

Abstract
Background In 2003, Taiwan experienced a series of outbreaks of severe acute respiratory syndrome (SARS) and 1 laboratory-contamination accident. Here we describe a new phylogenetic analytical method to study the sources and dissemination paths of SARS-associated coronavirus (SARS-CoV) infections in Taiwan
Methods A phylogenetic analytical tool for combining nucleotide sequences from 6 variable regions of a SARS-CoV genome was developed by use of 20 published SARS-CoV sequences; and this method was validated by use of 80 published SARS-CoV sequences. Subsequently, this new tool was applied to provide a better understanding of the entire complement of Taiwanese SARS-CoV isolates, including 20 previously published and 19 identified in this study. The epidemiological data were integrated with the results from the phylogenetic tree and from the nucleotide-signature pattern
Results The topologies of phylogenetic trees generated by the new and the conventional strategies were similar, with the former having better robustness than the latter, especially in comparison with the maximum-likelihood trees: the new strategy revealed that during 2003 there were 5 waves of epidemic SARS-CoV infection, which belonged to 3 phylogenetic clusters in Taiwan
Conclusions The new strategy is more efficient than its conventional counterparts. The outbreaks of SARS in Taiwan originated from multiple sources
Severe acute respiratory syndrome (SARS) is caused by SARS-associated coronavirus (SARS-CoV) [1–4]. The first known outbreak of SARS occurred in China’s Guangdong province during November 2002 [5]. By 7 August of the following year, SARS had spread to >30 countries, affecting 8096 people and resulting in 774 deaths worldwide [6]. In Taiwan, the first SARS case was diagnosed on 14 March 2003 [7, 8]. This index case involved a Taiwanese businessman who had visited Guangdong province during 5–21 February of that year. After he returned to Taiwan, he transmitted the disease to his wife, his son (SARS-CoV strain TW1), and the doctor who treated his son (SARS-CoV strain TW3)
On 15 March, 7 employees of a Taiwanese construction company flew from Hong Kong to Beijing; 4 of them developed SARS symptoms on 26 March, several days after returning to Taiwan [9]. Also on 26 March, a man residing at the Amoy Gardens housing complex in Hong Kong flew to Taiwan; the following day, he took a train from Taipei to Taichung City to visit his younger brother. The visitor returned to his Hong Kong home on 28 March after having experienced fever during the preceding evening. His younger brother (patient TWC), who developed symptoms on 31 March, became Taiwan’s first SARS-related fatality
On 6 April, a Taiwanese woman (patient TW-HP1) suffering from fever and coughing that continued for several days visited the emergency room (ER) at municipal hospital HP; she was transferred to another hospital on 9 April. Seven HP employees, including a laundry worker who was identified as the index case, eventually developed SARS, resulting, on 24 April, in a shutdown of all operations of hospital HP [7]. In all, 137 probable SARS cases and 26 HP-related fatalities resulted from this single nosocomial infection. Patient TW-HP1 had not traveled outside Taiwan during the preceding 12 months, but, on 27 March, she and the visitor from Hong Kong had taken the same train; their seats were in separate cars (numbers 3 and 5), yet the train ride constitutes their only possible point of contact
On 28 April, the government of Taiwan imposed mandatory quarantines on all air travelers from China, Hong Kong, Singapore, Macau, and Toronto; however, nosocomial SARS infections continued to be reported in many hospitals islandwide [5]. The hospitals that experienced the most-severe outbreaks are listed in figure 1; in the present study, they are referred to by initials—“HP,” “JC,” “KC,” “GD,” and “YM.” According to Taiwan’s Center for Disease Control (CDC), 346 of the 664 probable SARS cases that have been reported to the World Health Organization (WHO) were confirmed by reverse-transcriptase polymerase chain reaction (RT-PCR) and/or neutralizing-antibody tests [10]. Previously, Yeh et al. had studied the molecular epidemiology of SARS infection in Taiwan and had concluded that the origin of the Taiwanese SARS epidemic was mainly either Hong Kong or Guangdong, rather than Beijing [11]; in addition, they found that the SARS-CoV isolated from the younger brother (i.e., patient TWC) of the visitor from Hong Kong was not clustered with other isolates from hospital HP [11, 12]. Because a complete genome sequence from the nasopharyngeal aspirate from patient TWC (strain TC1) was available, we decided to reexamine both (1) the source of infection at hospital HP and (2) the paths of dissemination of SARS among hospitals in Taiwan
Figure 1.

