

Abstract
Overactivation of the mitogen-activated protein kinase (MAPK) pathway is an important driver of many human cancers. First line, FDA-approved therapies targeting MAPK signalling, which include BRAF and MEK inhibitors, have variable success across cancers, and a significant number of patients quickly develop resistance. In recent years, a number of preclinical studies have reported alternative methods of overcoming resistance, which include promoting apoptosis, modulating autophagy, and targeting mitochondrial metabolism. This review summarizes mechanisms of resistance to approved MAPK-targeted therapies in BRAF-mutated cancers and discusses novel preclinical approaches to overcoming resistance.
1. Introduction
The mitogen-activated protein kinase (MAPK) cascade plays a critical role in cell survival, proliferation, and differentiation [1]. The components of the MAPK pathway are a highly conserved and ubiquitously expressed family of enzymatic kinases that phosphorylate many different target substrates [2, 3]. MAPK components are part of the large tiered phosphorylation cascade that includes RAS, RAF, MEK, and ERK kinases [4–6]. This tiered organization provides flexibility and adaptability, allowing a broad range of higher-order kinases to respond to the environment and control cellular function [7, 8]. Through transduction of signals from extracellular stimuli to downstream effector proteins within the cell, the MAPK pathway plays a major role in nearly every cellular process [1, 9].
In healthy tissue, activation of the MAPK pathway arises from a variety of intracellular and extracellular stimuli, including metabolic stress, DNA damage, cytokines, and growth factors [9]. Typically, the MAPK pathway is stimulated by growth factors binding to receptor tyrosine kinases (RTKs). RTKs including epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor receptor (VEGFR) converge downstream onto MAPK [10–13]. Notably, hormone stimulation may also activate the MAPK pathway through the progesterone receptor (PgR) and estrogen receptor (ER) [14–16]. Progestin-bound PgR promotes rapid ER alpha/Src association to activate MAP [16]. Hormone-triggered MAPK signalling events have been well summarized by Giovannelli et al. [17]. In addition, stress-activated MAP kinases modulate the activity of several nuclear receptors, including androgen receptor (AR), estrogen receptor (ER), glucocorticoid receptor (GR), peroxisome proliferator-activated receptor (PPAR), and retinoic acid receptor (RAR) [18]. Overall, MAPK signalling is important for growth, development, and cell turnover across many tissue types. Canonical MAPK signalling results from membrane receptor stimulation that activates the small GTPase, RAS, leading to a kinase cascade that ultimately phosphorylates extracellular signal-related kinases (ERK) (Figure 1(a)) [19–22]. ERK has widespread cellular effects, activating target proteins in both the cytoplasm, including RSK and MNK (Figure 1(a)), and the nucleus, including c-JUN, MYC, and ELK1 (Figure 1(b)) [23–27]. ERK-MAPK signalling promotes cell survival, proliferation, and motility [28–31]. Notably, crosstalk between MAPK components and other pathways can enhance the effects of MAPK signalling and increase cell proliferation and oncogenic transformation [18].
Figure 1.
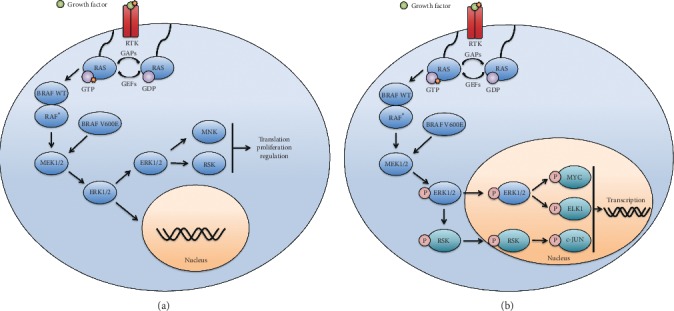
Mitogen-activated protein kinase (MAPK) pathway regulates nuclear and cytoplasmic activities. (a) Membrane receptor stimulation activates RAS GTPase which phosphorylates and activates RAF ⟶ MEK ⟶ ERK. BRAF forms homo- or heterodimers with other RAF-family proteins (ARAF or CRAF)∗ leading to MEK activation. BRAFV600E is constitutively active and phosphorylates MEK independent of RAS activation and dimerization. ERK-specific phosphorylation regulates its localization. Cytoplasmic ERK regulates RSK and MNK to modulate cellular function including transcription, proliferation, and invasion. (b) Phosphorylated ERK may phosphorylate RSK, which can translocate to the nucleus. In the nucleus, other transcription factors are recruited to promote expression of growth and prosurvival proteins.
Constitutive activation of the MAPK pathway, through overstimulation of receptors, RAS activation, or uncontrolled kinase activity, drives many human cancers [32]. Overactivation of BRAF, a RAF-family protein kinase and component of MAPK, is one of the most common events resulting in aberrant MAPK signalling [33]. BRAF is frequently mutated from GAG to GTG resulting in a valine to glutamic acid transition at amino acid position 600 in the activation loop of the BRAF kinase domain (BRAFV600E) [33–35]. This mutation forms a salt bridge between glutamic acid 600 and lysine 507 to promote an active, closed kinase conformation and facilitate catalysis [36]. In addition, the BRAFV600E mutation destabilizes the hydrophobic interactions between the aspartic acid-phenylalanine-glycine (DFG) motif and the P-loop to promote the DFG motif to adopt an active inconformation resulting in autoactivation of the monomeric form of the BRAF kinase [37, 38]. The BRAFV600E mutation constitutively activates the MAPK pathway independent of RAS stimulation and is the most common activating BRAF mutation [39–41]. However, other point mutations, gene fusions, splice site variants, and gene amplifications also lead to constitutive BRAF activity [42–45]. Mutations in BRAF are seen across many cancers including, but not limited to thyroid, melanoma, colon, squamous cell and hairy cell leukemia, and CNS-related malignancies [33, 46, 47]. Consequently, inhibiting BRAF kinase signalling is an attractive target, which can benefit patients across different cancer types (Table 1).
Table 1.
Summary pharmacological interventions for BRAFV600E mutated cancers✝.
| Compound | Target | Pathway | Cancer type∗ |
|---|---|---|---|
| Vemurafenib | BRAF | MAPK | CRC/G/M/T |
| Dabrafenib | BRAF | MAPK | CRC/M |
| Encorabenib | BRAF | MAPK | CRC/M |
| Trametinib | MEK | MAPK | CRC/M/PDA |
| Binimetinib | MEK | MAPK | M |
| Navitoclax | BCL-2/BCL-XL/BCLW | BH3 mimetic | M/T |
| ABT-737 | BCL-2 | BH3 mimetic | M |
| A-1210477 | MCL-1 | BH3 mimetic | M |
| Compound 1+ | SH2 | STAT3 | M |
| Hydroxychloroquine | Unknown | Autophagy | CRC/M/PDA |
| Lys05 | Lysosome | Lysosomal autophagy | M |
| Chloroquine | Unknown | Autophagy | CRC/M/PDA |
| Temozolomide | DNA | DNA replication | M |
| KP1339/IT-139 | GRP78 | ER homeostasis | MT |
| GSK2606414 | PERK | UPR# | M |
| A443654.3 | AKT | PI3K/AKT/mTOR | CC |
| MK2206 | AKT | PI3K/AKT/mTOR | CRC |
| LY294002 | PI3K | P3K/AKT/mTOR | CRC |
| GDC0941 | PI3K | P3K/AKT/mTOR | CRC |
| PPP++ | IGF-1R | PI3K/AKT/mTOR | M |
| Iso-orientin | Unknown | PI3K/AKT/Mitochondria | HBC |
| TM+++ | Copper | Angiogenesis/inflammation | CRC |
| Ibuprofen | COX1/2 | NSAID## | MT |
| Naproxen | COX1/2 | NSAID## | MT |
| Celecoxib | COX-2 | NSAID## | SCC |
| Etomoxir | CPT1A | Lipid oxidation | M/P |
| Phenformin | AMPK | Metabolic regulator | M |
| β-sitosterol | Complex I | ETC### | M |
| SR4 | Proton uncoupler | Mitochondria | M |
| Niclosamide | Proton uncoupler | Mitochondria | M |
∗CC = cholangiocarcinoma; CRC = colorectal carcinoma; G = glioma; HBC = hepatoblastoma cancer; M = melanoma; MT = multiple tumours; P = prostate; PDA = pancreatic ductal adenocarcinoma; PST = panel of solid tumours; SCC = squamous cell carcinoma; T = thyroid. +Compound 1 = quinoxaline-2,3-diylbis (methylene) dicarbamimidoselenoate dihydrobromide. ++PPP = cyclolignan propodophyllin. ++TM = tetrathiomolybdate. #UPR = unfolded protein response. ##NSAID = nonsteroidal anti-inflammatory. ###ETC = electron transport chain. ✝This is not a comprehensive list of compounds that target alternative mechanisms of BRAFi resistance and covers therapies that are referenced in this review.
