

Development of vaccines against hepatitis C virus (HCV), a major cause of cirrhosis and cancer, has been stymied by a lack of animal models. The recent discovery of an HCV-like rodent hepacivirus (RHV) enabled the development of such a model in rats. This platform recapitulates HCV hepatotropism and viral chronicity necessary for vaccine testing. Currently, there are few descriptions of RHV-specific responses and why they fail to prevent persistent infection in this model. Here, we show that RHV-specific CD8 T cells, while induced early at high magnitude, do not develop into functional effectors capable of controlling virus. This defect was partially alleviated by short-term treatment with an HCV antiviral. Thus, like HCV, RHV triggers dysfunction of virus-specific CD8 T cells that are vital for infection resolution. Additional study of this evasion strategy and how to mitigate it could enhance our understanding of hepatotropic viral infections and lead to improved vaccines and therapeutics.
KEYWORDS: animal models, hepatitis C virus, hepacivirus, T cells, antivirals, antiviral agents, vaccines
ABSTRACT
Immune-competent animal models for the hepatitis C virus (HCV) are nonexistent, impeding studies of host-virus interactions and vaccine development. Experimental infection of laboratory rats with a rodent hepacivirus isolated from Rattus norvegicus (RHV) is a promising surrogate model due to its recapitulation of HCV-like chronicity. However, several aspects of rat RHV infection remain unclear, for instance, how RHV evades host adaptive immunity to establish persistent infection. Here, we analyzed the induction, differentiation, and functionality of RHV-specific CD8 T cell responses that are essential for protection against viral persistence. Virus-specific CD8 T cells targeting dominant and subdominant major histocompatibility complex class I epitopes proliferated considerably in liver after RHV infection. These populations endured long term yet never acquired antiviral effector functions or selected for viral escape mutations. This was accompanied by the persistent upregulation of programmed cell death-1 and absent memory cell formation, consistent with a dysfunctional phenotype. Remarkably, transient suppression of RHV viremia with a direct-acting antiviral led to the priming of CD8 T cells with partial effector function, driving the selection of a viral escape variant. These data demonstrate an intrinsic abnormality within CD8 T cells primed by rat RHV infection, an effect that is governed at least partially by the magnitude of early virus replication. Thus, this model could be useful in investigating mechanisms of CD8 T cell subversion, leading to the persistence of hepatotropic pathogens such as HCV.
IMPORTANCE Development of vaccines against hepatitis C virus (HCV), a major cause of cirrhosis and cancer, has been stymied by a lack of animal models. The recent discovery of an HCV-like rodent hepacivirus (RHV) enabled the development of such a model in rats. This platform recapitulates HCV hepatotropism and viral chronicity necessary for vaccine testing. Currently, there are few descriptions of RHV-specific responses and why they fail to prevent persistent infection in this model. Here, we show that RHV-specific CD8 T cells, while induced early at high magnitude, do not develop into functional effectors capable of controlling virus. This defect was partially alleviated by short-term treatment with an HCV antiviral. Thus, like HCV, RHV triggers dysfunction of virus-specific CD8 T cells that are vital for infection resolution. Additional study of this evasion strategy and how to mitigate it could enhance our understanding of hepatotropic viral infections and lead to improved vaccines and therapeutics.
INTRODUCTION
The hepatitis C virus (HCV) chronically infects ∼71 million people worldwide and is a leading cause of severe progressive liver diseases, such as cirrhosis and hepatocellular carcinoma (1, 2). The recent development of highly effective direct-acting antivirals (DAA) has revolutionized chronic HCV therapy, eliminating infection in >95% of treated individuals (3, 4). However, global control of HCV is unlikely to be achieved by treatment alone (3, 5). Indeed, the majority of HCV infections are undiagnosed and occur in resource-limited countries where necessary surveillance programs do not exist. Furthermore, DAA therapy does not confer long-lived immunity, and many cured patients remain at risk for reinfection (6, 7). Additional roadblocks for treatment include the high cost of DAAs and the potential for drug resistance (3). Therefore, the development of a prophylactic vaccine is of critical importance for HCV eradication. Little progress has been made in this area, however, due to the absence of immunocompetent small-animal models. Chimpanzees, the only species besides humans fully permissive to HCV infection, are no longer available for research, and immunocompetent mouse models that support HCV replication and persistence have yet to be developed (8).
Recently, a number of HCV surrogate models have been developed using newly identified animal homologs (9–12). The most promising of these is a rodent hepacivirus (RHV) isolated from feral Norway rats (Rattus norvegicus). RHV is a genetic relative of HCV and recapitulates several important virological features. These include similar genomic structure and polyprotein processing, the presence of two liver-tropic microRNA-122 binding sites within the viral 5′ untranslated region, and sensitivity to DAA treatment (10, 13). In laboratory rats, RHV causes chronic hepatotropic viral infections mimicking HCV persistence in humans. The utility of the RHV rat model for vaccine development was recently demonstrated by two studies showing the successful prevention of virus persistence after prophylactic immunization with adenoviral vectors expressing RHV nonstructural proteins (14, 15). T cells were critical to vaccine success in this setting, as the depletion of CD4 or CD8 T cells resulted in prolonged or persistent infection in vaccinated animals (14), reminiscent of findings obtained from the chimpanzee model (16, 17).
Despite the promise of the RHV rat model, the underlying mechanisms of RHV persistence have yet to be determined. In particular, there is currently little understanding of RHV-specific immune responses and why they fail to terminate persistent viremia. For the spontaneous resolution of acute HCV infections, which occurs in a minority of infected individuals, the maintenance of an effective virus-specific CD8 T cell response is deemed necessary (18–20). During HCV persistence, however, these responses are subverted by viral escape or become functionally exhausted due to persistent antigenic stimulation (18–20). The extent to which these or similar mechanisms exist for RHV are not fully clear. Initial studies assessing CD8 T cell immunity in RHV-infected rats revealed limited or absent cytokine responses following peptide stimulation (14, 15). Direct visualization of CD8 T cells targeting a subdominant epitope in the RHV NS3 protein by class I tetramer staining confirmed the early expansion of responses in infected liver, but these were only marginally detectable and quickly disappeared, possibly by physical deletion (14). Whether this pattern of immunity is representative of other CD8 T cell responses induced by RHV infection is unclear.
In the present study, we characterized the RHV-specific CD8 T cell response in rats using two MHC class I tetramers targeting novel epitopes identified within the E1 and NS5B viral proteins. We demonstrate that these responses, despite vigorously expanding and enduring within infected liver, failed to develop antiviral effector function. Their inability to apply selective pressure on persistent virus was reflected by an absence of mutational escape at major histocompatibility complex (MHC) class I epitopes. Remarkably, the suppression of early RHV replication via treatment with a highly potent DAA facilitated the development of CD8 T cells with partial effector function, driving the selection of an escape variant. These results suggest that high levels of early viral replication contribute to the priming of RHV-specific CD8 T cells that lack the capacity to control and eliminate persistent infection.
RESULTS
RHV infection drives expansion of antiviral CD8 T cells.