Epidemiological curve of confirmed cases of severe acute respiratory syndrome (SARS) in Taiwan, which Taiwan’s Center for Disease Control validated by use of either reverse-transcriptase polymerase chain reaction or serological test. Arrows indicate dates of outbreaks of nosocomial infection in different hospitals and of diagnoses of SARS in several key patients
On the evening of 10 December 2003, a medical researcher who regularly worked in a biosafety level–4 laboratory started to feel feverish [13]. Although he had recently spent 4 days (7–10 December) in Singapore, an epidemiological investigation indicated that he had contracted SARS from a laboratory-contamination accident on 6 December. According to his description, the SARS-CoV isolates that he had handled in the laboratory included strain HKU-39849 [14] and other clinical isolates that have not been well characterized. The nasopharyngeal aspirate from this medical researcher was included in the present study
The size of the SARS-CoV genome has been measured as 29.7 kb [15, 16]. A comparative analysis of 14 SARS-CoV isolates has identified 2 distinct genotypes, which can be differentiated on the basis of 4 single-nucleotide variations (SNVs)—C:C:G:C versus T:T:T:T—in the variable regions of the SARS-CoV genome [17]. Furthermore, the genotype with the T:T:T:T SNVs has been associated with infections originating in Hotel M in Hong Kong [8, 17]. Currently, phylogenetic analyses of SARS-CoV require complete genome sequences [5, 11, 17]; however, because of the limited number of specimens available for complete genome sequencing, some researchers have used the SARS-CoV spike gene for this purpose; but most results have been less than satisfactory [18, 19]. Therefore, the objectives of the present study were (1) to use 20 complete SARS-CoV sequences to combine several variable regions of a SARS-CoV for phylogenetic analysis, in order to develop a simpler tool; (2) to use a different set of sequences from 80 SARS-CoV isolates to validate the method, by 3 different phylogenetic analytical methods; and (3) to apply this proposed tool to elucidate the origin and paths of dissemination of SARS-CoV infections, on the basis of samples collected from hospitals in Taiwan
Patients, Materials, and Methods
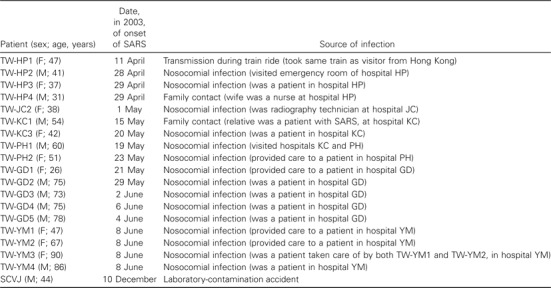
PatientsSerum, sputum, or throat-swab specimens from 19 patients with SARS associated with 5 nosocomial infections and 1 laboratory-contamination incident were collected for the present study. For each patient, data on the date at onset of the disease and on possible sources of infection were gathered by trained interviewers (table 1). We also downloaded, for our phylogenetic-tree analyses, 20 complete Taiwanese SARS-CoV sequences from GenBank; for 10 of these, SARS-CoV strains have been described elsewhere [11]
Table 1.
Demographic data and possible sources of infection in cases of severe acute respiratory syndrome (SARS) that were used for molecular epidemiological study
Epidemiological investigationTo track the origins of SARS cases in Taiwan, we used SARS-treatment reports written by physicians and submitted by their respective hospitals to Taiwan’s CDC. We also sent trained interviewers to all hospitals that reported nosocomial infections, to gather additional assessment data. We based our investigation on the WHO definition of SARS [20]
Analysis of SARS-CoV sequence variation, and selection of variable regions for phylogenetic analysisComplete nucleotide sequences from 20 SARS-CoV isolates available from GenBank were aligned by the BioEdit program [21]; the sequence variations were analyzed by the SimPlot program (http://sray.med.som.jhmi.edu/RaySoft/SimPlot). Genomic sequences from civet-cat SARS-CoV strains SZ-3 and SZ-16 were used as standards for comparison [19]. Sequence-variation–distance plots were generated by use of an 800-bp window, a 200-bp step, and a Jukes-Cantor correction. Initially, 4 variable regions (SC18, SC22-23, SC27, and SC28), which contain SNVs of different SARS-CoV genotypes [17], were combined for phylogenetic analysis. Because the resultant phylogenetic tree was not satisfactory, we added another 2 variable regions (SC10 and SC20) based on the sequence-variation plot (figure 2)
Figure 2.

