

ABSTRACT
Mutations in the splicing factor 3b subunit 1 (SF3B1) gene create a neomorphic protein that disrupts RNA splicing, but the downstream consequences of this missplicing are unclear. Our recent study of isogenic human cells demonstrated that SF3B1MUT induces reprogramming of energy metabolism and a targetable vulnerability to deprivation of the nonessential amino acid serine.
Keywords: SF3B1, spliceosome, serine, PHGDH, metabolism
Initially found in >70% of myelodysplastic syndromes (MDS), spliceosome mutations have now been described in many hematologic and solid malignancies.1–3 The splicing factor 3b subunit 1 (SF3B1) gene is the most widely mutated, occurring in MDS, acute myeloid leukemia (AML), chronic lymphocytic leukemia, melanoma, breast carcinoma, pancreas adenocarcinoma, and many other cancers. SF3B1 is a member of the U2 small nuclear ribonucleoprotein (U2snRNP) complex, essential for branch point sequence recognition in pre-mRNA. Mutant SF3B1 is a neomorphic protein that disrupts the usage of thousands of splice junctions, leading to altered expression of hundreds of genes, affecting dozens of cellular pathways.1,3 Given the variety of affected processes, it has been a challenge to understand what missplicing events are physiologically impactful in SF3B1MUT cells.
To understand consequences of SF3B1MUT missplicing, we recently characterized the transcriptome and proteome of SF3B1MUT human isogenic cells.4 This analysis showed enrichment of metabolic genes in SF3B1MUT cells, including a decrease in mitochondrial complex III of the electron transport chain (ETC), essential for cellular respiration. This was mediated through missplicing and downregulation of its assembly factor, ubiquinol-cytochrome c reductase complex assembly factor 1 (UQCC1), as re-expression of this protein was able to rescue complex III levels. SF3B1MUT also decreased cellular respiration, reduced citric acid cycle metabolites, and misspliced and downregulated other metabolic enzymes of the mitochondria – dihydrolipoamide S-succinyltransferase (DLST) and methylmalonyl-CoA mutase (MUT) (Figure 1). This reprogramming of mitochondrial metabolism bears particular relevance to the form of MDS in which SF3B1 is most frequently (>85% of cases) mutated: MDS with ring sideroblasts (MDS-RS).5 This disease is characterized by dysplastic erythroblasts (the “sideroblasts”) with iron-overloaded mitochondria (the “rings”). Interestingly, some forms of congenital sideroblastic anemia (CSA) are characterized by mutations in ETC genes, causally implicating impaired cellular respiration in ring sideroblast formation.6 In this context, our results suggest that ETC disruption by SF3B1MUT may contribute to the sideroblastic anemia of MDS-RS. Consistent with this, data from 40 years ago showed that in MDS-RS patients, granulocytes (SF3B1MUT in >85% of cases) had reduced ETC activity when compared to healthy controls, while ETC activity of lymphocytes (unmutated in MDS-RS) was similar between groups.7 Hsu et al also recently reported a decrease in metabolically active mitochondria in induced pluripotent stem cells derived from SF3B1MUT MDS.8 It will be interesting for future studies to determine how missplicing events, such as in UQCC1, DLST or ABCB7 (the latter downregulated by SF3B1MUT and also a cause of CSA), drive impairment of cellular respiration and ring sideroblast formation in SF3B1MUT MDS.5
Figure 1.
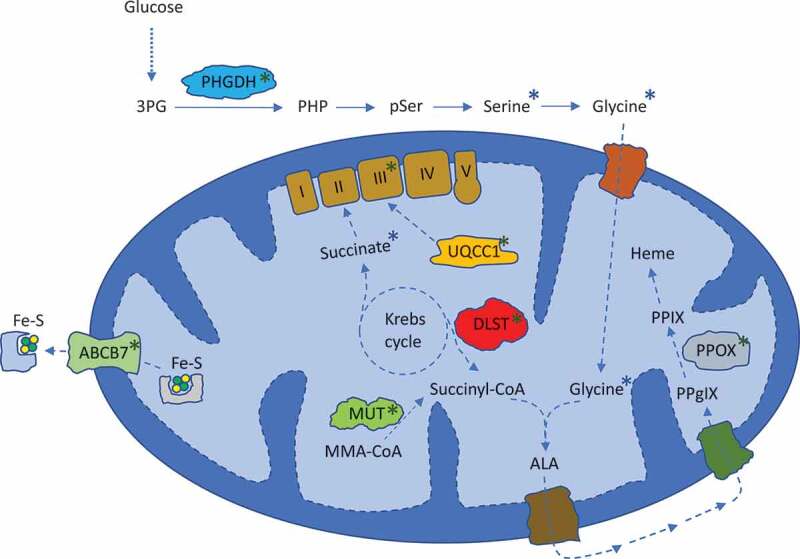
Metabolic pathways affected by mutant SF3B1. Blue asterisks indicate metabolites that were found to be downregulated by SF3B1MUT. Green asterisks show metabolic proteins found to be misspliced and downregulated by SF3B1MUT. 3PG = 3-phosphoglycerate. PHP = phosphohydroxypyruvate. pSer = phosphoserine. I, II, III, IV, V = mitochondrial complexes. Fe-S = iron-sulfur complex. MMA = methylmalonyl. ALA = aminolevulinic acid. PPgIX = protoporphyrinogen IX. PPIX = protoporphyrin IX. PHGDH = phosphoglycerate dehydrogenase. UQCC1 = ubiquinol-cytochrome c reductase complex assembly factor 1. DLST = dihydrolipoamide S-succinyltransferase. MUT = methylmalonyl-CoA mutase. ABCB7 = ATP binding cassette subfamily B member 7. PPOX = protoporphyrinogen oxidase.
