

Abstract
The use of biotin or biotin-containing reagents is an essential component of many protein purification and labeling technologies. Owing to its small size and high affinity to the avidin family of proteins, biotin is a versatile molecular handle that permits both enrichment and purity that is not easily achieved by other reagents. Traditionally, the use of biotinylation to enrich for proteins has not required the detection of the site of biotinylation. However, newer technologies for discovery of protein–protein interactions, such as APEX and BioID, as well as some of the click chemistry-based labeling approaches have underscored the importance of determining the exact residue that is modified by biotin. Anti-biotin antibody-based enrichment of biotinylated peptides (e.g., BioSITe) coupled to LC–MS/MS permit large-scale detection and localization of sites of biotinylation. As with any chemical modification of peptides, understanding the fragmentation patterns that result from biotin modification is essential to improving its detection by LC–MS/MS. Tandem mass spectra of biotinylated peptides has not yet been studied systematically. Here, we describe the various signature fragment ions generated with collision-induced dissociation of biotinylated peptides. We focused on biotin adducts attached to peptides generated by BioID and APEX experiments, including biotin, isotopically heavy biotin, and biotin-XX-phenol, a nonpermeable variant of biotin-phenol. We also highlight how the detection of biotinylated peptides in high-throughput studies poses certain computational challenges for accurate quantitation which need to be addressed. Our findings about signature fragment ions of biotinylated peptides should be helpful in the confirmation of biotinylation sites.
Keywords: mass spectrometry, post-translational modifications, marker ions, protein–protein interactions
Graphical Abstract

INTRODUCTION
Biotinylation of proteins is an often-used molecular handle that allows for robust purification of proteins. The strong interaction between biotin and streptavidin (Kd ≈ 10−15 M) makes it a very attractive and effective method for purification of chemically or enzymatically biotinylated proteins.1 Biotinylation is a rare post-translational modification (PTM) in nature, especially in eukaryotic cells, as there are less than 10 reported endogenously biotinylated proteins thus far.2 Biotinylation of endogenous proteins is catalyzed by biotin-ligases which activate biotin to form biotinyl 5′ adenylate and transfer the biotin specifically to ε-amino group of lysine side chains. A number of technologies that take inspiration from these biotin-ligases have recently been developed. Using engineered biotin ligases, these technologies enable labeling of proteins in very close proximity which allows analysis of protein–protein interactions and mapping of subcellular proteomes.3–6 The two most prominent examples of these technologies are the BioID and APEX systems. BioID leverages the biotin ligase, BirA*, which catalyzes creation of the reactive biotin-AMP leading to biotinylation of proximal lysine side chains. The APEX system uses a small monomeric peroxidase, APEX2, to covalently label proximal tyrosine residues with biotin through the oxidation of biotin-phenol.7
With the advent of these technologies, the detection of biotinylated proteins became essential. Many different methods have been used to detect proteins in APEX and BioID experiments but none of which were designed to detect actual biotinylation of proteins. Rather, these methods relied upon stringent sample preparation conditions to enrich biotinylated proteins and were not developed for detection of biotinylation itself. The main reasons being the low abundance of biotinylated peptides within the vast pool of nonbiotinylated peptides and the retention of biotinylated peptides on the capture reagent—streptavidin beads—which preclude the detection of biotinylated peptides in traditional LC–MS/MS experiments. Recent studies by our group8 as well as by others9 have led to development of a strategy that we have designated Biotinylation Site Identification Technology (BioSITe), which demonstrated the use of anti-biotin antibodies for the efficient and reproducible enrichment of biotinylated peptides for high-throughput identification of biotinylated peptides and site mapping. Antibody-based enrichment of biotinylated peptides thus ruled out most of the drawbacks encountered by the use of streptavidin or other avidin family proteins for biotinylation site mapping. Finally, the site of biotinylation on labelled proteins provided an additional level of information about protein–protein interactions and membrane protein topology.