To date, three RHV class I epitopes have been identified in the Lewis rat (RT1-Al background). The NS31487 epitope, which was recently incorporated into a functional RT1-Al tetramer, is subdominant and elicits a transient CD8 T cell response upon RHV infection (14). Attempts to construct functional tetramers for the more dominant NS3971 and NS4A1578 epitopes that escaped during persistent infection of a vaccinated rat (14) were not successful (data not shown). Therefore, we sought to identify additional class I epitopes that could be targeted for RT1-Al tetramer construction. Because CD8 T cells appear to be functionally blunted during the infection of naive rats (14, 15), we vaccinated animals with a recombinant adenovirus expressing the NS3-5B viral proteins and then challenged them with RHV. This immunization approach yields protective immunity in most rats, and resolution of acute viremia after challenge is associated with a strong recall of CD8 T cell responses (14, 15). By screening these responses against peptides with predicted binding affinity for RT1-Al (21), we identified four novel class I epitopes within the E1, E2, and NS5B proteins (Fig. 1A and B and Table 1). The detection of responses against the envelope proteins was unexpected, since these antigens were not encoded by vaccines and, thus, must have been expanded by replicating virus. Minimal peptide sequences were determined for two of the identified epitopes, E1191 and NS5B2511, and used to construct class I tetramers. These reagents readily stained epitope-specific populations in liver (Fig. 2A, right) and, therefore, were used to longitudinally track antiviral T cell responses after infection. As previously observed (14), RHV established a high-titer (108 to 1010 genomes/ml) persistent infection lasting >90 days that occurred without concurrent elevations in plasma alanine transaminase activity (Fig. 2A, left). RHV viremia was remarkably stable during infection, indicating a lack of effective antiviral immunity. Within 2 weeks, high frequencies of CD8 T cells targeting both epitopes expanded within liver (Fig. 2A). The E1191-specific response was dominant and reached a peak frequency of 8.7% by day 14 postinfection (p.i.) before contracting sharply (>90%) during days 14 to 90 p.i. (Fig. 2A, left). The NS5B2511-specific response was less robust but noticeably more durable. Intrahepatic NS5B2511-specific cells expanded to 1.1% by day 14 p.i. and then declined partially before reaching a peak frequency of 1.8% by day 90 to 97 p.i. (Fig. 1A, left). Similar tetramer response kinetics were observed in blood and spleen, albeit at substantially reduced frequencies (Fig. 2B), consistent with a hepatotropic immune response. Importantly, this pattern of CD8 T cell immunity was not shaped by loss of antigenic stimulation via viral escape, since all class I epitope sequences were intact throughout chronic infection (Fig. 2C).
FIG 1.

Identification of novel RHV class I epitopes. Seven- to 10-week-old Lewis rats were vaccinated with 5 × 108 IFU adenovirus expressing the RHV NS3-5B proteins. Two or 3 weeks later, animals were challenged with 106 genomes of RHV intravenously. On days 17 to 56 postinfection, CD8 T cell recall responses were analyzed in liver. (A) Course of viremia in three representative vaccinees that cleared RHV infection after challenge. (B, left) Percentage of CD8 T cells producing IFN-γ following 5-h stimulation with the indicated peptides (10 μg/ml). Representative results from a single animal are shown. (Right) Amino acid sequences of reactive peptides. Boldface indicates predicted RT1-Al binding nonamers.
TABLE 1.
RHV class I and II epitopesa
| Polyprotein locationb | Amino acid sequence | MHC class I/II |
|---|---|---|
| E1191–199 | SAFGTVARF | I |
| E2439–456 | SAGWTNLACYGQKGPFLP | I |
| NS3974–982 | SICVIGTPL | I |
| NS31299–1316 | IQKGRHLIFQTSKSHCDN | II |
| NS31425–1442 | IVPDACIYEAFDSGLAYF | II |
| NS31446–1463 | PAEVATHLSFYHNQVGLP | II |
| NS31481–1498 | YVQSNYLEMMKNRVDSYT | II |
| NS31487–1495 | YTYLYAAQY | I |
| NS31502–1519 | AAQYQLAKAEGAMAPNDN | II |
| NS4A1578–1586 | CVFMAIDLF | I |
| NS4B1747–1764 | MVGHAFLTYGSATSACLV | II |
| NS5A2248–2265 | MELLREYETSNDHVPKED | II |
| NS5B2486–2503 | SGKTEIVKTLYSKLEEGI | II |
| NS5B2511–2519 | CVMPKIETF | I |
| NS5B2552–2569 | VEKMVLGQIGPKTVKAVC | II |
| NS5B2559–2576 | QIGPKTVKAVCGDAYGFV | I |
Epitopes are RT1-Al (class I) or RT1-B/Dl (class II), restricted according to the MHC background of Lewis rats. Boldface indicates novel epitopes identified in this study.
FIG 2.

Early expansion of intrahepatic CD8 T cells targeting RHV after infection. Seven- to 10-week-old Lewis rats were infected with 106 genomes of RHV intravenously. Infection course and CD8 T cell immunity were tracked until 90 to 97 days p.i. (A, left) Plasma viremia, ALT levels, and percentage of liver-infiltrating CD8 T cells targeting the E1191 or NS5B2511 epitope, as determined by class I tetramer staining. (Right) Representative flow plots at day 14 p.i. showing frequency of intrahepatic CD8 T cells that bind the E1191 and NS5B2511 tetramers. Combined data from two independent experiments with n = 2 to 3 rats per time point are shown (means ± standard errors of the means [SEM]). (B) Frequency of CD8 T cells targeting the E1191 and NS5B2511 epitopes in blood and spleen, as determined by class I tetramer staining. (C) Sequence evolution of RHV class I epitopes. Consensus sequences were determined by direct PCR sequencing. The frequencies of rats infected with virus containing the indicated sequences are shown.
RHV-specific CD8 T cells lack cytokine effector functions.
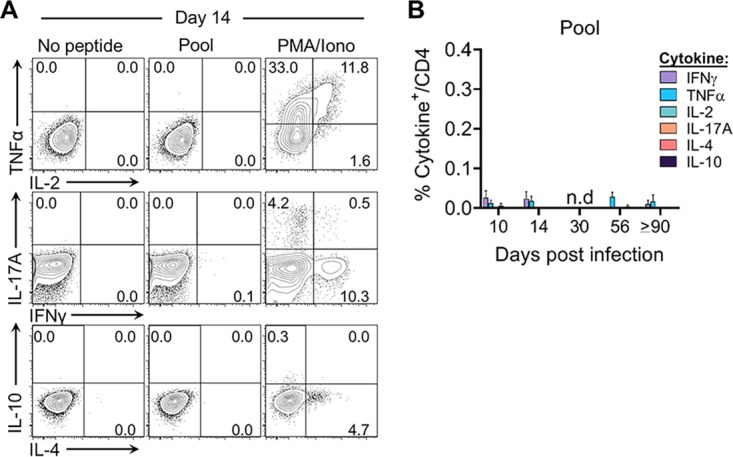
We next characterized the functional capacity of these RHV-specific CD8 T cell responses by intracellular cytokine-staining assays. Intrahepatic leukocyte preparations were stimulated with single or pooled class I epitopes (Table 1) and quantified for the production of classical (gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α], and interleukin-2 [IL-2]) and nonclassical (IL-17A, IL-4, and IL-10) antiviral cytokines (Fig. 3A). At all time points assessed, CD8 T cells failed to produce cytokine upon antigenic stimulation with the E1191 or NS5B2511 epitope (Fig. 3B). A similar functional deficit was observed following stimulation with pooled class I epitopes (Fig. 3B). Importantly, this apparent defect was not uniquely attributable to ongoing immune suppression driven by the liver microenvironment (22), since IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay responses were similarly lacking in blood and spleen (Fig. 3C) despite the presence of virus-specific cells within these compartments (Fig. 2B). It is also worth noting that CD4 T helper cell activity against virus was reduced in or absent from liver after infection (Fig. 4A and B), in line with previous data (14).
