

Introduction
Sleep is an evolutionarily conserved and powerful drive found widely in the animal kingdom (Lesku et al., 2008). Although sleep loss impairs brain, immune, and metabolic function, its complete functions are still unknown (Everson et al., 1989; Greene and Siegel, 2004). One possible function of sleep is that it promotes brain development (Roffwarg et al., 1966). The amount of sleep is greatest during ages when the brain is rapidly developing. In addition, sleep amounts are maximal and/or change dramatically in terms of organization and regulation during critical periods of synaptic plasticity (Frank, 2017). Sleep and sleep loss have been shown to influence critical period plasticity, supporting a role for sleep in brain development (Frank et al., 2001) and suggesting that abnormal sleep in early life may lead to abnormal development.
Autism Spectrum Disorder (ASD) is the most prevalent neurodevelopmental disorder in the United States and it is estimated that insomnia affects 44-86% of the ASD population (Maxwell-Horn and Malow, 2017; Souders et al., 2017). Sleep problems are predictive of severity of ASD core symptoms, social deficits and repetitive behaviors, and associated behavioral issues such as tantrums and aggression (Tudor et al., 2012; Cohen et al., 2014; Veatch et al., 2017; Mazurek et al., 2019). Sleep problems impact the quality of life of both ASD individuals and their caregivers, thus it is important to understand why they are so prevalent. One possibility is that the underlying cellular and molecular abnormalities causal to ASD also produce abnormal sleep (i.e., abnormal sleep is one of many outcomes of an underlying problem). Considering the potential role of sleep in brain development and that early sleep disruption has been shown to impair social bonding in animal models (Jones et al., 2019), it is also possible that abnormal sleep plays a causal role in ASD. A third non-mutually exclusive possibility is that there is an interaction between the genetic variants linked to ASD and the role of sleep early in life, which influences both the presence of sleep problems and the severity of ASD.
In this review, we explore the role of sleep in early life as a causal factor in ASD. First, we review fundamental steps in mammalian sleep ontogeny and regulation and how sleep influences brain development. Next, we summarize current knowledge gained from studying sleep in animal models of ASD. Ultimately, our goal is to highlight the importance of understanding the role of sleep in brain development and the use of animal models to provide mechanistic insight into the origin of sleep problems in ASD.
Fundamentals of mammalian sleep ontogeny
Changes in sleep and wake expression
The amount of sleep changes as an organism develops. In all mammals there are a number of conserved changes that occur in sleep in the perinatal period. These include changes in time spent in different sleep states, changes in sleeping brain activity, and changes in sleep and wake consolidation. Sleep states as defined using electroencephalographic recordings (EEG, see Box1) do not emerge in rats until postnatal days 12-16; and are preceded by relatively undifferentiated states termed active sleep (AS) and quiet sleep (QS) which are mostly behaviorally defined. An adultlike sleep cycle with EEG defined sleep states emerges between the second and third week of life in rodents (Frank and Heller, 1997) and around 3 months in humans (Roffwarg et al., 1966). Figure 1 shows the amount of time spent in each brain state as a function of age in both humans and rats. Neonates can spend up to fifty percent of their total time asleep in REMS (Roffwarg et al., 1966). The percentage of REMS that humans obtain in a day is largest in infant years and sharply diminishes before adulthood (Iglowstein et al., 2003). In contrast, the amount of NREMS increases slightly in the early years but remains more constant through one’s lifetime (Roffwarg et al., 1966; Frank and Heller, 1997).
Box 1. Fundamentals of mammalian sleep.
Definition of Sleep:
Sleep is a state of reduced mobility and responsiveness to stimuli that can be easily reversed and when prevented organisms try to recover it.
Definition of sleep states:
In mammals, sleep states are traditionally defined by measuring brain activity using electroencephalogram (EEG) and muscle activity using electromyogram (EMG) (Siegel, 2009):
Wakefulness, EEG patterns of high frequency and low amplitude.
Rapid-eye-movement sleep (REMS), brain activity patterns similar to wake along with myotonia from the neck down.
Non rapid-eye-movement sleep (NREMS), brain activity patterns with low frequency and high amplitude oscillations.
Figure 1.
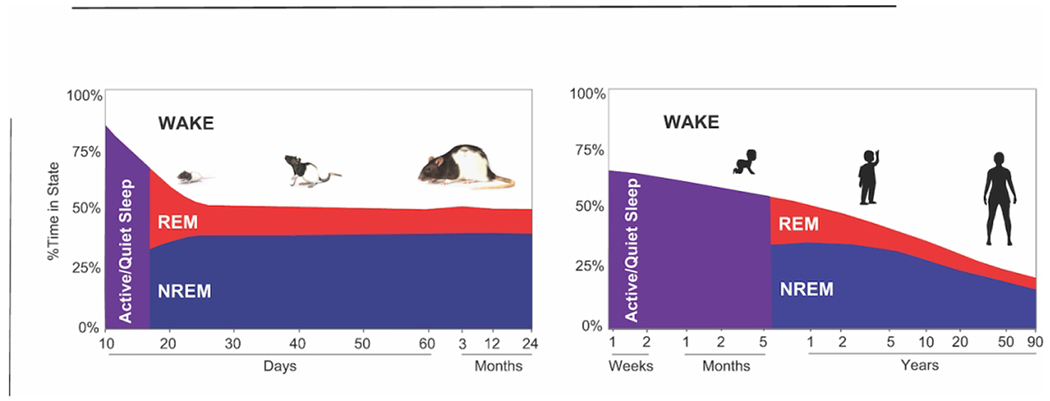
Sleep time as a function of age. Percent time over 24 hours spent in wakefulness (white), rapid eye movement sleep (REMS; red) and non-rapid eye movement sleep (NREMS; blue) on the y-axis. In purple: age ranges in which sleep states are not identifiable using EEG. The x-axis represents age. On the left, sleep time across the lifespan in rats, summarizing studies at the following age ranges: P9 (Seelke and Blumberg, 2008), P12-P60 (Frank and Heller, 1997), 3-12 months (Zepelin et al., 1972). On the right, sleep time across the lifespan in humans, adapted from (Roffwarg et al., 1966).
Maturation of sleep regulatory mechanisms
Sleep is under both circadian and homeostatic control. Circadian rhythms determine the timing of sleep and wake in a 24-hour period while the sleep homeostat regulates the need to sleep (sleepiness) as a function of time spent awake (Achermann and Borbély, 2017). The power in the low frequency range of EEG recordings in NREMS, known as delta power, is a well-known correlate of sleep (Achermann and Borbély, 2017). In adults, delta power increases proportionately to the amount of time spent awake and decreases with subsequent NREMS (recovery sleep). However, the adult sleep regulatory mechanisms are not present at birth and mature at different rates. In rats, the circadian regulation of sleep does not begin to emerge until postnatal day 17 (P17). This is followed by the development of sleep homeostasis. For example, the increase in delta power in response to sleep deprivation does not appear until P24 in rats as shown in Figure 2 (Frank et al., 2016). Thus, the ability to regulate sleep in response to both time of day and sleep need emerge after the rapid decline in sleep amounts (in particular REMS) during early life and after both cortical neurogenesis and myelination are mostly completed. This suggests that problems regulating sleep need (falling asleep when sleepy) would likely emerge later in life, since the sleep regulatory mechanisms as defined in adults are not present until near weaning.
Figure 2. Timeline of development of sleep homeostasis in rats.
