

Host cells mount a response to curb virus replication in infected cells and prevent spread of virus to neighboring, as yet uninfected, cells. The NF-κB family of proteins is important for the cell to mediate this response. In this study, we show that in cells infected with mammalian reovirus, NF-κB is inactive. Further, we demonstrate that NF-κB is rendered inactive because virus infection results in reduced levels of upstream intermediaries (called IKKs) that are needed for NF-κB function. Based on previous evidence that active NF-κB limits reovirus infection, we conclude that inactivating NF-κB is a viral strategy to produce a cellular environment that is favorable for virus replication.
KEYWORDS: NF-κB, innate immunity, reovirus
ABSTRACT
Viruses commonly antagonize innate immune pathways that are primarily driven by nuclear factor kappa B (NF-κB), interferon regulatory factor (IRF), and the signal transducer and activator of transcription proteins (STAT) family of transcription factors. Such a strategy allows viruses to evade immune surveillance and maximize their replication. Using an unbiased transcriptome sequencing (RNA-seq)-based approach to measure gene expression induced by transfected viral genomic RNA (vgRNA) and reovirus infection, we discovered that mammalian reovirus inhibits host cell innate immune signaling. We found that, while vgRNA and reovirus infection both induce a similar IRF-dependent gene expression program, gene expression driven by the NF-κB family of transcription factors is lower in infected cells. Potent agonists of NF-κB such as tumor necrosis factor alpha (TNF-α) and vgRNA failed to induce NF-κB-dependent gene expression in infected cells. We demonstrate that NF-κB signaling is blocked due to loss of critical members of the inhibitor of kappa B kinase (IKK) complex, NF-κB essential modifier (NEMO), and IKKβ. The loss of the IKK complex components prevents nuclear translocation and phosphorylation of NF-κB, thereby preventing gene expression. Our study demonstrates that reovirus infection selectively blocks NF-κB, likely to counteract its antiviral effects and promote efficient viral replication.
IMPORTANCE Host cells mount a response to curb virus replication in infected cells and prevent spread of virus to neighboring, as yet uninfected, cells. The NF-κB family of proteins is important for the cell to mediate this response. In this study, we show that in cells infected with mammalian reovirus, NF-κB is inactive. Further, we demonstrate that NF-κB is rendered inactive because virus infection results in reduced levels of upstream intermediaries (called IKKs) that are needed for NF-κB function. Based on previous evidence that active NF-κB limits reovirus infection, we conclude that inactivating NF-κB is a viral strategy to produce a cellular environment that is favorable for virus replication.
INTRODUCTION
The mammalian innate immune response is an effective response to viral intrusion. The primary mechanism of innate control of virus infection is the production of antiviral cytokines. Paracrine signaling by these cytokines establishes an antiviral state in neighboring, uninfected cells, making them refractory to virus infection and limiting dissemination of virus in the host (1). Expression of these cytokines is under the control of two major transcription factors, nuclear factor kappa B (NF-κB) and interferon regulatory factor 3 (IRF3) (2). NF-κB and IRF3 are activated downstream of a signal that is initiated by extracellular sensing of pathogen-associated molecules, during transit through cellular uptake pathways, or within the cell (2). For RNA viruses, cell surface or endosomal sensing of the genomic material via Toll-like receptors (TLRs) and cytoplasmic sensing via the RIG-I-like receptors (RLRs) are two major mechanisms of pathogen recognition (2). Animal models and cell lines lacking any component of the signaling module—sensor, transcription factors, or cytokines—are typically more susceptible to viral infections than those with an intact immune response (3–6).
Because these initial stages of the innate immune response are so effective at limiting viral replication, most viruses have evolved one or more mechanisms to limit either the production or activity of these antiviral cytokines (7, 8). Frequently, viruses antagonize the immune response by sequestering, degrading, or inactivating one or more cellular components that are required for a cytokine-based antiviral response (7, 8). Targeting transcription factor function is a commonly used viral strategy, perhaps because transcription factors serve a critical node that controls the expression of multiple antiviral molecules. Among these, NF-κB can control the function of a wide variety of proinflammatory chemokines and cytokines (9). The NF-κB transcription factor family is composed of five different subunits that function as homo- or heterodimers. The classical NF-κB complex (here referred to as NF-κB), composed of p65 and p50 subunits, is a critical regulator of antiviral gene expression (9). In an inactive state, it is sequestered in the cytoplasm by the inhibitor of κB (IκB) proteins (9). NF-κB transcriptional activity is regulated by the inhibitor of kappa B kinase (IKK) complex, a protein complex which causes the degradation of IκB (9). The IKK complex, which is composed of IKKα, IKKβ, and the NF-κB essential modifier (NEMO), phosphorylates IκB, which leads to its ubiquitination and degradation. Freed from its inhibitor, NF-κB translocates to the nucleus, binds to DNA, and initiates gene expression. The transactivation function of NF-κB also requires IKK-mediated phosphorylation of the p65 subunit (10).
Mammalian orthoreovirus (reovirus) is a double-stranded RNA (dsRNA) virus which replicates in the cytoplasm of the host cell (11). Like most other viruses, reovirus pathogenesis is influenced by NF-κB signaling (12). In a newborn mouse model, NF-κB plays an antiviral role in the heart. In comparison to wild-type mice, NF-κB p50−/− mice exhibit higher viral titers, tissue damage, and cell death, indicating that NF-κB is antiviral in this context. This outcome is due, at least in part, to an inability of p50−/− mice to produce beta interferon (IFN-β). Despite the fact that the viral genomic RNA remains within the reovirus core, the current model posits that the innate immune response is initiated when genomic RNA from incoming virions is sensed by the RLRs RIG-I and MDA5 (9, 13). The sensing of the RNA leads to the activation of IRF3 and NF-κB, which lead to the production of IFN and other inflammatory cytokines (14). Whether reovirus actively limits this antiviral response has not been extensively scrutinized.
In this study, we investigated whether reovirus inhibits innate immune signaling following infection. Using transcriptome sequencing (RNA-seq), we found that NF-κB activity was inhibited in infected cells following treatment with multiple agonists, including viral genomic RNA (vgRNA) and tumor necrosis factor alpha (TNF-α). We discovered that this inhibition was due to reduced cellular levels of the IKK components IKKβ and NEMO. Loss of the IKK complex led to inhibition of NF-κB nuclear translocation and consequent blockade of its transactivation function. Blockade of viral gene expression prevented IKK loss, suggesting that events in viral replication after cell entry are required for IKK loss and NF-κB inhibition. This study highlights a previously unknown mechanism by which reovirus infection blunts the host innate immune response.