One important additional output from BioSITe experiments is the set of tandem mass spectra acquired from fragmentation of biotinylated peptides. This allows one to distinguish peptides that contain biotin from a complex mixture of unmodified peptides by observing signature or diagnostic fragment ions. PTM-containing peptides have been known to retain unique signature fragment ions during tandem mass spectrometry, which unequivocally identify a given mass spectrum as originating from a specific PTM-modified peptide. For example, phosphotyrosine peptides generates a unique phosphotyrosine immonium ion at m/z 216.04257 by internal fragmentation.10 In addition, specific signature ions have been systematically elucidated for several other PTMs including acetylation11 (m/z 143.118 and 126.092), formylation12 (m/z 112.076), and methylation13 (m/z 143.129, 115.087, 112.087, 74.071 and 70.065). These findings of signature ions and fragmentation characteristics can improve overall PTM site localization, optimize search algorithms and accelerate instrument method development.14 Most of these unique fragment ions are used by site localization algorithms such as phosphoRS15 and AScore.16 Unlike other PTMs, fragmentation of biotinylated peptides has not been studied in detail. Biotinylated peptides are generally identified through dynamic modification of lysine side chains by biotinylation (+226.078 Da) during database searches against reference protein database. Biotinylation at lysine residue results in missed cleavage C-terminal to the modified lysine, which further confirms the presence of lysine biotinylation. Biotinylation of lysine is localized by the presence of a 354 Da mass difference, i.e., biotinylated lysine, between consecutive b- and y-ion fragments. The immonium ion of biotinylated lysine (m/z 327.185) is also used by some search algorithms, which indicates the presence of biotinylated lysine in the identified peptide. Watson et al.17 studied fragmentation of free biotin as a metabolite and reported two fragment ions, dehydrobiotin (m/z 227.085) and its derivative due to loss of thioformaldehyde (m/z 181.093). Fluorophosphinate-linked biotin has been utilized for activity-based profiling of serine hydrolases, and Schoper et al. reported m/z 329.3 for biotinamide with 1 linker arm, m/z 312.2 for loss of amine from m/z 329.3, and m/z 227.2 for dehydrobiotin as marker fragments of fluorophosphinate-biotinylated peptides.18 There is a recent report on signature fragment ions specific to tyrosine biotinylation using biotin-phenol, a biotin analogue.8 These ions were identified as m/z 227.085 for dehydrobiotin, m/z 497.222 for immonium ion of biotinylated tyrosine, and m/z 480.195 for loss of NH3 from immonium ion of biotinylated tyrosine. As no other study has reported signature fragment ions of biotinylated peptides using biotin, there is a need to systematically examine the specificity of signature ions and to identify additional diagnostic fragment ions that are generated upon fragmentation of biotinylated peptides. In this study, we examined the MS/MS spectra of peptides obtained from BioID and APEX experiments, including biotin, isotopically heavy biotin, and biotin-XX-phenol,19 a nonpermeable variant of biotin-phenol experiments, for presence of any signature or diagnostic fragment ions and their derivatives. We have systematically studied these signature fragment ions in biotinylated-bovine serum albumin standard protein as well as in high-throughput biotinylated peptides data acquired using BioSITe method. In addition, we also studied fragment ions of peptides modified with biotin-XX-phenol on tyrosine residues with delta mass of 587.314 Da through the APEX method.
METHODS
Generation of MS/MS Spectra from Biotinylated BSA.
Biotinylated BSA standard was purchased from Sigma-Aldrich (St. Louis, MO) and subjected to in-solution trypsin digestion. Briefly, the samples were reduced using 10 mM dithiothreitol at 37°C for 1 h followed by alkylation using 30 mM iodoacetamide at room temperature in the dark. The samples were then in-solution digested using sequencing grade trypsin (Promega Corporation, Madison, WI) at 1:50 enzyme to substrate ratio overnight at 37°C. The peptide digest was cleaned using C18 stage tips followed by LC–MS analysis on an Orbitrap Fusion Lumos using alternate HCD/CID fragmentation method. Both MS and MS/MS scans were acquired in the Orbitrap mass analyzer at 120000 and 30000 resolution, respectively. The raw MS data were searched against UniProt bovine database including common contaminants using Proteome Discoverer software suite (version 2.2, Thermo Scientific, San Jose, CA). Trypsin was used as a protease allowing up to three missed cleavages. MS and MS/MS mass error tolerance were set at 10 ppm and 0.02 Da, respectively. HCD and CID spectra were searched using different Sequest nodes within the same search workflow. Oxidations of methionine and biotinylation of lysine were set as variable modifications. whereas carbamidomethylation of cysteine was set as a fixed modification. The identified peptides were filtered at 1% false discovery rate. The frequency of fragment ions at low m/z was calculated using in-house scripts as described below.
Enrichment of Biotinylated Peptides Using BioSITe.
Anti-biotin antibody was used for efficient capture of biotinylated peptides from BirA-p190 and BirA-p210 BaF3 cells. Briefly, BaF3 cells either containing BirA-p190 or BirA-p210 constructs were incubated with biotin for 18 h and then lysed in 8 M urea buffer followed by protein estimation using BCA assay. A 10 mg protein sample was reduced using 10 mM DTT followed by alkylation using 30 mM IAA and digested with trypsin overnight at 37°C. The peptide mixture was cleaned using C18 SepPak cartridge followed by lyophilization. The lyophilized peptides were then used for pull-down of biotinylated peptides using anti-biotin antibody as described previously.8 The enriched biotinylated peptides were analyzed on an Orbitrap Fusion Lumos mass spectrometer using a 120 min method in a high–high mode with HCD fragmentation as described above. We also enriched biotinylated peptides from BirA-KRAS construct in HEK293 cells. APEX2 was performed using transgenic mouse primary neurons expressing transmembrane domain-APEX2 construct. The cells were labeled with biotin-XX-phenol (BxxP) and were harvested in urea lysis buffer followed by protein digestion and BioSITe-based enrichment of biotinylated peptides as described earlier. The raw data was searched using Sequest search algorithm against mouse RefSeq protein database containing appropriate BirA-p190 and BirA-p210 sequences and common contaminants in Proteome Discoverer software suite (version 2.2, Thermo Fisher Scientific). Search parameters included trypsin as protease allowing maximum of 3 missed cleavages. MS1 and MS2 mass tolerances were set to 10 ppm and 0.02 Da, respectively. Carbamidomethylation of Cysteine was used as fixed modification while oxidation of methionine and biotinylation of lysine as variable modifications. The identified peptides and proteins were filtered at 1% false discovery rate using decoy database searches. The resulting search results were used for further bioinformatics analysis. Above mentioned pipeline was also followed for experiments involving heavy-biotin (d4) and SILAC (K8R10) technology with respective addition of variable modifications in database searches.