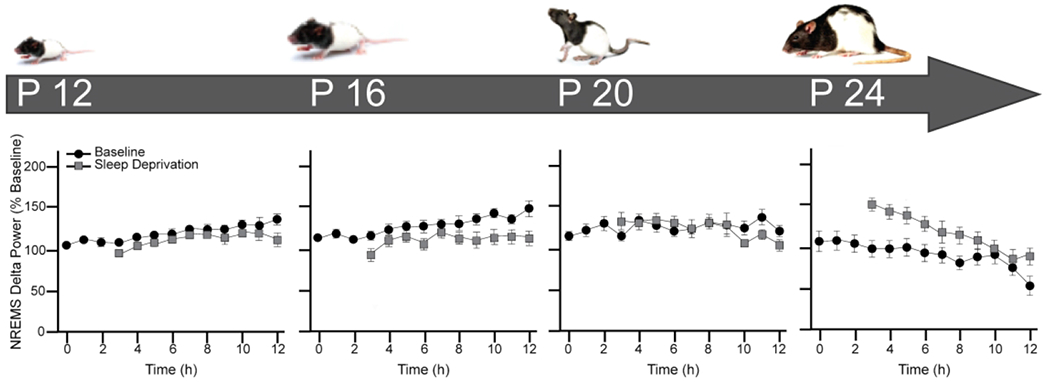
On top, age from birth: postnatal days 12 (P12), 16 (P16), 20 (P20) and 24 (P24) Below, graphs of changes in non-rapid eye movement sleep (NREMS) delta power (0.5-4.0Hz) after 3 hours of sleep deprivation (SD) corresponding to the different post-weaning ages (P12-P24) depicted above in developing rats. Mean ± SEM NREMS delta power is expressed as percent of the 12 hours mean NREMS delta power value before sleep deprivation at hours 0-3. Hour 0 represents the first hour of light period. Data originally reported by (Frank and Heller, 1997).
The role of sleep in brain development
Functional studies provide direct evidence that sleep is important for the developing brain and in particular for synaptic plasticity. Sleep has been shown to enhance cortical plasticity during the visual critical period (Frank et al., 2001). This appears to be highly dependent on REMS (Dumoulin Bridi et al., 2015). Other studies, as summarized in Table 1, have shown that REMS deprivation prolongs developmentally regulated cortical plasticity in situ. REMS has also been shown to increase synaptic pruning in adolescent mice (Li et al., 2017). Last, early life sleep deprivation has been shown to influence social bonding in adulthood (Jones et al., 2019). Overall these studies provide evidence that sleep contributes to brain development and that lack of sleep during early life may have long lasting effects on human behavior.
Table 1.
Summary of functional studies that support the role of sleep in brain development or developmental cortical plasticity. Studies are divided into those that analyze the role of NREMS or REMS.
| NREMS | REMS |
|---|---|
| NREMS enhances synaptic plasticity in the cortex of kittens (Marcos G. Frank, Issa, and Stryker 2001) | One week of REMS deprivation in immature rats prolongs the critical period for developmentally regulated synaptic plasticity (Shaffery et al. 2002) |
| Chronic sleep deprivation causes a decrease in hippocampal volume in adolescent rats (Novati et al. 2011) | REMS enhances experience-dependent plasticity in developing cerebral cortex in kittens (Dumoulin Bridi et al, 2015b) |
| REMS prunes new formed postsynaptic dendritic spines in mouse motor cortex during development and after motor learning (Li et al. 2017) | |
| Pharmacological sleep deprivation in rats 1-3 weeks of age results in behavioral changes and sleep disturbances into adulthood (Mirmiran 1986b) Early-life sleep disruption impairs social bonding. (Jones et el. 2019) | |
Insomnia in Autism Spectrum Disorder
Today, one in 59 children are diagnosed with ASD at age 8 (Baio et al., 2018) and it is estimated that up to 86% of this population suffers from at least one reported sleep problem (Maxwell-Horn and Malow, 2017; Souders et al., 2017). Individuals with ASD often sleep less and have problems falling and staying asleep. Insomnia in children is defined as a sleep latency longer than 30 minutes and/or frequent night awakenings that reduces sleep efficiency (the amount of time actually spent asleep in a night) and interferes with daytime functioning (Owens and Mindell, 2011). Insomnia prevalence in ASD ranges between 40 to 86% (Johnson et al., 2009; Richdale and Schreck, 2009; Maxwell-Horn and Malow, 2017; Souders et al., 2017) which is 2 to 3 times more prevalent than in typical development (Owens and Mindell, 2011). The majority of data regarding prevalence of insomnia in ASD has been collected using sleep questionnaires. However, more recent studies using actigraphy and polysomnography (PSG, which uses EEG, EMG, and heart rhythm monitoring) support the findings that ASD individuals have shorter sleep time, longer sleep latency, and decreased sleep efficiency relative to typical development (Elrod and Hood, 2015; Singh and Zimmerman, 2015). Sleep problems are predictive of severity of ASD (Tudor et al., 2012; Cohen et al., 2014; Veatch et al., 2017; Mazurek et al., 2019) and greatly impact the quality of life of both ASD individuals and their caregivers. Studies have shown that sleep problems in individuals with ASD also alter parents sleep and increase stress (Meltzer, 2008). However, why insomnia is so prevalent in ASD is unknown.
Is there a link between abnormal sleep development and ASD?
Many neurodevelopmental processes altered in ASD, such as neurogenesis, neuronal migration, synaptogenesis and synaptic plasticity (Gilbert and Man, 2017), occur at ages when sleep is the predominant brain state. In rats, neurogenesis starts at embryonic day 9.5 and is complete by P15 while myelination occurs between P10 and P20 (Semple et al., 2013); coinciding with the emergence of EEG identifiable NREMS and REMS and the maturation of sleep homeostasis. Another important process for brain development and ASD pathophysiology is the balance between excitatory and inhibitory neurotransmission, in particular the role of the neurotransmitter GABA (Coghlan et al., 2012). Abnormal GABA neurotransmission as mechanism for ASD pathogenesis has been widely studied in rodent models of Fragile X syndrome (FXS). These models exhibit extensive dampening of the GABAergic system (Paluszkiewicz et al., 2011). Although GABA is the main inhibitory neurotransmitter in the adult brain, it can be excitatory in the immature central nervous system. This paradoxical action of GABA may help developing neurons fire together, form synapses and cortical networks. The developmental switch in GABA polarity is estimated to occur in rats in the second postnatal week (Ben-Ari et al., 2007) and has been shown to be delayed in a mouse model of FXS (He et al., 2014). Inhibitory synaptogenesis continues after the switch in polarity and is characterized by a rapid increase in synapse number and maturation that terminates around the end of the 4th postnatal week in rats (De Felipe et al., 1997). As reviewed above, all key stages of sleep ontogeny occur between the second and fourth week of life in rodents. In other words, between the GABA switch and the end of inhibitory synaptogenesis.
The timing of these events and the established role of GABA as the primary neurotransmitter of sleep promoting nuclei (Schwartz and Kilduff, 2015) raises the possibility that the development of inhibitory input into the cortex and the role of sleep in brain development are related. GABAergic interneuron development has been shown to be sensitive to sleep disruption in both kittens and young voles (Hogan et al., 2001; Jones et al., 2019). The role of sleep in forms of developmental synaptic plasticity dependent upon inhibitory circuits is well established (Frank et al., 2001). In summary, events required for normal brain development, which are altered in ASD, occur when sleep amounts are at their highest and when all fundamental aspects of sleep ontogeny and maturation occur. Therefore, sleep may have a causal role in network dysfunction in ASD. The mechanisms, however, are still unknown. The use of animal models holds great promise to uncover the mechanisms that link sleep, development and ASD.