FIG 3.

RHV-specific CD8 T cells fail to produce effector cytokines. Seven- to 10-week-old Lewis rats were infected with 106 genomes of RHV intravenously. Virus-specific CD8 T cells were assessed for effector function by cytokine-staining assay until 90 to 97 days p.i. (A) Representative flow plots at day 14 p.i. showing percentage of intrahepatic CD8 T cells that stain positive for classical (IFN-γ, TNF-α, and IL-2) and nonclassical (IL-17A, IL-10, and IL-4) antiviral cytokines. Cells were stimulated for 5 h with the E1191 or NS5B2511 epitope (10 μg/ml), a pool of class I and II epitopes (5 μg/ml each; Table 1), or no peptide or PMA plus ionomycin (PMA/Iono) as negative and positive controls, respectively. (B) Percentage of CD8 T cells producing cytokine at the indicated days p.i. Combined data from two independent experiments of n = 2 to 3 rats per time point are shown (means ± SEM). n.d., not detected. (C) Number of ELISpot assay IFN-γ spot-forming cells (SFCs) following 40 to 48 h of stimulation with the E1191 or NS5B2511 epitope (10 μg/ml). Combined data from two independent experiments of n = 2 to 3 rats per time point are shown (means ± SEM). n.d., not detected.
FIG 4.
RHV-specific CD4 T cell cytokine responses after infection. Seven- to 10-week-old Lewis rats were infected with 106 genomes of RHV intravenously. Virus-specific CD4 T cells were assessed for effector function by cytokine-staining assay until 90 to 97 days p.i. (A) Representative flow plots at day 14 p.i. showing percentage of intrahepatic CD4 T cells that stain positive for classical (IFN-γ, TNF-α, and IL-2) and nonclassical (IL-17A, IL-10, and IL-4) antiviral cytokines. Cells were stimulated for 5 h with a pool of class I and II epitopes (5 μg/ml each; Table 1) or with no peptide or PMA plus ionomycin (PMA/Iono) as negative and positive controls, respectively. (B) Percentage of CD4 T cells producing cytokine at the indicated days p.i. Combined data from two independent experiments of n = 2 to 3 rats per time point are shown (means ± SEM). n.d., not detected.
Differentiation of RHV-specific CD8 T cells.
To understand the differentiation of RHV-specific CD8 T cells and gain insights into the nature of their impairment, we performed a detailed analysis of their phenotype during infection. Using the class I tetramers described above, we monitored for changes in the expression of various T cell activation and differentiation markers on liver-infiltrating cells (Fig. 5A and B). Because rapid clonal expansion is a hallmark of the primary CD8 T cell response to viruses (23), we first assessed the expression of the Ki-67 proliferation marker. Compared to uninfected naive (CD62L+) CD8 T cells, which were largely negative, >90% of E1191- and NS5B2511-specific cells were Ki-67 positive at early time points (days 10 to 14 p.i.) (Fig. 3B). The expression of Ki-67 declined sharply to near baseline levels by day 30 p.i. and remained there through 90 to 97 days p.i. This early proliferation was coupled with the upregulation of granzyme B, suggesting the acquisition of effector properties. Most granzyme B expression was observed at earlier time points, but a small minority of virus-specific cells retained expression through 90 days p.i. Surprisingly, the increased expression of CD25 (IL-2Rα) was not observed on cells during clonal expansion. However, the upregulation of this receptor is often transient and, therefore, could have been missed during this sampling period. Virus-experienced but not naive cells also upregulated the C-type lectin receptor CD161, which is thought to be a molecular signature of activated CD8 T cells possessing gut- or liver-homing properties (24). By comparison, the expression of the chemokine receptor CXCR3 was dynamic. Expression was lost during days 10 to 14 p.i., followed by slow reexpression into later stages of infection, a pattern that might be consistent with an effector-to-memory transition (23, 25). Costimulatory receptor expression (CD27 and CD28) was uniformly high throughout infection, except for intermediate expression of CD28 on the NS5B2511-specific population, which indicates a more terminally differentiated state (26).
FIG 5.

Phenotypic differentiation of RHV-specific CD8 T cells. Seven- to 10-week-old Lewis rats were infected with 106 genomes of RHV intravenously, and virus-specific CD8 T cells were profiled for phenotypic changes by multiparametric flow cytometry. Responses were analyzed until 90 to 97 days p.i. (A) Representative flow plots showing percentage of intrahepatic E1191-specific CD8 T cells expressing the indicated markers at 14 days p.i. Total CD8 T cells are shown underlayed in black for comparison. (B) Percentage of E1191- and NS5B2511-specific CD8 T cells expressing the indicated markers. Baseline expression values at day 0 p.i. were assessed on naive (CD62L+) CD8 T cells from uninfected rats. Combined data from two independent experiments of n = 2 to 3 rats per group are shown (means ± SEM).
Most importantly, we also assessed the expression of markers associated with memory cell maturation and exhaustion. The reacquisition of CD127 (IL-7Rα) expression is an important identifier of CD8 T cells destined to populate the memory pool (27). We observed near-complete downregulation of CD127 upon RHV-specific CD8 T cell activation with no subsequent reexpression at later time points. In contrast, the upregulation of the coinhibitory receptor PD-1, a classic indicator of T cell exhaustion during chronic viral infection (28), occurred early after activation and remained persistently elevated on ∼40 to 60% of tetramer-positive cells. Nonuniform expression of PD-1 was notable, considering the level of dysfunction displayed by these populations. This contrasts with chronic HCV infection, where nearly all virus-specific CD8 T cells infiltrating the liver express high levels of PD-1 (29, 30).
In summary, the primary CD8 T cell response to RHV was characterized by a rapid phase of proliferation and expansion that yielded a large pool of tetramer-positive cells possessing molecular attributes of an effector phenotype. The contraction of responses, in contrast to acute viral infections, was not associated with transition to memory or loss of coinhibitory receptor expression, which is consistent with an exhausted or dysfunctional phenotype.
Rapid viral suppression induces RHV-specific CD8 T cells with partial effector function.
The liver is now recognized to be a front-line immunological organ with a bias toward immune tolerance induction (22). This tolerogenic property ensures immunological unresponsiveness toward harmless microbes and food-derived antigens delivered from the gut but likely contributes to the persistence of hepatotropic pathogens, such as HCV. A key factor thought to govern the fate of liver immune responses (effective versus ineffective) is the level of antigen expression. For instance, studies in mice have shown that CD8 T cells primed by high levels of hepatocyte-expressed antigen are more dysfunctional than those activated under conditions of low expression (31–34).