Analysis of nucleotide-sequence variation of 20 isolates of human severe acute respiratory syndrome–associated coronavirus (SARS-CoV), by the SimPlot program. Two civet-cat SARS-CoV strains were used as references. The X-axis indicates the nucleotide location of the SARS-CoV genome; the Y-axis indicates the rate of nucleotide-sequence variation. Sequence differences are plotted with a window of 800 nt and 200-nt steps. The 6 variable regions—SC10, SC18, SC20, SC22-23, SC27, and SC28—used for the combined phylogenetic analyses are in boldface type
Comparison of conventional and proposed strategies for phylogenetic analysisThree methods—neighbor joining (NJ), Fitch and Wagner parsimony (Pars), and maximum likelihood (ML)—were used for comparison of the conventional (i.e., complete genome) and proposed strategies. The proposed strategy for phylogenetic analysis entailed the deletion of conserved domains and the combination of minimal variable regions. The MEGA2 and Phylip3.6 software packages were used to construct the phylogeny [22–24]; and 80 SARS-CoV sequences downloaded from the GenBank database were used for the comparison. A bootstrap analysis of 100 replicates was used to compare the robustness of the NJ and Pars trees generated by the conventional strategy versus that of the NJ and Pars trees generated by the proposed strategy [25]. For the ML method, the P value for branch length was calculated by use of the hidden Markov model, before the conventional and proposed strategies were compared [26]
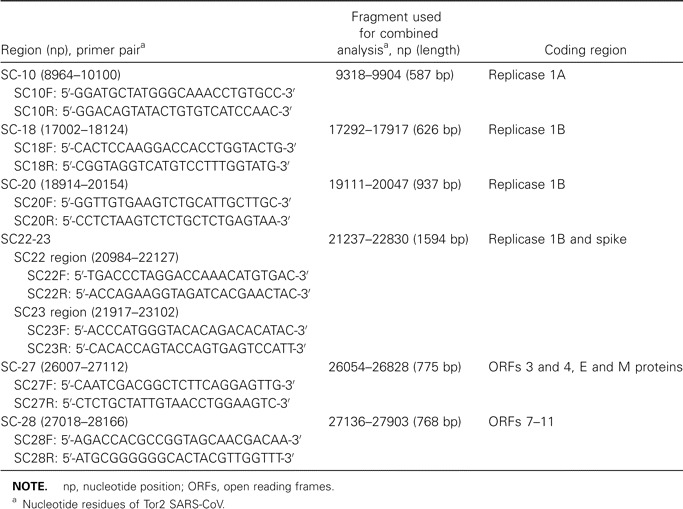
RT-PCR and sequencingRNA was extracted from serum, sputum, or throat-swab specimens by QIAamp viral-RNA minikits (Qiagen). RT-PCR primer pairs for the 6 SARS-CoV variable regions are listed in table 2. RT-PCR was performed in a single-tube reaction (Qiagen) using primers from each region. The PCR thermocycler program consisted of predenaturing for 4 min at 95°C; 35 cycles of denaturing for 30 s at 95°C, annealing for 30 s at 52°C, and initial extension at 68°C for 3 min; and final extension at 68°C for 10 min. The PCR product was gel-purified for DNA sequencing by use of an ABI PRISM 3700 DNA Analyzer (Applied Biosystems). For the laboratory-contamination case, multiple primers [17] were used for complete genome sequencing. Superscript III RT (Invitrogen) was used for production of cDNA, and a RACE kit (Roche) was used to amplify the 5′ and 3′ ends of cDNA. Both strands of the PCR product were analyzed, and the resultant sequences were assembled by use of the SeqMan II software package (version 5.0; Lasergene)
Table 2.
Primers used for reverse-transcriptase–polymerase chain reaction analysis of 6 variable regions of the severe acute respiratory syndrome–associated coronavirus (SARS-CoV) genome
Analysis of the nucleotide-signature patterns of different waves of epidemic SARS infectionTo perform the analysis of nucleotide-signature patterns, representative SARS-CoV strains with complete sequences were chosen from each wave of epidemic SARS infection in Taiwan. In addition, cases that were of unclear origin were also included in the analysis. The nucleotide sequence of SARS-CoV strain Urbani [16] was used as a prototype for the comparison
Results
Development of a proposed strategy for phylogenetic analysis of SARS-CoV isolatesPairwise comparisons were used to analyze sequence variations among 20 SARS-CoV isolates. The results show that the 3′ region of the viral genome had the highest sequence variation, especially near the junction of replicase 1b and the spike genes (figure 2). Six variable regions—SC10, SC18, SC20, SC22-SC23, SC27, and SC28—were chosen for testing the proposed strategy for phylogenetic-tree analysis; the total length of the sequences in these 6 regions was 5287 nt. Using 3 phylogenetic analytical methods, we compared, for 80 SARS-CoV isolates, the topology and robustness of the phylogenetic tree generated by the proposed strategy, which uses only these 6 regions, versus the topology and robustness of the tree generated by the conventional strategy, which uses the complete genome sequences. As shown in figure 3, the topology of the NJ tree generated by use of the complete genome sequence was almost identical to that of the NJ tree generated by use of a combination of the 6 variable regions; similar results were obtained for the Pars trees and the ML trees (data not shown)
Figure 3.