Sleep studies in animal models of ASD
Animal models are a useful tool to understand mechanisms underlying core symptoms and co-morbid problems in ASD (Crawley, 2012). Clinically valid animal models are those that have both construct and face validity. Construct validity requires that the animal model is generated with the same underlying biological cause, e.g. the targeted mutations in the animal model replicate the causal genetic variant in the patient population. Face validity ensures that the model’s behavior is analogous to the symptoms of the human disease (Chadman et al., 2009). To study sleep problems in ASD we need to use models with both construct and face validity. We focused on sleep and circadian activity studies performed in rodent models of mutations in genes that have been linked to ASD according to the Simons Foundation Autism Research Initiative (SFARI) gene database, and examined whether they showed reduced sleep time, fragmented sleep and longer latency to fall asleep. In Table 2, we report the animal models in which sleep studies were performed using EEG & EMG, the standard for analyzing sleep in mammals. There is a great amount of variability in sleep phenotypes among the different ASD rodent models. For example, the mutant rat models of CDFE (Cntnap2) show longer waking periods whereas the mutant mice show fragmented wakefulness (Thomas et al., 2017). The Neuroligin-1 knockout mice report decreased wakefulness whereas the Neuroligin-2 knockout mice have increased wakefulness and the Neuroligin-3 mutant mice have no changes in sleep state (El Helou et al., 2013; Liu et al., 2017; Thomas et al., 2017; Seok et al., 2018). It is also important to note that based on the SFARI gene evidence scores, not all genes have the same strength of evidence linking them to ASD which could be a factor affecting the variability in data. In Table 3, we summarize published findings performed using activity monitoring to study sleep and circadian rhythms. The results were categorized by studies performed in standard light:dark conditions or studies performed in the absence of light to investigate the endogenous activity of the circadian clock. Most rodent models display total reduced activity, which could be indicative of reduced sleep. However, that could simply reflect reduced mobility. Therefore, it is hard to draw conclusions regarding sleep from studies that only use actigraphy in rodents.
Table 2.
Summary of published studies using EEG recordings to measure sleep in rodent models of ASD. Only genes and copy number variants (CNVs) reported in the Simons Foundation Autism Research Initiative gene database (SFARI GENE) are reported. Out of the 304 mouse/rat CNV/gene models reported, 22 have been used to study sleep using EEG. SFARI evidence scores categorize genes based on evidence supporting a gene’s relevance to ASD risk. Categories defined by SFARI database are as follows: 1 (High Confidence), 2 (Strong Candidate), 3 (Suggestive Evidence), 4 (Minimal Evidence), 5 (Hypothesized), S (Syndromic) (Wisor et al., 2002; Dudley et al., 2003; Lee et al., 2004; Anderson et al., 2005; Colas et al., 2005; Lonart et al., 2008; Kimura et al., 2010; Papale et al., 2010, 2013; El Helou et al., 2013; Johnston et al., 2014; Zhou et al., 2014; Ehlen et al., 2015; Kalume et al., 2015; Kumar et al., 2015; Jaaro-Peled et al., 2016; Tatsuki et al., 2016; Angelakos et al., 2017; Diessler et al., 2017; Dittrich et al., 2017; Holst et al., 2017; Liu et al., 2017; Thomas et al., 2017, n.d.; Boone et al., 2018; Seok et al., 2018; Ingiosi et al., 2019; Lu et al., 2019).
| CNV/Gene | PMID | SFARI Evidence Score | Total sleep time | Sleep fragmentation | Latency to sleep |
|---|---|---|---|---|---|
| 16p11.2 (Mouse) | 27739237 | N/A | Increased wakefulness, decreased NREMS | Consolidation of wakefulness | Not reported |
| 16p11.2 (Mouse) | 30541142 | N/A | Increased wakefulness, decreased REMS& NREMS | Decreased REMS bout duration and number | Not reported |
| Fmr1 (Mouse) | 29775702 | S | Decreased REMS | Decreased number of REMS bouts | Not reported |
| Shank3 (Mouse) | 30973326 | 1, S | Decreased total sleep time during light and dark | Decreased REMS bouts at baseline and shorter NREMS and REMS bouts in the dark period | Increased latency to NREMS following SD |
| Mecp2 (Mouse) | 25018705 | 1 | No difference across 24 hours. Increased wakefulness in dark period | Shorter REMS bouts in light period, longer REMS bouts in dark period | Increased latency to NREMS at lights on |
| Ube3a (Mouse) | 26446213 | 1 | Decreased REMS | Decreased number of REMS bouts. Fragmented NREMS during dark period | Not reported |
| Ube3a (Mouse) | 15921919 | 1 | Decreased NREMS | Increased wakefulness and NREMS bouts. Decreased duration of bouts in dark period | Not reported |
| Cacna1c (Mouse) | 25845695 | 1 | No difference in total sleep time. Decreased REMS after SD | Increased number and shorter NREMS bouts | Not reported |
| Rai1 (Mouse) | 28548639 | 1 | No difference over 24 hours. Decreased wakefulness in light | Not reported | Not reported |
| Rims1 (Mouse) | 18495360 | 1 | Decreased NREMS in dark period. Decreased REMS in 24 hours | Decreased number of NREMS and REMS bouts | Not reported |
| Scn1a (Mouse) | 25766678 | 1 | No difference | Increased wakefulness bouts | Not reported |
| Scn1a | 23311867 | 1 | Increased wakefulness, decreased NREMS and REMS in dark period. No difference after SD | No difference | Not reported |
| Scn8a (Mouse) | 20353942 | 1 | Decreased wakefulness in dark period. Increased NREMS. Decreased REMS in liqht period | Decreased number and duration of REMS bouts during the light period | Not reported |
| Cntnap2 (Mouse) | 28364455 | 2, S | No difference in time in state | Fragmentated wake | Decreased latency to sleep at lights on |
| Cntnap2 (Rat) | 28364455 | 2, S | No difference in time in state | Consolidation of wakefulness and REMS | No difference in latency to NREMS. Increased latency to REMS lights on |
| Cacna1h (Mouse) | 26996081 | 2 | Decrease sleep duration | Not reported | Not reported |
| Disc1 (Mouse) | 28720848 | 2 | Decreased NREMS, increased REMS | Increased REMS and wakefulness bouts. Decreased REMS bout duration | No difference |
| Disc1 (Mouse) | 27354230 | 2 | Increased wakefulness and NREMS No difference after 2 h SD | Not reported | Not Reported |
| Gabrb3 (Mouse) | 12419540 | 2 | Decreased REMS during light | Decreased number of REMS bouts during light period | Not reported |
| Nlgn3 (Mouse) | 28385162 | 2 | No difference in time in state | No sleep fragmentation | Not reported |
| Nlgn3 (Rat) | 28958035 | 2 | Decreased NREMS, increased REMS during light period | Longer wake and REMS bouts. No difference in NREMS bouts | No difference in latency to NREMS o REMS at light onset |
| Nlgn1 (Mouse) | 23716671 | 3 | Decreased wakefulness, increased NREMS. No difference after SD | No difference | No difference at baseline or after SD |
| Camk2a (Mouse) | 19455148 | 3, S | Decreased NREMS during the light period, increased REMS during the dark period. Decreased NREMS and increased REMS after SD | Not reported | Not reported |
| Cacna1g (Mouse) | 15677322 | 3 | Decreased NREMS | Fragmentation of NREMS | Increased latency to NREMS at light onset |
| Cacna1g (Mouse) | 15601764 | 3 | Decreased NREMS in light period | Longer wakefulness bouts during NREMS | No difference |
| Csnk1e (Mouse) | 24744456 | 3 | Increased REMS in the dark period | Decreased NREMS bouts in light period. Increased REMS bouts in dark period | Not reported |
| Grm5 (Mouse) | 28980941 | 3 | Increased NREMS,decreased REMS and wakefulness during light period. Increased wakefulness in dark period after SD | Not reported | Not reported |
| Nlgn2 (Mouse) | 30231918 | 4 | Increased wakefulness, decreased REMS. Decreased NREMS in dark | Consolidation of NREMS bouts in light period and consolidation of wakefulness in dark period | Not reported |
| Npas2 (Mouse) | 12843397 | 4 | Decreased NREMS and REMS in dark period | Not reported | Not reported |
Table 3.