Given that CD8 T cells are immediately silenced upon activation by RHV infection, we reasoned that this outcome may be driven by the magnitude of early virus replication in the liver. To test this, we reduced RHV replication levels by treating rats subcutaneously with the pangenotypic HCV DAA sofosbuvir, a potent inhibitor of hepaciviral NS5B polymerase activity. Treatment was initiated on day 2 p.i. before the first appearance of CD8 T cell responses (Fig. 2A) and maintained for twelve straight days (10 mg/daily). Consistent with previous findings (10, 13), sofosbuvir was remarkably effective at inhibiting RHV replication. Serum viremia declined by a factor of 105 within days of treatment initiation and became mostly undetectable by day 10 p.i. (Fig. 6A). Breakthrough viremia was observed in two rats, however, by day 14 p.i. Direct PCR sequencing of recovered virus from both revealed a single-amino-acid substitution within the NS5B coding sequence (S2654T) that quickly reverted to the wild type following treatment cessation in one of the animals (Table 2). Interestingly, this mutation is identical to the HCV S282T resistance-associated substitution that infrequently emerges in treated patients (35, 36), indicating a conserved mechanism of antiviral resistance.
FIG 6.

Early DAA treatment partially ameliorates RHV-specific CD8 T cell dysfunction. Seven- to 10-week-old Lewis rats were infected with 106 genomes of RHV intravenously. Starting at day 2 p.i., rats were treated daily with 10 mg sofosbuvir subcutaneously for twelve days. Following treatment, half of the rats were analyzed immediately at day 14 p.i. for recovery of liver CD8 T cell immunity. The remaining half were assessed for immunity and infection outcome at day 63 p.i. (A) Serum RHV RNA and ALT levels. Shading indicates timing of sofosbuvir treatment. ALT data show results from n = 6 to 12 rats per time point (means ± SEM). (B, left) Percentage of intrahepatic CD8 T cells targeting the E1191 and NS5B2511 epitopes as determined by class I tetramer staining. (Right) Representative flow plots showing frequency of CD8 T cells that bind the E1191 or NS5B2511 tetramer at day 14 p.i. (C) Percentage of CD8 T cells producing IFN-γ or TNF-α after 5 h of stimulation of the E1191 or NS5B2511 epitope (10 μg/ml) or a pool of class I and II epitopes (5 μg/ml each; Table 1) at the indicated days p.i. (D) Percentage of CD4 T cells producing IFN-γ or TNF-α after 5 h of stimulation of a pool of class I and II epitopes (5 μg/ml each; Table 1) at the indicated days p.i. (E) Comparison of frequency of E1191-specific CD8 T cells expressing the indicated markers at day 14 p.i. between untreated control (Fig. 5B) and sofosbuvir-treated rats. (F) Sequence evolution of E1191 and NS5B2511 epitopes during RHV infection. Consensus sequences were determined by direct PCR sequencing. Frequencies of rats infected with virus containing the indicated sequences are shown. (G) Percentage of intrahepatic CD8 T cells from immune rat producing IFN-γ upon 5 h of stimulation with titrated concentrations of the E1191 epitope or peptide containing the F199L mutation. Panels B to E show data from n = 6 rats per group (means ± SEM). n.d., not detected. ***, P < 0.001; *, P < 0.05; ns, not significant as determined by Student's t test.
TABLE 2.
NS5B polymerase mutations associated with sofosbuvir resistancea
| Rat ID and day p.i. | Plasma viral load (genomes/ml)b | Substitution |
|
|---|---|---|---|
| Nucleotidec | Amino acidd | ||
| R352e | |||
| 2 | 3.35 × 108 | ||
| 10 | <LOD | Not determined | Not determined |
| 14 | 9.95 × 106 | T8445A | S2654T |
| R362 | |||
| 2 | 2.64 × 107 | ||
| 10 | <LOD | Not determined | Not determined |
| 14 | 2.84 × 105 | T8445A | S2654T |
| 21 | 9.35 × 109 | ||
Boldface indicates sofosbuvir treatment.
Limit of detection (LOD) of RT-PCR assay is 1,875 genomes/ml.
Nucleotide position according to full-length RHV genome (accession no. KX905133).
Amino acid position according to the start of the RHV polyprotein (accession no. AQV09561; protein cleavage sites predicted in reference 10).
Rat R352 was euthanized at day 14 prior to treatment cessation and virus rebound.
As predicted, CD8 T cell responses were dramatically altered by short-term reduction in viral titers. At day 14 p.i., massive frequencies (∼30%) of RHV-specific CD8 T cells targeting the E1191 but not the NS5B2511 epitope expanded in liver (Fig. 6B); interestingly, we observed in some animals two populations that differed in their tetramer binding affinity (Fig. 4B, right). In sharp contrast to untreated rats (Fig. 3), these responses were capable of producing antiviral cytokine upon peptide stimulation (Fig. 6C). The frequency of functional cells, however, remained >98% lower than that revealed by class I tetramer staining (Fig. 6B versus C), indicating that most responding cells remained dysfunctional. The functional recovery of CD8 T cells was largely restricted to the E1191 epitope alone, since pooled epitope stimulation did not significantly increase the magnitude of the virus-specific cytokine response, although there was a clear trend upwards (Fig. 6C). Notably, partial recovery of CD8 T cell functions was not accompanied by a substantial improvement in CD4 T helper cell activity (Fig. 6D) or fluctuations in serum ALT levels (Fig. 6A). However, there was a significant decrease in PD-1 expression on E1191-specific CD8 T cells at day 14 p.i. compared to that of untreated control rats (Fig. 6E), suggesting a role for coinhibitory receptor signaling in mediating immune dysfunction in this model. Other phenotypic changes associated with increased functionality included increased CXCR3 and modestly decreased granzyme B expression (Fig. 6E).
Despite the induction of responses with partial effector function, persistent viremia rapidly rebounded following treatment cessation (Fig. 6A). Interestingly, the resurgence of viremia did not reimpose the silencing effect upon E1191-specific CD8 T cells, since these responses were detectable by both tetramer and functional assays at day 63 p.i., although at sharply reduced levels compared to that at the earlier time point (Fig. 6B and C). In contrast, low frequencies of CD8 T cells recognizing the NS5B2511 epitope were only detectable by class I tetramer staining (Fig. 6B and C), consistent with these cells likely having been primed by resurgent, high-level viremia. To determine how RHV was able to evade the E1191-specific CD8 T cell response, we sequenced persistent virus for evolution at this class I epitope. At day 21 p.i., immediately after virus rebound, a single-amino-acid substitution was identified at the P9 anchor position (F199L) in 4/6 rats (Fig. 6F). This variant became dominant in 6/6 rats by day 63 p.i., while the NS5B2511 epitope, which lacked immune pressure, remained intact (Fig. 6F). Importantly, testing of variant F199L peptide for T cell recognition confirmed viral escape at this epitope (Fig. 6G). Thus, while lowering viral replication primed CD8 T cells with partial effector function, RHV was able to utilize a previously unneeded mechanism to efficiently evade them.
DISCUSSION
The discovery and subsequent characterization of RHV was a significant advance for the HCV field, facilitating the development of a comparative rat model that can be efficiently utilized for studies of protective immunity and vaccine testing (10, 14, 15, 37). The most attractive feature of this model is RHV’s ability to spontaneously persist in a fully immunocompetent environment, a trait not yet achieved for HCV mouse models (8, 11). Defining the virological and immunological mechanisms that underlie this capability is important for understanding potential similarities and differences with HCV and also could yield novel insights into failed immunity relevant to other persistent viruses, especially those possessing hepatotropism. Therefore, this study was undertaken to determine the nature and fate of CD8 T cell responses induced against RHV and why they fail to provide requisite control of persistent infection. Our results indicate that immune dysfunction is the major mechanism of RHV evasion from antiviral CD8 T cells, although mutational escape can occur when immunity is indirectly stimulated via antiviral intervention.