RESULTS
NF-κB-dependent gene expression is blocked in reovirus-infected cells.
The genomic dsRNA within reovirus particles serves as the pathogen-associated molecular pattern that activates the innate immune response via RIG-I and MDA5 (15–17). To determine if reovirus infection modifies this response, we compared the host cell response in L929 cells following transfection of vgRNA with the response following infection with reovirus strain T3A. Using RNA-seq analyses, we found that viral RNA transfection induced the expression of 978 genes (Fig. 1A and B). For these analyses, we considered only those genes whose expression was increased by >4-fold (log2 fold change [log2FC] > 2) and that were identified with a false-discovery rate (FDR) of <0.05 to be significantly different. We used iRegulon, which predicts transcriptional regulators for a similarly expressed gene set by providing a normalized enrichment score (NES) (18). A high NES for a given transcription factor indicates that many of the genes in a set are likely regulated by that transcription factor. We used this program to identify which transcription factors most likely regulate the genes induced following treatment with vgRNA. We found that, of the 978 genes induced by vgRNA, the highest NES scores were assigned to NF-κB and IRF, with scores of 4.0 and 10.0, respectively (Fig. 1C). Predictably, reovirus infection induced a similar gene expression profile. In total, 65% of the 978 genes induced by vgRNA were also induced by reovirus. When we used iRegulon to predict the transcription factors that regulate the genes induced by reovirus infection, we found that, while genes regulated by IRF were enriched in this list (NES, 12.0), genes regulated by NF-κB were not. Surprisingly, the NES for NF-κB fell below the 3.0 cutoff, indicating that NF-κB target genes were not enriched in the set of genes induced by reovirus infection (Fig. 1C).
FIG 1.

Reovirus strain T3A inhibits NF-κB-dependent gene expression. (A) ATCC L929 cells were transfected with 0.5 μg of vgRNA. At 7 h following transfection, total RNA was extracted and subjected to RNA-seq analyses. A volcano plot showing genes whose expression was induced by >4-fold (log2 fold change [log2FC] > 2) and with a false-discovery rate (FDR) of <0.05 in comparison to that in mock-infected cells are shown within the box. (B, C) ATCC L929 cells were transfected with 0.5 μg of vgRNA for 7 h or infected with 10 PFU/cell of reovirus strain T3A for 20 h. Total RNA was extracted and subjected to RNA-seq analyses. (B) Heat map comparing expression of the genes shown in the boxed region of panel A following vgRNA transfection and T3A infection. (C) Scatterplot comparing expression of the genes shown in the boxed region of panel A following vgRNA transfection and T3A infection. Black dots denote genes that were not expressed significantly differently in the two treatments. Red dots represent genes that were expressed to a significantly lower extent in T3A-infected cells. iRegulon analyses of both sets of genes is also shown. NES, normalized enrichment score. (D, E) ATCC L929 cells were adsorbed with phosphate-buffered saline (PBS) (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 20 h, cells were transfected with 0.5 μg viral RNA for 7 h. Total RNA was extracted and subjected to RNA-seq analyses. (D) Heat map comparing expression of the genes shown in the boxed region of panel A following vgRNA transfection of mock-infected and T3A-infected cells. (E) Scatterplot comparing expression of the genes shown in the boxed region of panel A following vgRNA transfection of mock-infected and T3A-infected cells. Black dots denote genes that are not expressed significantly differently in the two treatments. Red dots represent genes that are expressed to a significantly lower extent in T3A-infected cells transfected with vgRNA. iRegulon analyses of both sets of genes are also shown.
Of the 978 genes induced by vgRNA described above, 35% (339 of 978) were expressed to a lower extent in reovirus-infected cells. Using iRegulon, we predicted that NF-κB target genes were enriched in this set, as the NES for NF-κB increased from 4.0 to 4.9 (Fig. 1C). In agreement with the results we show in Fig. 1A and B, these data suggest that NF-κB and IRF transcription factor families are regulated differently in cells transfected with RNA and in cells infected with reovirus. Thus, the observed differences in the gene expression profiles of RNA-transfected and reovirus-infected cells are not related to differences in RNA sensing. Instead, this difference may be because reovirus fails to activate the NF-κB signaling pathway or because it has evolved a mechanism to block NF-κB signaling.
To distinguish between these possibilities, we determined whether reovirus infection inhibits vgRNA-induced NF-κB activation. Toward this end, we compared if gene expression in uninfected cells transfected with vgRNA differed from that in infected cells transfected with vgRNA. As described above, vgRNA transfection of uninfected cells induces expression of 978 genes (Fig. 1A). In infected cells, however, vgRNA failed to induce 13% of these genes (133 of 978) (Fig. 1D and E). We used iRegulon to predict that the most likely transcriptional regulator of genes whose expression was inhibited by reovirus infection was NF-κB, with an NES of 6.1. These data allow us to conclude that reovirus blocks NF-κB-dependent gene expression, even in the presence of a potent agonist such as vgRNA.
vgRNA and reovirus infection activate NF-κB downstream via a common set of sensors that detect RNA (15–17, 19). To determine if the inhibitory effect of reovirus on NF-κB-dependent gene expression is only restricted to viral RNA-induced gene expression, we used TNF-α, a potent stimulator of NF-κB signaling. RNA-seq analyses of uninfected cells treated with TNF-α, using the same criteria described above, led to the upregulation of 32 transcripts (Fig. 2A). In contrast, TNF-α had no significant effect on gene expression in cells infected with T3A (Fig. 2B to D). These data indicate that infection of cells with T3A results in blockade of NF-κB-dependent transcription. vgRNA and TNF-α initiate NF-κB signaling via distinct routes. Therefore, our analyses suggest that reovirus blocks NF-κB signaling at a common downstream step.
FIG 2.