Bioinformatics Analysis for Identification of Signature Fragment Ions.
First, instrument raw files were converted to mzML format using msConvert.20 Peptide identification details from database searches including their modifications were taken and their corresponding MS/MS spectra were extracted from mzML files. Using an in-house developed python programming language script, we tabulated a list of fragment ions with m/z ≤400 and ion intensity of at least 5% of base peak from these MS/MS spectra. We derived their frequency by searching in MS/MS spectrum of biotinylated and non-biotinylated peptide spectrum matches. We then sorted the MS/MS fragment ion masses from high-to-low frequency in biotinylated peptides. MS/MS fragment ions matching to amino acid derivatives such as neutral losses and immonium ions were filtered out. Remaining MS/MS fragment ions were selected for high prevalence in biotinylated peptides by comparing against their frequency in non-biotinylated peptide data. Additionally, intensity of selected fragment ions was extracted. Different thresholds of MS/MS tolerance levels between 5 and 20 ppm were tested to estimate the sensitivity of selected fragments to consider in biotinylated versus non-biotinylated peptide fragment spectra. Selected fragment ions were further validated by surveying their prevalence in many other independent BioSITe experiments. Similar analysis was also carried out to identify the biotinylation-specific fragment ions in MS/MS spectra of BioSITe experiments involving heavy-biotin or SILAC labeling.
RESULTS AND DISCUSSION
Fragment ions of biotin as a compound17 and signature ions of tyrosine or serine biotinylation using biotin derivatives such as biotin-phenol8 or fluorophosphinate-biotin18 have been previously reported. However, diagnostic ions pertaining to lysine biotinylation or biotin analogues in used in various labeling strategies have not been investigated in detail. Here, we investigated diagnostic fragment ions formed due to collision induced dissociation of peptides modified with biotin and biotin-XX-phenol enriched using an anti-biotin antibody approach that we have recently developed, which facilitates detection of such ions. We also studied isotopically heavy biotin as those approaches are getting popular. Figure 1 shows the structures of biotin (1A), heavy-biotin (1B) and biotin-XX-phenol (1C.). Heavy-biotin (+4 Da) contains four deuterium atoms denoted as “D” in Figure 1B.
Figure 1.

Structure of biotin (A), heavy-biotin (B), and biotin-XX-phenol (C) and signature fragment ions of lysine biotinylated peptides biotinylated lysine immonium ion, ImKbio at m/z 327.185, its derivative, ImKbio- NH3 at m/z 310.158 and dehydrobiotin at m/z 227.085 (D).
Identification of Signature Fragment Ions from Lysine Biotinylated Peptides.
We investigated tandem mass spectrometry datasets that we previously generated using BioSITe experiments as described for de novo identification of signature ions that are commonly observed upon fragmentation of lysine biotinylated peptides with delta mass of 226.078 Da.8 We observed three distinct ions that were predominant in most MS/MS spectra of identified biotinylated peptides at m/z 327.185, 310.158, and 227.085. Structural investigation of these fragment ions showed that they are products of biotinylated lysines - immonium ion of biotinylated lysine (ImKbio) at m/z 327.185 and its derivative formed due to loss of ammonia (ImKbio-NH3) at m/z 310.158 and loss of water from biotin fragment ion (dehydrobiotin) at m/z 227.085. Figure 1D shows structures of these signature ions due to fragmentation of lysine biotinylated peptide with collision induced dissociation. Figure 2 shows representative MS/MS spectra from four different biotinylated peptides, AGDSLMVMIAMkMEHTIK from protein methylcrotonoyl-CoA carboxylase subunit alpha, mitochondrial (Mccc1) (Figure 2A), A VGTQALSGAGLLkMFNK from protein sorting nexin-1 (Snx1) (Figure 2B), KFFNkEFLSKPTV from protein cytosolic phospholipase A2 (Pla2g4a) (Figure 2C), and GLVkVNDkEVSDR from protein breakpoint cluster region protein (Bcr) (Figure 2D). We studied the specificity of these signature fragment ions in lysine biotinylated peptides. We found that 99% and 97% lysine biotinylated peptides from BirA-p190 and BirA-KRAS BioSITe datasets showed presence of the ion at m/z 310.158 (Table 1, Figure 3A). This indicates high specificity of m/z 310.158 ion and presence of lysine biotinylation in these peptides. On the other hand, fragment ions m/z 227.085 and m/z 327.185 were found in 88% and 38% of identified biotinylated peptides respectively, from BirA-p190 and in 80% and 14%, respectively, from BirA-KRAS dataset (Unpublished data). We also assessed the frequency distribution of combination of these fragment ions and found that m/z 227.085 and m/z 310.158 fragment ions were found in 87% of identified lysine biotinylated peptides in BirA-p190 dataset, while combinations of m/z 310.158/327.185 and m/z 227.085/327.185 were found in 29% of lysine biotinylated peptides (Table 2). This suggests that more than one signature fragment ion can be efficiently used to confirm lysine biotinylation. We observed similar trend in BirA-KRAS dataset (Table 2). MaxQuant software allows specifying immonium ions as diagnostic signatures along with any custom modifications added to the Andromeda search configurations. Database searches were carried out for BirA-p190 BioSITe dataset using Andromeda with and without diagnostic ions information for biotin modification. We noticed a significant (p-value = 6.4e–10) gain in Andromeda scores for biotinylated peptides when searched with diagnostic ion information as show in Figure 3B,C.