Summary of published studies using activity monitoring in 12:12 hours light:dark cycle and in the absence of light to measure sleep and circadian rhythms in rodent models of ASD. Only genes and copy number variants (CNVs) reported in the Simons Foundation Autism Research Initiative gene database (SFARI GENE) are reported. Out of the 304 mouse/rat CNV/gene models reported, 17 have been used to study circadian rhythms. SFARI evidence scores categorize genes based on evidence supporting a gene’s relevance to ASD risk. Categories defined by SFARI database are as follows: 1 (High Confidence), 2 (Strong Candidate), 3 (Suggestive Evidence), 4 (Minimal Evidence), 5 (Hypothesized), S (Syndromic) (Zheng et al., 2001; Dudley et al., 2003; Meredith et al., 2006; Kozlov et al., 2007; Zhang et al., 2008; Sahar et al., 2011; Han et al., 2012; Wither et al., 2012; Lacaria et al., 2013, 2013, 2013 p.1; Ehlen et al., 2015; Kalume et al., 2015; Li et al., 2015; Tsuchiya et al., 2015; Jaaro-Peled et al., 2016; Diessler et al., 2017; Lipton et al., 2017; Saré et al., 2017; Ingiosi et al., 2019).
| CNV/Gene | PMID | SFARI Evidence | Activity monitoring in 12:12 h light:dark cycle | Activity monitoring in the absence of light |
|---|---|---|---|---|
| 17p11.2 (Mouse) | 23703963 | N/A | Reduced total running wheel activity | Reduced free running period length. Reduced total running wheel activity |
| Shank3 (Mouse) | 30973326 | 1, S | Reduced amplitude of running wheel activity | Reduced amplitude of running wheel activity with normal period length |
| Fmr1 (Mouse) | 28919851 | S | Shorter sleep duration in light period under homecage activity monitoring | Not recorded |
| Fmr1 (Mouse) | 18589395 | S | Fmr1 and Fxr2 knock out mice show no difference in wheel running compared to wild types. Fmr1/Fxr2 double knock out mice show loss of rhythmic activity | Fmr1 and Fxr2 knock out mice show shorter-free running period in constant dark and loss of rhythmic activity. Fmr1/Fxr2 double knock our mice show loss rhythmic activity |
| Tsc1andTsc2 (Mouse) | 28746872 | 1 | No difference in wheel running activity | Shorter free running period with no change in circadian amplitude or overall activity |
| Ube3a (Mouse) | 26446213 | 1 | No difference in circadian period or overall activity | No difference in circadian period or overall activity |
| Mecp2 (Mouse) | 26456390 | 1 | No difference in circadian rhythm | No difference in period length. Decrease in wheel running activity |
| Mecp2 (Mouse) | 25779967 | 1 | Reduced total wheel running activity, reduced alpha length and increased fragmentation of wheel running | Reduced total wheel running activity. Increase in-free running period length, decreased alpha length and increased fragmentation of wheel running |
| Mecp2 (Mouse) | 22523589 | 1 | Decrease in activity bout length but no difference in number of bouts (home cage activity monitoring) | Not reported |
| Rai1 (Mouse) | 28548639 | 1 | Decreased overall running wheel activity, decreased running wheel activity in the light period | Increase phase advance and increase in alpha length |
| Rai1 (Mouse) | 23703963 | 1 | Decrease total running wheel activity | Decreased period length and decreased total running wheel activity |
| Scn1a (Mouse) | 22223655 | 1 | Delayed phase of activity onset. Reduced total wheel running activity | Increased period length. Reduced total wheel running activity |
| Scn1a (Mouse) | 25766678 | 1 | Reduced total wheel running activity | Reduced total wheel running activity |
| Disc1 (Mouse) | 27354230 | 2 | Decreased activity during the dark period | No difference in circadian period |
| Magel2 (Mouse) | 17893678 | 2, S | No difference in wheel running activity | Reduced amplitude of running wheel activity with reduced activity in the subjective dark period |
| Cd38 (Mouse) | 21937766 | 3 | Reduced total locomotor activity | Shorter period length |
| Kcnma1 (Mouse) | 16845385 | 3 | Decreased amplitude of locomotor activity | Increase in period length. Decreased amplitude of locomotor activity |
| Npas2 (Mouse) | 12843397 | 4 | Increased running wheel activity during the dark period | Shorter period length |
| Per1 & Per2 (Mouse) | 11389837 | 4 | No difference in wheel running activity | PER1 mutants have a shorter period length. PER1 mutants and PER1/PER2 double mutants have no circadian rhythm |
| Rasd1 (Mouse) | 23703963 | 5 | No difference in wheel running | Female mice displayed reduced free running period lengths |
Overall, among all studies that looked at sleep in rodent models using EEG, very few show reduced sleep time, sleep fragmentation and longer latency to fall asleep. The following rodent models display reduced sleep time: 16p11.2, Shank3, Fmr1, Mecp2, Ube3a, Rims1, Scn1a, Scn8a, Disc1, Gabrb3, Nrlgn3, Camk2a, Cacna1g, Nlgn2, Npas2. From those, only 5 (Shank3, Ube3a, Scn1a, Disc1, Cacna1g) reported increased sleep fragmentation. In addition, only 3 (Shank3, Nlgn3, Cacna1g) looked at sleep latency and only 2 (Shank3, Cacna1g) show an increase in latency to fall asleep. There are two possible non-mutually exclusive explanations regarding the lack of prevalence of sleep problems in ASD rodent models relative to what has been observed in the clinical population: 1) some genes linked to ASD are not linked to sleep problems 2) some of the ASD animal models do have face validity but either the sleep phenotype or some of the relevant features in regards to ASD were not evaluated.
To find animal models with both face and construct validity with the goal of unraveling the mechanisms behind sleep problems in ASD, it is important that standardized protocols and consistent criteria be used to study sleep. Figure 3 outlines an ideal protocol for conducting sleep studies in rodents. A basic protocol of sleep phenotyping needs to include at least 48 hours of continuous EEG and EMG recordings. Sleep states (NREMS, REMS and wake) should be accurately determined, which cannot be done using automated software at present. Proper quantification of sleep parameters, including time in state, sleep fragmentation, latency to sleep and spectral power, should be performed using properly powered statistics. A 24-hour baseline sleep study should be performed to assess the spontaneous sleep patterns of the animals. This should be followed up by a 24-hour sleep deprivation study. The latter should include a period of sleep deprivation (5-6 hours) and recovery sleep for the remainder of the 24 hours, to assess the animal’s homeostatic response. A key component of sleep phenotyping that is missing from most studies is the study of the sleep homeostatic response. Increase latency to fall asleep is a commonly reported sleep problem in ASD, yet many of the animal models in Table 2 do not report latency to fall asleep or do so without performing sleep deprivation. Sleep latency should be evaluated following sleep deprivation. The reason is that latency can only be compared across animals if the prior wake history is the same (Mang and Franken, 2015). Sleep latency is also a marker for sleep homeostasis and should decrease with increased sleep pressure. Therefore, it is possible that some of the rodent models in Table 2 will show longer latencies to fall asleep, if the homeostatic response to sleep deprivation was evaluated.