Vemurafenib, a kinase inhibitor, was first used in a phase 1 clinical trial for BRAFV600E-mutated metastatic melanoma in 2010 and was approved for use in 2011 [48, 49]. Despite improvements in overall survival, the majority of patients treated with BRAF inhibitors develop resistance and disease progression after 6-7 months [50, 51]. In 2018, the FDA approved the BRAF/MEK combination encorafenib plus binimetinib for BRAFV600E- and BRAFV600K-mutated metastatic melanoma based on more durable results from a Phase 3 clinical trial [52]. Combining BRAF and MEK inhibitors has improved the average progression-free survival of metastatic melanoma from 5.8 to 9.4 months but many patients still suffer from resistance to combination therapy [50]. Despite successes in melanoma, vemurafenib has variable efficacy in other BRAFV600E-mutated cancers such as thyroid, colorectal, and glial, with only a small fraction of patients responding [53–55]. While somewhat controversial, histological subtype, microsatellite instability, and other genetic alterations are proposed to contribute to variable responses to vemurafenib across cancers [55, 56].
Resistance to BRAF inhibition may be intrinsic or acquired, which is respectively observed as either no response to initial therapy or a response and later resistance to therapy. In this review, we will focus on the acquired resistance mechanisms precipitated by BRAF inhibition and alternative therapies that may overcome acquired resistance. Acquired resistance is a cellular alteration in addition to the BRAFV600E mutation that facilitates tumour cells to grow despite BRAF inhibitor (BRAFi) therapy. Reactivation of MAPK signalling is the most common mechanism of acquired resistance to BRAFi therapy across cancer types [57]. However, recent studies have identified that overexpression of antiapoptotic genes, stimulation of autophagy, adenosine monophosphate protein kinase (AMPK) activation, alterations in the tumour microenvironment, and changes in metabolic flux also promote resistance and treatment failure [58–65]. Each section of this review describes the normal role, the aberrant activity resulting in acquired resistance, and potential approaches to targeting and overcoming these resistance mechanisms. Here, we review cellular adaptations that promote resistance to anti-MAPK therapy and summarize novel preclinical approaches to improving long-term patient responses across cancer types. We summarize recent promising preclinical therapies in this review; however, it is important to note that this is not a comprehensive list (Table 1).
2. Targeting Apoptosis in Cancer
2.1. Antiapoptotic BCL-2 Family Proteins Promote Resistance
Apoptosis, or programmed cell death, is a tumour suppression mechanism that is critical to healthy tissue homeostasis [66]. This program is a downstream effector pathway of the MAPK signalling cascade (Figure 2(a)) [67]. Apoptotic pathways are driven by the caspase protease family, which are proteolytically activated by either the intrinsic or extrinsic apoptotic pathway [68]. The intrinsic pathway (the “stress” or “mitochondrial” pathway) is mediated by pro- and antiapoptotic members of the B-cell lymphoma-2 (BCL-2) protein family and is implicated in tumourigenesis [69, 70]. Proapoptotic BCL-2 Associated X (BAX) and BCL-2 Antagonist Killer (BAK) form homo- and heterodimers stimulate the release of mitochondrial cytochrome c [71–73]. Cytosolic cytochrome c subsequently associates with apoptotic protease-activating factor (APAF-1) to form the apoptosome, which cleaves and activates proapoptotic caspases [74]. Apoptosis is modulated in part by antiapoptotic members of the BCL-2 protein family, which inhibit BAX and BAK activity and promote cell survival [75, 76]. Conversely, BH3-only proteins, such as BAD and BIM, stimulate cell death by activating BAX and BAK [77–79].
Figure 2.
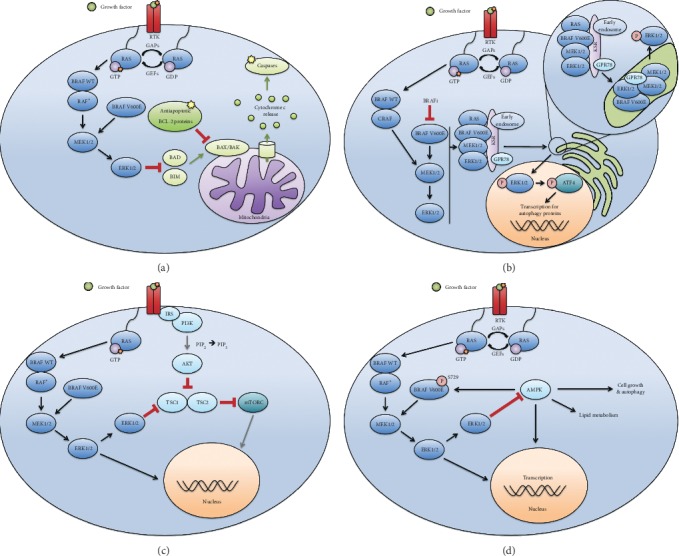
Noncanonical functions of MAPK are emerging targets for BRAFV600E-mutated cancers. (a) MAPK mediates apoptosis by inhibiting BH3-only proteins that activate proapoptotic BAX and BAK proteins. BCL-2 proteins inhibit caspase-mediated apoptosis by sequestering BAX and BAK to prevent release of mitochondrial cytochrome c. (b) MAPK proteins associate with KSR2, GRP78, and endosomes to localize to the ER. Translocation to the ER is required for ERK reactivation, ATF4 phosphorylation, and subsequent autophagy. (c) MAPK negatively regulates PI3K/AKT/mTOR through TSC1 dimers. The Insulin receptor substrate (IRS) recruits PI3K to the membrane resulting in activation and conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4, 5-triphosphate (PIP3). The second messenger PIP3 promotes the activation of AKT. AKT inhibits the dimerization and activation of TSC1 and TSC2, leading to mTOR activation. (d) Downstream ERK inhibits the metabolic sensor AMPK, which modulates the MAPK pathway via phosphorylation of serine 729 on BRAF, likely representing a regulatory feedback loop. AMPK promotes catabolism, including lipid breakdown and autophagy.
Evading apoptosis is an important hallmark of cancer, and the success of therapy depends, in large part, on its ability to induce cell death [80]. Many cancers, including breast, pancreatic, and endometrial cancer, evade apoptosis through upregulation of antiapoptotic proteins, including those in the BCL-2 protein family [81–84]. BCL-1, myeloid-cell leukemia (MCL-1), and B-cell lymphoma extralarge (BCL-XL) are antiapoptotic BCL-2 proteins frequently overexpressed in human cancer and protect cells against apoptosis under normal conditions in breast, prostate, and cholangiocarcinoma cancer cells [85–87]. In BRAFV600E-mutated cancers, MCL-1 has been shown to be aberrantly upregulated [58, 59]. Furthermore, constitutively active MAPK signalling phosphorylates and inactivates BAD and BIM, two proapoptotic BH3-only proteins (Figure 2(a)) [88, 89].
In recent years, co-opting proapoptotic pathways had been of interest in several cancer types, including melanoma and thyroid carcinoma [60, 90]. BH3 mimetics, single-agent small molecule inhibitors that have proapoptotic effects similar to BH3-only proteins, have shown efficacy in melanoma clinical trials for reducing tumour size [91]. Combining drugs that target proapoptotic BCL-2 family proteins with BRAF inhibitors in BRAFV600E-mutated cancers sensitizes melanoma cells to apoptosis but has not been shown to reverse resistance that is acquired over time [60]. However, Jeong et al. showed BRAFi therapy increased BCL-XL and BCL-2 expression in BRAFV600E-mutated human thyroid cancer cells [90]. Treating the same cells with BH3 mimetic, navitoclax, promoted apoptosis and growth inhibition compared with vemurafenib or navitoclax alone [90].
2.2. STAT3 is an Alternative Mechanism to Activate AntiaApoptotic Pathways
STAT proteins constitute a diverse array of transcription factors and are activated by a variety of extracellular signals, including epidermal growth factor (EGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), granulocyte-colony stimulating factor (G-CSF), interleukin-6 (IL-6), and insulin-like growth factor (IGF) [92–97]. STAT3 is activated via tyrosine phosphorylation and subsequently translocates to the nucleus where it activates transcription of genes involved in cell cycle progression including CCND1, c-MYC, and FOXM1 [98–100]. Interestingly, STAT3 also activates antiapoptotic BCL-2 family proteins, highlighting its dual involvement in prosurvival and apoptotic pathways [101].
In addition to BCL-2 family proteins, STAT3 represents an attractive target of antiapoptotic therapies. BRAFV600E mutations are highly abundant in advanced-stage melanoma and colorectal cancer (CRC) [102, 103]. In tumours with acquired resistance to BRAFi therapy, STAT3 activation is associated with a poor prognosis [104, 105]. Alcolea recently developed and tested an organoseleneium compound, which reduced melanoma cell viability, suppressed proliferation, induced apoptosis, inhibited STAT3, and induced cell cycle inhibitor p21 [106]. This compound did not decrease phosphorylation of ERK1/2 or other relevant kinases, suggesting that it suppresses cell death through an alternative mechanism [106]. Dysregulation of apoptotic pathways may serve to increase the metastatic potential of BRAFV600E-mutated cancer cells [107, 108]. These data suggest that combining STAT3 inhibitors with BRAFi may be an effective way to overcome resistance in cancers that have failed BRAFi monotherapy.
