

ABSTRACT
The oncogenic fusion transcription factor TCF3-HLF identifies an aggressive subtype of acute lymphoblastic leukemia. TCF3-HLF imposes a malignant program by activation of lineage-specific oncogenic enhancers. Among critical cofactors of the TCF3-HLF complex we identified EP300, which functional inhibition results in potent anti-leukemic activity by interference with the specific gene expression.
KEYWORDS: Leukemia, drug resistance, chimeric transcription factor, TCF3-HLF, enhancer
Acute lymphoblastic leukemia (ALL) caused by the malignant transformation of lymphoid precursor cells is the most common cancer in children and adolescents. Despite impressive progress with ALL therapy, the salvage of a subset of patients with the high-risk disease remains challenging. Gene fusions generated by chromosomal translocations constitute frequent driver events and define molecular subgroups in ALL.1 As these fusions often affect transcription factors (TFs), new approaches to target oncogenic transcriptional activity more specifically should be developed.2
The chimeric TF TCF3-HLF generated by the t(17;19)(q22;p13) translocation occurs in approximately 1% of childhood B-lineage ALL and defines a subtype of ALL that represents a paradigm of resistant disease.3 The TCF3-HLF fusion consists of the transactivation domains of the TCF3, which drives lymphoid development, fused to the DNA-binding and dimerization domains of HLF. In mice, Hlf is expressed in multipotent hematopoietic progenitors, required for the maintenance of the stem cell pool under stress conditions and was identified as one of six TFs that can reprogram of committed lymphoid cells to induced hematopoietic stem cells (HSC). However, the modeling of TCF3-HLF in mice using various strategies did not result in leukemia. Notably, TCF3-HLF expression in hematopoietic stem/progenitor cells was embryonically lethal, whereas expression in B-cell progenitors induced hyposplenia and lymphopenia.4 These imply that the genetic context of the cell of origin and/or cooperative genetic events that suppress cell death must be critical to drive leukemogenesis. We identified accompanying genetic lesions that are in general common in ALL, including frequent deletion of PAX5 or of other B-cell differentiation gene, of CDKN2A/B and activation mutation of signaling pathways driving proliferation.3 Thus, the transcriptional program driven by TCF3-HLF must be driving the features conferring such a resistant phenotype. We employed a combined functional genomic and proteomic approach to dissect the transcriptional dependencies in TCF3-HLF positive leukemia in the original cellular context and gene dosage (Figure 1).5
Figure 1.
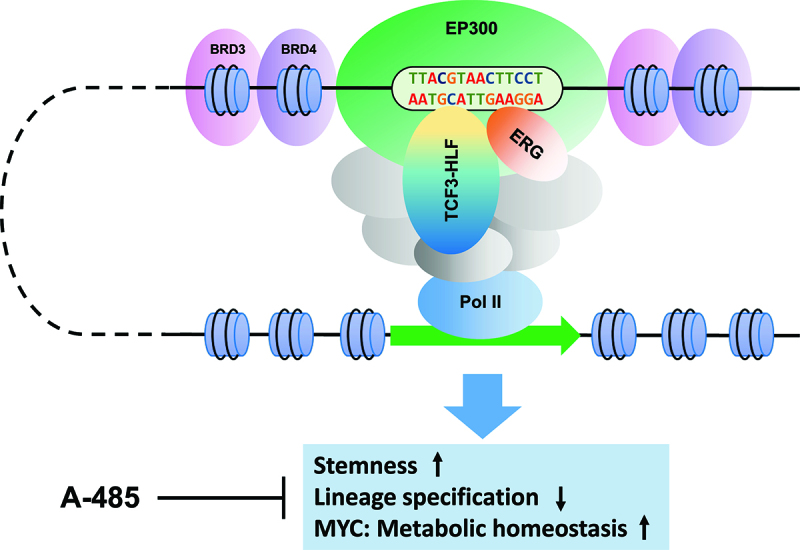
The leukemogenic activity of TCF3-HLF fusion. The TCF3-HLF fusion recruits enhancer associated apparatus including ERG and EP300 to activate leukemia critical enhancers driving cellular identity and self-renewal. Inhibition of EP300 with a catalytic EP300 inhibitor, A-485, interferes the functional output of TCF3-HLF and reduce leukemia cell survival.
We first demonstrated the dependence of the leukemia cells on TCF3-HLF via CRISPR dropout experiments in a patient-derived cell line in vitro and patient-derived xenografts (PDXs) in vivo. Time-course analysis of the transcriptome in this system enabled to detect the gene expression signature of TCF3-HLF activity. This signature was enriched with stem-cell and myeloid lineage features, including the 17-gene signature that was identified to predict a higher risk of relapse in acute myeloid leukemia.6 The target signature is further dominated by MYC and the expression of MYC-associated metabolic pathways genes. In fact, TCF3-HLF induce MYC expression by direct activation of its enhancer, but also by repression of the expression of ubiquitin ligase FBXW7, which regulates MYC among other targets by ubiquitin-dependent protein degradation. Thus, TCF3-HLF imposed a malignant cellular identity enabling both self-renewal and cell growth.