Figure 3. Outline of experimental design for sleep studies in rodent models.
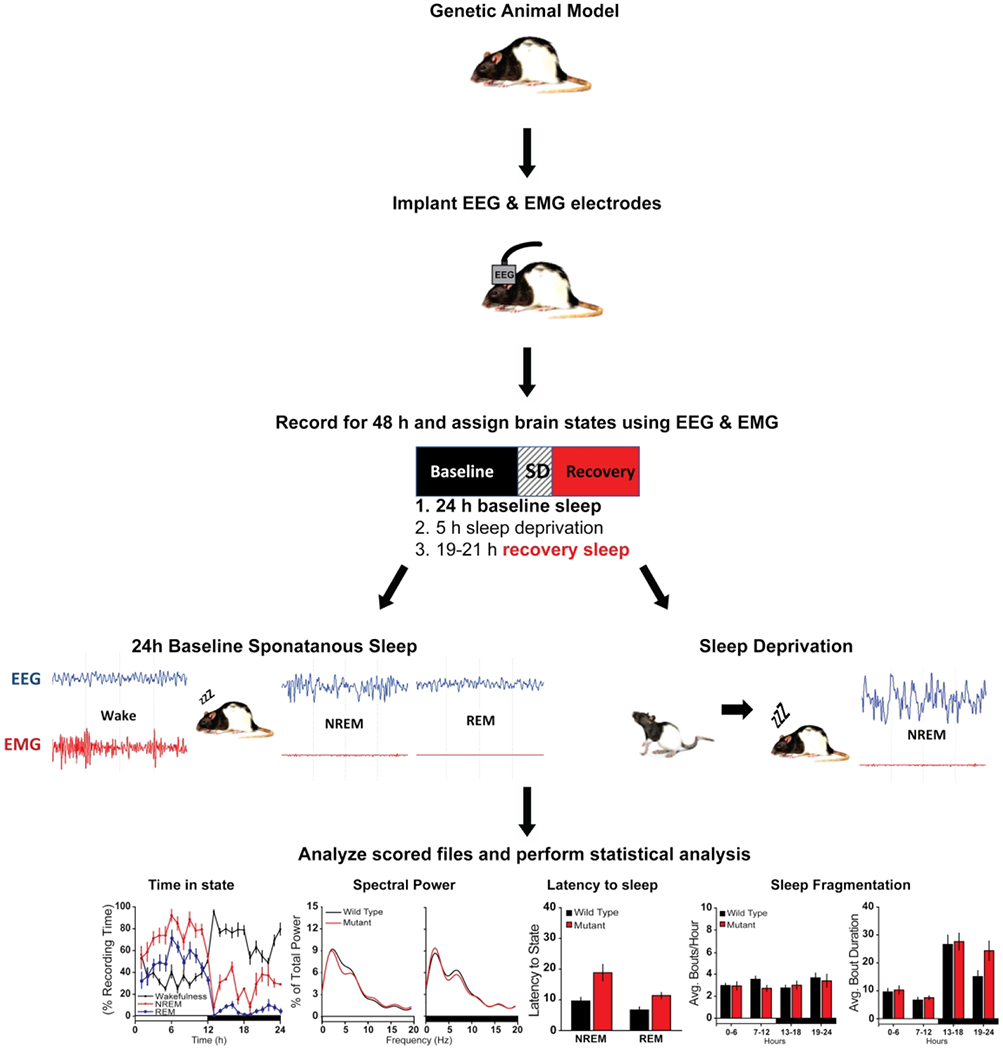
Mice are implanted with electroencephalographic (EEG) and electromyographic (EMG) electrodes under anesthesia. Four EEG electrodes are placed contralaterally over frontal (2) and parietal (2) cortices, and two EMG electrodes are inserted in the nuchal muscles. Mice are allowed 5 days of recovery from surgery prior to habituation to the recording environment. After one-week habituation, mice undergo 24 hours of undisturbed baseline EEG and EMG recording starting at light onset. The next day, mice are sleep deprived (SD) for 5 hours via gentle handling beginning at light onset. Mice are then allowed 19 hours undisturbed recovery sleep. EEG and EMG data are collected via a lightweight, counterbalanced cables, amplified, and digitized at 200 Hz using acquisition software. Wakefulness and sleep states are determined by visual inspection of the EEG waveform, EMG activity, and fast Fourier transform (FFT) analysis by an experimenter blinded to the genotype. Data is scored as wakefulness, NREMS, or REMS with 4 second resolution bins. Sleep parameters are quantified from the scored data and statistically analyzed. Required parameters for sleep characterization are as follows: time in state (wake, NREMS and REMS, baseline and SD day), spectral power for each state (baseline and SD day), latency to sleep following SD, sleep fragmentation (number of bouts in each state per hours, baseline and SD). Standardized protocol adapted from (Ingiosi et al., 2019).