3. Dual Roles of Autophagy in Cancer as Potential Therapeutic Targets
Autophagy is a process by which cells recycle the cytoplasmic material for energy use or biosynthesis of macromolecules. Importantly, autophagy plays dual roles in either promoting or preventing cell death. In healthy cells, autophagic processes are upregulated in response to stress, allowing for cell survival [109]. Accordingly, cells lacking autophagic capabilities cannot adapt to stressed environments and have a lower apoptotic threshold [110]. Despite its critical function in cell survival, autophagy also plays a role in cell death. In some cases, cells may overstimulate autophagy to catabolize indispensible cellular components, leading to “autophagic cell death” (also known as “Type II apoptosis”) [111]. High numbers of autophagosomes are often seen in dying cells, and there is evidence that autophagy alone is sufficient for cell death under certain conditions [112]. Pathway crosstalk between autophagy and apoptosis is explained in part by Beclin-1, a BH3-only protein that is critical in autophagy and interacts with antiapoptotic proteins BCL-2 and BCL-XL [113–115]. Proapoptotic caspase-mediated cleavage of Beclin-1 inhibits its autophagic effects and stimulates apoptosis through mitochondrial release of cytochrome c [115].
In relation to cancer, autophagy is complex and multifaceted. Similar to healthy tissue, autophagy plays dual roles in either promoting or inhibiting cancer cell death. Some evidence suggests that inhibition of autophagic processes can contribute to tumourigenesis [116, 117]. For example, the mammalian target of rapamycin (mTOR) negatively regulates autophagy, and consequently, inhibition of mTOR leads to cell death [118]. Similarly, stimulation of autophagy through other mechanisms, including intermittent fasting and AMPK activation, have been shown to inhibit tumour growth and selectively promote cancer cell death [119, 120]. However, in cancer, autophagic processes may be modulated to promote survival in conditions that would otherwise trigger programmed cell death [121]. During nutrient deprivation or metabolic stress, cells stimulate autophagy to survive, adapt to their environment, and evade cell death, which may result in oncogenesis by preventing senescence [122, 123]. Notably, cancer cells may co-opt normal autophagic processes to survive stressors in their microenvironment, suppress p53 function, and increase mitochondrial respiration [124–126]. In starved conditions, K-RAS-mutated cells become addicted to autophagy, allowing them to retain functional mitochondria [126]. RAS-mutated cells with abnormal autophagosomes accumulate defective mitochondria and have decreased oxygen consumption [126]. Interestingly, autophagy, mitophagy, and mitochondrial metabolism are attenuated during tumour formation, but can increase to maintain energy homeostasis and promote tumour survival in a stressed environment [126]. Taken together, autophagy serves dual functions; it either promotes apoptosis or cell survival. These functions are modulated based on the stage of tumour development, indicating that autophagy-targeted treatments should be tailored to the cancer phenotype.
In BRAFV600E-mutated cancer cells, autophagy may be upregulated as a protective mechanism in response to cellular stressors [61, 127, 128]. Recently, there have been preclinical studies using autophagy inhibitors in combination with BRAFi for BRAFV600E-mutated cancer cells [62, 129]. Kinsey et al. showed that inhibiting the RAF ⟶ MEK ⟶ ERK signalling cascade in K-RAS-driven cancers stimulated autophagy through activation of AMPK, a key energy sensor and metabolic regulator [62, 130]. In low glucose conditions, AMPK phosphorylates ULK1 at two serine residues leading to autophagosome formation and initiation of autophagy [130]. Conversely, under conditions of high nutrient availability, active mTOR phosphorylates ULK1 at a different serine residue, preventing its interaction with AMPK [130]. In preclinical models, the MEK1/2 inhibitor, trametinib, in combination with an autophagy inhibitor, chloroquine, demonstrated synergy in pancreatic ductal adenocarcinoma, colorectal carcinoma, and melanoma patient-derived xenograft (PDX) models with RAS and BRAFV600E mutations [62].
Autophagy serves as an adaptive drug resistance mechanism in BRAFV600E-mutated cancers [61]. Ojha et al. showed that in melanoma, BRAFi therapy induces an ER stress response that stimulates autophagy and promotes cell survival. This stress response is mediated by protein-protein interactions between MAPK components (including BRAFV600E), chaperone protein GRP78, scaffolding protein KSR2, ER translocase SEC61, and early endosomes (Figure 2(b)) [129]. Activation of the ER stress response results in components of the MAPK pathway translocating to the ER via association with GRP78, KSR2, and SEC61 [129]. This translocation is required for ERK reactivation and subsequent stimulation of cytoprotective autophagy via ATF4 phosphorylation [129]. Accordingly, expression of mutant ATF4 has been shown to improve cellular sensitivity to MAPK inhibition in vivo [129]. Ryabaya et al. have shown that GRP78 blockade inhibits the ER stress-mediated autophagy and promotes apoptosis through caspase 7 to sensitize melanoma cells to temozolomide [131]. KP1339/IT-139, a GRP78 inhibitor, demonstrated significant anticancer activity in a Phase I clinical trial and is an attractive potential treatment for BRAFi-resistant tumours [132]. Taken together, these data provide rationale for clinical studies combining antiautophagic agents with BRAF and MEK inhibitors.
4. AKT, Copper Complexes, and Arachidonic Acid Metabolism Are Inflammatory Targets in BRAFV600E-Mutated Cancer
Lipid signalling in the phosphoinositide 3-kinase (PI3K)-AKT pathway plays a critical role in differentiation, cytoskeletal rearrangement, vesicle trafficking, growth, and mounting inflammatory responses [133, 134]. Membrane inositol phospholipids (PI) are modified by different classes of PI3K. PI3K has different isoforms, Class I, Class II, and Class III, which selectively phosphorylate membrane PIs [135, 136]. Importantly, Class I PI3Ks convert phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2 or “PIP2”) to phosphatidylinositol-3,4,5-triphosphate (P(3,4,5)P3 or “PIP3”) [137, 138]. The PIP3 second messenger has many functions but classically activates the downstream kinase, AKT, which negatively regulates TSC1 and TSC2 heterodimers to modulate mTORC1 activity (Figure 2(b)) [139].
Inflammation and cancer are related bidirectionally, with high levels of chronic inflammation conferring an increased risk for tumour development [140, 141]. Conversely, cancer has been shown to alter surrounding tissue to suppress cancer-killing immune cells and promote chronic, proresolving inflammation [142, 143]. Tumour-infiltrating lymphocytes and macrophages constitute a significant population of cells in the microenvironment and modulate response to treatment [144, 145]. Cytokines associated with chronic inflammation, including TNF-α, TGF-β, and IL-6, remodel the microenvironment and promote tumourigenesis in part through generation of reactive oxygen species (ROS) [65, 146, 147]. The PI3K-AKT-mTOR signalling axis remodels surrounding tumour-stromal cells, playing an important role in tumour-infiltrating lymphocyte activity and oncogenesis [119]. PI3K signalling is often upregulated in BRAFV600E-mutated cells that are resistant to BRAFi therapy [148–151]. Isoforms of PI3K play important roles in lymphocyte chemotaxis and NK cell extravasation into tumour-stromal tissue [152, 153]. In addition, PI3K upregulates mTOR signalling, further driving oncogenesis (Figure 2(c)) [154]. Preclinical studies have successfully targeted PI3K signalling in combination with BRAF/MEK inhibitors in BRAFV600E-mutated colorectal cancer and melanoma cells [155–157]. BRAFV600E melanoma cells treated to resistance with monotherapy BRAFi are sensitive to IGF-1R/PI3K and MEK inhibitors [155]. While single-agent vemurafenib is effective in less than 10% of BRAFV600E-mutated CRCs, the addition of PI3K inhibitors shows synergistic growth inhibition in drug-resistant CRC cells [156]. Jiang et al. showed BRAFi resistance increases tumour PD-L1 expression, which can be overcome by combined MEK and PI3K inhibition [157]. PI3K inhibitors in combination with vemurafenib have shown promising results in early clinical trials [158, 159]. In summary, PI3K inhibitors in combination with other treatments offer potential therapeutic benefit for BRAFV600E-mutated cancers resistant to monotherapy BRAFi.