From ChIP-seq studies we identified 484 TCF3-HLF binding regions mostly in enhancers, including 84 regions in the most active enhancers termed ‘super-enhancers’ (SEs).7 These corresponded to regions that are normally active in stem and progenitor cells but not in lymphoid cells. Using a saturation mutagenesis screen approach, we identified 10 high-confident TCF3-HLF regions that are critical for leukemia cell survival. Remarkably, one of these regions was in a MYC enhancer within the “Blood Enhancer Cluster”, which is critical for blood development in mice and occupied by Hlf.8,9 Therefore, TCF3-HLF activates HSC-associated enhancers by hijacking HLF sites to drive the malignant program.
Understanding the TCF3-HLF transcriptional complex composition may provide specific ways to target its oncogenic activity. As predicted by a characteristic TF motif grammar juxtaposing ETS and HLF binding sites at enhancers, we uncovered that ERG is recruited by TCF3-HLF at such sites including MYC enhancer. CRISPR knockdown of ERG dramatically attenuated MYC expression and resulted in leukemia dropout in PDXs in vivo. Proteomic profiling of the endogenous TCF3-HLF complex further identified ~90 proteins in this interactome. Targeted functional validation with CRISPR identify druggable components with signaling functions and the histone acetyltransferase p300 (EP300) among the most significant hits for ALL survival. Given the role of EP300 as a transcriptional cofactor in enhancer function, we verified its functional interaction with TCF3-HLF. Knockdown of TCF3-HLF abolished EP300 binding at TCF3-HLF occupied enhancers with a marked decrease of the active chromatin marker, H3K27ac, at their flanking sites. Pharmacologic interference with EP300 using a novel catalytic inhibitor, A-485,10 resulted in the inactivation of TCF3-HLF binding enhancers and specifically interfering with the TCF3-HLF driven gene expression program. In particular, A-485 suppressed the transcription of MYC and showed potent anti-leukemic activity as a single agent in PDXs in vivo. This provides proof of concept evidence that specific interference with the oncogenic activity of chimeric TFs may also be achievable by targeting components of its complex. Although there is no early clinical data to our knowledge of A-485, mice appear to tolerate well when exposure to this agent. Further investigation on the combination effect of this inhibitor with a standard treatment regimen may identify synergistic activity using in vivo models.
The importance of oncogenic fusion proteins in cancer has been widely recognized.2 Concerted action will be needed in order to generate data at sufficient resolution for a larger number of fusion TFs in order to identify common features that are amenable to drug repurposing or new development. The National Cancer Institute funds a consortium with the Cancer Moonshot Initiative to study such chimeric TFs as drivers in childhood cancer, which together many other projects will provide a fertile ground for the discovery of specific ways to target the disease at its source.
Funding Statement
This work was supported by the Swiss Cancer League [KFS-3526-08-2014; KFS-4237-08-2017]; the Swiss National Research Foundation SNF [310030-133108 and 323530-164223]; the foundation ‘Kinderkrebsforschung Schweiz’, the ‘Krebsliga Zurich’, the Novartis Foundation of Biomedical Research, the clinical research focus program ‘Human Hemato-Lymphatic Diseases’ of the University of Zurich, the Iten-Kohaut Stiftung, the Panacée Foundation, the Innovative Medicines Initiative project ULTRA-DD [FP07/2007-2013, grant no. 115766], the German Federal Office for Radiation Protection [grant St.Sch. 3611S70014]; and by the Max Planck Society.
Acknowledgments
We are grateful to team members for scientific discussions and apologize to many colleagues whose work could not be cited due to space constraints.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Inaba H, Greaves M, Mullighan CG.. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1–3. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mertens F, Johansson B, Fioretos T, Mitelman F.. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- 3.Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, Bornhauser B, Gombert M, Kratsch C, Stutz AM, et al. Genomics and drug profiling of fatal tcf3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9):1020–1029. doi: 10.1038/ng.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duque-Afonso J, Smith KS, Cleary ML. Conditional expression of e2a-HLF induces b-cell precursor death and myeloproliferative-like disease in knock-in mice. PLoS One. 2015;10(11):e0143216. doi: 10.1371/journal.pone.0143216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, Mouttet B, Warnatz H-J, Risch T, Rietmann F, Frommelt F, Ngo QA, Dobay MP, Marovca B, Jenni S, et al. The leukemogenic tcf3-HLF complex rewires enhancers driving cellular identity and self-renewal conferring ep300 vulnerability. Cancer Cell. 2019;36:630–644.e9. doi: 10.1016/j.ccell.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 7.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. A myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553(7689):515–520. doi: 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- 9.Wahlestedt M, Ladopoulos V, Hidalgo I, Sanchez Castillo M, Hannah R, Sawen P, Wan H, Dudenhoffer-Pfeifer M, Magnusson M, Norddahl GL, et al. Critical modulation of hematopoietic lineage fate by hepatic leukemia factor. Cell Rep. 2017;21(8):2251–2263. doi: 10.1016/j.celrep.2017.10.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. Discovery of a selective catalytic p300/cbp inhibitor that targets lineage-specific tumours. Nature. 2017;550(7674):128–132. doi: 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]