Figure 2.

Representative MS/MS spectra of lysine biotinylated peptides, AGDSLMVMIAMkMEHTIK from protein methylcrotonoyl-CoA carboxylase subunit α, mitochondrial (Mccc1) (A), AVGTQALSGAGLLkMFNK from protein sorting nexin-1 (Snx1) (B), KFFNkEFLSKPTV from protein cytosolic phospholipase A2 (Pla2g4a) (C) and GLVkVNDkEVSDR from protein breakpoint cluster region protein (Bcr) (D). showing the signature fragment ions - biotinylated lysine immonium ion, ImKbio at m/z 327.185, its derivative, ImKbio- NH3 at m/z 310.158 and dehydrobiotin at m/z 227.085.
Table 1.
Frequency Distribution of Signature Fragment Ions from Lysine Biotinylated Peptides Identified from BioSITe Experiments
| total | m/z 227.085 | m/z 310.158 | m/z 327.185 | |
|---|---|---|---|---|
| BirA-p190 | 3813 | 3337 (88%) | 3798 (99%) | 1443 (38%) |
| BirA-KRAS | 1008 | 809 (80%) | 992 (98%) | 144 (14%) |
Figure 3.

Relative intensity of signature fragment ions, ImKbio (m/z, 327.158), ImKbiotin-NH3 at m/z 310.158, and dehydrobiotin at m/z 227.085 of identified lysine biotinylated peptides (A), distribution of Andromeda score for identified biotinylated peptides with/without inclusion of diagnostic ions for MaxQuant database searches (B), quantitative BioSITe experiment with relative intensity of signature fragment ions for light and heavy biotinylated peptides (C), and signature fragment ions from tyrosine biotinylated peptides with BxxP (D); ImYbxxp (m/z, 723.388), ImYbxxp-NH3 at m/z 706.361, biotin-XX at m/z, 453.251, biotin-X at m/z 340.168, and dehydrobiotin at m/z 227.085
Table 2.
Frequency Distribution of Combination of Signature Fragment Ions from Lysine Biotinylated Peptides Identified from BioSITe Experiments
| m/z | |||||
|---|---|---|---|---|---|
| 227.085 and 310.158 | 310.158 and 327.185 | 227.085 and 327.185 | all | ||
| BirA-p190 | no. of peptides | 3337 (87%) | 1443 (43%) | 1431 (43%) | 1431 (43%) |
| BirA-KRAS | no. of peptides | 809 (80%) | 144 (17%) | 140 (17%) | 140 (17%) |
Assessment of Signal Intensity Distribution of Signature Fragment Ions of Lysine Biotinylated Peptides.
One of the properties of diagnostic fragment ions is that they are often quite predominant. Therefore, we investigated the relative signal intensity distribution of the three signature fragment ions, ImKbio, ImKbio-NH3, and dehydrobiotin, across various BioSITe datasets. We found that ImKbio-NH3 showed median ion intensity of 75% that of base peak from identified lysine biotinylated peptides (Figure 3A) indicating the presence of intense ions in identified lysine biotinylated peptides. On the other hand, signal intensities of fragment ions m/z 327.185 and m/z 227.158 relative to base peak were found to be less than 10%. To assess the impact of collision energy on signal intensity of these signature fragment ions, we carried out collision energy ramping experiment using either CID or HCD using biotinylated BSA digest. BSA digest was analyzed in DDA mode using alternate HCD/CID method with varying normalized collision energy (NCE) from NCE = 20 to NCE = 60 (Table 3). As a result, 423 and 284 lysine biotinylated peptides were confirmed by HCD and CID at NCE = 20, respectively. Interestingly, we confirmed that the highest number of biotinylated peptides were confirmed in NCE = 55 by the CID method. In the case of CID, the lowest number of peptides that were confirmed in NCE = 20. The number of confirmed biotinylated peptides were gradually increased as the energy ramped to NCE = 35. There were no specific patterns post NCE = 35 with consistent number of biotinylated peptides (Supplementary Figure 1A). In the case of HCD, it was noted that increased pattern in the number of confirmed biotinylated peptides from NCE = 20 and NCE = 25 with 423 and 442 peptides, respectively. We observed a dramatic increase in the intensity of signature ions including m/z 227.085 and m/z 310.158 with increased collision energy in HCD mode (Supplementary Figure 1B). Understandably, due to over-fragmentation, there was consistent decrease in the intensity of b- and y-ions with a lower number of confirmed biotinylated peptides. Although we see intense signature fragment ions at higher NCE, there is a trade-off for identification of biotinylated peptides.