Summary and future directions
The mechanisms underlying insomnia in ASD are still not known. Animal models are a promising tool to answer this question. To date, from the 305 rodent models of CNVs or genes considered to be linked to ASD according to the SFARI database, sleep was studied using EEG in only 22. However, most models do not show the key features of insomnia: longer latency to fall asleep, increased sleep fragmentation and reduced sleep time. As outlined in Table 2, 15 rodent models of ASD report decreases in either NREMS or REMS time, from which 5 reported fragmented sleep and 2 show increased latency to sleep, although only one study evaluated sleep latency following sleep deprivation. Therefore at present, there is only one ASD rodent model that has both construct and face validity to study sleep in ASD (Ingiosi et al., 2019). Nonetheless, can we learn something in regards to mechanisms linking sleep to ASD from some of the known mechanisms underlying the genetically determined ASD models that show reduced sleep time (14 in total)? Several interesting patterns emerge that possibly point to mechanisms by which sleep, and brain development may interact in ASD. First, a good proportion of the mouse models also present with learning and memory impairments. The relationship between sleep and learning is well documented. Sleep deprivation impairs learning and memory by inhibiting translation (Vecsey et al., 2012; Tudor et al., 2016). Sleep on the other hand promotes translation, and protein synthesis during sleep is required to consolidate cortical plasticity in vivo (Seibt et al., 2012). Aberrant protein synthesis has been proposed as one of the molecular pathways leading to ASD (Kelleher and Bear, 2008). Aberrant protein synthesis has been thoroughly characterized in the Fmr1 model. Ube3A is also known to influence protein turnover. Mecp2, Fmr1, Npas2 and Ube3A are all transcriptional/epigenetic regulators that can control how activity influences gene expression and subsequently protein levels. Although the role of epigenetics in early brain development as well as learning and memory is well established, how sleep may influence those processes has not yet been explored in depth. Therefore, the relationship between sleep, chromatin regulation and ASD is an interesting new direction of inquiry. Second, the majority of the genes are involved in synaptic function (Shank3, Disc1, Rims1, Gabrb3, Nrlgn2, Nrlgn3, Camk2a, Scn1a, Scn8a and Cacna1g). This is not surprising given both the established role of sleep in early life in synaptic plasticity and its disfunction in ASD. These “synaptic” genes include several classes of molecules, such as scaffolding proteins (Shank3, Disc1), adhesion molecules (Nrlgn 2 and 3), vesicle release (Rims1) ion channels (Scn1a, Scn8a, Cacna1g) and signaling molecules (Camk2a), indicating that sleep could influence all levels of synapse formation (or vice versa). Interestingly, only one neurotransmitter receptor mutant shows reduced sleep time: the beta subunit of the GABA a receptor (Gabrb3). As we discussed earlier, there’s a strong case to be made for the role of GABA in early development, sleep regulation and ASD. Therefore, it is reasonable to propose that GABA mediated neurotransmission is an important component of the mechanism underlying insomnia in ASD. Third, according to the Allen Brain Institute: Developing Mouse Brain Atlas, all above mentioned genes except Disc1 reach peak expression before P14. Consequently, sleep is the predominant brain state and the main source of spontaneous brain activity when these genes are maximally expressed in the brain. This is true for synaptic components and transcriptional regulators alike. This raises the question of whether sleep is required for defining the role of these genes in the establishment of synapses and networks, since activity-dependent regulation at P14 is sleep-dependent regulation. Last, it is important to highlight the distinction between sleep and circadian effects. The gene lists between Table 2 and 3 are largely non-overlapping, mostly due to a lack of study of both phenomena in the same animal models. In cases in which both have been studied there’s no clear correlation between the phenotypes. For example, Shank3, Disc1, Mep2 and Scn1a mutants sleep less, but show a reduction in total activity and no disruption in the timing of the activity. Ube3a mutants show changes in activity while Npas2 mutants show an increase. Mecp2 and Fmr1 mutants are the only ones that show a change in the timing of the activity, which would be considered a true circadian effect. These findings suggest that sleep and circadian regulating mechanisms involve a different but partially overlapping set of genes. Indeed, research supports that circadian disruption is prevalent in ASD independently of insomnia, with dysregulation in expression of circadian genes and melatonin being commonly reported (Karaivazoglou and Assimakopoulos, 2018; Carmassi et al., 2019). Overall, even though the number of rodent ASD models for which sleep has been studied is small, those studies already provide several future directions of inquiry in regard to the mechanism linking sleep, brain development and ASD.
There are important caveats that need to be taken into account when interpreting the available literature on sleep in ASD rodent models. First, all sleep phenotyping was performed using adult animals, usually only male. Since sleep is functionally important for brain development, sleep in rodent models of ASD should also be analyzed from a developmental perspective. Studies of sleep in developing animals, looking into the time periods in which homeostatic sleep regulation emerges as outlined earlier in this review, could tell us when and how these mechanisms begin to go wrong in ASD. Thus, studies should be conducted as early as possible in development so that the origins and trajectory of sleep problems can be discovered. Although there is a gender bias in ASD diagnosis, it is not clear whether there is a gender bias regarding insomnia in ASD, so in animal studies both sexes should be analyzed. Second, the role of most high-confidence ASD risk genes in terms of sleep regulation has not yet been explored. Therefore, it is possible that our understanding of the functional pathways that underlie the common mechanisms between sleep, development and ASD will change once that happens. Last, most of the animal models used to study ASD are models of syndromic forms of ASD, which makes up a small proportion of ASD cases. ASD is known to have a strong genetic component (Bai et al., 2019). However, that includes a variety of types of genetic variation in addition to genetically identifiable syndromes, including de novo single nucleotide and copy number variants, as well as polygenic risk that results from a combination of rare and common variants (Weiner et al., 2017). Therefore, the use of animal models that mimic rare genetic mutations to understand mechanism is inherently limited. Nonetheless further study of sleep in animal models of ASD holds great promise to understand this link and potentially find the cause and treatments for insomnia on the Autism Spectrum.
Significance statement:
Insomnia affects up to 86% of individuals with Autism Spectrum Disorder, predicts severity of core symptoms and impacts quality of life of caregivers. In this article we highlight how understanding the role of sleep in brain development and using animal models to study mechanisms is a promising avenue to uncover the origin of sleep problems in the spectrum.
Acknowledgments:
National Institute of Neurological Disorders and Stroke K01NS104172
Footnotes
Conflict of interest statement: The authors declare no competing financial interests.
References
- Achermann P, Borbély AA (2017) Principles and Practice of Sleep Medicine (eds Kryger M, Roth T, & Dement W.), 5th ed. Philadelphia: Elsevier; Available at: https://www.elsevier.com/books/principles-and-practice-of-sleep-medicine/9781416066453. [Google Scholar]
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S (2005) Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A 102:1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelakos CC, Watson AJ, O’Brien WT, Krainock KS, Nickl-Jockschat T, Abel T (2017) Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res Off J Int Soc Autism Res 10:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D et al. (2019) Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J et al. (2018) Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. Morb Mortal Wkly Rep Surveill Summ Wash DC 2002 67:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa J-L, Tyzio R, Khazipov R (2007) GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87:1215–1284. [DOI] [PubMed] [Google Scholar]