Using MHC class I tetramers incorporating novel epitopes, we directly measured the expansion and contraction of CD8 T cell responses to RHV infection. In contrast to HCV, which typically takes 8 to 12 weeks to trigger an adaptive immune response (18), RHV infection resulted in a massive influx of virus-specific CD8 T cells into the acutely infected liver. Responses were first measurable at day 10 p.i. and peaked days later, followed by sharp contraction and maintenance at low levels despite no control of viremia. Since class I epitope sequences were intact, the contraction of these responses was likely programmed rather than a result of reduced antigen stimulation facilitated by mutational escape (38). Detection of virus-specific CD8 T cells late into infection is notable, since these populations were thought to be deleted (14). How these responses are sustained and whether there is a memory-like population that fulfills this role (39–41) will need to be determined. Furthermore, using these tetramers, it will be important to compare CD8 T cells induced by RHV versus vaccine to identify molecular features associated with lasting immunity against an HCV-like virus.
Consistent with recent reports (14, 15), RHV-specific CD8 T cells were nonfunctional, as measured by conventional cytokine assays. This is in sharp contrast to most HCV infections in humans, where acute viremic control strongly correlates with the onset of functional T cell immunity (18–20). Although killing capacity was not quantified in this study, the lack of viral evolution at MHC class I epitopes, which can occur after prophylactic vaccination (14, 15), remarkable stability of RHV viremia, and the absence of ALT elevations supports the notion that RHV-specific CD8 T cell responses are incapable of elaborating key antiviral effector functions in this model. The ability of vaccines to induce virus-specific CD8 T cells that do not acquire this functional defect following RHV infection (14, 15) suggests that such approaches will prove effective against HCV.
Chronic viral infections such as HCV are associated with T cell dysfunction or exhaustion, a distinct differentiation state characterized by altered effector cell functionality and poor memory formation (28). Phenotypically, RHV-specific CD8 T cells showed several features consistent with such a state. Despite apparent progression through normal expansion and contraction phases, RHV-specific CD8 T cells failed to form immunological memory, as determined by reacquisition of CD127 expression. For HCV, early expression of this memory precursor marker on virus-specific CD8 T cells was linked to spontaneous resolution of infection in the chimpanzee model (42). RHV-specific CD8 T cells also persistently upregulated the coinhibitory receptor PD-1, a classic signature of T cell exhaustion (28). However, it is notable that PD-1 expression was not uniform on CD8 T cells infiltrating the liver, even at later time points, when antigen stimulation had been prolonged. This contrasts with HCV and could represent a subtle difference between species, although the expression of other coinhibitory receptors will need to be assessed. Given their profound impairment, it would be of interest to assess whether blockade of PD-1 signaling can rescue CD8 T cell functions (29, 30, 43). Furthermore, as noted above, the identification of exhausted CD8 T cells with stem cell-like characteristics (39–41) would be an important validation of this model.
Because of the strong conservation among hepacivirus proteins (9), RHV is sensitive to multiple HCV DAAs (10, 13). This afforded us the unique opportunity to test whether the magnitude of early RHV replication in liver influences virus-specific CD8 T cell dysfunction, as has been observed in mouse studies assessing the impact of hepatocyte antigen expression (31–34). Remarkably, short-term treatment with the pangenotypic DAA sofosbuvir, targeting the NS5B polymerase, resulted in the generation of RHV-specific CD8 T cells with partial cytokine function and reduced PD-1 expression. The downregulation of PD-1 could underpin the improved functionality observed here or simply reflect the reduced levels of antigen provided by treatment. Two lines of evidence indicate this functional reversal was mediated during the priming phase of the response. First, treatment was initiated well before CD8 T cells first appeared by class I tetramer staining. Second, the rebound of viremia after treatment cessation did not resilence CD8 T cell functions, as evidenced by both ongoing cytokine activity and the selection of an escape variant. However, these data do not explain how reduction in virus facilitated this effect. Antigen-expressing hepatocytes can directly activate CD8 T cells in vivo, although this is considered largely nonproductive (22, 44). Therefore, one possibility is that reduction in virus decreased the tolerance-inducing potential of antigen-expressing hepatocytes. Alternatively, smaller numbers of virally infected hepatocytes may have increased the likelihood that at least some CD8 T cells engaged with conventional antigen-presenting cells possessing effective stimulatory properties (44). The presence of two virus-specific populations with an apparent difference in tetramer binding affinity may support this notion.
The absence of CD8 T cells targeting the NS5B2511 epitope immediately following DAA treatment was an unexpected finding and not easily explained. Since this population was eventually detected by class I tetramer staining late into infection after viremia had rebounded to high magnitude, it is probable that the stimulation of this response requires a level of antigen availability that was not met during therapy, whereas a lesser threshold was crossed for the more dominant E1191 epitope. This may reflect the immunodominance hierarchy or be due to differences in structural versus nonstructural antigen processing and presentation.
A well-defined feature of sofosbuvir is its high barrier to HCV treatment resistance. However, in rare cases, the emergence of strains bearing the major NS5B S282T resistance-associated substitution have been described (35, 36). Remarkably, we found an identical amino acid substitution (S2654T) in viral variants recovered from two sofosbuvir-treated rats showing breakthrough viremia. The pace at which resistance developed in these two cases (<2 weeks of treatment) was noteworthy and suggests that RHV possesses more flexibility at this region than HCV or that combination DAA treatment is needed to more firmly suppress viral replication and decrease the likelihood of acquiring resistance mutations. Although we did not cure RHV infection in this study, our results are encouraging that this could be accomplished through combination DAA therapy or the inclusion of therapeutics targeting microRNA-122 (10, 13, 45). This would provide a unique opportunity to assess vaccination and immune recovery in the postcure period, a critical question not currently addressable by other animal models of chronic viral infection (3, 5).
The lack of strong CD4 help in liver, as noted here and elsewhere (14), could represent an additional mechanism of CD8 T cell dysfunction in this model. CD4 T cell help is critical for optimal maturation of CD8 T cell responses to persistent viruses (46) and are required for the elimination of HCV (16, 47, 48) and RHV (14). CD4 T cells could initiate a successful response during priming or by secreting essential cytokines that promote CD8 T cell effector functions (46). Interestingly, we found that the resolution of RHV infection following prophylactic vaccination induces functional CD8 T cell responses against viral proteins not targeted by immunization. This indicates that CD8 T cells primed by replicating virus can, in fact, acquire effector capacity when triggered under a different immunological context, an effect that may have been mediated by the addition of CD4 T cell help via vaccine. Surprisingly, DAA treatment had no clear restorative effect upon CD4 T cell help despite the improved functionality of RHV-specific CD8 T cells. Reasons for this differential responsiveness to treatment are unknown but could be related to differences in liver antigen presentation for each T cell subset. The inability to restore this arm of cellular immunity may explain why most RHV-specific CD8 T cells remained dysfunctional after treatment. The development of RHV class II tetramers will be important for assessing mechanisms that silence CD4 help in this model.