Table 3.
Number of PSMs and Peptides Identified in CID and HCD Fragmentation Mode with Varying Normalized Collision Energy (NCE)
| activation | NCE (%) | 20 | 25 | 30 | 35 | 40 | 45 | 50 | 55 | 60 |
|---|---|---|---|---|---|---|---|---|---|---|
| CID | no. of PSMs | 721 | 830 | 836 | 956 | 820 | 1064 | 1298 | 1164 | 1287 |
| no. of peptides | 259 | 260 | 264 | 262 | 275 | 297 | 275 | 266 | 275 | |
| HCD | no. of PSMs | 934 | 922 | 854 | 864 | 693 | 530 | 428 | 238 | 145 |
| no.of peptides | 303 | 279 | 273 | 256 | 237 | 180 | 121 | 83 | 57 |
Signature Fragment Ions in Heavy Biotin-Labeled Peptides.
Isotopically labeled heavy biotin has been successfully utilized by our group for quantitation of lysine biotinylated peptides.7 Light and heavy biotinylated peptides showed delta mass of +4 Da (Table 4, Figure 3C, and 4A). Parts B and C of Figure 4 show representative MS/MS spectra of light and heavy biotin-labeled peptide at lysine residue, SQSTSEQEkR, respectively, from breakpoint cluster region protein bcr. The y-ion series (y2 to y8) in MS/MS spectrum of heavy biotin-labeled peptide shows clear shifting +4 Da. We looked for the presence of signature fragment ions in heavy biotinylated peptides. As a result, 4 Da heavier versions of signature fragment ions were observed in heavy biotinylated peptides at m/z 231.110, m/z 314.184 and m/z 331.210 (Figure 4). In the dataset of BirA-p190 and BirA-p210 containing light and heavy biotinylated peptides, we observed that the intensity distribution of heavy versions of signature fragment ions was similar to that of light biotin signature fragment ions. We also observed that signature fragment ions from both light and heavy biotinylated peptides were found in MS/MS spectra of each of the light and heavy biotin labeled peptides with ≥3 charge states. This is due co-fragmentation of these peptides owing to relatively low delta mass of +4 Da and formation of multiply charged peptides due to trypsin inability to cleave at biotinylated lysine residues resulting in peptides with more missed cleavages. At present, only +4 Da heavier version of biotin is commercially available and the co-fragmentation would be minimized by using at least +6 to +10 Da heavier version of biotin. Table 5 lists the various delta masses to be used for database searches of light biotin (+226.078 Da), heavy biotin (+230.031 Da) and light biotin used along with 2plex or 3-plex SILAC reagents (K4–m/z 230.031, K6–m/z 232.013 and K8–m/z 234.092 Da). Supplementary Figure 2 shows representative MS and MS/MS spectra of light and heavy-labeled SILAC peptide, GPQVALkGSR from Neuroblast differentiation-associated protein (AHNAK). The signature fragment ions 310.158 from light peptide and 316.158 from K6-heavy peptide signature fragment ions identified in these labeling situations are also listed in Table 5.
Table 4.
Frequency Distribution of Signature Fragment Ions from Lysine Heavy-Biotinylated Peptides Identified from BioSITe Experiments
| total | 231.110 Da | 314.184 Da | 331.210 Da | |
|---|---|---|---|---|
| BirA-p190 | 2364 | 1828 (77%) | 2342 (99%) | 395 (17%) |
Figure 4.

MS1 spectrum showing light and heavy biotin labeled peptide indicating delta mass of +4 Da (A). MS/MS spectrum of light biotinylated lysine-containing peptide (B) and heavy biotin labeled peptide (C) showing light and heavy (+4 Da) version of biotinylated lysine immonium ion, ImKbio at m/z 327.185 and 331.214 and its derivatives, ImKbio-NH3 at m/z 310.158 and 314.178.
Table 5.