In addition to classic pathway inhibition, other anti-inflammatory drugs have been tested preclinically with promising results. Copper is a tightly regulated cofactor for a wide variety of enzymes and is an essential micronutrient [160]. Copper dysregulation is seen in a variety of chronic diseases including Wilson's disease and Alzheimer's disease [161, 162]. Inflammatory ROS is seen with high serum copper [160, 163–166]. Elevated serum copper levels are correlated with poor survival in CRC and drug resistance in other tumour types [167]. In BRAFV600E-mutated lung cancer and melanoma cells, copper has been shown to enhance MEK1 phosphorylation of ERK1/2 through formation of a MEK1-copper complex [168]. Therapeutics targeting copper-driven ROS sequester block the uptake or chelate copper to mitigate ROS production [160, 167, 168]. Metallothionine and glutathione can protect against copper-driven ROS by sequestering it in the cytosol [160]. Copper uptake into the cell can be inhibited by targeting the CTR1 receptor and disrupting the MEK1-copper binding site to decrease ERK1/2 phosphorylation and downstream signalling [168]. Copper chelation therapy with tetrathiomolybdate is used in Wilson's disease and is being studied in cancers due to its antiangiogenic and anti-inflammatory properties [167]. In BRAFi resistant CRC cells, copper chelation therapy has been shown to decrease proliferation, survival, migration, and clonogenic potential [167]. These data highlight alternative approaches to overcoming resistance and developing drugs that address multiple cellular adaptations that contribute to oncogenesis (i.e., metabolism, generation of ROS, and oncogenic transformation).
One of the most widely used classes of drugs, nonsteroidal anti-inflammatory drugs (NSAIDs), has the potential to decrease the proliferative capacity of cancer [169, 170]. NSAIDs target cyclooxygenase (COX) enzymes, which play important roles in fever, pain, and inflammation. Membrane phospholipids are converted into arachidonic acid (AA) by either membrane phospholipase A2 (mPLA2) or soluble phospholipase A2 (sPLA2) [171–173]. AA is converted into lipid signalling molecules including prostaglandins, thromboxanes, and prostacyclins by COX1 and COX-2 isoenzymes [170]. In healthy tissues, COX1 is expressed and constitutively active, functioning as a “housekeeping” protein. Under ordinary conditions, COX1 produces thromboxane, prostacyclin, and prostaglandin E2, which serve normal functions in platelet aggregation and gastrointestinal cytoprotection. By contrast, COX-2 expression is induced during inflammation. COX-2 plays a critical role in mounting immune responses by producing prostaglandin E2, leading to fever and increased blood vessel permeability [170]. Classic inhibitors of COX1/2, including ibuprofen and naproxen, and more recent selective COX-2 inhibitors, including celecoxib, have been employed in clinical practice [174]. Targeting BRAF and the COX-2 orthogonal pathway could benefit patients with resistance to BRAFi drugs. Escuin-Ordinas et al. describe how celecoxib prevents the development of BRAFi-induced secondary cutaneous squamous cell carcinomas (cuSCCs) that result from paradoxical BRAF activation. In cuSCC cells, vemurafenib alone increased phosphorylation of ERK, a response that was decreased significantly by celecoxib. Furthermore, trametinib, a MEK inhibitor, reduced cuSCC development less efficiently than celecoxib. The authors suggest that reduced prostaglandin synthesis resulting from inhibition of COX-2 decreases the development of secondary cuSCCs [175]. The results of this study may inform us that targeting an orthogonal metabolic pathway could improve therapeutic efficacy of BRAFi therapy in BRAFV600E-mutated tumours. These data provide rationale for future investigations into inhibiting inflammatory processes in BRAFV600E-mutated cancers to overcome resistance to BRAFi therapy.
5. Energy Homeostasis in Resistant BRAFV600E-Mutated Cancer as a New Avenue for Therapy
Metabolic efficiency is frequently optimized to produce maximum ATP per molecule of glucose. To achieve maximum efficiency, healthy cells will metabolize glucose through oxidative phosphorylation in the mitochondria in the presence of oxygen. The flux through these metabolic pathways will be tightly regulated to meet the metabolic demands of any cell, modulate reactive oxygen formation, and keep metabolites at homeostatic abundancies through anaplerotic and cataplerotic reactions [176].
In 1931, Otto Warburg was awarded the Nobel Prize in Medicine and Physiology for his proposal that cancer cells metabolize glucose in the presence of oxygen, a process known as aerobic glycolysis [177–179]. This observation was named the Warburg Effect and has remained the classic paradigm of cancer metabolism [177, 180]. However, recent evidence suggests certain cancer types can uptake and oxidize lipids [181, 182]. Lipid catabolism is regulated by AMPK, a nutrient sensor of normal and cancer cells. AMPK phosphorylates and inactivates acetyl-CoA carboxylase, which impedes fatty acid synthesis and promotes lipid utilization by beta-oxidation in the mitochondria (Figure 2(d)) [183]. In fact, a recent study by Aloia has shown that upregulation of fat oxidation plays a role in the adaptive response to MAPK inhibition [184]. Particularly, the pharmacological blockade of CPT1A (the rate-limiting step for fat oxidation) reactivated glycolysis in MAPKi-treated melanoma cells. This compensatory increase in glycolytic flux in response to CPT1A inhibition has already been described and highlights the flexibility of cancer cell metabolism to promote resistance [185]. Thus, the concomitant inhibition of CPT1A, glycolysis, and MAPK synergistically inhibited tumour cell growth in vitro and in BRAFV600E-mutated melanoma mouse models [184].
Interestingly, AMPK phosphorylates BRAF at serine 729 [186]. Shen et al. showed that this phosphorylation leads to decreased MAPK signalling by preventing BRAF association with CRAF and KSR1 in BRAF wild type cells [186]. Consequently, AMPK presents an attractive therapeutic target for BRAFi-resistant tumours. However, the relationship between BRAF and AMPK is less clear in the context of BRAFV600E mutations. Ritt et al. demonstrated that mutating serine 729 in BRAFV600E cells did not affect MEK activation or transformation potential [187]. By contrast, mutating serine 729 in cells with intermediate BRAF kinase activity led to increased MEK activation [187]. While more investigation into the mechanism of AMPK regulation of BRAFV600E-mutated cells is necessary, AMPK inhibitors have shown efficacy in combination with BRAFi therapy [188]. Melanoma and CRC studies indicate that cancer cells increase their tolerance to MAPK pathway inhibition by activating AMPK-mediated autophagy [63, 189]. Consistent with these findings, a preclinical study by Yuan et al. demonstrated reduced drug resistance in melanoma cells treated with AMPK and BRAF inhibitor therapy compared with single-agent vemurafenib [188]. In contrast, a recent study found that BRAF inhibitors in combination with biguanide and phenformin (AMPK activators and complex I inhibitors) induced tumour regression [64]. Taken together, these studies merit further investigation into the relationship between BRAF and AMPK.
Metabolic reprogramming events may play a crucial role in tumourigenesis, drug resistance, and metastases [190]. A study on slow-cycling, chemotherapy-resistant BRAFV600E-mutated melanoma showed oxidative phosphorylation enzymes were upregulated, and consequently, their inhibition resulted in cell death [191]. Several compounds targeting mitochondrial respiration have been studied preclinically with promising results [192–194]. Molecular profiling shows enzymes involved in oxidative phosphorylation are enriched in melanoma brain metastases [193]. In a metastatic melanoma PDX study, β-sitosterol, an electron transport chain complex I inhibitor, effectively reduced oxidative phosphorylation and prevented the development of brain metastases [192]. Moreover, β-sitosterol increased the generation of ROS and consequently induced apoptosis [192]. Treatment with an oxidative phosphorylation inhibitor improved survival and decreased the development of brain metastases in BRAF/MEK inhibitor-resistant mice [193]. Uncoupling agents, which dissipate the proton gradient in the mitochondrial intermembrane space, have also inhibited tumour growth in vemurafenib-resistant melanoma PDX models [194]. Mechanistic studies indicate that uncouplers modulate mTOR and AMPK and induce apoptosis without perturbing MAPK signalling [194]. These data indicate that the metabolic profile of drug-resistant cancer cells clearly differs from drug sensitive subpopulations. Metabolic differences in lipid metabolism can be modulated therapeutically to overcome BRAFi resistance. The current paradigm and our understanding of the Warburg effect in cancer may be overlooking metabolic rewiring, such as lipid metabolism, and other adaptations in cancer.
6. Conclusions
Preclinical data on agents that work synergistically with MAPK therapy offer promising avenues for future clinical studies. Apoptosis, autophagy, and metabolism are particularly interesting areas of research and present opportunities for targeting nongenomic cancer transformations. Understanding the interconnectivity and regulation of these cellular processes will provide insight into the efficacy of targeted therapeutics in the context of tumour development, invasion, progression, and metastasis. So far, preclinical data show BRAF/MEK inhibitors in combination with alternative therapies are novel and efficacious approaches to overcoming BRAFi resistance. There have been advances in overcoming acquired resistance to BRAFi across cancers. However, it is important to acknowledge there are some leading fields, including quickly advancing therapies in melanoma, which could be beneficial for other tumour types and translated across cancers. Future research must elucidate mechanisms in which BRAFi-resistant cancers alter protein-protein interactions and their metabolism in order to develop rational targets for BRAFV600E-mutated cancers. Understanding the unique metabolic and autophagic profiles of BRAFV600E-mutated tumours across cancer types and disease stages will help to advance the development of therapeutics with lower toxicity than conventional treatments.