Neighbor-joining (NJ) trees of 80 isolates of severe acute respiratory syndrome–associated coronavirus (SARS-CoV) that were downloaded from GenBank, by use of (left) the conventional strategy, which uses the complete genome sequence, and (right) the proposed strategy, which uses sequences from 6 SARS-CoV variable regions. Numbers at nodes are bootstrap values (%), and those at 3 nodes are circled: node a, between civet-cat SARS-CoV and human SARS-CoV; node b, between SARS-CoV subgroups A1 (early phase) and A2 (middle phase); and node c, between SARS-CoV subgroups A and B (late phase) [5]. The scale bar indicates the genetic distance, estimated on the basis of Kimura’s 2-parameter substitution model [24]
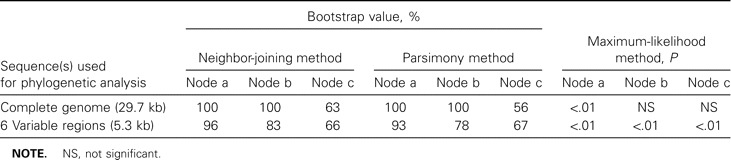
To compare the robustness of trees generated by the proposed method versus that of trees generated by the conventional method, we focused on the bootstrap values for 3 bifurcation nodes between SARS-CoV clusters: node “a,” between civet-cat SARS-CoV and human SARS-CoV; node “b,” between SARS-CoV subgroups A1 and A2; and node “c,” between SARS-CoV subgroups A and B. In the NJ trees, the bootstrap values for nodes a, b, and c were, respectively, 100%, 100%, and 63% for the conventional method versus 96%, 83%, and 66% for the proposed method (figure 3); similar results were obtained for the Pars trees generated by these 2 methods (table 3). For the ML tree generated by the proposed method, all P values for bifurcation nodes between different clusters were <.01; for the ML trees generated by the conventional method, the only node with a P value <.01 occurred at the bifurcation node between civet-cat SARS-CoV and human SARS-CoV (table 3)
Table 3.
Bootstrap and P values for trees generated by use of the complete genome and by use of 6 variable regions of sequences of severe acute respiratory syndrome–associated coronavirus, by 3 traditional analytical methods
Phylogenetic analysis of SARS-CoV infections in Taiwan The proposed method is useful for study of the molecular epidemiology of SARS-CoV strains, both because of its compatibility and because it can obtain results by using fewer specimens; in addition, it is more economical and less time-consuming. We applied this tool to better understand the entire complement of Taiwanese SARS-CoV isolates, including the 20 that had been published previously and the additional 19 identified in the present study. Epidemiological information, nasopharyngeal aspirates, and serum samples were collected from 18 patients with SARS who were treated at 6 Taiwanese hospitals (table 1). The SARS-CoV from a laboratory worker (patient SCVJ) who contracted SARS during December 2003 was sequenced and analyzed. An NJ tree was constructed, and the results showed that all 39 Taiwanese SARS-CoV isolates belonged to subgroup B and could be divided into 3 clusters—B1, B2, and B3
Integration of the results of the phylogenetic analyses and the epidemiological information (figure 4) indicated that there were 5 waves of epidemic SARS-CoV infection in Taiwan during 2003. The first wave, during early March, was composed of 1 imported case, 2 cases of transmission between family members (strain TW1), and 1 nosocomial infection (strain TW3); in cluster B1, both SARS-CoV strain TW1 and SARS-CoV strain TW3 were clustered with other SARS-CoV strains linked to Hotel M in Hong Kong [8, 17]. The second wave (strain TW5) consisted of 4 Taiwanese individuals who contracted the disease as they flew from Hong Kong to Beijing [9] and who then carried it back to Taiwan during mid-March. The third wave, which began in late March, consisted of an infection that occurred on a train (patient TW-HP1), a case of transmission between family members (strain TC1), and multiple nosocomial infections (all “TW-HP” patients and patients TW-GD1, TW-GD-3, and TW-GD4). In cluster B2, all the SARS-CoV isolates mentioned above clustered with the SARS-CoV isolates from Amoy Gardens (strains CUHK-AG01, CUHK-AG02, and CUHK-AG03) [27], with a bootstrap value of 71%. The fourth wave, which started during late April and ended in mid-June, contained SARS-CoV isolates not only from hospitals JC, KC, PH, GD (patients TW-GD2 and TW-GD5), and YM but also from sporadic community outbreaks (strains TW10 and TW11), and all of these SARS-CoV isolates belonged to cluster B3, with a bootstrap value of 71%. The fifth wave occurred during early December and began with a laboratory-contamination case, patient SCVJ. It clustered with both HKU-39849, a SARS-CoV strain used in the laboratory [14], and TWC, another strain from Taiwan [12]. Sequence-variation rates for the strain from patient SCVJ versus strain TWC and for the strain from patient SCVJ versus strain HKU-39849 were, respectively, 0.01% (3/29,756) and 0.04% (12/29,756). In addition, a 24-nt deletion occurred at nucleotide position 26132–26155, resulting in both a frameshift of open reading frame 4 and a deletion of 8 aa residues within the small-envelope glycoprotein
Figure 4.