Mass List of Accurate Mass of Signature Fragment Ions Generated Using Various Biotinylation Reagents
| experiment | Δ mass (Da) for db searches | signature fragment ions (m/z) | ||
|---|---|---|---|---|
| dehydrobiotin | ImKbio | ImKbio-NH3 | ||
| biotin | 226.078 | 227.085 | 327.185 | 310.158 |
| heavy biotin | 230.031 | 231.110 | 331.210 | 314.184 |
| SILAC K4 + biotin | 230.031 | 227.085 | 331.210 | 314.184 |
| SILAC K6 + biotin | 232.013 | 227.085 | 333.210 | 316.172 |
| SILAC K8 + biotin | 234.092 | 227.085 | 334.196 | 318.168 |
| dyhydrobiotin | ImYBxxp | ImYBxxp-NH3 | ||
| biotin-XX-phenol | 587.314 | 227.085 | 723.388 | 706.361 |
| biotin-XX | biotin-X | |||
| 453.251 | 340.168 | |||
Signature Fragment Ions Generated Using Biotin-XX-phenol.
While biotin-phenol used for APEX proximity-labeling is membrane-permeabl, the highly reactive biotin-phenoxyl radicals generated by this enzyme are not enabling compartment-specific biotinylation and proteomic mapping of intracellular membrane-bound organelles21 Recently, a membrane-impermeable variant of biotin-phenol, BxxP, was described.19 BxxP contains a long, polar polyamide linker rendering it membrane impermeant enabling cell surface restricted proteomic analysis. The utility of BxxP was initially demonstrated using horseradish peroxidase, which displays higher activity compared to APEX2 and is predicted to have a larger labeling radius. HRP-BxxP biotinylation was used to map the protein composition of the synaptic cleft, a ~20 nm wide extracellular compartment that mediates communication between neurons. In this study, we demonstrate BxxP is also a suitable substrate for APEX2-mediated labeling of surface proteomes. We generated BxxP-labeled proteins using HEK cells expressing surface-targeted APEX2 (TM-APEX2). The BxxP-modified peptides were identified by using variable modification of tyrosine (+587.314 Da). We investigated the resulting BxxP modified peptides for the presence of signature fragment ions. Due to longer chain structure of biotin-XX-phenol, we identified a number of signature fragment ions that result from the many liable amide bonds in the polyamide linker. These include ions at m/z 723.388, 706.361, 453.251, 340.168, and 227.085 (Table 5). After structural analysis of these fragment ions based on the m/z values, we observed that these signature fragment ions were formed due to fragmentation of tyrosine biotinylated residue (Figure 5). Representative MS/MS spectra of tyrosine biotinylated peptides, ESQAyYDGRR from major prion protein precursor (Prnp) (Figure 6A), IIELVPDGAPyITCITK from sodium/potassium-transporting ATPase subunit β−3 (Atp1b3) (Figure 6B), GFQIyDGPIHLTK from Transmembrane protein 2 (Tmem2) (Figure 6C) and YHySSATIPR from Transmembrane protein 178B (Tmem178b) (Figure 6D) clearly showing these unique fragment ions. The relative signal intensity distribution of the signature fragment ions from BxxP-modified peptides was investigated. As a result, we observed that dehydrobiotin (m/z 227.085) ion showed the median intensity of 80%, followed by ImYbxxp-NH3 (m/z 706.361) at 30% and biotin-X (m/z 340.168) at 25%. Another fragment ion, ImYbxxp (m/z 723.388) showed median the intensity of 10% (Figure 3D). Table 6 shows frequency distribution of signature fragment ions from Bxxp-modified peptides. More than 80% of identified Bxxp-modified peptides contain fragments 706.361 and 340.168.
Figure 5.