Acknowledgments
This work was supported by funding from the American Cancer Society RSG 16-256-01-TBE.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Kim E. K., Choi E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Gardner A. M., Vaillancourt R. R., Lange-Carter C. A., Johnson G. L. MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Molecular Biology of the Cell. 1994;5(2):193–201. doi: 10.1091/mbc.5.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu C., Liu R, Zhang Q, Chen X, Qian Y, Fang W. The diversification of evolutionarily conserved MAPK cascades correlates with the evolution of fungal species and development of lifestyles. Genome Biology and Evolution. 2017;9(2):311–322. doi: 10.1093/gbe/evw051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graves J. D., Campbell J. S., Krebs E. G. Protein serine/threonine kinases of the MAPK cascade. Annals of the New York Academy of Sciences. 1995;766(1):320–343. doi: 10.1111/j.1749-6632.1995.tb26684.x. [DOI] [PubMed] [Google Scholar]
- 5.Ahn N. G. The MAP kinase cascade. Discovery of a new signal transduction pathway. Reversible Protein Phosphorylation in Cell Regulation. 1993;127-128:201–209. doi: 10.1007/bf01076771. [DOI] [PubMed] [Google Scholar]
- 6.Seger R., Krebs E. G. The MAPK signaling cascade. The FASEB Journal. 1995;9(9):726–735. doi: 10.1096/fasebj.9.9.7601337. [DOI] [PubMed] [Google Scholar]
- 7.Vaillancourt R. R., Gardner A. M., Johnson G. L. B-Raf-dependent regulation of the MEK-1/mitogen-activated protein kinase pathway in PC12 cells and regulation by cyclic AMP. Molecular and Cellular Biology. 1994;14(10):6522–6530. doi: 10.1128/mcb.14.10.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin L.-L., Wartmann M., Lin A. Y., Knopf J. L., Seth A., Davis R. J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 9.Burotto M., Chiou V. L., Lee J.-M., Kohn E. C. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–3456. doi: 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chadee D. N., Kyriakis J. M. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nature Cell Biology. 2004;6(8):770–776. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- 11.Yadav V., Zhang X., Liu J., et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. Journal of Biological Chemistry. 2012;287(33):28087–28098. doi: 10.1074/jbc.m112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graves L. M., Bornfeldt K. E., Sidhu J. S., et al. Platelet-derived growth factor stimulates protein kinase A through a mitogen-activated protein kinase-dependent pathway in human arterial smooth muscle cells. Journal of Biological Chemistry. 1996;271(1):505–511. doi: 10.1074/jbc.271.1.505. [DOI] [PubMed] [Google Scholar]
- 13.Doanes A. M., Hegland D. D., Sethi R., Kovesdi I., Bruder J. T., Finkel T. VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochemical and Biophysical Research Communications. 1999;255(2):545–548. doi: 10.1006/bbrc.1999.0227. [DOI] [PubMed] [Google Scholar]
- 14.Vicent G. P., Nacht A. S., Zaurín R., Ballaré C., Clausell J., Beato M. Minireview: role of kinases and chromatin remodeling in progesterone signaling to chromatin. Molecular Endocrinology. 2010;24(11):2088–2098. doi: 10.1210/me.2010-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Migliaccio A., Di Domenico M., Castoria G., et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. The EMBO Journal. 1996;15(6):1292–1300. doi: 10.1002/j.1460-2075.1996.tb00471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Migliaccio A., Piccolo D., Castoria G., et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. The EMBO Journal. 1998;17(7):2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giovannelli P., Di Donato M., Giraldi T., Migliaccio A., Castoria G., Auricchio F. Targeting rapid action of sex steroid receptors in breast and prostate cancers. Front in Bioscience. 2011;16:2224–2232. doi: 10.2741/3849. [DOI] [PubMed] [Google Scholar]
- 18.Plotnikov A., Zehorai E., Procaccia S., Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research. 2011;1813(9):1619–1633. doi: 10.1016/j.bbamcr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 19.Colicelli J. Human RAS superfamily proteins and related GTPases. Science’s STKE. 2004;2004(250):p. RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Der C. J., Krontiris T. G., Cooper G. M. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proceedings of the National Academy of Sciences. 1982;79(11):3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harvey J. J. An unidentified virus which causes the rapid production of tumours in mice. Nature. 1964;204(4963):1104–1105. doi: 10.1038/2041104b0. [DOI] [PubMed] [Google Scholar]
- 22.Qi M., Elion E. A. MAP kinase pathways. Journal of Cell Science. 2005;118(16):3569–3572. doi: 10.1242/jcs.02470. [DOI] [PubMed] [Google Scholar]
- 23.Doehn U., Hauge C., Frank S. R., et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Molecular Cell. 2009;35(4):511–522. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waskiewicz A. J. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. The EMBO Journal. 1997;16(8):1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulverer B. J., Kyriakis J. M., Avruch J., Nikolakaki E., Woodgett J. R. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353(6345):670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 26.Sears R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & Development. 2000;14(19):2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruzalegui F. H., Cano E., Treisman R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene. 1999;18(56):7948–7957. doi: 10.1038/sj.onc.1203362. [DOI] [PubMed] [Google Scholar]
- 28.Sahai E., Olson M. F., Marshall C. J. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. The EMBO Journal. 2001;20(4):755–766. doi: 10.1093/emboj/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vial E., Sahai E., Marshall C. J. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4(1):67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 30.Mavria G., Vercoulen Y., Yeo M., et al. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell. 2006;9(1):33–44. doi: 10.1016/j.ccr.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 31.Marshall C. J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 32.Prior I. A., Lewis P. D., Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Research. 2012;72(10):2457–2467. doi: 10.1158/0008-5472.can-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies H., Bignell G. R., Cox C., et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 34.Haling J. R., Sudhamsu J., Yen I., et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26(3):402–413. doi: 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Thevakumaran N., Lavoie H., Critton D. A., et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nature Structural & Molecular Biology. 2015;22(1):37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 36.Bollag G., Hirth P., Tsai J., et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulikakos P. I., Persaud Y., Janakiraman M., et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan P. T. C., Garnett M. J., Roe S. M., et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 39.Kim S.-J., Lee K. E., Myong J. P., et al. BRAFV600E mutation is associated with tumor aggressiveness in papillary thyroid cancer. World Journal of Surgery. 2012;36(2):310–317. doi: 10.1007/s00268-011-1383-1. [DOI] [PubMed] [Google Scholar]
- 40.Klempner S. J., Gershenhorn B., Tran P., et al. BRAFV600E mutations in high-grade colorectal neuroendocrine tumors may predict responsiveness to BRAF-MEK combination therapy. Cancer Discovery. 2016;6(6):594–600. doi: 10.1158/2159-8290.cd-15-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mar V. J., Liu W., Devitt B., et al. The role ofBRAFmutations in primary melanoma growth rate and survival. British Journal of Dermatology. 2015;173(1):76–82. doi: 10.1111/bjd.13756. [DOI] [PubMed] [Google Scholar]
- 42.Turner J., Couts K., Sheren J., et al. Kinase gene fusions in defined subsets of melanoma. Pigment Cell & Melanoma Research. 2017;30(1):53–62. doi: 10.1111/pcmr.12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner J. A., Bemis J. G. T., Bagby S. M., et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene. 2019;38(8):1296–1308. doi: 10.1038/s41388-018-0514-7. [DOI] [PubMed] [Google Scholar]
- 44.Baitei E. Y., Zou M., Al-Mohanna F., et al. Aberrant BRAFsplicing as an alternative mechanism for oncogenic B-Raf activation in thyroid carcinoma. The Journal of Pathology. 2009;217(5):707–715. doi: 10.1002/path.2496. [DOI] [PubMed] [Google Scholar]
- 45.Marranci A. The landscape of BRAF transcript and protein variants in human cancer. Molecular Cancer. 2017;16(1):p. 85. doi: 10.1186/s12943-017-0645-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen Y., Xing M., Mambo E., et al. BRAF mutation in papillary thyroid carcinoma. JNCI Journal of the National Cancer Institute. 2003;95(8):625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 47.Falini B., Martelli M. P., Tiacci E. BRAF V600E mutation in hairy cell leukemia: from bench to bedside. Blood. 2016;128(15):1918–1927. doi: 10.1182/blood-2016-07-418434. [DOI] [PubMed] [Google Scholar]
- 48.Flaherty K. T., Puzanov I., Kim K. B., et al. Inhibition of mutated, activated BRAF in metastatic melanoma. New England Journal of Medicine. 2010;363(9):809–819. doi: 10.1056/nejmoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim G., McKee A. E., Ning Y.-M., et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clinical Cancer Research. 2014;20(19):4994–5000. doi: 10.1158/1078-0432.ccr-14-0776. [DOI] [PubMed] [Google Scholar]
- 50.Long G. V., Stroyakovskiy D., Gogas H., et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. New England Journal of Medicine. 2014;371(20):1877–1888. doi: 10.1056/nejmoa1406037. [DOI] [PubMed] [Google Scholar]