Neighbor-joining phylogenetic tree of 39 Taiwanese severe acute respiratory syndrome (SARS)–associated coronavirus isolates, generated by the proposed strategy. The 5 waves of epidemic SARS infection that are related to the 3 phylogenetic clusters are described to the right. Numbers at nodes are bootstrap values (%). The scale bar indicates genetic distance, estimated on the basis of Kimura’s 2-parameter substitution model [24]
As shown in figure 5, there were 27 SNVs and 1 dinucleotide deletion among 14 SARS-CoV isolates; 12 of the 27 SNVs were nonsynonymous changes. Distinctive SNVs were identified for each wave of epidemic SARS infection in Taiwan—at nucleotide position 3165 for wave 1; at nucleotide positions 3852, 11493, and 26477 for both wave 3 and wave 4; and at nucleotide positions 26203 and 27812 for wave 4. The nucleotide-signature pattern between the strain from patient SCVJ (the laboratory-contamination case) and strain TWC consisted of 2 SNVs (at nucleotide position 16325–26600) and 1 dinucleotide deletion (at nucleotide position 27808–27809)
Figure 5.
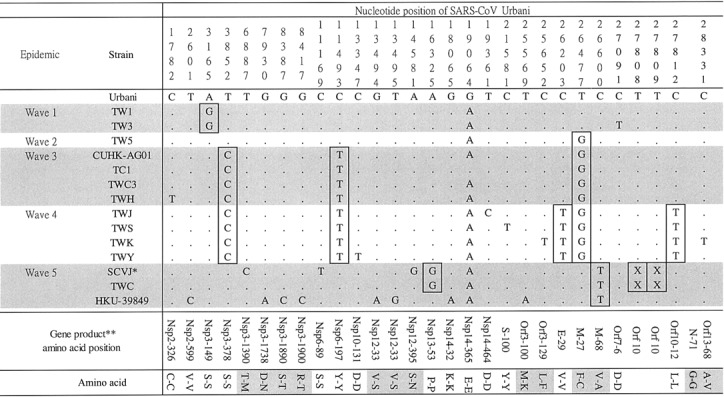
Signature patterns of single-nucleotide variations in 5 waves of epidemic severe acute respiratory syndrome–associated coronavirus (SARS-CoV) infection in Taiwan. Nucleotides are numbered on the basis of the complete genome sequence of SARS-CoV strain Urbani [16]. Deletions are denoted by X’s. *, Isolate with 24-nt deletion at nucleotide position 26132–26155; **, amino acid residues of SARS-CoV nonstructural protein (Nsp) and open reading frame (Orf) [16]
Discussion
When, for phylogenetic analysis, we combined sequences from different variable regions of the SARS-CoV genome, we assumed that no dual infection or recombination between the SARS-CoV subgroups had occurred. Because SARS is an acute infectious disease, the odds that it will be contracted from 2 subgroups are very low. Furthermore, the strategy of combining different genomic regions for phylogenetic analysis has been used in molecular epidemiological investigations of the origins of HIV-2. In those studies, the evolutionary history of a simian immunodeficiency virus/HIV-2 lineage was reconstructed by use of a combination of partial gag and env sequences; the method increased the accuracy of the phylogenetic analysis [28]
It is noteworthy that the SimPlot analysis demonstrated that the 3′ region of the viral genome, especially near the junction of replicase 1b and the spike genes (encoding protein S), had the greatest sequence variation (figure 2). The S protein is considered to be the most important target for the humoral and cellular immune responses to SARS-CoV [15, 16]
The 3 algorithms most commonly used in molecular phylogenetic analyses are the NJ, ML, and Pars methods; we used all 3 to obtain tree topologies. In terms of robustness, a bootstrap value of 70% is often cited as a cutoff for a reliable cluster [25]. Although the bootstrap values for nodes a and b in the NJ and Pars trees generated by the conventional method were higher than those in the NJ and Pars trees generated by the proposed method, the difference was not statistically significant. In contrast, the bootstrap values for node c in the Pars trees generated by the conventional and proposed methods were 56% and 67%, respectively, and the corrresponding bootstrap values in the NJ trees were 63% and 66%. This finding was confirmed by the ML trees: the P value for node c in the tree generated by the conventional method was not statistically significant, and that for node c in the tree generated by the proposed method was <.01. Accordingly, the tree generated by the proposed method was more reliable; also, because the proposed method requires only 7 RT-PCR reactions to perform the analysis, it is less time-consuming and more efficient than the conventional method. To facilitate other laboratories’ future molecular epidemiological studies of outbreaks of SARS, we have made the nucleotide-sequence–alignment file of 80 SARS-CoV reference strains available on our center’s Web site (http://www.ym.edu.tw/aids/Molepi/)