- Boone CE, Davoudi H, Harrold JB, Foster DJ (2018) Abnormal Sleep Architecture and Hippocampal Circuit Dysfunction in a Mouse Model of Fragile X Syndrome. Neuroscience 384:275–289. [DOI] [PubMed] [Google Scholar]
- Carmassi C, Palagini L, Caruso D, Masci I, Nobili L, Vita A, Dell’Osso L (2019) Systematic Review of Sleep Disturbances and Circadian Sleep Desynchronization in Autism Spectrum Disorder: Toward an Integrative Model of a Self-Reinforcing Loop. Front Psychiatry 10 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6581070/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadman KK, Yang M, Crawley JN (2009) Criteria for validating mouse models of psychiatric diseases. Am J Med Genet B Neuropsychiatr Genet 150B:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ (2012) GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev 36:2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Conduit R, Lockley SW, Rajaratnam SM, Cornish KM (2014) The relationship between sleep and behavior in autism spectrum disorder (ASD): a review. J Neurodev Disord 6 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4271434/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas D, Wagstaff J, Fort P, Salvert D, Sarda N (2005) Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol Dis 20:471–478. [DOI] [PubMed] [Google Scholar]
- Crawley JN (2012) Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin Neurosci 14:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe J, Marco P, Fairén A, Jones EG (1997) Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb Cortex N Y N 1991 7:619–634. [DOI] [PubMed] [Google Scholar]
- Diessler S, Kostic C, Arsenijevic Y, Kawasaki A, Franken P (2017) Rai1 frees mice from the repression of active wake behaviors by light. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich L, Petese A, Jackson WS (2017) The natural Disc1-deletion present in several inbred mouse strains does not affect sleep. Sci Rep 7:5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL (2003) Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science 301:379–383. [DOI] [PubMed] [Google Scholar]
- Dumoulin Bridi MC, Aton SJ, Seibt J, Renouard L, Coleman T, Frank MG (2015) Rapid eye movement sleep promotes cortical plasticity in the developing brain. Sci Adv 1:e1500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlen JC, Jones KA, Pinckney L, Gray CL, Burette S, Weinberg RJ, Evans JA, Brager AJ, Zylka MJ, Paul KN, Philpot BD, DeBruyne JP (2015) Maternal Ube3a Loss Disrupts Sleep Homeostasis But Leaves Circadian Rhythmicity Largely Intact. J Neurosci Off J Soc Neurosci 35:13587–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Helou J, Bélanger-Nelson E, Freyburger M, Dorsaz S, Curie T, La Spada F, Gaudreault P-O, Beaumont É, Pouliot P, Lesage F, Frank MG, Franken P, Mongrain V (2013) Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci U S A 110:9974–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod MG, Hood BS (2015) Sleep differences among children with autism spectrum disorders and typically developing peers: a meta-analysis. J Dev Behav Pediatr JDBP 36:166–177. [DOI] [PubMed] [Google Scholar]
- Everson CA, Bergmann BM, Rechtschaffen A (1989) Sleep deprivation in the rat: III. Total sleep deprivation. Sleep 12:13–21. [DOI] [PubMed] [Google Scholar]
- Frank MG (2017) Sleep and plasticity in the visual cortex: more than meets the eye. Curr Opin Neurobiol 44:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Heller HC (1997) Development of diurnal organization of EEG slow-wave activity and slow-wave sleep in the rat. Am J Physiol-Regul Integr Comp Physiol 273:R472–R478. [DOI] [PubMed] [Google Scholar]
- Frank MG, Issa NP, Stryker MP (2001) Sleep Enhances Plasticity in the Developing Visual Cortex. Neuron 30:275–287. [DOI] [PubMed] [Google Scholar]
- Frank MG, Ruby NF, Heller HC, Franken P (2016) Development of Circadian Sleep Regulation in the Rat: A Longitudinal Study Under Constant Conditions. Sleep 40 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6251512/ [Accessed August 4, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Man H-Y (2017) Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front Cell Neurosci 11:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene R, Siegel J (2004) Sleep. NeuroMolecular Med 5:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Yu FH, Schwartz MD, Linton JD, Bosma MM, Hurley JB, Catterall WA, de la Iglesia HO (2012) Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A 109:E368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Nomura T, Xu J, Contractor A (2014) The Developmental Switch in GABA Polarity Is Delayed in Fragile X Mice. J Neurosci 34:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan D, Roffwarg HP, Shaffery JP (2001) The effects of 1 week of REM sleep deprivation on parvalbumin and calbindin immunoreactive neurons in central visual pathways of kittens. J Sleep Res 10:285–296. [DOI] [PubMed] [Google Scholar]
- Holst SC, Sousek A, Hefti K, Saberi-Moghadam S, Buck A, Ametamey SM, Scheidegger M, Franken P, Henning A, Seifritz E, Tafti M, Landolt H-P (2017) Cerebral mGluR5 availability contributes to elevated sleep need and behavioral adjustment after sleep deprivation. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglowstein I, Jenni OG, Molinari L, Largo RH (2003) Sleep duration from infancy to adolescence: reference values and generational trends. Pediatrics 111:302–307. [DOI] [PubMed] [Google Scholar]
- Ingiosi A, Schoch H, Wintler TP, Singletary KG, Righelli D, Roser L, Medina E, Risso D, Frank MG, Peixoto L (2019) Shank3 Modulates Sleep and Expression of Circadian Transcription Factors Kim E, ed. eLife 8:e42819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Altimus C, LeGates T, Cash-Padgett T, Zoubovsky S, Hikida T, Ishizuka K, Hattar S, Mongrain V, Sawa A (2016) Abnormal wake/sleep pattern in a novel gain-of-function model of DISC1. Neurosci Res 112:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KP, Giannotti F, Cortesi F (2009) Sleep patterns in autism spectrum disorders. Child Adolesc Psychiatr Clin N Am 18:917–928. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Ammanuel S, O’Driscoll C, Wozniak A, Naidu S, Kadam SD (2014) Twenty-four hour quantitative-EEG and in-vivo glutamate biosensor detects activity and circadian rhythm dependent biomarkers of pathogenesis in Mecp2 null mice. Front Syst Neurosci 8:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CE, Opel RA, Kaiser ME, Chau AQ, Quintana JR, Nipper MA, Finn DA, Hammock EAD, Lim MM (2019) Early-life sleep disruption increases parvalbumin in primary somatosensory cortex and impairs social bonding in prairie voles. Sci Adv 5 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6353622/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, Catterall WA (2015) Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis 77:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaivazoglou K, Assimakopoulos K (2018) Circadian Dysregulation in Young Children with Autism Spectrum Disorder.
- Kelleher RJ, Bear MF (2008) The autistic neuron: troubled translation? Cell 135:401–406. [DOI] [PubMed] [Google Scholar]
- Kimura M, Müller-Preuss P, Lu A, Wiesner E, Flachskamm C, Wurst W, Holsboer F, Deussing JM (2010) Conditional corticotropin-releasing hormone overexpression in the mouse forebrain enhances rapid eye movement sleep. Mol Psychiatry 15:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov SV, Bogenpohl JW, Howell MP, Wevrick R, Panda S, Hogenesch JB, Muglia LJ, Van Gelder RN, Herzog ED, Stewart CL (2007) The imprinted gene Magel2 regulates normal circadian output. Nat Genet 39:1266–1272. [DOI] [PubMed] [Google Scholar]
- Kumar D, Dedic N, Flachskamm C, Voulé S, Deussing JM, Kimura M (2015) Cacna1c (Cav1.2) Modulates Electroencephalographic Rhythm and Rapid Eye Movement Sleep Recovery. Sleep 38:1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaria M, Gu W, Lupski JR (2013) Circadian abnormalities in mouse models of Smith-Magenis syndrome: evidence for involvement of RAI1. Am J Med Genet A 161A:1561–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim D, Shin H-S (2004) Lack of delta waves and sleep disturbances during non-rapid eye movement sleep in mice lacking alpha1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A 101:18195–18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesku JA, Roth TC, Rattenborg NC, Amlaner CJ, Lima SL (2008) Phylogenetics and the correlates of mammalian sleep: a reappraisal. Sleep Med Rev 12:229–244. [DOI] [PubMed] [Google Scholar]