- 51.Long G. V., Stroyakovskiy D., Gogas H., et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. The Lancet. 2015;386(9992):444–451. doi: 10.1016/s0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 52.Dummer R., Ascierto P. A., Gogas H. J., et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. The Lancet Oncology. 2018;19(5):603–615. doi: 10.1016/s1470-2045(18)30142-6. [DOI] [PubMed] [Google Scholar]
- 53.Brose M. S., Cabanillas M. E., Cohen E. E. W., et al. Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. The Lancet Oncology. 2016;17(9):1272–1282. doi: 10.1016/s1470-2045(16)30166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kopetz S., Desai J., Chan E., et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. Journal of Clinical Oncology. 2015;33(34):4032–4038. doi: 10.1200/jco.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaley T. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. Journal of Clinical Oncology. 2018;35 doi: 10.1200/JCO.2018.78.9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.BRAF/MEK combo active across tumor types. Cancer Discovery. 2019;9(8) doi: 10.1158/2159-8290.CD-NB2019-074. [DOI] [PubMed] [Google Scholar]
- 57.Carlino M. S., Gowrishankar K., Saunders C. A. B., et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Molecular Cancer Therapeutics. 2013;12(7):1332–1342. doi: 10.1158/1535-7163.mct-13-0011. [DOI] [PubMed] [Google Scholar]
- 58.Mukherjee N. BH3 mimetics induce apoptosis independent of DRP-1 in melanoma. Cell Death & Disease. 2018;9(9):p. 907. doi: 10.1038/s41419-018-0932-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haass N. K., Schumacher U. Melanoma never says die. Experimental Dermatology. 2014;23(7):471–472. doi: 10.1111/exd.12400. [DOI] [PubMed] [Google Scholar]
- 60.Wroblewski D., Mijatov B., Mohana-Kumaran N., et al. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis. 2013;34(2):237–247. doi: 10.1093/carcin/bgs330. [DOI] [PubMed] [Google Scholar]
- 61.Ma X.-H., Piao S.-F., Dey S., et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. Journal of Clinical Investigation. 2014;124(3):1406–1417. doi: 10.1172/jci70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinsey C. G., Camolotto S. A., Boespflug A. M., et al. Protective autophagy elicited by RAF ⟶ MEK ⟶ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nature Medicine. 2019;25(4):620–627. doi: 10.1038/s41591-019-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sueda T. BRAF V600E inhibition stimulates AMP-activated protein kinase-mediated autophagy in colorectal cancer cells. Science Reports. 2016;6:p. 18949. doi: 10.1038/srep18949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan P., Ito K., Perez-Lorenzo R., et al. Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proceedings of the National Academy of Sciences. 2013;110(45):18226–18231. doi: 10.1073/pnas.1317577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Landskron G. Chronic inflammation and cytokines in the tumor microenvironment. Journal of Immunology Research. 2014;2014:p. 19. doi: 10.1155/2014/149185.149185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baar M. P. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1):132–147. doi: 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan L., Wang J., Xiao H., Wu W., Wang Y., Liu X. MAPK signaling pathways regulate mitochondrial-mediated apoptosis induced by isoorientin in human hepatoblastoma cancer cells. Food and Chemical Toxicology. 2013;53:62–68. doi: 10.1016/j.fct.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 68.Hengartner M. O. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 69.Hockenbery D., Nuñez G., Milliman C., Schreiber R. D., Korsmeyer S. J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348(6299):334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 70.Scorrano L. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300(5616):135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 71.Mikhailov V., Mikhailova M., Degenhardt K., Venkatachalam M. A., White E., Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria. Journal of Biological Chemistry. 2003;278(7):5367–5376. doi: 10.1074/jbc.m203392200. [DOI] [PubMed] [Google Scholar]
- 72.Tan C., Dlugosz P. J., Peng J., et al. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. Journal of Biological Chemistry. 2006;281(21):14764–14775. doi: 10.1074/jbc.m602374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mandal T. Assembly of Bak homodimers into higher order homooligomers in the mitochondrial apoptotic pore. Science Reports. 2016;6 doi: 10.1038/srep30763.30763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou M., Li Y., Hu Q., et al. Atomic structure of the apoptosome: mechanism of cytochromec- and dATP-mediated activation of Apaf-1. Genes & Development. 2015;29(22):2349–2361. doi: 10.1101/gad.272278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dlugosz P. J., Billen L. P., Annis M. G., et al. Bcl-2 changes conformation to inhibit Bax oligomerization. The EMBO Journal. 2006;25(11):2287–2296. doi: 10.1038/sj.emboj.7601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Llambi F., Moldoveanu T., Tait S. W. G., et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Molecular Cell. 2011;44(4):517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Czabotar P. E., Westphal D., Dewson G., et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152(3):519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 78.Moldoveanu T., Grace C. R., Llambi F., et al. BID-induced structural changes in BAK promote apoptosis. Nature Structural & Molecular Biology. 2013;20(5):589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hockings C. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death & Disease. 2015;105:p. 6. doi: 10.1038/cddis.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanahan D., Weinberg R. A. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 81.Pidgeon G. P., Barr M. P., Harmey J. H., Foley D. A., Bouchier-Hayes D. J. Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. British Journal of Cancer. 2001;85(2):273–278. doi: 10.1054/bjoc.2001.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oehler M. K., Norbury C., Hague S., Rees M. C., Bicknell R. Adrenomedullin inhibits hypoxic cell death by upregulation of Bcl-2 in endometrial cancer cells: a possible promotion mechanism for tumour growth. Oncogene. 2001;20(23):2937–2945. doi: 10.1038/sj.onc.1204422. [DOI] [PubMed] [Google Scholar]
- 83.Cho H. J., Kim J. K., Kim K. D., et al. Upregulation of Bcl-2 is associated with cisplatin-resistance via inhibition of Bax translocation in human bladder cancer cells. Cancer Letters. 2006;237(1):56–66. doi: 10.1016/j.canlet.2005.05.039. [DOI] [PubMed] [Google Scholar]
- 84.Dong J., Zhao Y.-P., Zhou L., Zhang T.-P., Chen G. Bcl-2 upregulation induced by miR-21 via a direct interaction is associated with apoptosis and chemoresistance in MIA PaCa-2 pancreatic cancer cells. Archives of Medical Research. 2011;42(1):8–14. doi: 10.1016/j.arcmed.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 85.Gillett C. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Research. 1994;54(7):1812–1817. [PubMed] [Google Scholar]
- 86.Kobayashi S., Werneburg N. W., Bronk S. F., Kaufmann S. H., Gores G. J. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128(7):2054–2065. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 87.Lebedeva I. Bcl-xL in prostate cancer cells: effects of overexpression and down-regulation on chemosensitivity. Cancer Research. 2000;60(21):6052–6060. [PubMed] [Google Scholar]
- 88.Eisenmann K. M., VanBrocklin M. W., Staffend N. A., Kitchen S. M., Koo H. M. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Research. 2003;63(23):8330–8337. [PubMed] [Google Scholar]
- 89.Goldstein N. B., Johannes W. U., Gadeliya A. V., et al. Active N-Ras and B-Raf inhibit anoikis by downregulating bim expression in melanocytic cells. Journal of Investigative Dermatology. 2009;129(2):432–437. doi: 10.1038/jid.2008.227. [DOI] [PubMed] [Google Scholar]
- 90.Jeong J. H., Oh J. M., Jeong S. Y., Lee S.-W., Lee J., Ahn B.-C. Combination treatment with the BRAFV600E inhibitor vemurafenib and the BH3 mimetic navitoclax for BRAF-mutant thyroid carcinoma. Thyroid. 2019;29(4):540–548. doi: 10.1089/thy.2018.0511. [DOI] [PubMed] [Google Scholar]
- 91.Mukherjee N., Schwan J. V., Fujita M., Norris D. A., Shellman Y. G. Alternative treatments for melanoma: targeting BCL-2 family members to De-Bulk and kill cancer stem cells. Journal of Investigative Dermatology. 2015;135(9):2155–2161. doi: 10.1038/jid.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y., van Boxel-Dezaire A. H. H., Cheon H., Yang J., Stark G. R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proceedings of the National Academy of Sciences. 2013;110(42):16975–16980. doi: 10.1073/pnas.1315862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Udayakumar T. S., Stratton M. S., Nagle R. B., Bowden G. T. Fibroblast growth factor-1 induced promatrilysin expression through the activation of extracellular-regulated kinases and STAT3. Neoplasia. 2002;4(1):60–67. doi: 10.1038/sj.neo.7900207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y.-Z., Wharton W., Garcia R., Kraker A., Jove R., Pledger W. J. Activation of Stat3 preassembled with platelet-derived growth factor β receptors requires Src kinase activity. Oncogene. 2000;19(17):2075–2085. doi: 10.1038/sj.onc.1203548. [DOI] [PubMed] [Google Scholar]
- 95.Tian S., Lamb P., Seidel H., Stein R., Rosen J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood. 1994;84(6):1760–1764. doi: 10.1182/blood.v84.6.1760.1760. [DOI] [PubMed] [Google Scholar]
- 96.Gao S. P., Mark K. G., Leslie K., et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. Journal of Clinical Investigation. 2007;117(12):3846–3856. doi: 10.1172/jci31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zong C. S., Chan J., Levy D. E., Horvath C., Sadowski H. B., Wang L.-H. Mechanism of STAT3 activation by insulin-like growth factor I receptor. Journal of Biological Chemistry. 2000;275(20):15099–15105. doi: 10.1074/jbc.m000089200. [DOI] [PubMed] [Google Scholar]