Reovirus strain T3A inhibits TNF-α-stimulated NF-κB-dependent gene expression. (A) ATCC L929 cells were treated with 10 ng/ml TNF-α. At 1 h following treatment, total RNA was extracted and subjected to RNA-seq analyses. A volcano plot showing genes whose expression is induced by 4-fold (log2FC > 2) and which had an FDR of <0.05 in comparison to that in untreated cells are shown within the box. (B, C, D) ATCC L929 cells were adsorbed with 10 PFU/cell of T3A. Following incubation at 37°C for 20 h, cells were treated with 0 or 10 ng/ml TNF-α for 1 h. Total RNA was extracted from cells and was subjected to RNA-seq analyses. (B) Scatterplot comparing expression of the genes shown in the boxed region of panel A following infection with T3A with or without TNF-α. A trendline showing linear regression and coefficient of determination is shown. (C) Scatterplot comparing expression of the genes shown in the boxed region of panel A following TNF-α treatment of mock-infected and T3A-infected cells. A trendline showing linear regression and coefficient of determination is shown. (D) Heat map comparing expression of the genes shown in the boxed region of panel A following TNF-α treatment of mock-infected and T3A-infected cells. Expression of the same set of genes in T3A-infected cells is also shown.
IKK activity is diminished in reovirus-infected cells.
To verify our RNA-seq analyses, we measured the capacity of vgRNA and TNF-α to induce the expression of an NF-κB target gene in reovirus-infected cells using reverse transcription-quantitative PCR (RT-qPCR). For these experiments, we monitored the transcript levels of IκBα, an NF-κB target gene. Because the IκBα protein inhibits NF-κB nuclear translocation, its expression serves as a feedback inhibitor of NF-κB activity (20). Consistent with our RNA-seq data, we found that reovirus inhibits IκBα expression to a significant extent following treatment with either agonist (vgRNA or TNF-α) (Fig. 3A and B). Because the effects of reovirus on both NF-κB agonists were equivalent, we used TNF-α for the remainder of our experiments. TNF-α treatment of cells should promote nuclear translocation of p65. We measured nuclear p65 levels in mock-infected and reovirus-infected cells treated with TNF-α. As expected, TNF-α treatment of mock-infected cells resulted in an accumulation of p65 in the nucleus within 1 h (Fig. 3C). Prior infection with T3A prevented TNF-α-driven accumulation of p65 in the nucleus. These data agree with previous evidence indicating that degradation of the IκBα protein is blocked in reovirus-infected cells (21). IκBα degradation is initiated by the phosphorylation of IκBα by the IKK complex, which leads to polyubiquitination and subsequent degradation of IκBα by the proteasome (9). Thus, the reduction in nuclear p65 levels in T3A-infected cells treated with TNF-α may be due to an absence of sufficient levels of active IKKs. In addition to IκBα, the IKK complex also phosphorylates p65 at Ser536 prior to nuclear translocation (10). IKK-mediated p65 Ser536 phosphorylation is critical for NF-κB-dependent gene expression and is considered a measure for IKK activity (10). To determine if IKK activity is compromised in T3A-infected cells, we assessed the capacity of TNF-α to promote p65 phosphorylation at Ser536. While TNF-α potently induced p65 phosphorylation in mock-infected cells, both basal and TNF-α-induced p65 phosphorylation were dramatically reduced in T3A-infected cells (Fig. 3D). Thus, in reovirus-infected cells, p65 nuclear translocation and phosphorylation, both of which require the IKK complex, are inhibited. These data suggest that reovirus may inhibit NF-κB-dependent gene expression due to inactivity of the IKK complex.
FIG 3.

Reovirus inhibits NF-κB signaling upstream of gene expression. (A) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 20 h, cells were transfected with vgRNA and incubated for 7 h. RNA was extracted from cells, and levels of IκBα mRNA relative to those in a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control were measured using RT-qPCR. IκBα expression in mock-infected cells treated with agonist vgRNA was set to 100%. Gene expression of each replicate, the mean value, and standard deviation (SD) are shown. ***, P < 0.001 by Student’s t test in comparison to mock-infected cells transfected with vgRNA. (B) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 20 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. RNA was extracted from cells and levels of IκBα mRNA relative to those in a GAPDH control were measured using RT-qPCR. IκBα expression in mock-infected cells treated with the agonist TNF-α was set to 100%. Gene expression of each replicate, the mean value, and SD are shown. **, P < 0.01 by Student’s t test in comparison to mock-infected cells transfected with vgRNA. (C) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 24 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. Nuclear extracts were immunoblotted using antiserum specific for p65 or for PSTAIR loading control. (D) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 24 h, cells were treated with 20 μM proteasome inhibitor PSI for 1 h (to prevent turnover of proteins regulated by TNF-α signaling), then with 10 ng/ml TNF-α for 30 min. Whole-cell extracts were immunoblotted with antisera specific for p65, p65 Ser536 phosphorylation, and PSTAIR.
Levels of IKKβ and NEMO are diminished following reovirus infection.
To determine the basis of IKK inactivity following infection with T3A, we sought to determine the levels of IKKβ and NEMO, key IKK components that are required for NF-κB activation following TNF-α treatment. We found that levels of IKKβ and NEMO are dramatically lower at 12 and 24 h following infection with T3A (Fig. 4A and B). In contrast, levels of an upstream signaling protein, RIP1, were unaffected by T3A infection. Similarly, levels of NF-κB constituents p50 and p65 also remained constant. These data indicate that T3A-mediated diminishment in levels of IKKβ and NEMO likely contributes to a reduction in IKK activity and resultant blockade of NF-κB in infected cells. Because IKKβ is the catalytic component of the IKK complex that is required for IκBα and p65 Ser536 phosphorylation, we used IKKβ levels as a surrogate to monitor the mechanism by which IKK activity is diminished following infection.
FIG 4.

Reovirus infection causes a decrease in IKKβ and NEMO levels. (A, B) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 12 or 24 h, whole-cell extracts were immunoblotted with antisera specific to p50, p65, IKKβ, NEMO, RIP1, and PSTAIR. (C) ATCC L929 cells were absorbed with PBS (mock), 10 PFU/cell of T3A, or 10 PFU/cell of T1L. Following incubation at 37°C for 24 h, whole-cell extracts were immunoblotted with antisera specific to IKKβ and PSTAIR. Reo, reovirus.