- Li Q, Loh DH, Kudo T, Truong D, Derakhshesh M, Kaswan ZM, Ghiani CA, Tsoa R, Cheng Y, Sun YE, Colwell CS (2015) Circadian rhythm disruption in a mouse model of Rett syndrome circadian disruption in RTT. Neurobiol Dis 77:155–164. [DOI] [PubMed] [Google Scholar]
- Li W, Ma L, Yang G, Gan W (2017) REM sleep selectively prunes and maintains new synapses in development and learning. Nat Neurosci 20:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Boyle LM, Yuan ED, Hochstrasser KJ, Chifamba FF, Nathan A, Tsai PT, Davis F, Sahin M (2017) Aberrant Proteostasis of BMAL1 Underlies Circadian Abnormalities in a Paradigmatic mTOR-opathy. Cell Rep 20:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Grace KP, Horner RL, Cortez MA, Shao Y, Jia Z (2017) Neuroligin 3 R451C mutation alters electroencephalography spectral activity in an animal model of autism spectrum disorders. Mol Brain 10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Tang X, Simsek-Duran F, Machida M, Sanford LD (2008) The role of active zone protein Rab3 interacting molecule 1 alpha in the regulation of norepinephrine release, response to novelty, and sleep. Neuroscience 154:821–831. [DOI] [PubMed] [Google Scholar]
- Lu H-C, Pollack H, Lefante JJ, Mills AA, Tian D (2019) Altered sleep architecture, rapid eye movement sleep, and neural oscillation in a mouse model of human chromosome 16p11.2 microdeletion. Sleep 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang GM, Franken P (2015) Genetic Dissection of Sleep Homeostasis In: Sleep, Neuronal Plasticity and Brain Function (Meerlo P, Benca RM, Abel T, eds), pp 25–63 Current Topics in Behavioral Neurosciences. Berlin, Heidelberg: Springer; Available at: 10.1007/7854_2013_270. [DOI] [PubMed] [Google Scholar]
- Maxwell-Horn A, Malow BA (2017) Sleep in Autism. Semin Neurol 37:413–418. [DOI] [PubMed] [Google Scholar]
- Mazurek MO, Dovgan K, Neumeyer AM, Malow BA (2019) Course and Predictors of Sleep and Co-occurring Problems in Children with Autism Spectrum Disorder. J Autism Dev Disord 49:2101–2115. [DOI] [PubMed] [Google Scholar]
- Meltzer LJ (2008) Brief report: sleep in parents of children with autism spectrum disorders. J Pediatr Psychol 33:380–386. [DOI] [PubMed] [Google Scholar]
- Meredith AL, Wiler SW, Miller BH, Takahashi JS, Fodor AA, Ruby NF, Aldrich RW (2006) BK calcium-activated potassium channels regulate circadian behavioral rhythms and pacemaker output. Nat Neurosci 9:1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens JA, Mindell JA (2011) Pediatric insomnia. Pediatr Clin North Am 58:555–569. [DOI] [PubMed] [Google Scholar]
- Paluszkiewicz SM, Martin BS, Huntsman MM (2011) Fragile X Syndrome: The GABAergic System and Circuit Dysfunction. Dev Neurosci 33:349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Makinson CD, Christopher Ehlen J, Tufik S, Decker MJ, Paul KN, Escayg A (2013) Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia 54:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Paul KN, Sawyer NT, Manns JR, Tufik S, Escayg A (2010) Dysfunction of the Scn8a voltage-gated sodium channel alters sleep architecture, reduces diurnal corticosterone levels, and enhances spatial memory. J Biol Chem 285:16553–16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richdale AL, Schreck KA (2009) Sleep problems in autism spectrum disorders: prevalence, nature, & possible biopsychosocial aetiologies. Sleep Med Rev 13:403–411. [DOI] [PubMed] [Google Scholar]
- Roffwarg HP, Muzio JN, Dement WC (1966) Ontogenetic Development of the Human Sleep-Dream Cycle. Science 152:604–619. [DOI] [PubMed] [Google Scholar]
- Sahar S, Nin V, Barbosa MT, Chini EN, Sassone-Corsi P (2011) Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging 3:794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saré RM, Harkless L, Levine M, Torossian A, Sheeler CA, Smith CB (2017) Deficient Sleep in Mouse Models of Fragile X Syndrome. Front Mol Neurosci 10 Available at: https://www.frontiersin.org/articles/10.3389/fnmol.2017.00280/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MD, Kilduff TS (2015) THE NEUROBIOLOGY OF SLEEP AND WAKEFULNESS. Psychiatr Clin North Am 38:615–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelke AMH, Blumberg MS (2008) The Microstructure of Active and Quiet Sleep as Cortical Delta Activity Emerges in Infant Rats. Sleep 31:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, Frank MG (2012) Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol CB 22:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ (2013) Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106–107:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok BS, Cao F, Bélanger-Nelson E, Provost C, Gibbs S, Jia Z, Mongrain V (2018) The effect of Neuroligin-2 absence on sleep architecture and electroencephalographic activity in mice. Mol Brain 11:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Zimmerman AW (2015) Sleep in Autism Spectrum Disorder and Attention Deficit Hyperactivity Disorder. Semin Pediatr Neurol 22:113–125. [DOI] [PubMed] [Google Scholar]
- Souders MC, Zavodny S, Eriksen W, Sinko R, Connell J, Kerns C, Schaaf R, Pinto-Martin J (2017) Sleep in Children with Autism Spectrum Disorder. Curr Psychiatry Rep 19:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuki F, Sunagawa GA, Shi S, Susaki EA, Yukinaga H, Perrin D, Sumiyama K, Ukai-Tadenuma M, Fujishima H, Ohno R, Tone D, Ode KL, Matsumoto K, Ueda HR (2016) Involvement of Ca(2+)-Dependent Hyperpolarization in Sleep Duration in Mammals. Neuron 90:70–85. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Schwartz MD, Saxe MD, Kilduff TS (2017) Sleep/Wake Physiology and Quantitative Electroencephalogram Analysis of the Neuroligin-3 Knockout Rat Model of Autism Spectrum Disorder. Sleep 40. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Schwartz MD, Saxe MD, Kilduff TS (n.d.) Cntnap2 Knockout Rats and Mice Exhibit Epileptiform Activity and Abnormal Sleep-Wake Physiology. Sleep 40 Available at: https://academic.oup.com/sleep/sleep/article/2661545/<italic>Cntnap2</italic>. [DOI] [PubMed] [Google Scholar]
- Tsuchiya Y, Minami Y, Umemura Y, Watanabe H, Ono D, Nakamura W, Takahashi T, Honma S, Kondoh G, Matsuishi T, Yagita K (2015) Disruption of MeCP2 attenuates circadian rhythm in CRISPR/Cas9-based Rett syndrome model mouse. Genes Cells Devoted Mol Cell Mech 20:992–1005. [DOI] [PubMed] [Google Scholar]
- Tudor JC, Davis EJ, Peixoto L, Wimmer ME, van Tilborg E, Park AJ, Poplawski SG, Chung CW, Havekes R, Huang J, Gatti E, Pierre P, Abel T (2016) Sleep deprivation impairs memory by attenuating mTORC1-dependent protein synthesis. Sci Signal 9:ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor ME, Hoffman CD, Sweeney DP (2012) Children With Autism: Sleep Problems and Symptom Severity. Focus Autism Dev Disabil 27:254–262. [Google Scholar]
- Veatch OJ, Sutcliffe JS, Warren ZE, Keenan BT, Potter MH, Malow BA (2017) Shorter sleep duration is associated with social impairment and comorbidities in ASD. Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/aur.1765. [DOI] [PMC free article] [PubMed]
- Vecsey CG, Peixoto L, Choi JHK, Wimmer M, Jaganath D, Hernandez PJ, Blackwell J, Meda K, Park AJ, Hannenhalli S, Abel T (2012) Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol Genomics 44:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DJ et al. (2017) Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 49:978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, DeLorey TM, Homanics GE, Edgar DM (2002) Sleep states and sleep electroencephalographic spectral power in mice lacking the beta 3 subunit of the GABA(A) receptor. Brain Res 955:221–228. [DOI] [PubMed] [Google Scholar]
- Wither RG, Colic S, Wu C, Bardakjian BL, Zhang L, Eubanks JH (2012) Daily rhythmic behaviors and thermoregulatory patterns are disrupted in adult female MeCP2-deficient mice. PloS One 7:e35396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepelin H, Whitehead WE, Rechtschaffen A (1972) Aging and sleep in the albino rat. Behav Biol 7:65–74. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, Albrecht U, Tamanini F, Meijer JH, Oostra BA, Nelson DL (2008) Fragile X-Related Proteins Regulate Mammalian Circadian Behavioral Rhythms. Am J Hum Genet 83:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC (2001) Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell 105:683–694. [DOI] [PubMed] [Google Scholar]
- Zhou L, Bryant CD, Loudon A, Palmer AA, Vitaterna MH, Turek FW (2014) The circadian clock gene Csnk1e regulates rapid eye movement sleep amount, and nonrapid eye movement sleep architecture in mice. Sleep 37:785–793, 793A–793C. [DOI] [PMC free article] [PubMed] [Google Scholar]