- 98.Saxena N. K., Vertino P. M., Anania F. A., Sharma D. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. Journal of Biological Chemistry. 2007;282(18):13316–13325. doi: 10.1074/jbc.m609798200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kiuchi N., Nakajima K., Ichiba M., et al. STAT3 is required for the gp130-mediated full activation of the c-mycGene. The Journal of Experimental Medicine. 1999;189(1):63–73. doi: 10.1084/jem.189.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mencalha A. L. Forkhead box M1 (FoxM1) gene is a new STAT3 transcriptional factor target and is essential for proliferation, survival and DNA repair of K562 cell line. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0048160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhattacharya S., Ray R. M., Johnson L. R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochemical Journal. 2005;392(2):335–344. doi: 10.1042/bj20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimada Y. BRAF V600E and SRC mutations as molecular markers for predicting prognosis and conversion surgery in stage IV colorectal cancer. Science Reports. 2019;9(1):p. 2466. doi: 10.1038/s41598-019-39328-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amin M. B., Greene F. L., Edge S. B., et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA: A Cancer Journal for Clinicians. 2017;67(2):93–99. doi: 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 104.Kusaba T. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncology Reports. 2006;15(6):1445–1451. [PubMed] [Google Scholar]
- 105.Zhuang L., Lee C. S., Scolyer R. A., et al. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Modern Pathology. 2007;20(4):416–426. doi: 10.1038/modpathol.3800750. [DOI] [PubMed] [Google Scholar]
- 106.Alcolea V. Identification of a novel quinoxaline-isoselenourea targeting the STAT3 pathway as a potential melanoma therapeutic. International Journal of Molecular Sciences. 2019;20(3) doi: 10.3390/ijms20030521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knudson A. G. Two genetic hits (more or less) to cancer. Nature Reviews Cancer. 2001;1(2):157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 108.Knudson A. G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parzych K. R., Klionsky D. J. An overview of autophagy: morphology, mechanism, and regulation. Antioxidants & Redox Signaling. 2014;20(3):460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mariño G., Niso-Santano M., Baehrecke E. H., Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nature Reviews Molecular Cell Biology. 2014;15(2):81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Y., Shoji-Kawata S., Sumpter R. M., et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proceedings of the National Academy of Sciences. 2013;110(51):20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bialik S., Dasari S. K., Kimchi A. Autophagy-dependent cell death—where, how and why a cell eats itself to death. Journal of Cell Science. 2018;131(18) doi: 10.1242/jcs.215152. [DOI] [PubMed] [Google Scholar]
- 113.Maiuri M. C., Le Toumelin G., Criollo A., et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. The EMBO Journal. 2007;26(10):2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oberstein A., Jeffrey P. D., Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex. Journal of Biological Chemistry. 2007;282(17):13123–13132. doi: 10.1074/jbc.m700492200. [DOI] [PubMed] [Google Scholar]
- 115.Wirawan E. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death & Disease. 2010;16:p. 1. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang X. H., Jackson S., Seaman M., et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 117.Mathew R., Karp C. M., Beaudoin B., et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rad E., Murray J. T., Tee A. R. Oncogenic signalling through mechanistic target of rapamycin (mTOR): a driver of metabolic transformation and cancer progression. Cancers. 2018;10(1) doi: 10.3390/cancers10010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hafner C., Reichle A., Vogt T. New indications for established drugs: combined tumor-stroma-targeted cancer therapy with PPARγ agonists, COX-2 inhibitors, mTOR antagonists and metronomic chemotherapy. Current Cancer Drug Targets. 2005;5(6):393–419. doi: 10.2174/1568009054863591. [DOI] [PubMed] [Google Scholar]
- 120.Buzzai M., Jones R. G., Amaravadi R. K., et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Research. 2007;67(14):6745–6752. doi: 10.1158/0008-5472.can-06-4447. [DOI] [PubMed] [Google Scholar]
- 121.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Reviews Cancer. 2012;12(6):401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boya P., Gonzalez-Polo R.-A., Casares N., et al. Inhibition of macroautophagy triggers apoptosis. Molecular and Cellular Biology. 2005;25(3):1025–1040. doi: 10.1128/mcb.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Y., Wang X. D., Lapi E., et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proceedings of the National Academy of Sciences. 2012;109(33):13325–13330. doi: 10.1073/pnas.1120193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Degenhardt K., Mathew R., Beaudoin B., et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huo Y., Cai H., Teplova I., et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discovery. 2013;3(8):894–907. doi: 10.1158/2159-8290.cd-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guo J. Y., Chen H.-Y., Mathew R., et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & Development. 2011;25(5):460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu Y. L. Noxa upregulation by oncogenic activation of MEK/ERK through CREB promotes autophagy in human melanoma cells. Oncotarget. 2014;5(22):11237–11251. doi: 10.18632/oncotarget.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao Y., Wang W., Min I., et al. BRAF V600E-dependent role of autophagy in uveal melanoma. Journal of Cancer Research and Clinical Oncology. 2017;143(3):447–455. doi: 10.1007/s00432-016-2317-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ojha R., Leli N. M., Onorati A., et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discovery. 2019;9(3):396–415. doi: 10.1158/2159-8290.cd-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim J., Kundu M., Viollet B., Guan K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ryabaya O., Prokofieva A, Khochenkov D, Abramov I, Zasedatelev A, Stepanova E. Inhibition of endoplasmic reticulum stress-induced autophagy sensitizes melanoma cells to temozolomide treatment. Oncology Reports. 2018;40(1):385–394. doi: 10.3892/or.2018.6430. [DOI] [PubMed] [Google Scholar]
- 132.Burris H. A. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: a first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open. 2016;1(6) doi: 10.1136/esmoopen-2016-000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vanhaesebroeck B., Welham M. J., Kotani K., et al. p110, a novel phosphoinositide 3-kinase in leukocytes. Proceedings of the National Academy of Sciences. 1997;94(9):4330–4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wymann M. P., Pirola L. Structure and function of phosphoinositide 3-kinases. Biochimica et Biophysica Acta. 1998;1436(1-2):127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 135.Linassier C. Molecular cloning and biochemical characterization of a drosophila phosphatidylinositol-specific phosphoinositide 3-kinase. Biochemical Journal. 1997;321(3):849–856. doi: 10.1042/bj3210849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.MacDougall L. K., Domin J., Waterfield M. D. A family of phosphoinositide 3-kinases in Drosophila identifies a new mediator of signal transduction. Current Biology. 1995;5(12):1404–1415. doi: 10.1016/s0960-9822(95)00278-8. [DOI] [PubMed] [Google Scholar]
- 137.Hawkins P. T., Jackson T. R., Stephens L. R. Platelet-derived growth factor stimulates synthesis of Ptdlns(3,4,5)P3 by activating a Ptdlns(4,5)P2 3-OH kinase. Nature. 1992;358(6382):157–159. doi: 10.1038/358157a0. [DOI] [PubMed] [Google Scholar]
- 138.Poyner D. R., Jackson T. R., Hawkins P. T. Stimulation of phosphatidylinositol-3-kinase by insulin-like growth factor 1 and other agonists. Biochemical Society Transactions. 1992;20(2):p. 140S. doi: 10.1042/bst020140s. [DOI] [PubMed] [Google Scholar]
- 139.Sarbassov D. D., Ali S. M., Sabatini D. M. Growing roles for the mTOR pathway. Current Opinion in Cell Biology. 2005;17(6):596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 140.Divella R., De Luca R., Abbate I., Naglieri E., Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. Journal of Cancer. 2016;7(15):2346–2359. doi: 10.7150/jca.16884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Abu-Remaileh M., Bender S., Raddatz G., et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Research. 2015;75(10):2120–2130. doi: 10.1158/0008-5472.can-14-3295. [DOI] [PubMed] [Google Scholar]
- 142.Gallina G., Dolcetti L., Serafini P., et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. Journal of Clinical Investigation. 2006;116(10):2777–2790. doi: 10.1172/jci28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Colegio O. R., Chu N.-Q., Szabo A. L., et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Denkert C., Loibl S., Noske A., et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. Journal of Clinical Oncology. 2010;28(1):105–113. doi: 10.1200/jco.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- 145.Jinushi M., Chiba S., Yoshiyama H., et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proceedings of the National Academy of Sciences. 2011;108(30):12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dethlefsen C., Højfeldt G., Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Research and Treatment. 2013;138(3):657–664. doi: 10.1007/s10549-013-2488-z. [DOI] [PubMed] [Google Scholar]