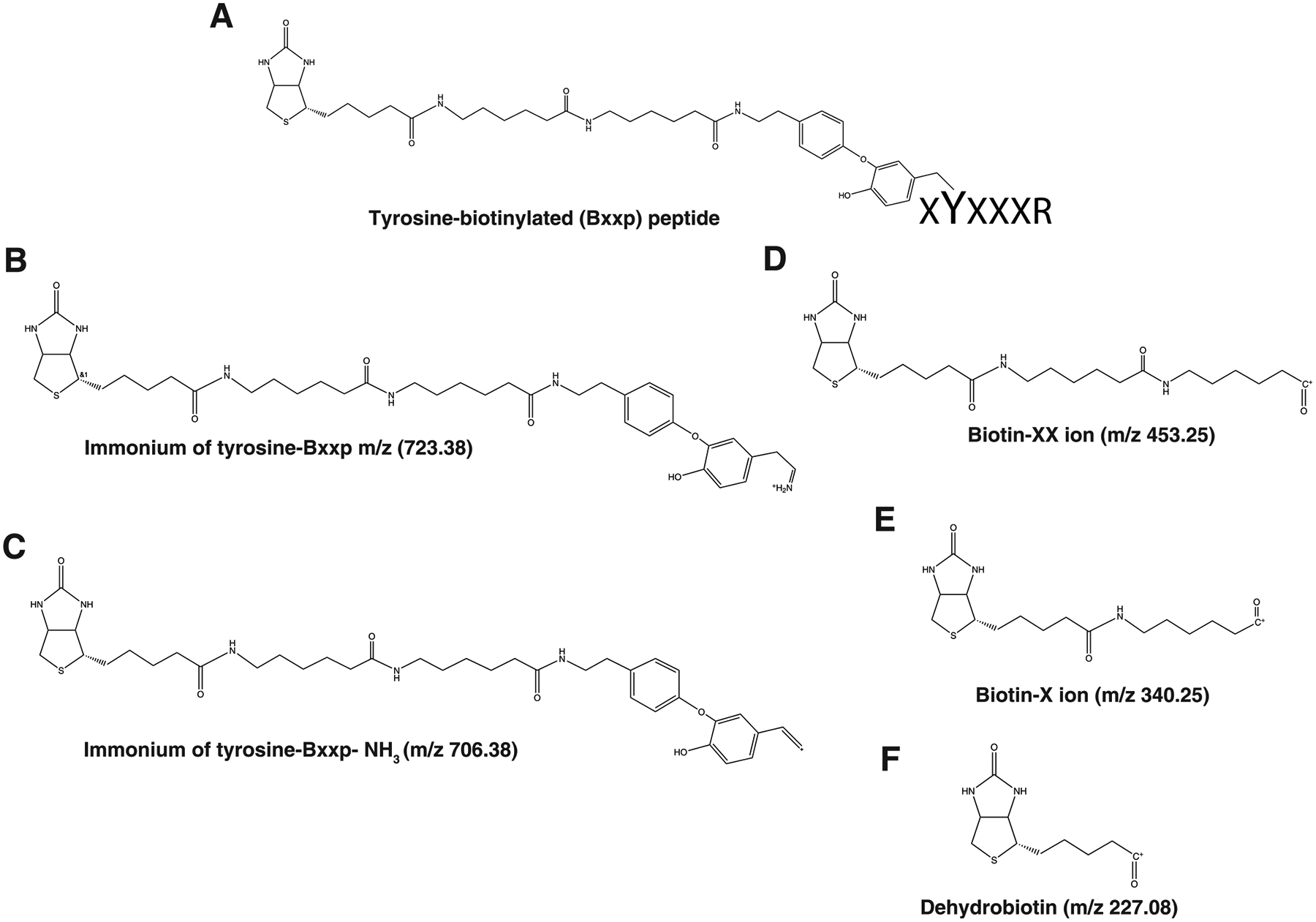
Structure of signature fragment ions identified in tyrosine biotinylated peptides with biotin-XX-phenol, ImYbio (m/z, 723.388), ImYbio- NH3 at m/z 706.361 and dehydrobiotin at m/z 227.085.
Figure 6.

Representative MS/MS spectra of tyrosine biotinylated peptides, ESQAyYDGRR from major prion protein precursor (Prnp) (A), IIELVPDGAPyITCITK from Sodium/potassium-transporting ATPase subunit beta-3 (Atp1b3) (B), GFQIyDGPIHLTK from transmembrane protein 2 (Tmem2) (C) and YHySSATIPR from transmembrane protein 178B (Tmem178b) (D)-biotinylated tyrosine immonium ion, ImYbio at m/z 723.388, its derivative, ImYbio-NH3 at m/z 706.361 and dehydrobiotin at m/z 227.085.
Table 6.
Frequency Distribution of Signature Fragment Ions from Bxxp-Modified Tyrosine Peptides Identified from Apex2 BioSITe Experiment
| m/z | no. of PSMs | no.of peptides |
|---|---|---|
| 227.085 | 883 (99%) | 464 (99%) |
| 310.159 | 56 (6%) | 45 (10%) |
| 340.168 | 826 (93%) | 436 (93%) |
| 453.251 | 113 (13%) | 77 (16%) |
| 706.361 | 782 (88%) | 411 (88%) |
| 723.388 | 473 (53%) | 260 (56%) |
Quantitation of Biotinylated Peptides.
Challenges in quantitative analysis of biotinylation have recently been realized in experiments involving labeling of samples for comparative analysis that employ heavy-biotin or SILAC approaches. Quantitative studies using heavy-biotin to label one sample against unlabeled sample in a BioSITe experiment requires two dynamic modifications (+226.078 Da and +230.031 Da) to be specified in database searches. In case of SILAC experiments (K4R6 and/or K8R10), detection of biotinylation in proteins requires multiple dynamic modifications (+226.078 Da, K4+226.078 Da, K8+226.078 Da) to be specified during database searches to enable identify and quantify them across the study conditions. Current proteomics software platforms including MaxQuant22 and Proteome Discoverer (Thermo Scientific) do not accurately quantitate peptides labeled with variable modifications such as biotinylation, acetylation, methylation on lysine residues coupled with SILAC approach using heavy lysine. Sequest and MaxQuant search engines do identify the biotinylated peptides along with SILAC modifications on lysine given that specific modifications needs to be added for lysine residue. For example, along with biotin (226.08 Da) modification on lysine, SILAC modifications such as +4 Da or +6 Da or +8 Da (depending upon the labeling scheme) need to be added. For cases wherein heavy lysine is modified with biotin, biotin +SILAC modifications are required to identify such peptides. We have observed that due to dual modifications on lysine residues biotinylated peptides containing both biotin and SILAC modification on same lysine were not quantified by MaxQuant and/or Proteome Discoverer. Moreover, identification of biotinylated peptides under these circumstances is also limited by the number of variable modifications that a database search engine generally allows. These issues require attention so that high-throughput confident identification as well as quantification of biotinylated peptides for accurate site mapping would become a routine exercise in studies involving protein–protein interactions.