In comparison to T3A, reovirus strain T1L blocks NF-κB less efficiently (22). To determine if the efficiency of blockade of NF-κB correlated with loss of IKKβ, we compared IKKβ levels following infection by T3A and T1L. We found that IKKβ levels following infection with T3A were significantly lower than that following infection with T1L. These data suggest a link between IKKβ levels and NF-κB function (Fig. 4C).
Reovirus gene expression is required for the loss of IKKβ.
To determine the stage of the reovirus replication cycle that is required for the loss of the IKK complex and the inhibition of NF-κB, we treated cells with ribavirin, which diminishes viral gene expression (23). We found that ribavirin treatment prevented T3A-mediated loss of IKKβ (Fig. 5A). Consistent with this, in cells treated with ribavirin, T3A was no longer able to prevent TNF-α-driven nuclear accumulation of p65 (Fig. 5B). Furthermore, ribavirin treatment also reduced the capacity of T3A to block NF-κB-dependent gene expression (Fig. 5C). Together, these experiments indicate that one or more viral proteins produced following virus infection or a specific event in viral replication trigger the loss of IKKβ.
FIG 5.
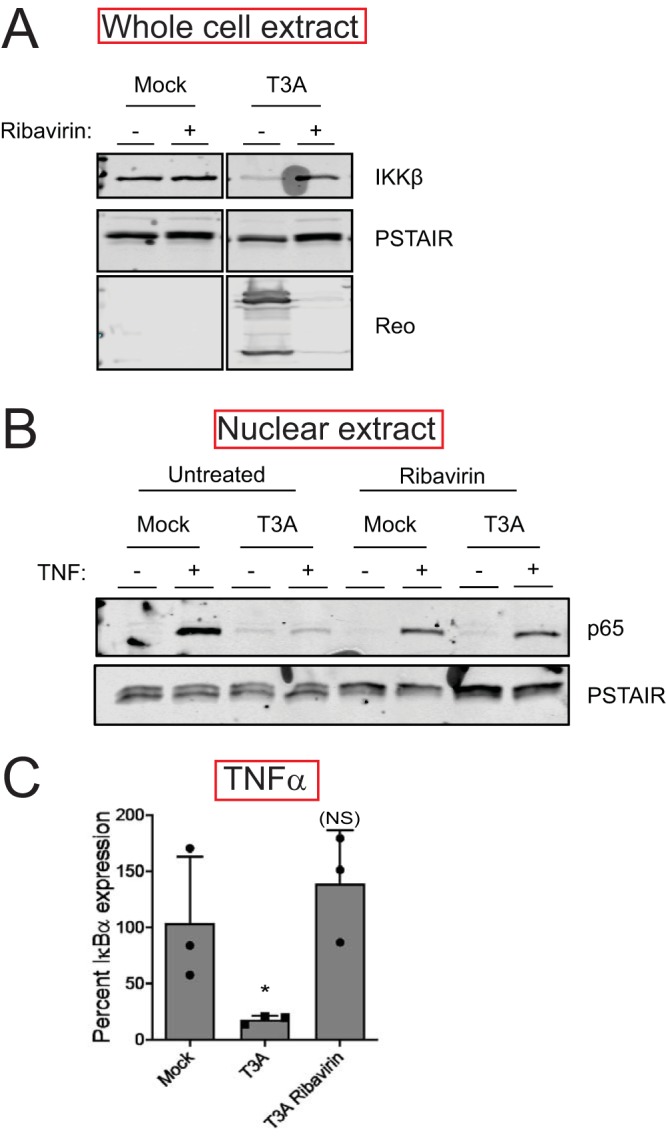
Reovirus gene expression is required for loss of IKKβ. (A) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A in the presence or absence of 200 μM ribavirin. Following incubation at 37°C for 24 h, whole-cell extracts were immunoblotted with antisera specific for IKKβ, PSTAIR, and reovirus. (B) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A in the presence or absence of 200 μM ribavirin. Following incubation at 37°C for 24 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. Nuclear extracts were immunoblotted using antiserum specific for p65 or PSTAIR. (C) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A in the presence or absence of 200 μM ribavirin. Following incubation at 37°C for 24 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. RNA was extracted from cells, and IκBα gene expression was measured using RT-qPCR. Gene expression in mock-infected cells treated with TNF-α was set to 100%. Gene expression of each replicate, the mean value, and SD are shown. *, P < 0.05; NS, P > 0.05 by Student’s t test in comparison to mock-infected cells treated with TNF-α.
Reovirus-induced IKKβ loss occurs posttranslationally.
To ascertain the mechanism of the lowered cellular levels of IKKβ, we measured steady-state levels of IKKβ mRNA in reovirus-infected cells using RT-qPCR. We found that levels of IKKβ mRNA in mock-infected and reovirus-infected cells were similar, indicating that IKKβ protein levels are not decreased due to lower transcriptional activity or lower stability of the IKKβ transcript (Fig. 6A). This finding was corroborated in our RNA-seq data (not shown). In uninfected cells treated with the protein synthesis inhibitor cycloheximide, IKKβ levels did not decrease until 24 h posttreatment. Because IKKβ loss occurs with significantly faster kinetics in reovirus-infected cells, we think that the decrease in IKKβ levels is not a result of the effect of reovirus infection on host translation. These data suggest that IKKβ levels are controlled posttranslationally (Fig. 6B). A common mechanism of protein turnover in cells is through the use of acid-dependent proteases present in lysosomal and autophagic vesicles. To test the role of these proteases, we treated cells with a weak base, ammonium chloride (AC), which prevents acidification of these compartments (24). Because reovirus infection is affected by AC, we initiated infection with an in vitro-generated entry intermediate called an infectious subvirion particle (ISVP) that bypasses inhibition by AC (25). Preventing acid-dependent protease activity did not block the capacity of ISVPs to diminish IKKβ levels, indicating that lysosomal or autophagic degradation does not contribute to IKK loss (Fig. 6C). This result is in contrast to those of a previous study, which suggested that TRIM29 expression in reovirus-infected alveolar macrophages leads to turnover of NEMO via lysosomal degradation (26). Another mechanism of turnover in cells is via the proteasome. Unexpectedly, treatment of cells with effective concentrations of a proteasome inhibitor (PSI) diminished reovirus infection (Fig. 6D). Two other proteasome inhibitors, MG132 and epoxomicin, also diminished reovirus infection (not shown). The effect of proteasome inhibitors on infection precludes us from directly testing the role of the proteasome in IKKβ turnover. Based on these data, we conclude that reovirus infection leads to IKK loss through a posttranslational mechanism, likely degradation via a nonlysosomal pathway.