In conclusion, our results indicate that RHV infection triggers the activation and expansion of virus-specific CD8 T cells that, in contrast to HCV (18), do not mature into antiviral effectors capable of exerting selective pressure on circulating virus genomes. Importantly, we have shown that this defect in CD8 T cell maturation is at least partially determined by priming of responses in a setting of high antigen load, although even when functional responses are generated through therapeutic means, the high mutational capacity of RHV facilitates immune escape. Thus, RHV can employ multiple mechanisms to evade CD8 T cell immunity depending on the immunological context, with viral escape mainly serving as a backup strategy to the clearly more dominant approach of inducing functional impairment. This differs with HCV, where both of these mechanisms are fully operant.
MATERIALS AND METHODS
Animals.
Female Lewis rats were purchased from Charles River Laboratories and housed within the Animal Vivarium Core of the Abigail Wexner Research Institute. All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Nationwide Children’s Hospital prior to study initiation.
Virus infections.
In vivo generation and recovery of infectious RHV from a complete genome clone has been described (10, 14). Rats were infected with 106 genomes of RHV via tail vein following careful restraint.
Class I epitope sequencing.
DNA fragments containing the coding sequences of all RHV class I epitopes were amplified from recovered viral cDNA using the Q5 high-fidelity polymerase (New England Biolabs) and subsequently sequenced (Eurofins Genomics) using nested sequencing primers. All primer sequences used can be found in Table 3.
TABLE 3.
RHV primer sequences
| Name | Sequence (5′→3′) |
|---|---|
| C-NS2-1-F1 | CGAGGCGTTTCCGCTGTAA |
| C-NS2-1-F2 | CGAGGCGTTTCCGCTGTAAACC |
| C-NS2-1-R1 | CACTTGCTCGCAGATGACACA |
| C-NS2-1-R2 | CTTGCTCGCAGATGACACAGCC |
| C-NS2-2-F1 | CTGCACTGAGCTTTCCTGCAT |
| C-NS2-2-F2 | CTGCACTGAGCTTTCCTGCATGCA |
| C-NS2-2-R1 | CAGTTGTYAGCCAGGGYGTAGT |
| C-NS2-2-R2 | GCCAGGGYGTAGTGCCACA |
| NS34-1-F1 | GGCAAGACGGTCAGAGC |
| NS34-1-F2 | GACGGTCAGAGCGGGCAT |
| NS34-1-R1 | GCCTCAGGTATCTGGGC |
| NS34-1-R2 | CCTCAGGTATCTGGGCTCC |
| NS34-3-F1 | GCCAACGATCTGAGGGC |
| NS34-3-F2 | CCAACGATCTGAGGGCAGC |
| NS34-3-R1 | CGCCTGCAAGGGTCATAAC |
| NS34-3-R2 | CTGCAAGGGTCATAACCGTCTC |
| NS34-4-F1 | GCTTGGAGGGGCCTTTC |
| NS34-4-F2 | CTTGGAGGGGCCTTTCAGG |
| NS34-4-R1 | GCAGCCCCAAAGACAGC |
| NS34-4-R2 | CCCAAAGACAGCAGCGCC |
| NS5-3-F1 | CAGCTTACACTCCAGCAGCTG |
| NS5-3-F2 | GCTTACACTCCAGCAGCTGGGA |
| NS5-3-R1 | GTGCTGTCAAAGCACACGGT |
| NS5-3-R2 | GTGCTGTCAAAGCACACGGTGTC |
| NS5-4-F1 | CTGTGTGCGGGGATGCATA |
| NS5-4-F2 | CTGTGTGCGGGGATGCATATGG |
| NS5-4-R1 | GTGGGTTGTAGCCCTTTCC |
| NS5-4-R2 | GTGGGTTGTAGCCCTTTCCCTC |
| NS5-5-F1 | CAGTGGCCATGAAGCGCAT |
| NS5-5-F2 | GTGGCCATGAAGCGCATGGG |
| NS5-5-R1 | GGTGGTAAGAGTTGGAGGTTG |
| NS5-5-R2 | GTGGTAAGAGTTGGAGGTTGAGGG |
Vaccinations.
The recombinant adenovirus serotype 5 vector containing the coding sequence of RHV NS3-NS5B genes (Ad-NS) has been described (14). For all vaccinations, rats were injected in the quadriceps muscle with 5 × 108 infectious units (IFU) of Ad-NS. For prophylactic vaccinations, rats were challenged 2 to 3 weeks later with RHV.
DAA treatment.
The direct-acting antiviral sofosbuvir was kindly provided by Gilead Sciences as a crystalline solid and stored at room temperature until use. For the treatment of RHV infection, rats were administered 10 mg sofosbuvir subcutaneously in 0.5 ml phosphate-buffered saline (PBS) daily for 12 days (60 to 70 mg/kg of body weight).
Viral RNA quantification.
Reverse transcription-PCR (RT-PCR) quantification of plasma or serum viral loads was performed as described previously (14), with a few modifications. In brief, viral RNA was extracted using the Quick-RNA viral kit (Zymo Research), followed by GoScript reverse transcription (Promega) via random hexamer priming. Viral genomes were subsequently quantified using the PowerUP SYBR green master mix (Applied Biosystems) on a StepOnePlus RT-PCR system (Applied Biosystems). A linearized plasmid containing the RHV-encoded NS3 gene was used for the construction of a standard curve.
Plasma ALT levels.
Quantification of plasma ALT levels was performed using the MaxDiscovery alanine transaminase enzymatic kit (Bioo Scientific).
Peptides.
All peptides were synthesized by Genemed Synthesis, Inc. Peptides were reconstituted at 1 or 10 mg/ml in 10% dimethyl sulfoxide-water solutions and stored at −80°C until use. The final concentration of each peptide in all functional assays was 5 or 10 μg/ml unless otherwise specified.
Identification of RT1-Al-restricted epitopes.
Minimal 9-mer epitopes with high potential for presentation by the Lewis RT1-Al MHC class I molecule were predicted using the online SYFPEITH online server (21). Eighteen-mer peptides containing the predicted epitope were tested for immunogenicity in functional response assays using recovered lymphocytes from vaccinated rats that previously cleared RHV (14). In two cases, positive responses were confirmed by testing of the minimal predicted epitope.
Cell isolation and culture.
Rats were humanely euthanized in a CO2 inhalation chamber, followed by blood and tissue collection. For the removal of blood leukocytes, livers were perfused with PBS via the inferior vena cava prior to organ collection. Peripheral blood mononuclear cells were isolated from EDTA-treated blood by Ficoll-Paque density gradient centrifugation (GE Healthcare). For single-cell splenocyte preparations, spleens were gently disrupted through a 100-μm cell strainer in PBS containing 2.5% fetal bovine serum (FBS). Hepatic leukocytes were purified by gently disrupting livers through a metal sieve, followed by gradient centrifugation in a 37.5% Percoll (GE Healthcare) solution (14). Residual erythrocytes in preparations were lysed by 3-min treatment with ACK solution (Gibco). For all functional assays, cells were cultured in RPMI 1640 medium containing GlutaMAX and HEPES (Gibco) and supplemented with 10% FBS (Gibco) and 55 μM 2-mercaptoethanol (Gibco).
IFN-γ ELISpot assay.