CONCLUSIONS
Several known post-translational modifications (PTMs) are known to generate characteristic fragment ions during tandem mass spectrometry. Bioinformatics analysis for the identification of such diagnostic ions requires inspecting PTM enriched high-resolution mass spectrometry datasets. High prevalence of PTM fragment ions against a background of unmodified peptide counterparts in the same experiments might reveal specific fragment signatures. Well-characterized fragment ion signatures will assist in validation of the PTM in the peptide fragmentation spectra. But identification of such PTM signatures requires a deep analysis of many independent PTM datasets as well as tools and methods to mine the datasets. BioSITe enables unbiased identification of protein–protein interactions in which proteins proximal to the bait protein undergo biotinylation. In this study, we carried out data analysis for biotin signatures by surveying for the most frequently observed fragment ions in the peptide fragmentation spectra. Our study demonstrated the presence of various signature fragment ions for peptides modified with biotin and biotin-XX-phenol. Many of these fragment ions are observed to be highly specific to biotinylated peptides. These signature fragment ions are valuable in the assessment of identification and confirmation of biotinylation on peptides as well as site mapping. Further work on this is required for utilization of these signature fragment ions in various search algorithms for confident identification of biotinylated peptides in an automated fashion.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Wellcome Trust/DBT India Alliance Margdarshi Fellowship grant IA/M/15/1/502023 awarded to A.P.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.9b00024.
Supporting Figures 1 and 2 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chaiet L; Wolf FJ The Properties of Streptavidin, a Biotin-Binding Protein Produced by Streptomycetes. Arch. Biochem. Biophys 1964, 106, 1–5. [DOI] [PubMed] [Google Scholar]
- (2).Cronan JE Jr. Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J. Biol. Chem 1990, 265 (18), 10327–33. [PubMed] [Google Scholar]
- (3).Roux KJ; et al. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 2012, 196 (6), 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim DI; et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27 (8), 1188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lobingier BT; et al. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169 (2), 350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ramanathan M; et al. RNA-protein interaction detection in living cells. Nat. Methods 2018, 15 (3), 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lam SS; et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12 (1), 51–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kim DI; et al. BioSITe: A Method for Direct Detection and Quantitation of Site-Specific Biotinylation. J. Proteome Res 2018, 17 (2), 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Udeshi ND; et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods 2017, 14 (12), 1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Steen H; et al. Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode. Anal. Chem 2001, 73 (7), 1440–8. [DOI] [PubMed] [Google Scholar]
- (11).Trelle MB; Jensen ON Utility of immonium ions for assignment of epsilon-N-acetyllysine-containing peptides by tandem mass spectrometry. Anal. Chem 2008, 80 (9), 3422–30. [DOI] [PubMed] [Google Scholar]
- (12).Kelstrup CD; et al. Analytical utility of mass spectral binning in proteomic experiments by SPectral Immonium Ion Detection (SPIID). Mol. Cell. Proteomics 2014, 13 (8), 1914–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Matthiesen R; et al. VEMS 3.0: algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J. Proteome Res 2005, 4 (6), 2338–47. [DOI] [PubMed] [Google Scholar]
- (14).Kim MS; Zhong J; Pandey A Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics 2016, 16 (5), 700–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Taus T; et al. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res 2011, 10 (12), 5354–62. [DOI] [PubMed] [Google Scholar]
- (16).Beausoleil SA; et al. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 2006, 24 (10), 1285–92. [DOI] [PubMed] [Google Scholar]
- (17).Watson DG A rough guide to metabolite identification using high resolution liquid chromatography mass spectrometry in metabolomic profiling in metazoans. Comput. Struct. Biotechnol. J 2013, 4, e201301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schopfer LM; et al. Characteristic mass spectral fragments of the organophosphorus agent FP-biotin and FP-biotinylated peptides from trypsin and bovine albumin (Tyr410). Anal. Biochem 2005, 345 (1), 122–32. [DOI] [PubMed] [Google Scholar]
- (19).Loh KH; et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 2016, 166 (5), 1295–1307.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Adusumilli R; Mallick P Data Conversion with ProteoWizard msConvert. Methods Mol. Biol 2017, 1550, 339–368. [DOI] [PubMed] [Google Scholar]
- (21).Rhee HW; et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339 (6125), 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26 (12), 1367–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.