FIG 6.

(A) ATCC L929 cells were infected with PBS (mock) or 10 PFU/cell of T3A for 20 h. RNA was extracted from cells, and levels of IKKβ mRNA relative to those in a GAPDH control were measured using RT-qPCR. IKKβ expression in mock-infected cells was set to 1. Gene expression of each replicate, the mean value, and SD are shown. (B) ATCC L929 cells were left untreated or were treated with 10 μg/ml cycloheximide for indicated times. Samples were immunoblotted with antisera specific to IKKβ and PSTAIR. (C) ATCC L929 cells were adsorbed with PBS (mock) or 3 PFU/cell of T3A ISVPs in the presence or absence of 20 mM ammonium chloride. Following incubation at 37°C for 16 or 24 h, whole-cell extracts were immunoblotted with antisera specific for IKKβ, PSTAIR, and reovirus. (D) ATCC L929 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A in the presence or absence of 20 μM PSI proteasome inhibitor. Following incubation at 37°C for 12 or 16 h, whole-cell extracts were immunoblotted with antisera specific for PSTAIR and reovirus.
IKK overexpression restores NF-κB signaling in reovirus-infected cells.
To define whether blockade of NF-κB by T3A also occurred in other cell lines, we measured the capacity of T3A to influence TNF-α-induced expression of IκBα using RT-qPCR in HEK293 cells. Analogously to our observation in L929 cells, TNF-α failed to induce IκBα gene expression in T3A-infected HEK293 cells (Fig. 7A). Phosphorylation of p65 at Ser536 (Fig. 7B) and its nuclear translocation following TNF-α treatment were also inhibited (Fig. 7C). Additionally, we noted a decrease in IKKβ levels in comparison to those in mock-infected cells (Fig. 7B). To determine if ectopic overexpression of the IKK complex restores NF-κB signaling in reovirus-infected cells, we transfected cells with constructs expressing tagged forms of IKKβ and NEMO. Overexpression of these constructs was sufficient to induce NF-κB signaling in mock-infected cells, as indicated by p65 Ser536 phosphorylation (Fig. 7D). We found that upon infection of cells with T3A, no decrease in IKKβ levels was observed. Correspondingly, IKK overexpression-induced p65 phosphorylation remained unaffected by reovirus infection. These data further indicate a link between IKKβ levels and NF-κB activity in reovirus-infected cells. Thus, we conclude that diminishment in IKKβ and NEMO levels is a key mechanism of reovirus-induced blockade of NF-κB signaling.
FIG 7.

IKK overexpression overcomes reovirus-mediated blockade of NF-κB signaling. (A) HEK293 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 24 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. RNA was extracted from cells, and IκBα gene expression was measured using RT-qPCR. Gene expression in mock-infected cells treated with TNF-α was set to 100%. Gene expression of each replicate, the mean value, and SD are shown. *, P < 0.05 by Student’s t test in comparison to mock-infected cells treated with TNF-α. (B) HEK293 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 24 h, cells were treated with proteasome inhibitor PSI for 1 h, then 10 ng/ml TNF-α for 30 min. Whole-cell extracts were immunoblotted using antisera specific for IKKβ, p65 Ser536 phosphorylation, p65, and PSTAIR. (C) HEK293 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following incubation at 37°C for 24 h, cells were treated with 10 ng/ml TNF-α and incubated for 1 h. Nuclear extracts were immunoblotted using antisera specific for p65 or PSTAIR. (D) HEK293 cells were transfected with vectors expressing Flag-tagged IKKβ and NEMO. Following incubation at 37°C for 24 h, HEK293 cells were adsorbed with PBS (mock) or 10 PFU/cell of T3A. Following an additional incubation at 37°C for 24 h, whole-cell extracts were immunoblotted using antisera specific for FLAG, p65, p65 Ser536 phosphorylation, reovirus, and PSTAIR.
DISCUSSION
In this study, we sought to evaluate if reovirus alters the cellular response to infection. Using RNA-seq, which allowed us to examine changes in the global transcriptional landscape, we found that IRF target genes were induced to a similar extent in cells infected with reovirus and in cells transfected with vgRNA. NF-κB target genes, however, were expressed to a much lower extent in infected cells than in cells transfected with vgRNA. Moreover, exogenous NF-κB agonists failed to induce an NF-κB-dependent gene expression program in infected cells. These data indicate that NF-κB activity is blocked in infected cells. We found that this blockade of NF-κB-dependent gene expression is caused by a loss of IKKβ and NEMO, two critical components of the IKK complex. We propose that reovirus inhibits NF-κB to counter its antiviral effects and produce a cellular environment that is conducive for replication.
We show here that reovirus infection inhibits NF-κB signaling (Fig. 1 to 3). In contrast, previous studies demonstrate that reovirus infection leads to the activation of NF-κB (27). We think that this apparent discrepancy is related to differences in the timing of when NF-κB is activated and inhibited in reovirus-infected cells. Reovirus-induced activation of NF-κB occurs early following infection. While canonical NF-κB signaling requires IKKβ and NEMO, reovirus-induced NF-κB activation requires an unusual combination of IKKα and NEMO (28). Recent studies demonstrating a requirement for mitochondrial antiviral-signaling protein (MAVS) for NF-κB activation indicate that vgRNA initiates this response (17). While other work suggests roles for reovirus μ1 and μ2 proteins in activating NF-κB, it is unclear if this effect is through controlling the exposure of vgRNA to cytoplasmic sensors or via another mechanism (29–31). Regardless, innate immune activation early in infection does not require viral gene expression (17, 32). In contrast, our studies presented here indicate that viral gene expression is required for blockade of NF-κB (Fig. 5). Thus, detection of viral RNA activates NF-κB early in infection, and expression of one or more viral gene products following establishment of infection results in blockade NF-κB, limiting further signaling through this pathway. Biphasic regulation of NF-κB was also previously suggested (21, 22). Our work presented here provides an explanation for this phenomenon. Differences in the capacity of strains T3A and T1L to inhibit NF-κB are genetically linked to the genome segment encoding the reovirus attachment protein σ1 (22). We suspect that differences in IKKβ loss following infection with T3A and T1L also are controlled by differences in the properties of their σ1 proteins. Because σ1 properties impact the efficiency of infection and the kinetics of viral gene expression (33), we think that the genetic link between σ1 and NF-κB is indirect and the viral factor responsible for diminishment of IKK levels and blockade of NF-κB signaling remains unknown.
