton of the botrydial family of natural products,[1b] with two members of this class successfully synthesized in a concise fashion. Furthermore, mechanistic studies provide a sound rationale for the success of the sequence.
Our investigations commenced using alkenyl triflate 17 as the initial test substrate, searching for competent oxygen nucleophiles and ligands in the presence of catalytic Pd-(OAc)2. As shown in Table 1, when Trixiephos[11] was used as the ligand at a 30 mol% loading along with 10 mol% of Pd(OAc)2, only trace product was obtained with NaOAc or KOAc as the oxygen source (entries 1 and 2). By contrast, when the commercially available and highly nucleophilic n-Bu4NOAc[12] was used, 18 was obtained in 61% yield based on NMR analysis (Entry 3). Pleasingly, a decrease in the ligand loading to 15 mol% led to a further increase in yield to 86% (Entry 4); any additional attenuation in loading level resulted in lower conversion, as did efforts to use less than 3.0 equivalents of n-Bu4NOAc. Further ligand screening under these parameters ultimately revealed that t-BuMephos[13] afforded the highest yield of 18 (95%, Entry 9). Finally, the reaction temperature could also be lowered to 90 °C with no erosion in material throughput (Entry 10).
Table 1:
Condition screening for a Pd-based cyclization–functionalization.[a]
 |
|||
|---|---|---|---|
| Entry | Ligand (loading) | Acetate source | Yield [%][b] |
| 1 | Trixiephos (30 mol%) | NaOAc | – |
| 2 | Trixiephos (30 mol%) | KOAc | 4 |
| 3 | Trixiephos (30 mol%) | n-Bu4OAc | 61 |
| 4 | Trixiephos (15 mol%) | n-Bu4OAc | 86 |
| 5 | Johnphos (15 mol%) | n-Bu4OAc | 76 |
| 6 | t-BuDavephos (15 mol%) | n-Bu4OAc | 82 |
| 7 | t-BuXantphos (15 mol%) | n-Bu4OAc | 52 |
| 8 | dtbpf (15 mol%) | n-Bu4OAc | 22 |
| 9 | t-BuMephos (15 mol%) | n-Bu4OAc | 95 |
| 10[c] | t-BuMephos (15 mol%) | n-Bu4OAc | 96 (95) |
Reaction conditions unless otherwise noted: 17 (0.2 mmol), Pd-(OAc)2 (0.02 mmol), ligand (0.03 mmol), acetate source (0.6 mmol), toluene (2 mL), 130°C.
Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard with the yield in parentheses reflecting the yield of isolated product.
Reaction temperature was 90°C.
Having identified this optimal condition set, we sought next to explore the scope of compatible oxygen nucleophiles using substrate 17 with a range of tetrabutylammonium salts. As shown in Table 2, a wide variety of such salts formed from benzoic acids with electron-donating and electron-withdrawing substituents proved successful, irrespective of their overall patterning, leading to the formation of products 19–31. In nearly all cases the yields were high (> 70%), although they were more modest for the two most electron-rich analogs leading to 21 and 22. For salts with halogen substituents, only those that were not prone to oxidative addition with Pd0, such as -F and -Cl, proved competent. Gratifyingly, aliphatic acid salts with a tetrabutylammonium counterion worked as well to afford 32–36 smoothly in high yield. Finally, investigations with non-carboxylic acid-based nucleophiles revealed that phenols were not competent unless the pKa of their acidic proton was in the range of a typical carboxylic acid, as is true for pentafluorophenol (pKa = 5.5 in H2O),[14] whose salt afforded a 78% yield of 37. 3,5-Ditrifluoromethylphenol (pKa = 8.26 in H2O),[15] by contrast, did not lead to any desired product. A very modest yield was observed using the salt of the acidic N-oxide HOBt in the reaction leading to 38. Critical for all these successful cases listed in Table 2 was that the salts (while easily prepared) had to be scrupulously dry, an important issue, as some proved to be hygroscopic if not stored properly (see Supporting Information for detailed procedure).
Table 2:
 |
Reaction conditions unless otherwise noted: 17 (0.1 mmol), Pd-(OAc)2 (0.01 mmol), t-BuMephos (0.015 mol), acetate source (0.3 mmol), toluene (1 mL), 90°C, 5 h.
Yields shown are of isolated products.
Reaction temperature was 130°C.