The number of IFN-γ-secreting cells was enumerated using an enzymatic staining rat ELISpot assay kit (U-Cytech). Cells were plated in duplicate at 2 × 105 per well and stimulated with viral peptides, or with medium alone or 5 μg/ml concanavalin A (Sigma-Aldrich) as negative and positive controls, respectively, for 40 to 48 h. The number of spot-forming cells was measured using an automatic counter (Immunospot). The total number of spot-forming cells was determined by subtracting the mean number of spots in negative-control wells from that of experimental wells. A positive response was defined as ≥3 times the response of negative-control wells prior to normalization.
Flow cytometry.
Surface and intracellular staining was performed as described below. All stainings were performed in 50 μl buffer in 96-well round-bottom plates at 4°C. Events were collected on a BD Fortessa flow cytometer following compensation with UltraComp eBeads (Invitrogen). Data were analyzed using FlowJo v7.6.5 (TreeStar).
Antibodies.
The following fluorophore-conjugated monoclonal antibodies reactive to rat lymphocytes, from BioLegend, BD Biosciences, eBioscience, and Miltenyi Biotec, were used: CD3-VioGreen, CD4-peridinin chlorophyll protein (PerCP)-eFluor710, CD8-BV786, CD27-BV650, CD28-phycoerythrin (PE)-Vio770, CD25-PerCP-eFluor710, CD161-PerCP-eFluor710, CD62L-BUV395, CXCR3-fluorescein isothiocyanate (FITC), CD127-PE, granzyme B-AF700, Ki67-AF700, IFN-γ-AF647, IFN-γ-FITC, TNF-α–PE, IL-17A–PE–Cy7, IL-4–PE, and IL-10–AF647. Unconjugated monoclonal antibodies against rat PD-1 and IL-2 were obtained from Invitrogen and rapidly conjugated against PE and allophycocyanin (APC), respectively, using corresponding kits (Abcam).
Tetramer analysis.
Biotinylated rat MHC class I RT1-Al monomers specific for the RHV-encoded E1191 and NS5B2511 peptides were obtained from the NIH Tetramer Core Facility. Monomers were tetramerized with streptavidin-APC (Prozyme) by a standard protocol. Both reagents were validated and titrated against lymphocytes from immune animals prior to use in experiments. For the analysis of virus-specific populations, cells were stained with tetramer (1:100) for 30 min at 4°C, stained with antibody against CD3, CD8, CD27, CD28, CD161, CD62L, CXCR3, or CD127, stained with the LIVE/DEAD near-IR cell dye (Invitrogen), fixed and permeabilized using the transcription factor staining buffer set (eBioscience), and then intracellularly labeled with antibodies against granzyme B or Ki-67.
Intracellular cytokine analysis.
For the measurement of intracellular cytokine production, 106 hepatic leukocytes were plated in a 96-well round-bottom plate in 200 μl complete medium containing GolgiPlug (BD Biosciences). Cells were stimulated for 5 h with peptide or with medium alone or phorbol myristate acetate (PMA)-ionomycin (BioLegend) as negative and positive controls, respectively. Following incubation, cells were stained with antibodies against CD3, CD4, and CD8 (20 min), stained with the LIVE/DEAD near-IR cell dye (Invitrogen), fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences), and then intracellularly labeled with antibody against IFN-γ, TNF-α, IL-2, IL-17A, IL-4, or IL-10. Percent response was calculated by subtracting the frequency of cytokines positive in the negative control from that of cells stimulated with viral peptides. A positive response was defined as ≥3 times the response of the negative control prior to normalization.
Statistics.
All statistical determinations described were calculated using a two-tailed Student's t test of unequal variance.
ACKNOWLEDGMENTS
We thank the NIH Tetramer Core Facility for the production of rat RT1-Al tetramers, Gilead Sciences for the provision of sofosbuvir, and Arash Grakoui and Victoria M. Velazquez for thoughtful discussions.
This study was supported by funding from the NIAID (R01 AI137567) and the Abigail Wexner Research Institute at Nationwide Children’s Hospital. A.S.H. was supported by an NIAID Ruth L. Kirschstein National Research Service Award predoctoral fellowship (F30 AI143060).
REFERENCES
- 1.Hoofnagle JH. 2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2017. Global hepatitis report 2017. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Bartenschlager R, Baumert TF, Bukh J, Houghton M, Lemon SM, Lindenbach BD, Lohmann V, Moradpour D, Pietschmann T, Rice CM, Thimme R, Wakita T. 2018. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: considerations for scientists and funding agencies. Virus Res 248:53–62. doi: 10.1016/j.virusres.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker CM. 2017. Designing an HCV vaccine: a unique convergence of prevention and therapy? Curr Opin Virol 23:113–119. doi: 10.1016/j.coviro.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi C, Butt ZA, Wong S, Buxton J, Islam N, Yu A, Darvishian M, Gilbert M, Wong J, Chapinal N, Binka M, Alvarez M, Tyndall M, Krajden M, Janjua N. 2018. Hepatitis C virus reinfection after successful treatment with direct-acting antiviral therapy in a large population-based cohort. Hepatology 68:1007–1014. doi: 10.1016/j.jhep.2018.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Callendret B, Eccleston HB, Hall S, Satterfield W, Capone S, Folgori A, Cortese R, Nicosia A, Walker CM. 2014. T-cell immunity and hepatitis C virus reinfection after cure of chronic hepatitis C with an interferon-free antiviral regimen in a chimpanzee. Hepatology 60:1531–1540. doi: 10.1002/hep.27278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. 2013. Animal models for hepatitis C. Curr Top Microbiol Immunol 369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 9.Hartlage A, Cullen J, Kapoor A. 2016. The strange, expanding world of animal hepaciviruses. Annu Rev Virol 3:53–75. doi: 10.1146/annurev-virology-100114-055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trivedi S, Murthy S, Sharma H, Hartlage AS, Kumar A, Gadi S, Simmonds P, Chauhan LV, Scheel TKH, Billerbeck E, Burbelo PD, Rice CM, Lipkin WI, Vandergrift K, Cullen JM, Kapoor A. 2017. Viral persistence, liver disease and host response in hepatitis C-like virus rat model. Hepatology 68:435–448. doi: 10.1002/hep.29494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Billerbeck E, Wolfisberg R, Fahnoe U, Xiao JW, Quirk C, Luna JM, Cullen JM, Hartlage AS, Chiriboga L, Ghoshal K, Lipkin WI, Bukh J, Scheel TKH, Kapoor A, Rice CM. 2017. Mouse models of acute and chronic hepacivirus infection. Science 357:204–208. doi: 10.1126/science.aal1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheel TK, Kapoor A, Nishiuchi E, Brock KV, Yu Y, Andrus L, Gu M, Renshaw RW, Dubovi EJ, McDonough SP, Van de Walle GR, Lipkin WI, Divers TJ, Tennant BC, Rice CM. 2015. Characterization of nonprimate hepacivirus and construction of a functional molecular clone. Proc Natl Acad Sci U S A 112:2192–2197. doi: 10.1073/pnas.1500265112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfisberg R, Holmbeck K, Nielsen L, Kapoor A, Rice CM, Bukh J, Scheel T. 2019. Replicons of a rodent hepatitis C model virus permit selection of highly permissive cells. J Virol 93:e00733-19. doi: 10.1128/JVI.00733-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartlage AS, Murthy S, Kumar A, Trivedi S, Dravid P, Sharma H, Walker CM, Kapoor A. 2019. Vaccination to prevent T cell subversion can protect against persistent hepacivirus infection. Nat Commun 10:1113. doi: 10.1038/s41467-019-09105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atcheson E, Li WQ, Bliss CM, Chinnakannan S, Heim K, Sharpe H, Hutchings C, Dietrich I, Nguyen D, Kapoor A, Jarvis MA, Klenerman P, Barnes E, Simmonds P. 10 August 2019. Use of an outbred rat hepacivirus challenge model for design and evaluation of efficacy of different immunization strategies for hepatitis C virus. Hepatology doi: 10.1002/hep.30894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, Murthy KK, Rice CM, Walker CM. 2003. HCV persistence and immune evasion in the absence of memory T cell help. Science 302:659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 17.Shoukry NH, Grakoui A, Houghton M, Chien DY, Ghrayeb J, Reimann KA, Walker CM. 2003. Memory CD8(+) T cells are required for protection from persistent hepatitis C virus infection. J Exp Med 197:1645–1655. doi: 10.1084/jem.20030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker CM. 2010. Adaptive immunity to the hepatitis C virus. Adv Virus Res 78:43–86. doi: 10.1016/B978-0-12-385032-4.