NF-κB is an effective proinflammatory signaling pathway that curbs infection. Pathogens therefore have evolved mechanisms to limit its activity (34, 35). Among these, targeting upstream activators of the IKK complex is not uncommon. Infection with human coronavirus causes a loss of both IKKβ and NEMO through an unknown mechanism (36). The murine cytomegalovirus (MCMV) protein M45 targets NEMO for autophagolysosomal degradation (37). Shigella, an intracellular pathogen, secretes an effector with E3 ligase activity which targets NEMO for proteasomal degradation (38). In addition to degradation, pathogens also sequester the IKK complex (influenza A virus [IAV] NS1 protein) or prevent its activation (human cytomegalovirus [HCMV] and enterovirus) (39–41). Here, we show that reovirus infection leads to the loss of both IKKβ and NEMO (Fig. 4). Reovirus does not encode a protease, ruling out a direct effect of a viral protease on IKKβ. In the work presented here, we excluded the possibility that IKKβ levels were diminished as a consequence of blockade of host transcription and translation. Additionally, we showed that IKKβ is not degraded by proteases residing in lysosomes or autophagic compartments (Fig. 6). Because inhibition of proteasome activity led to reduction in viral gene expression and because viral gene expression is necessary for IKKβ loss, whether proteasomal degradation contributes to IKKβ remains an unanswered question and is a subject of our future studies. We also do not know why both IKKβ and NEMO are targeted by reovirus infection. It is possible that they are removed together because they reside in a complex. Another possibility is that targeting both IKKβ and NEMO ensures more complete inhibition of NF-κB.
A possible reason for reovirus to block NF-κB could be that NF-κB signaling limits reovirus infection (12). Which NF-κB target(s) control virus infection has not been identified. An obvious NF-κB target that could inhibit reovirus infection is IFN. However, consistent with previous work suggesting that certain mouse cell types do not require NF-κB for IFN production (42, 43), we found that vgRNA transfection-induced IFN expression occurred in an NF-κB-independent manner in the absence of infection (data not shown). Our RNA-seq analyses in reovirus-infected, vgRNA-transfected cells also showed that inhibition of NF-κB by reovirus did not affect IFN production (Fig. 1). In contrast with IFN, we found that the expression of several other chemokines and cytokines was inhibited in reovirus-infected cells. We hypothesize that one or more of these factors negatively regulates reovirus replication.
Two previous studies have suggested that reovirus limits the innate immune response. Reovirus can inhibit IFN production by sequestering IRF3 into viral factories (44). Additionally, reovirus can inhibit IFN signaling by nuclear sequestration of IRF9, which functions with STAT1 and STAT2 to promote expression of IFN-stimulated genes (45). While our work did not directly test these ideas, comparison of the gene expression profiles of reovirus-infected and vgRNA-transfected cells and iRegulon analyses of these data indicate that reovirus does not inhibit expression of IRF and STAT target genes (Fig. 1). Because these previous studies were performed in different cell types and used different reovirus strains, we propose that reovirus has evolved multiple mechanisms to dampen the innate immune response. Our study presented here unveils one such mechanism.
MATERIALS AND METHODS
Cells and viruses.
Murine L929 cells (ATCC CCL-1) were maintained in Eagle’s minimal essential medium (MEM) (Lonza) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. Spinner-adapted L929 cells (obtained from T. Dermody’s laboratory) were maintained in Joklik’s MEM (Lonza) supplemented to contain 5% FBS, 2 mM l-glutamine, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 25 ng/ml of amphotericin B. HEK293 cells (obtained from M. Marketon’s laboratory) were maintained in Dulbecco’s modified essential medium (DMEM; Lonza) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. Spinner-adapted L929 cells were used for cultivating and purifying viruses and for plaque assays. ATCC L929 cells and HEK293 cells were used for all experiments to assess cell signaling. No differences were observed in permissivity between ATCC L929 cells and spinner-adapted L929 cells. A laboratory stock of T3A (obtained from T. Dermody’s laboratory) was used for infections. Infectious viral particles were purified by Vertrel XF extraction and CsCl gradient centrifugation (46). Viral titer was determined by a plaque assay using spinner-adapted L929 cell with chymotrypsin in the agar overlay. To generate ISVPs, purified T3A virions (2 × 1012 particles/ml) were digested with 200 μg/ml Nα-p-tosyl-l-lysine chloromethyl ketone (TLCK)-treated chymotrypsin (Worthington Biochemical) in a total volume of 100 μl for 20 min at 32°C. After 1 h, the reaction mixtures were incubated for 20 min on ice and quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). The generation of ISVPs was confirmed by SDS-PAGE and Coomassie brilliant blue staining.
Antibodies and reagents.
Polyclonal antisera raised against T3D and T1L that have been described previously (47) were used to detect viral proteins in T3A-infected cells. Rabbit antisera specific for IKKβ and p65 Ser536 phosphorylation-specific antibody were purchased from Cell Signaling (catalog no. 8943 and 3033), and rabbit antisera specific for p65 and NEMO were purchased from Santa Cruz Biotechnology (catalog no. sc-372 and sc-8330). Mouse antisera specific for PSTAIR and FLAG was purchased from Sigma-Aldrich (catalog no. P7962 and F-3165), mouse antiserum specific for RIP1 was purchased from BD Biosciences (catalog no. 610458), and Alexa Fluor-conjugated anti-mouse IgG and anti-rabbit IgG secondary antibodies were purchased from LI-COR. TNF-α was purchased from Sigma and used at a concentration of 10 ng/ml. PSI proteasome inhibitor was purchased from Millipore (catalog no. 53-916) and used at a concentration of 20 μM. Ribavirin was purchased from Sigma-Aldrich (catalog no. R9644) and used at a concentration of 200 μM. Ammonium chloride was used at a concentration of 20 mM. Cycloheximide was purchased from EMD Millipore (catalog no. 239764) and was used at a concentration of 10 μg/ml.