We next investigated the scope of the alkenyl triflate. Gratifyingly, starting materials with different core skeletons performed effectively under the optimal conditions using n-Bu4NOAc as the terminating nucleophile to deliver a range of fused bicyclic products in high yields and as single diastereomers. Of note, the ring size of the starting alkenyl triflate and the nature and position of substituents on the periphery of that ring showed no impact on reactivity (39–41, Table 3). Equally significant, a substrate with an oxygen linkage between the alkenyl triflate and terminating olefin acceptor led to product 42, containing both an acetate group as well as an ether ring. Moreover, an alkoxymethyl substituted olefin precursor led to the asymmetric formation of a new quaternary center in 43 in which the two oxygen centers were differentiated by the acetate group installed from the reaction. Substrates designed to undergo 6-exo-trig cyclization proved to be the most challenging, as the yield for these events, illustrated here for product 44, were consistently lower than for those formed through 5-exo-trig cyclizations. If intramolecular β-hydride elimination was possible, the reaction was unable to deliver product in any useful yield; one example is provided in the Supporting Information section.
Table 3:
The scope of the alkenyl triflate in the Pd-based cyclization.[a]
 |
Reaction conditions unless otherwise noted: substrate (0.1 mmol), Pd(OAc)2 (0.01 mmol), t-BuMephos (0.015 mol), n-Bu4NOAc (0.3 mmol), toluene (1 mL), 90°C, 5 h.
Given that final set of results, and in an effort to study the mechanism and deduce what parameters might be key for success of the sequence, deuterium-labeled 45 was prepared and subjected to the standard reaction conditions (Scheme 2). This transformation led to the formation of 46 in 95% yield as a single diastereomer. As such, the formation of a single product provided evidence that a radical mechanism was not at play. To establish the relative stereochemistry of the deuterium-containing carbon, 46 was subsequently treated with K2CO3 in MeOH. That operation afforded lactone 47 in 13% yield along with both recovered starting material and hydrolyzed, but non-cyclized material. Critically, nOe studies of 47 revealed that the deuterium atom was trans to the methyl group, indicating that the new C─O bond within 46 was formed through a stereoinvertive process; therefore, either an SN2-type substitution on intermediate 49 or an SN2′-type reaction with its equilibrium congener cyclopropane 50 would be required for its formation.[16] We believe the latter is more likely, with the generation of a strained ring potentially being a critical element for the reactivity needed for oxygen incorporation by giving the intermediate enhanced reactive character.[17] Evidence for the existence of cyclopropane intermediates was provided by substrate 51, with functionalization of the intermediate π-allyl species (52) proving to be more expedient than cyclopropane ring opening.
Scheme 2.

Key mechanistic analysis of the process using deuterated substrate 45. a) Pd(OAc)2 (10 mol%), t-BuMephos (15 mol%), n-Bu4NOAc (3.0 equiv), toluene, 90°C, 5 h, 95%. b) K2CO3 (5.0 equiv), MeOH, 12 h, 13%. c) Pd(OAc)2 (10 mol%), t-BuMephos (15 mol%), n-Bu4NOAc (3.0 equiv), toluene, 90°C, 5 h, 56%.
As a final demonstration of the power of the developed reaction, we attempted to utilize it for the preparation of natural products of types 2 and 3 (see Scheme 1). As shown in Scheme 3, we were able to access both botrydienal (3) as well as an aromatized variant (58). Those efforts began with ketone 54,[3] prepared in one step from (R)-pulegone. Subsequent conversion into 55 was achieved in a single pot by effecting oxycarbonylation with Mander’s reagent[18] followed by alkenyl triflate formation. With the stage now set for the key Pd-based cyclization, we found that while the reaction proceeded effectively using toluene (35% yield), a superior outcome was obtained using EtOAc instead, with 56 formed as a single diastereomer in 67% yield after 12 h. Subsequent hydrolysis with 1.05 equivalents of K2CO3 in MeOH at 23 °C over the course of 10 h afforded 57 in 60% yield; its oxidation with SeO2 in 1,4-dioxane at 110°C then completed a 5 step synthesis of the enantiomer of 10-oxodehydrodihydrobotrydial (58).[19] Intriguingly, if 56 was treated with an excess of K2CO3 (10 equiv) in MeOH under the same conditions, a free alcohol was generated instead which could be smoothly protected as a TBS ether. This specific protecting group proved essential to the subsequent SeO2-mediated allylic oxidation,[20] as the original acetate or its pivaloate, TIPS, or TMS-protected congeners generally afforded aromatized materials and/or uncharacterized side-products during that oxidation (including attempts with many other oxidants). Finally, dehydration of that newly installed allylic alcohol with the Burgess reagent[21] followed by reduction, standard protecting group removal, and oxidation completed the enantioselective total synthesis of botrydienal (3) in 9 steps overall.[22]
Scheme 3.