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SH, Rehermann B. 2014. Immune responses to HCV and other hepatitis viruses. Immunity 40:13–24. doi: 10.1016/j.immuni.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heim MH, Thimme R. 2014. Innate and adaptive immune responses in HCV infections. J Hepatol 61:S14–S25. doi: 10.1016/j.jhep.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 21.Schuler MM, Nastke MD, Stevanovikc S. 2007. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol 409:75–93. doi: 10.1007/978-1-60327-118-9_5. [DOI] [PubMed] [Google Scholar]
- 22.Kubes P, Jenne C. 2018. Immune responses in the liver. Annu Rev Immunol 36 36:247–277. doi: 10.1146/annurev-immunol-051116-052415. [DOI] [PubMed] [Google Scholar]
- 23.Cui WG, Kaech SM. 2010. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev 236:151–166. doi: 10.1111/j.1600-065X.2010.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Northfield JW, Kasprowicz V, Lucas M, Kersting N, Bengsch B, Bengsh B, Kim A, Phillips RE, Walker BD, Thimme R, Lauer G, Klenerman P. 2008. CD161 expression on hepatitis C virus-specific CD8+ T cells suggests a distinct pathway of T cell differentiation. Hepatology 47:396–406. doi: 10.1002/hep.22040. [DOI] [PubMed] [Google Scholar]
- 25.Kaech SM, Wherry EJ. 2007. Heterogeneity and cell-fate decisions in effector and memory CD8(+) T cell differentiation during viral infection. Immunity 27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahlers JD, Belyakov IM. 2010. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood 115:1678–1689. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 28.McLane LM, Abdel-Hakeem MS, Wherry EJ. 2019. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 37:457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 29.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, Altman JD, Rouse BT, Freeman GJ, Ahmed R, Grakoui A. 2007. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol 81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. 2007. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8(+) T cells associated with reversible immune dysfunction. J Virol 81:9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ochel A, Cebula M, Riehn M, Hillebrand U, Lipps C, Schirmbeck R, Hauser H, Wirth D. 2016. Effective intrahepatic CD8+ T-cell immune responses are induced by low but not high numbers of antigen-expressing hepatocytes. Cell Mol Immunol 13:805–815. doi: 10.1038/cmi.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tay SS, Wong YC, McDonald DM, Wood NAW, Roedige B, Sierro F, Mcguffog C, Alexander IE, Bishop GA, Gamble JR, Weninger W, McCaughan GW, Bertolino P, Bowen DG. 2014. Antigen expression level threshold tunes the fate of CD8 T cells during primary hepatic immune responses. Proc Natl Acad Sci U S A 111:E2540–E2549. doi: 10.1073/pnas.1406674111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krebs P, Scandella E, Odermatt B, Ludewig B. 2005. Rapid functional exhaustion and deletion of CTL following immunization with recombinant adenovirus. J Immunol 174:4559–4566. doi: 10.4049/jimmunol.174.8.4559. [DOI] [PubMed] [Google Scholar]
- 34.Lukens JR, Dolina JS, Kim TS, Tacke RS, Hahn YS. 2009. Liver is able to activate naive CD8(+) T cells with dysfunctional anti-viral activity in the murine system. PLoS One 4:e7619. doi: 10.1371/journal.pone.0007619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gane EJ, Metivier S, Nahass R, Ryan M, Stedman CA, Svarovskaia ES, Mo HM, Doehle B, Dvory-Sobol H, Hedskog C, Lin M, Brainard DM, Yang JC, McHutchison JG, Sulkowski M, Younes Z, Lawitz E. 2017. The emergence of NS5B resistance associated substitution S282T after sofosbuvir-based treatment. Hepatol Commun 1:538–549. doi: 10.1002/hep4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newsum AM, Molenkamp R, van der Meer JT, Rebers SP, Prins M, van der Valk M, Schinkel J. 2018. Persistence of NS5B-S282T, a sofosbuvir resistance-associated substitution, in a HIV/HCV-coinfected MSM with risk of onward transmission. J Hepatol 69:968–970. doi: 10.1016/j.jhep.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 37.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, Conte JM, Ng J, Garcia J, Bhuva NP, Lee B, Che X, Quan PL, Lipkin WI. 2014. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 5:e01933-14. doi: 10.1128/mBio.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badovinac VP, Porter BB, Harty JT. 2002. Programmed contraction of CD8(+) T cells after infection. Nat Immunol 3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 39.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, Held W. 2016. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 40.Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, Held W, Zehn D, Hofmann M, Thimme R. 2017. TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat Commun 8:15050. doi: 10.1038/ncomms15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu TQ, Ji Y, Moseman EA, Xu HFC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu WW, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1:eaai8593. doi: 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin EC, Park SH, Nascimbeni M, Major M, Caggiari L, de Re V, Feinstone SM, Rice CM, Rehermann B. 2013. The frequency of CD127(+) hepatitis C virus (HCV)-specific T cells but not the expression of exhaustion markers predicts the outcome of acute HCV infection. J Virol 87:4772–4777. doi: 10.1128/JVI.03122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, Sharpe AH, Dustin LB, Rice CM, Grakoui A, Ahmed R, Walker CM. 2013. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc Natl Acad Sci U S A 110:15001–15006. doi: 10.1073/pnas.1312772110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowen DG, Zen M, Holz L, Davis T, McCaughan GW, Bertolino P. 2004. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Investig 114:701–712. doi: 10.1172/JCI21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swain SL, McKinstry KK, Strutt TM. 2012. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol 12:136–148. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerlach JT, Diepolder HM, Jung MC, Gruener NH, Schraut WW, Zachoval R, Hoffmann R, Schirren CA, Santantonio T, Pape GR. 1999. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology 117:933–941. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- 48.Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, Aneja J, Reyor LL, Allen TM, Lohse AW, McGovern B, Chung RT, Kwok WW, Kim AY, Lauer GM. 2012. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med 209:61–75. doi: 10.1084/jem.20100388. [DOI] [PMC free article] [PubMed] [Google Scholar]