Infections.
Confluent monolayers of ATCC L929 or HEK293 cells were adsorbed with either phosphate-buffered saline (PBS) or reovirus at the indicated multiplicity of infection (MOI) at room temperature for 1 h, followed by incubation with medium at 37°C for the indicated time interval. All inhibitors were added to cells in medium after the 1-h adsorption period.
Analysis of host gene expression by RNA-seq.
Total RNA extracted using an Aurum total RNA minikit (Bio-Rad) was submitted to Indiana University’s Center for Genomics and Bioinformatics for cDNA library construction using a TruSeq stranded mRNA low-throughput (LT) sample prep kit (Illumina) following the manufacturer’s protocol. Sequencing was performed using an Illumina NextSeq 500 platform with a 75-bp sequencing module generating 38-bp paired-end reads. After the sequencing run, demultiplexing with performed with bcl2fastq v2.20.0.422. Sequenced reads were adapter trimmed and quality filtered using Trimmomatic v0.33 (48) with the cutoff threshold for average base quality score set at 20 over a sliding window of 3 bases. Reads shorter than 20 bases posttrimming were excluded (LEADING:20 TRAILING:20 SLIDINGWINDOW:3:20 MINLEN:20). Cleaned reads were mapped to the GRCm38.p6 mouse genome reference using STAR v2.5.2b (49). Read pairs aligning to each gene from GENCODE vM17 annotation were counted with strand specificity using the featureCounts tool from the subread package (50). Differential expression analysis was performed using DESeq2 version 1.12.3 (51).
iRegulon, a plugin to Cytoscape v3.7.1, was used to predict transcription factor activity based on a differentially expressed gene set (18). A maximum FDR value of motif similarity was set to 0.001. We used an NES value of 3.0 as the minimum cutoff for transcription factor enrichment.
RT-qPCR.
RNA was extracted from infected cells at various times after infection using an Aurum total RNA minikit (Bio-Rad). For RT-qPCR, 0.5 to 2 μg of RNA was reverse transcribed with the high-capacity cDNA reverse transcription (RT) kit (Applied Biosystems) using random hexamers. cDNA was subjected to PCR using SYBR Select mastermix and gene-specific primers (Applied Biosystems). Fold increases in gene expression with respect to that of control samples (indicated in each figure legend) were measured using the threshold cycle (ΔΔCT) method (52).
Preparation of cellular extracts.
For preparation of whole-cell lysates, cells were washed in phosphate-buffered saline (PBS) and lysed with 1× RIPA (50 mM Tris [pH 7.5], 50 mM NaCl, 1% TX-100, 1% deoxycholate, 0.1% SDS, and 1 mM EDTA) containing a protease inhibitor cocktail (Roche), 500 μM dithiothreitol (DTT), and 500 μM phenylmethylsulfonyl fluoride (PMSF), followed by centrifugation at 15,000 × g at 4°C for 15 min to remove debris. Nuclear extracts were prepared by lysing cells in a hypotonic lysis buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl, 0.5 mM DTT, and 0.5 mM PMSF for 15 min, subsequent addition of 0.5% NP-40, and 10 s of vortexing. After centrifugation at 10,000 × g at 4°C for 10 min, the nuclear pellet was washed with hypotonic lysis buffer and then resuspended in high-salt nuclear extraction buffer (25% glycerol, 20 mM HEPES, 0.42 M NaCl, 10 mM KCl, 1.5 mM MgCl, 0.5 mM DTT, and 0.5 mM PMSF) at 4°C for 1 h. Nuclear extracts were obtained following removal of the insoluble fraction by centrifugation at 12,000 × g at 4°C for 10 min.
Plasmid transfections.
Nearly confluent monolayers of HEK293 cells in 12-well plates were transfected with either 0.5 μg of empty vector or 0.25 μg each of FLAG-IKKβ or FLAG-NEMO expression vector using 1.5 μl Lipofectamine 2000 according to the manufacturer’s instructions. Transfected cells were incubated at 37°C for 24 h prior to infection to allow expression from the plasmids.
Immunoblot assay.
Protein concentrations were estimated using a DC protein assay from Bio-Rad. Equal amounts of protein were loaded, and the cell lysates or extracts were resolved by electrophoresis in 10% polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked for at least 1 h in blocking buffer (StartingBlock T20 [Tris-buffered saline (TBS)] blocking buffer) and incubated with antisera against p65 (1:1,000), p65 p-Ser536 (1:1,000), p50 (1:500), NEMO (1:500), IKKβ (1:1,000), RIP1 (1:1,000), reovirus (1:5,000), FLAG (1:1,000), and PSTAIR (1:5,000) at 4°C overnight. Membranes were washed three times for 5 min each with washing buffer (TBS containing 0.1% Tween 20) and incubated with a 1:20,000 dilution of Alexa Fluor-conjugated goat anti-rabbit Ig (for p65, p50, IKKβ, NEMO, and reovirus) or goat anti-mouse Ig (for PSTAIR, RIP1, and FLAG) in blocking buffer. Following three washes, membranes were scanned and quantified using an Odyssey infrared imager (LI-COR).
ACKNOWLEDGMENTS
We thank members of our laboratory and the Indiana University Virology community for helpful suggestions. We are also grateful to scientists in the Indiana University Center for Genomics and Bioinformatics.