In the present study, we used 39 Taiwanese SARS-CoV isolates, including 20 downloaded from the GenBank database, to trace the origin and the path of dissemination of SARS-CoV infections that occurred in Taiwan during 2003. Phylogenetic analyses demonstrated that the Taiwanese SARS-CoV strains were distributed in 3 clusters—B1, B2, and B3—which differ from clusters 1–3 reported by Yeh et al. [11]; the latter clusters were not defined on the basis of a bootstrap value, whereas we used a bootstrap value of 70% as the cutoff to define our clusters B1–B3. Therefore, our cluster B1 contains both cluster 1 and cluster 2 of Yeh et al., and their cluster 3 was divided into 2 clusters—B2 and B3—in our study
On the basis of the epidemiological data for the patients with SARS who were considered in the present study, it is clear that Taiwan experienced 5 waves of epidemic SARS infection during 2003. The first and second waves were in different phylogenetic clusters, suggesting that they had different origins. Although both the second and the third waves were in the B2 cluster, the SARS-CoV isolates in the third wave had their own nucleotide-signature pattern (figure 5). Neither the first wave nor the second wave led to serious outbreaks, but the third wave—originating with a resident of Amoy Gardens who visited Taiwan—led to 1 transmission on a train (strain TWC3), 1 case of transmission between family members (strain TC1), and nosocomial infections in at least 2 hospitals (HP and GD) in northern Taiwan. Only 1 nucleotide difference between strains TC1 and TWC3 was noted (figure 5). In addition, strain TWC3 and an Amoy Gardens strain, CUHK-AG01, shared an identical sequence, even though the woman who was the source of the isolate of strain TWC3 never left Taiwan at any time during the epidemic. An epidemiological investigation showed that the visitor from Amoy Gardens and this woman sat in different cars during the train ride. Because this is the first documented case in which there is molecular proof of transmission on a train, it raises the question of why only 1 passenger contracted the infection. Because it has been reported that SARS appears to be most infectious at 6–11 days after onset of illness and not during the first day of symptoms [29], we assumed that the visitor from Amoy Gardens was not highly infectious during the train ride, even though he developed symptoms during the same evening that he traveled from Taipei to Taichung
It is important to note that TWC, the SARS-CoV strain isolated from patient TWC, clustered with WHU, an isolate from Wuhan City, China, in cluster 1 but did not cluster with either strain TC1 (direct sequencing of a sample from patient TWC) or strain TWC3 (direct sequencing of a sample from patient TW-HP1) (figures 3 and 4). In addition, there was a 7-nt difference between TWC and TC1 (figure 5). If we assume that strain CUHK-AG01 represents the first-generation SARS-CoV in the transmission link, then both strain TC1 and strain TWC3 were the second-generation, and TWH was the third-generation. According to the SNV-based analysis (figure 5), the number of nucleotide changes in the SARS-CoV genome per number of intermediate hosts was extremely low (<1 nt change/host). Because it has also been shown that no or very limited nucleotide changes occur in SARS-CoV sequences from either cultures or primary clinical specimens [11, 30], we can tentatively conclude that strain TWC is a laboratory contaminant and did not originate from patient TC1
The origin of the fourth wave (cluster B3) is still unknown. The molecular epidemiological data suggest that it originated at hospital JC (patient TW-JC2) and then spread to hospitals KC, PH, GD, and YM, as well as to others. The epidemiological investigations also support this hypothesis: TW-PH1, a patient with SARS who was treated at hospital JC went to Kaohsiung City and received treatment at hospital KC, and, after being treated at hospital KC (table 1), went back to Penghu Island and was hospitalized in hospital PH, where he transmitted the disease to other health-care workers. As shown in figure 6, after combining the epidemiological data with the results of our phylogenetic-tree analysis, we conclude that the fourth wave of epidemic SARS infection progressed in 2 dissemination paths: the first path was from hospital JC to hospital KC in Kaohsiung and then to hospital PH on Penghu Island; the second path was from hospital JC to hospital GD and then to hospital YM. Two cases associated with community outbreaks in Taipei (strains TW10 and TW11) also belong to this cluster
Figure 6.