- 147.Nabors L. B., Suswam E., Huang Y., Yang X., Johnson M. J., King P. H. Tumor necrosis factor alpha induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Research. 2003;63(14):4181–4187. [PubMed] [Google Scholar]
- 148.Irvine M. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis. 2018;7(9):p. 72. doi: 10.1038/s41389-018-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nathanson K. L., Martin A.-M., Wubbenhorst B., et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436) Clinical Cancer Research. 2013;19(17):4868–4878. doi: 10.1158/1078-0432.ccr-13-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hu W. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death & Disease. 2013;441:p. 4. doi: 10.1038/cddis.2013.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Paraiso K. H. T., Xiang Y., Rebecca V. W., et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Research. 2011;71(7):2750–2760. doi: 10.1158/0008-5472.can-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Thomas M. S., Mitchell J. S., DeNucci C. C., Martin A. L., Shimizu Y. The p110γ isoform of phosphatidylinositol 3-kinase regulates migration of effector CD4 T lymphocytes into peripheral inflammatory sites. Journal of Leukocyte Biology. 2008;84(3):814–823. doi: 10.1189/jlb.0807561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Saudemont A., Garcon F., Yadi H., et al. p110 and p110 isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proceedings of the National Academy of Sciences. 2009;106(14):5795–5800. doi: 10.1073/pnas.0808594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Porta C., Paglino C., Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Frontiers Oncology. 2014;4:p. 64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Villanueva J., Vultur A., Lee J. T., et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mao M., Tian F., Mariadason J. M., et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clinical Cancer Research. 2013;19(3):657–667. doi: 10.1158/1078-0432.ccr-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jiang X., Zhou J., Giobbie-Hurder A., Wargo J., Hodi F. S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clinical Cancer Research. 2013;19(3):598–609. doi: 10.1158/1078-0432.ccr-12-2731. [DOI] [PubMed] [Google Scholar]
- 158.Yam C., Xu X., Davies M. A., et al. A multicenter phase I study evaluating dual PI3K and BRAF inhibition with PX-866 and vemurafenib in patients with advanced BRAF V600-mutant solid tumors. Clinical Cancer Research. 2018;24(1):22–32. doi: 10.1158/1078-0432.ccr-17-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.van Geel R. M. J. M., Tabernero J., Elez E., et al. A phase Ib Dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discovery. 2017;7(6):610–619. doi: 10.1158/2159-8290.cd-16-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pereira T. C. B., Campos M. M., Bogo M. R. Copper toxicology, oxidative stress and inflammation using zebrafish as experimental model. Journal of Applied Toxicology. 2016;36(7):876–885. doi: 10.1002/jat.3303. [DOI] [PubMed] [Google Scholar]
- 161.Cumings J. N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain. 1948;71(4):410–415. doi: 10.1093/brain/71.4.410. [DOI] [PubMed] [Google Scholar]
- 162.Squitti R., Simonelli I., Ventriglia M., et al. Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2014;38(4):809–822. doi: 10.3233/JAD-131247. [DOI] [PubMed] [Google Scholar]
- 163.Yang H., Liu C.-N., Wolf R. M., et al. Obesity is associated with copper elevation in serum and tissues. Metallomics. 2019;11(8):1363–1371. doi: 10.1039/c9mt00148d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yang T.-H., Yuan T.-H., Hwang Y.-H., Lian I.-B., Meng M., Su C.-C. Increased inflammation in rheumatoid arthritis patients living where farm soils contain high levels of copper. Journal of the Formosan Medical Association. 2016;115(11):991–996. doi: 10.1016/j.jfma.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 165.Squitti R., Mendez A. J., Simonelli I., Ricordi C. Diabetes and Alzheimer’s disease: can elevated free copper predict the risk of the disease? Journal of Alzheimer’s Disease. 2017;56(3):1055–1064. doi: 10.3233/jad-161033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Choo X. Y. Neuroinflammation and copper in Alzheimer’s disease. International Journal of Alzheimer’s Disease. 2013;2013:p. 12. doi: 10.1155/2013/145345.145345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baldari S. Effects of copper chelation on BRAF(V600E) positive colon carcinoma cells. Cancers. 2019;11(5) doi: 10.3390/cancers11050659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brady D. C., Crowe M. S., Turski M. L., et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509(7501):492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hanif R., Pittas A., Feng Y., et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochemical Pharmacology. 1996;52(2):237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 170.Smith W. L., DeWitt D. L., Garavito R. M. Cyclooxygenases: structural, cellular, and molecular biology. Annual Review of Biochemistry. 2000;69(1):145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 171.Kramer R. M., Hession C, Johansen B, et al. Structure and properties of a human non-pancreatic phospholipase A2. Journal of Biological Chemistry. 1989;264(10):5768–5775. [PubMed] [Google Scholar]
- 172.Seilhamer J. J., Pruzanski W., Vadas P., et al. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. Journal of Biological Chemistry. 1989;264(10):5335–5338. [PubMed] [Google Scholar]
- 173.Balsinde J., Winstead M. V., Dennis E. A. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Letters. 2002;531(1):2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 174.Kearney P. M., Baigent C., Godwin J., Halls H., Emberson J. R., Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302–1308. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Escuin-Ordinas H., Atefi M., Fu Y., et al. COX-2 inhibition prevents the appearance of cutaneous squamous cell carcinomas accelerated by BRAF inhibitors. Molecular Oncology. 2014;8(2):250–260. doi: 10.1016/j.molonc.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Naifeh J., Varacallo M. Biochemistry, aerobic glycolysis. StatPearls. 2019;41 [PubMed] [Google Scholar]
- 177.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 178.Warburg O., Geissler A.-W., Lorenz S. Notizen: Über die Erzeugung von normalem Stoffwechsel aus Krebsstoffwechsel durch Zusatz von Vitamin B1. Zeitschrift für Naturforschung B. 1970;25(5):p. 559. doi: 10.1515/znb-1970-0530. [DOI] [PubMed] [Google Scholar]
- 179.Warburg O., Wind F., Negelein E. The metabolism of tumors in the body. The Journal of General Physiology. 1927;8(6):519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hsu P. P., Sabatini D. M. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 181.Schlaepfer I. R., Rider L., Rodrigues L. U., et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Molecular Cancer Therapeutics. 2014;13(10):2361–2371. doi: 10.1158/1535-7163.mct-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang M., Di Martino J. S., Bowman R. L., et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discovery. 2018;8(8):1006–1025. doi: 10.1158/2159-8290.cd-17-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Park S. H. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. Journal of Applied Physiology. 1985;92(6):2475–2482. doi: 10.1152/japplphysiol.00071.2002. [DOI] [PubMed] [Google Scholar]
- 184.Aloia A. A fatty acid oxidation-dependent metabolic shift regulates the adaptation of BRAF-mutated melanoma to MAPK inhibitors. Clinical Cancer Research. 2019;25(22) doi: 10.1158/1078-0432.CCR-19-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schlaepfer I. R., Glodé L. M., Hitz C. A., et al. Inhibition of lipid oxidation increases glucose metabolism and enhances 2-deoxy-2-[18F]Fluoro-d-Glucose uptake in prostate cancer mouse xenografts. Molecular Imaging and Biology. 2015;17(4):529–538. doi: 10.1007/s11307-014-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shen C.-H., Yuan P., Perez-Lorenzo R., et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Molecular Cell. 2013;52(2):161–172. doi: 10.1016/j.molcel.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ritt D. A., Monson D. M., Specht S. I., Morrison D. K. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Molecular and Cellular Biology. 2010;30(3):806–819. doi: 10.1128/mcb.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yuan J., Ng W. H., Yap J., et al. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho-Ser-621 in the C terminus of CRAF. Journal of Biological Chemistry. 2018;293(37):14276–14284. doi: 10.1074/jbc.ra118.004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sanduja S., Feng Y., Mathis R. A., et al. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene. 2016;35(40):5295–5303. doi: 10.1038/onc.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Abildgaard C., Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends in Molecular Medicine. 2015;21(3):164–171. doi: 10.1016/j.molmed.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 191.Roesch A., Vultur A., Bogeski I., et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell. 2013;23(6):811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sundstrøm T., Prestegarden L., Azuaje F., et al. Inhibition of mitochondrial respiration prevents BRAF-mutant melanoma brain metastasis. Acta Neuropathol Commun. 2019;7(1):p. 55. doi: 10.1186/s40478-019-0712-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fischer G. M., Jalali A., Kircher D. A., et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 2019;9(5):628–645. doi: 10.1158/2159-8290.CD-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Figarola J. L. Bioenergetic modulation with the mitochondria uncouplers SR4 and niclosamide prevents proliferation and growth of treatment-naive and vemurafenib-resistant melanomas. Oncotarget. 2018;9(97):36945–36965. doi: 10.18632/oncotarget.26421. [DOI] [PMC free article] [PubMed] [Google Scholar]