Research reported in this publication was supported by funds from the National Institute of Allergy and Infectious Diseases under award number R01AI110637 (to P.D.) and by funds from the Indiana Clinical and Translational Sciences Institute under Clinical and Translational Sciences Award number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Mogensen TH, Paludan SR. 2001. Molecular pathways in virus-induced cytokine production. Microbiol Mol Biol Rev 65:131–150. doi: 10.1128/MMBR.65.1.131-150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 3.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 5.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 6.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 7.Fensterl V, Chattopadhyay S, Sen GC. 2015. No love lost between viruses and interferons. Annu Rev Virol 2:549–572. doi: 10.1146/annurev-virology-100114-055249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Sastre A. 2017. Ten strategies of interferon evasion by viruses. Cell Host Microbe 22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Lenardo MJ, Baltimore D. 2017. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. 2005. NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dermody TS, Parker JC, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM (ed), Fields Virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 12.O’Donnell SM, Hansberger MW, Connolly JL, Chappell JD, Watson MJ, Pierce JM, Wetzel JD, Han W, Barton ES, Forrest JC, Valyi-Nagy T, Yull FE, Blackwell TS, Rottman JN, Sherry B, Dermody TS. 2005. Organ-specific roles for transcription factor NF-κB in reovirus-induced apoptosis and disease. J Clin Invest 115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger AK, Hiller BE, Thete D, Snyder AJ, Perez E Jr, Upton JW, Danthi P. 2017. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J Virol 91:e02404-16. doi: 10.1128/JVI.02404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Donnell SM, Holm GH, Pierce JM, Tian B, Watson MJ, Chari RS, Ballard DW, Brasier AR, Dermody TS. 2006. Identification of an NF-κB-dependent gene network in cells infected by mammalian reovirus. J Virol 80:1077–1086. doi: 10.1128/JVI.80.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis E. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart JD, Holm GH, Boehme KW. 2018. Differential delivery of genomic double-stranded RNA causes reovirus strain-specific differences in interferon regulatory factor 3 activation. J Virol 92:e01947-17. doi: 10.1128/JVI.01947-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, Svetlichnyy D, Kalender Atak Z, Fiers M, Marine JC, Aerts S. 2014. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M Jr. 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baeuerle P, Baltimore D. 1988. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 21.Clarke P, Meintzer SM, Moffitt LA, Tyler KL. 2003. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J Biol Chem 278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- 22.Clarke P, Debiasi RL, Meintzer SM, Robinson BA, Tyler KL. 2005. Inhibition of NF-κ B activity and cFLIP expression contribute to viral-induced apoptosis. Apoptosis 10:513–524. doi: 10.1007/s10495-005-1881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rankin UT Jr, Eppes SB, Antczak JB, Joklik WK. 1989. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology 168:147–158. doi: 10.1016/0042-6822(89)90413-3. [DOI] [PubMed] [Google Scholar]
- 24.Galloway CJ, Dean GE, Marsh M, Rudnick G, Mellman I. 1983. Acidification of macrophage and fibroblast endocytic vesicles in vitro. Proc Natl Acad Sci U S A 80:3334–3338. doi: 10.1073/pnas.80.11.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol 61:2351–2361. doi: 10.1128/JVI.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, Lu H, Li XC, Liu YJ, Zhang Z. 2016. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol 17:1373–1380. doi: 10.1038/ni.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J Virol 74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. 2007. IκB kinase subunits alpha and gamma are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J Virol 81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stebbing RE, Irvin SC, Rivera-Serrano EE, Boehme KW, Ikizler M, Yoder JA, Dermody TS, Sherry B. 2014. An ITAM in a nonenveloped virus regulates activation of NF-κB, induction of beta interferon, and viral spread. J Virol 88:2572–2583. doi: 10.1128/JVI.02573-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 φ domain. PLoS Pathog 4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holm GH, Zurney J, Tumilasci V, Leveille S, Danthi P, Hiscott J, Sherry B, Dermody TS. 2007. Retinoic acid-inducible gene-I and interferon-β promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem 282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- 33.Hiller BE, Berger AK, Danthi P. 2015. Viral gene expression potentiates reovirus-induced necrosis. Virology 484:386–394. doi: 10.1016/j.virol.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santoro MG, Rossi A, Amici C. 2003. NF-κB and virus infection: who controls whom. EMBO J 22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, He S, Minassian A, Li J, Feng P. 2015. Recent advances on viral manipulation of NF-κB signaling pathway. Curr Opin Virol 15:103–111. doi: 10.1016/j.coviro.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poppe M, Wittig S, Jurida L, Bartkuhn M, Wilhelm J, Muller H, Beuerlein K, Karl N, Bhuju S, Ziebuhr J, Schmitz ML, Kracht M. 2017. The NF-κB-dependent and -independent transcriptome and chromatin landscapes of human coronavirus 229E-infected cells. PLoS Pathog 13:e1006286. doi: 10.1371/journal.ppat.1006286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, Hohenberg H, Ghazal P, Brune W. 2012. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog 8:e1002517. doi: 10.1371/journal.ppat.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C. 2010. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat Cell Biol 12:66–73. doi: 10.1038/ncb2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q, Zheng Z, Liu Y, Zhang Z, Liu Q, Meng J, Ke X, Hu Q, Wang H. 2016. 2C proteins of enteroviruses suppress IKKβ phosphorylation by recruiting protein phosphatase 1. J Virol 90:5141–5151. doi: 10.1128/JVI.03021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao S, Song L, Li J, Zhang Z, Peng H, Jiang W, Wang Q, Kang T, Chen S, Huang W. 2012. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol 14:1849–1866. doi: 10.1111/cmi.12005. [DOI] [PubMed] [Google Scholar]
- 41.Goodwin CM, Schafer X, Munger J. 2019. UL26 attenuates IKKβ-mediated induction of ISG expression and enhanced protein ISGylation during HCMV infection. J Virol 93. doi: 10.1128/JVI.01052-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basagoudanavar SH, Thapa RJ, Nogusa S, Wang J, Beg AA, Balachandran S. 2011. Distinct roles for the NF-κB RelA subunit during antiviral innate immune responses. J Virol 85:2599–2610. doi: 10.1128/JVI.02213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, Garcia-Sastre A, Balachandran S, Beg AA. 2010. NF-κB RelA subunit is crucial for early IFN-β expression and resistance to RNA virus replication. J Immunol 185:1720–1729. doi: 10.4049/jimmunol.1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanifer ML, Kischnick C, Rippert A, Albrecht D, Boulant S. 2017. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci Rep 7:10873. doi: 10.1038/s41598-017-11469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. 2009. Reovirus μ2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol 83:2178–2187. doi: 10.1128/JVI.01787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit15C.1. [DOI] [PubMed] [Google Scholar]
- 47.Wetzel JD, Chappell JD, Fogo AB, Dermody TS. 1997. Efficiency of viral entry determines the capacity of murine erythroleukemia cells to support persistent infections by mammalian reoviruses. J Virol 71:299–306. doi: 10.1128/JVI.71.1.299-306.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]