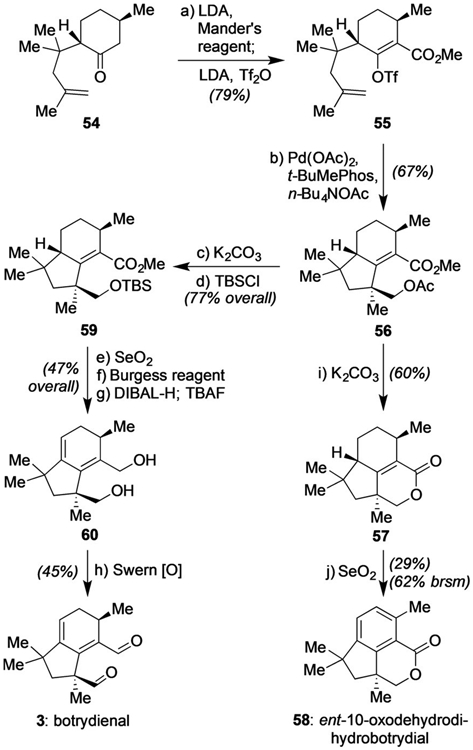
Total synthesis of botrydienal (3) and ent-10-oxodehydrodihydrobotrydial (58) through the Pd-catalyzed cyclization cascade. Reaction conditions: a) LDA (3.0 equiv), THF, −78 to 0°C, 1.5 h, then HMPA (3.0 equiv), Manders’ reagent (3.3 equiv), THF, −78°C, 1 h, then LDA (10 equiv), Tf20 (10 equiv), −78°C, 4 h, 79%. b) Pd(OAc)2 (10 mol%), t-BuMephos (15 mol%), n-Bu4NOAc (3.0 equiv), EtOAc, 90°C, 12 h, 67%. c) K2CO3 (10 equiv), MeOH, 23 °C, 10 h, 84%. d) TBSCl (1.5 equiv), imidazole (1.6 equiv), DMF, 23°C, 12 h, 92%. e) SeO2 (4.0 equiv), 1,4-dioxane, 110°C, 12 h, 49%, and 25% aromatization product. f) Burgess reagent (3.0 equiv), toluene, 80°C, 12 h, 96%. g) DIBAL-H (3.0 equiv), CH2Cl2, 0°C, 1 h, then TBAF (3.0 equiv), 3 h, 99%. h) (COCl)2 (20 equiv), DMSO (10 equiv), Et3N (30 equiv), CH2Cl2, −78°C, 3 h, 45%. i) K2CO3 (1.05 equiv), MeOH, 23 °C, 10 h, 60%. j) SeO2 (4 equiv), 1,4-dioxane, 110°C, 12 h, 29%, 62% (based on recovered starting material).
In conclusion, inspired by the homology of several natural product architectures and aiming to develop a unified approach to access their differentially functionalized variants, we were able to develop a one-pot Pd-catalyzed cyclization─oxygenation reaction featuring C─O bond formation from an unactivated C(sp3)─PdII intermediate in a Pd0/PdII catalytic cycle. Mechanistic studies have revealed this event to be a stereoinvertive process, with reaction probes revealing wide scope in substrate and nucleophile. Specific applications have ultimately afforded short and enantioselective total syntheses of two sesquiterpenes in the botrydial family. Further efforts to extend this reaction process and apply it to other complex molecules, as well as to develop additional means of terminating functionalization, are the subject of current endeavors.