Our study also identified another metabolic gene that was heavily misspliced and downregulated by SF3B1MUT: phosphoglycerate dehydrogenase (PHGDH), the gatekeeper enzyme controlling synthesis of the nonessential amino acid serine. We found that SF3B1MUT cells had lower baseline serine synthesis, as well as a reduced ability to increase its relative synthesis when deprived of exogenous serine. Accordingly, SF3B1MUT cells exhibited greatly decreased growth without exogenous serine, as compared to their wild type counterparts. This we observed in several contexts, including untransformed and transformed breast epithelial cell knockins, leukemia cell knockins, and AML cell lines from patients with naturally acquired SF3B1MUT (the latter cells simply died in the absence of exogenous serine). Overexpression of PHGDH – or supplementation with its reaction product, phosphohydroxypyruvate (PHP) – was able to rescue growth without exogenous serine, supporting deficient serine synthesis as a mechanism for this vulnerability. Since Maddocks et al showed in mice that a serine-free diet will drop serine levels by 60%, exert no clear toxicity, and can produce anticancer activity,9 we performed this intervention on two different naturally SF3B1MUT AML cell lines. These experiments showed that SF3B1MUT cancers in mice fed a serine-free diet grew significantly slower than that of mice given a serine-replete diet, demonstrating that this vulnerability could be exploited in vivo.
Our findings offer serine deprivation as a novel treatment strategy for SF3B1MUT cancers. Modulating serine availability in humans might be approached in several ways, including dietary serine restriction (dietary restriction of individual amino acids is commonplace in management of inborn errors of metabolism), development of a therapeutic serine-catabolizing enzyme (asparaginase is a backbone of acute lymphoblastic leukemia treatment), inhibition of serine transport (pharmacologic inhibition of cystine transport is under clinical investigation), and other modalities.10 Further insight into the mechanisms of serine auxotrophy in SF3B1MUT cells may also help such efforts. Though our data causally implicates decreased serine synthesis through PHGDH downregulation, other mechanisms may contribute, such as decreased cellular respiration, given that human cells increase their reliance on oxidative phosphorylation as a compensatory adaptation to serine starvation.9 Finally, it is worth noting that it remains largely unclear how spliceosome mutations confer a clonal advantage to cancer cells, so it is possible that metabolic reprogramming by SF3B1MUT has advantageous, and not just detrimental, effects. Future research is certainly needed to better understand the mechanisms and consequences of metabolic rewiring by SF3B1 mutations. Nonetheless, our study provides a novel and therapeutically relevant connection between two previously unacquainted processes in molecular and cellular oncology: mutant spliceosomes and metabolic reprogramming.
Funding Statement
This work was supported by the Eli Lilly and Company;U.S. Department of Defense [W81XWH-17-1-0035].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Darman RB, Seiler M, Agrawal A, Lim K, Peng S, Aird D, Bailey S, Bhavsar E, Chan B, Colla S, et al. Cancer-associated SF3B1 hotspot mutations induce Cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015;13:1–3. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 3.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:ncomms10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalton BW, Helmenstine E, Walsh N, Gondek LP, Kelkar DS, Read A, Natrajan R, Christenson ES, Roman B, Das S, et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J Clin Invest. 2019;130:4708–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, Giai V, Fernandez-Mercado M, Papaemmanuil E, Högstrand K, et al. The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia. 2013;27:889. doi: 10.1038/leu.2012.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ducamp S, Fleming MD.. The molecular genetics of sideroblastic anemia. Blood. 2019;133:59–69. doi: 10.1182/blood-2018-08-815951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki Y. Multiple enzymatic defects in mitochondria in hematological cells of patients with primary sideroblastic anemia. J Clin Invest. 1980;66:43–49. doi: 10.1172/JCI109833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu J, Reilly A, Hayes BJ, Clough CA, Konnick EQ, Torok-Storb B, Gulsuner S, Wu D, Becker PS, Keel SB, et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood. 2019;2018884338. doi: 10.1182/blood.2018884338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, Warren PP, White DM, Reid MA, Eschbacher JM, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med. 2015;7:289ra86–289ra86. doi: 10.1126/scitranslmed.aaa8103. [DOI] [PMC free article] [PubMed] [Google Scholar]