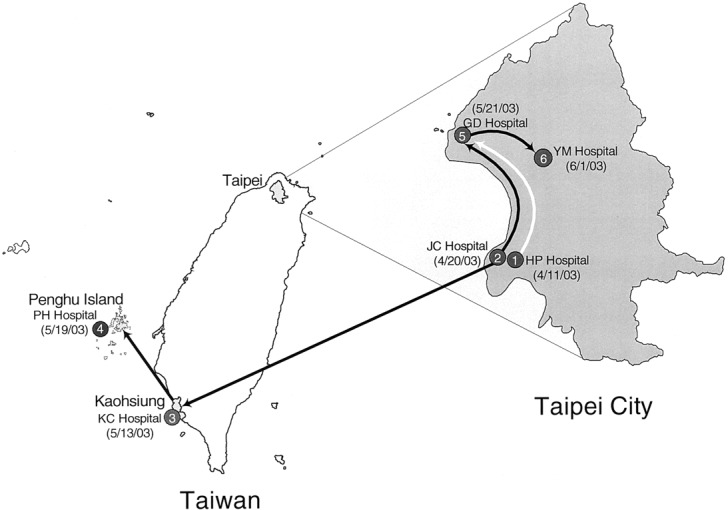
Two possible paths of dissemination—between hospitals on Taiwan Island and a hospital on Penghu Island—of severe acute respiratory syndrome–associated coronavirus (SARS-CoV) infection. Numbers in shaded circles indicate the temporal order of nosocomial infections; numbers in parentheses indicate dates at onset of the infections
With regard to the laboratory contamination, the laboratory researcher said that he had used SARS-CoV strain HKU-39849 for his experiment; however, our results indicate that the sequence in patient SCVJ is more closely related to that of strain TWC (figure 5). Because the researcher claimed that he had not obtained strain TWC from Taiwan’s CDC and that he had used many clinical SARS-CoV strains besides HKU-39849, we are continuing our investigation to confirm both (1) whether the virus that he had obtained from Taiwan’s CDC was in fact strain HKU-39849 or strain TWC and (2) which SARS-CoV strain that he handled was the source of the laboratory contamination. Because >30 imported SARS cases have been reported by Taiwan’s CDC but have not yet been analyzed, we plan to use the proposed tool to conduct further analyses, in an attempt to identify their origins. These contact histories will provide valuable information for the control of future SARS infections
Nucleotide-Sequence Accession Numbers
The SARS-CoV nucleotide sequences (6 sequences for each of 18 strains) identified during this research have been deposited in GenBank (accession numbers AY451856–AY451963). The reference sequences (accession numbers) used in our sequence-variation analysis were from the following 20 strains: Urbani (AY278741), CUHK-W1 (AY278554), TOR2 (AY274119), HKU-39849 (AY278491), BJ01 (AY278488), BJ02 (AY278487), BJ03 (AY278490), BJ04 (AY279354), GD01 (AY278489), TW1 (AY291451), TWC (AY321118), SIN2774 (AY283798), SIN2748 (AY283797), SIN2679 (AY283796), SIN2677 (AY283795), SIN2500 (AY283794), HSR1 (AY323977), CUHK-Su10 (AY282752), Frankfurt1 (AY291315), and GZ50 (AY304495). In addition, 58 SARS-CoV genomes from GenBank were used for comparisons of phylogenetic analyses using the conventional and proposed strategies: TW9 (AY502932), TW8 (AY502931), TW7 (AY502930), TW6 (AY502929), TW5 (AY502928), TW4 (AY502927), TW3 (AY502926), TW2 (AY502925), TW11 (AY502924), TW10 (AY502923), GZ02 (AY390556), ZS-C (AY395003), LC5 (AY395002), LC4 (AY395001), LC3 (AY395000), LC2 (AY394999), LC1 (AY394998), ZS-A (AY394997), ZS-B (AY394996), HSZ-Cc (AY394995), HSZ-Bc (AY394994), HZS2-C (AY394992), HZS2-Fc (AY394991), HZS2-E (AY394990), HZS2-D (AY394989), HZS2-Fb (AY394987), HSZ-Cb (AY394986), HSZ-Bb (AY394985), HSZ2-A (AY394983), GZ-C (AY394979), GZ-B (AY394978), NS-1 (AY508724), WHU (AY394850), ShanghaiQXC1 (AY463059), ShanghaiQXC2 (AY463060), GD69 (AY313906), FRA (AY310120), SoD (AY461660), Sino3-11 (AY485278), Sino1-11 (AY485277), CUHK-AG03 (AY345988), CUHK-AG02 (AY345987), CUHK-AG01 (AY345986), PUMC03 (AY357076), PUMC02 (AY357075), PUMC01 (AY350750), GZ50 (AY304495), TWC3 (AY362699), TWC2 (AY362698), ZMY 1, (AY351680), TWY (AP006561), TWS (AP006560), TWK (AP006559), TWJ (AP006558), TWH (AP006557), TC3 (AY348314), TC2 (AY338175), and TC1 (AY338174). Two civet-cat SARS-CoV strains (SZ3 [AY304495] and SZ16 [AY304488]) were used as the outgroup of the rooted trees [19]
Acknowledgments
We thank K.-C. Chen and I.-P. Shih (AIDS Prevention and Research Center, National Yang-Ming University, Taipei), for their technical assistance, and Profs. Donald E. Morisky and Kathleen Ahrens, for their help in editing the manuscript. This work was supported in part by the Genome Research Center of National Yang-Ming University. We dedicate this work to the physicians and nurses who died of SARS during 2003
Footnotes
Presented in part: International Symposium on the Prevention and Control of SARS and Avian Flu, Beijing, China, 20–22 August 2004
Financial support: National Research Program for Genomic Medicine, National Science Council (grants NSC92-2751-B010-001-Y and 93-0324-19-F-00-00-00-35); Veterans General Hospitals/University System of Taiwan (grant VGHUST94-P7-42); National Health Research Institutes (grant NHRI BS-092-PP-05)
References
- 1.Peiris JS, Lai ST, Poon LL, et al. Coronavirus as a possible cause of severe cause acute respiratory syndrome. Lancet. 2003;361:1319–25. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ksiazek TG, Erdman D, Goldsmith C, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Fouchier RA, Kuiken T, Schutten M, et al. Aetiology: Koch’s postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chinese SARS Molecular Epidemiology Consortium Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–9. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization . Geneva: WHO; 2004. Summary of probable SARS cases with onset of illness 2002 to 31 July 2003. Available at: http://www.who.int/csr/sars/country/table2004_04_21/en/. Accessed 21 April 2004. [Google Scholar]