Supplementary Material
Acknowledgements
We thank Dr. Antoni Jurkiewicz and Dr. C. Jin Qin for assistance with NMR and mass spectrometry, respectively. We also thank Mr. Kenneth DeBacker for the preparation and purification of some of the starting materials. Financial support for this work came from the University of Chicago, the National Institutes of Health (R01-GM132570) and Bristol–Myers Squibb (graduate fellowship to P.H.).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].For selected reviews, see: a) Mehta G, Srikrishna A, Chem. Rev 1997, 97, 671; [DOI] [PubMed] [Google Scholar]; b) Collado IG, Sánchez AJM, Hanson JR, Nat. Prod. Rep 2007, 24, 674. [DOI] [PubMed] [Google Scholar]
- [2].For isolation, see: a) Bohlmann F, Zdero C, Jakupovic J, Robinson H, King RM, Phytochemistry 1981, 20, 2239; [Google Scholar]; b) Coates RM, Ho Z, Klobus M, Wilson SR, J. Am. Chem. Soc 1996, 118, 9249; [Google Scholar]; For a review discussing the biosynthesis and chemical synthesis of the presilphiperfolanol family, in particular, see: c) Hong AY, Stoltz BM, Angew. Chem. Int. Ed 2014, 53, 5248; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014,126, 5350. [Google Scholar]
- [3].Hu P, Snyder SA, J. Am. Chem. Soc 2017, 139, 5007. [DOI] [PubMed] [Google Scholar]
- [4] a).Grigg R, Sansano JM, Santhakumar V, Sridharan V, Thangavelanthum R, Thornton-Pett M, Wilson D, Tetrahedron 1997, 53, 11803; [Google Scholar]; b) Overman LE, Abelman MM, Kucera DJ, Tran VD, Ricca DJ, Pure Appl. Chem 1992, 64, 1813. [Google Scholar]
- [5].For selected reviews on such reactions and subsequent functionalizations, see: a) Negishi E, Copéret C, Ma S, Liou S-Y, Liu F, Chem. Rev 1996, 96, 365; [DOI] [PubMed] [Google Scholar]; b) Grigg R, Sridharan V, J. Organomet. Chem 1999, 576, 65; [Google Scholar]; c) Petrone DA, Ye J, Lautens M, Chem. Rev 2016, 116, 8003; [DOI] [PubMed] [Google Scholar]; d) Ping Y, Li Y, Zhu J, Kong W, Angew. Chem. Int. Ed 2019,58, 1562; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 1576; [Google Scholar]; e) For selected examples of recent cyclization and functionalization chemistry, see: Burns B, Grigg R, Santhakumar V, Sridharan V, Steven P, Worakun T, Tetrahedron 1992, 48, 7297; [Google Scholar]; f) Newman SG, Lautens M, J. Am. Chem. Soc 2011,133, 1778; [DOI] [PubMed] [Google Scholar]; g) Liu H, Li C, Qiu D, Tong X, J. Am. Chem. Soc 2011, 133, 6187; [DOI] [PubMed] [Google Scholar]; h) Chen C, Hou L, Cheng M, Su J, Tong X, Angew. Chem. Int. Ed 2015, 54, 3092; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 3135; [Google Scholar]; i) Zheng H, Zhu Y, Shi Y, Angew. Chem. Int. Ed 2014, 53, 11280; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 11462; [Google Scholar]; j) Kong W, Wang Q, Zhu J, J. Am. Chem. Soc 2015, 137, 16028; [DOI] [PubMed] [Google Scholar]; k) Kong W, Wang Q, Zhu J, Angew. Chem. Int. Ed 2016, 55, 9714; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 9866; [Google Scholar]; l) Jiang Z, Hou L, Ni C, Chen J, Wang D, Tong X, Chem. Commun 2017, 53, 4270; [DOI] [PubMed] [Google Scholar]; m) Lu A, Ji X, Zhou B, Wu Z, Zhang Y, Angew. Chem. Int. Ed 2018, 57,3233; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 3287; [Google Scholar]; n) Hosoya Y, Kobayashi I, Mizoguchi K, Nakada M, Org. Lett 2019, 21, 8280. [DOI] [PubMed] [Google Scholar]
- [6].Fehlhaber H-W, Geipel R, Mercker H-J, Tschesche R, Welmar K, Schönbeck F, Chem. Ber 1974, 107, 1720. [Google Scholar]
- [7].Kimata T, Natsume M, Marumo S, Tetrahedron Lett 1985, 26, 2097. [Google Scholar]
- [8].a) For reviews, see: Handbook of Organopalladium Chemistry for Organic Synthesis (Ed.: Negishi E), Wiley, New York, 2003; [Google Scholar]; b) Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McDonald RI, Liu G, Stahl SS, Chem. Rev 2011, 111, 2981; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) He J, Wasa M, Chan KSL, Shao Q, Yu J-Q, Chem. Rev 2017,117, 8754; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu Y, Dong G, Chem. Sci 2018, 9,1424; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) For selected examples, see: Desai LV, Hull KL, Sanford MS, J. Am. Chem. Soc 2004, 126, 9542; [DOI] [PubMed] [Google Scholar]; g) Giri R, Liang J, Lei J, Li J, Wang D, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q, Angew. Chem. Int. Ed 2005, 44, 7420; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005,117, 7586; [Google Scholar]; h) Camasso NM, Pérez-Temprano MH, Sanford MS, J. Am. Chem. Soc 2014, 136, 12771. [DOI] [PubMed] [Google Scholar]