- 7.Lee ML, Chen CJ, Su IJ, et al. Severe acute respiratory syndrome—Taiwan, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:461–6. [PubMed] [Google Scholar]
- 8.Center for Disease Control and Prevention Update: outbreak of severe acute respiratory syndrome—worldwide, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:241–8. [PubMed] [Google Scholar]
- 9.Olsen SJ, Chang HL, Cheung TY-Y, et al. Transmission of the severe acute respiratory syndrome on aircraft. N Engl J Med. 2003;349:2416–22. doi: 10.1056/NEJMoa031349. [DOI] [PubMed] [Google Scholar]
- 10.Division of Surveillance and Investigation, Center for Disease Control, Taiwan . SARS probable cases in Taiwan—reclassified on 15 September, 2003. In: Su IJ, editor. Memoir of severe acute respiratory syndrome control in Taiwan. Taipei: Republic of China Center for Disease Control; 2004. pp. 6–10. [Google Scholar]
- 11.Yeh SH, Wang HY, Tsai CY, et al. Characterization of severe acute respiratory syndrome coronavirus genomes in Taiwan: molecular epidemiology and genome evolution. Proc Natl Acad Sci USA. 2004;101:2542–7. doi: 10.1073/pnas.0307904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lan YC, Liu HF, Shih YP, Yang JY, Chen HY. Phylogenetic analysis and sequence comparisons of structural and non-structural SARS coronavirus proteins in Taiwan. Infect Genet Evol. 2005;5:261–9. doi: 10.1016/j.meegid.2004.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Health Organization . Geneva: WHO; 2003. SARS case in laboratory worker in Taiwan, China. Available at: http://www.who.int/mediacentre/releases/2003/np26/en/ Accessed 11 March 2005. [Google Scholar]
- 14.Zeng FY, Chan CW, Chan MN, et al. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU-39849 (HK-39) Exp Biol Med. 2003;228:866–73. doi: 10.1177/15353702-0322807-13. [DOI] [PubMed] [Google Scholar]
- 15.Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 16.Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–9. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 17.Ruan YJ, Wei CL, Ling AE, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361:1779–85. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan Y, Peiris JS, Zheng B, et al. Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome. Lancet. 2004;363:99–104. doi: 10.1016/S0140-6736(03)15259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan Y, Zheng SJ, He YQ, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–8. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization . Geneva: WHO; 2003. Case definition for surveillance of severe acute respiratory syndrome (SARS) Available at: http://www.who.int/csr/sars/casedefinition/en/. Accessed 1 May 2003. [Google Scholar]
- 21.Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. [Google Scholar]
- 22.Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–5. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- 23.Felsenstein J. PHYLIP-Phylogeny Inference Package (version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- 24.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 25.Hillis DM, Bull JJ. An empirical test of bootstrapping confidence in phylogenetic analysis. Syst Biol. 1993;42:182–92. [Google Scholar]
- 26.Felsenstein J, Churchill GA. A hidden Markov model approach to variation among sites in rate of evolution. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 27.Chim SS, Tsui SK, Chan KC, et al. Genomic characterization of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong. Lancet. 2003;362:1807–8. doi: 10.1016/S0140-6736(03)14901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemey P, Pybus OG, Wang B, et al. Tracing the origin and history of the HIV-2 epidemic. Proc Natl Acad Sci USA. 2003;100:6588–92. doi: 10.1073/pnas.0936469100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng PKC, Wong DA, Tong LK, et al. Viral shedding patterns of coronavirus in patients with probable severe acute respiratory syndrome. Lancet. 2004;363:1699–70. doi: 10.1016/S0140-6736(04)16255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsui SK, Chim SS, Lo YM. Coronavirus genomic-sequence variations and the epidemiology of the severe acute respiratory syndrome. N Engl J Med. 2003;349:187–8. doi: 10.1056/NEJM200307103490216. [DOI] [PubMed] [Google Scholar]