- [9].Hu P, Chi HM, Debacker KC, Gong X, Keim JH, Hsu IT, Snyder SA, Nature 2019, 569, 703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].In perhaps the closest example to this process, the Mulzer group reported a Pd-catalyzed cyclization and C─O bond formation reaction on an advanced intermediate in their studies towards bielschowskysin. However, they utilized ligand-free conditions significantly different from those reported herein. In addition, their system tolerates halides, which are not compatible in our reaction. The authors proposed a distinct mechanism involving acetoxypalladation, cyclization, and β-Br elimination processes, all as a PdII-based system, not a Pd0/PdII cycle. However, it should be noted that since no mechanistic experiments were performed, a pathway akin to that presented herein cannot be ruled out: Himmelbauer M, Farcet J-B, Gapnepain J, Mulzer JA, Org. Lett 2013, 15, 3098.23724910 [Google Scholar]
- [11].a) For a review of the power of several ligands developed by the Buchwald group, see: Surry DS, Buchwald SL, Angew. Chem. Int. Ed 2008, 47, 6338; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 6438; [Google Scholar]; b) For uses of Trixiephos, see: Torraca KE, Kuwabe S-I, Buchwald SL, J. Am. Chem. Soc 2000, 122, 12907. [Google Scholar]
- [12].a) Intriguingly, the same oxygen source was the only one indicated as competent in early work by Shibasaki to functionalize activated π-allyl based systems: Kagechika K, Shibasaki M, J. Org. Chem 1991, 56, 4093; [Google Scholar]; b) Ohshima T, Kagechika L, Adachi M, Sodeoka M, Shibasaki M, J. Am. Chem. Soc 1996, 118, 7108. [Google Scholar]
- [13].For the development and use of t-BuMephos, see: Fox JM, Huang X, Chieffi A, Buchwald SL, J. Am. Chem. Soc 2000, 122, 1360. [Google Scholar]
- [14].Gasbarri C, Angelini G, RSC Adv. 2014, 4, 17840. [Google Scholar]
- [15].Kütt A, Movchun V, Rodima T, Dansauer T, Rusanov EB, Leito I, Kaljurand I, Koppel J, Pihl V, Koppel I, Ovsjannikov G, Toom L, Mishima M, Medebielle M, Lork E, Röschenthaler G-V, Koppel IA, Kolomeitsev AA, J. Org. Chem 2008, 73, 2607. [DOI] [PubMed] [Google Scholar]
- [16] a).Brown A, Grigg R, Ravishankar T, Thornton-Pett M, Tetrahedron Lett. 1994, 35, 2753; [Google Scholar]; b) Grigg R, Sakee U, Sridharan V, Sukirthalingam S, Thangavelauthum R, Tetrahedron 2006, 62, 9523. [Google Scholar]
- [17].McCormick JP, Barton DL, J. Org. Chem 1980, 45, 2566. [Google Scholar]
- [18] a).Mander LN, Sethi SP, Tetrahedron Lett. 1983, 24, 5425; [Google Scholar]; b) Mander LN, Shing TKM, Yeung YY, Lujan-Montelongo JA, Encyclopedia of Reagents for Organic Synthesis, Wiley, New York, 2018, 10.1002/047084289X.rm168.pub3. [DOI] [Google Scholar]
- [19].a) For its original isolation and characterization, see: Colmenares AJ, Durán-Patrón RM, Hernández-Galán R, Collado IG, J. Nat. Prod 2002, 65, 1724; [DOI] [PubMed] [Google Scholar]; b) For its first chemical synthesis as achieved over 9 steps, which includes a revision of its absolute configuration, see: Qiao C, Zhang W, Han J-C, Li C-C, Org. Lett 2016, 18, 4932.27619953 [Google Scholar]
- [20].Nicolaou KC, Petasis NA, Selenium in Natural Product Synthesis, CIS, Philadelphia, 1984. [Google Scholar]
- [21] a).Atkins GM, Burgess EM, J. Am. Chem. Soc 1968, 90, 4744; [Google Scholar]; b) Taibi P, Mobashery S, Hart AC, Encyclopedia of Reagents for Organic Synthesis, Wiley, New York, 2008, 10.1002/047084289X.rm095m.pub2. [DOI] [Google Scholar]
- [22].For the only additional reported synthesis of members of the class (targeting other compounds), see: Qiao C, Zhang W, Han J-C, Dai W-M, Li C-C, Tetrahedron 2019, 75, 1739. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.