

Abstract
Introduction:
The Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) consortia are two closely connected studies, involving multiple North American centers that evaluate both sporadic and familial frontotemporal dementia (FTD) participants and study longitudinal changes.
Methods:
We screened the major dementia-associated genes in 302 sporadic and 390 familial (symptomatic or at-risk) participants enrolled in these studies.
Results:
Among the sporadic patients, 16 (5.3%) carried chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN) pathogenic variants, whereas in the familial series we identified 207 carriers from 146 families. Of interest, one patient was found to carry a homozygous C9orf72 expansion, while another carried both a C9orf72 expansion and a GRN pathogenic variant. We also identified likely pathogenic variants in the TAR DNA binding protein (TARDBP), presenilin 1 (PSEN1), and valosin containing protein (VCP) genes, and a subset of variants of unknown significance in other rare FTD genes.
Discussion:
Our study reports the genetic characterization of a large FTD series and supports an unbiased sequencing screen, irrespective of clinical presentation or family history.
Keywords: C9orf72, familial, frontotemporal dementia, GRN, MAPT, sporadic
1 |. INTRODUCTION
Frontotemporal dementia (FTD) is the second most common neurodegenerative dementia in the younger population (<65 years), after Alzheimer’s disease (AD). It comprises a group of heterogeneous neurodegenerative syndromes caused by progressive degeneration of the temporal and frontal lobes, key brain areas for decision-making, behavioral control, emotion, and language.
FTD typically presents with changes in personality and behavior (behavioral variant of frontotemporal dementia [bvFTD]) or language impairment (primary progressive aphasias [PPAs] including non-fluent/agrammatic variant [nfvPPA] and semantic variant [svPPA]). Some FTD patients also develop motor symptoms, such as weakness or muscle wasting, characteristic of amyotrophic lateral sclerosis (ALS). In addition, there is an overlap with the atypical parkinsonian syndromes, progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS). In contrast to AD, FTD is more commonly observed in patients younger than 65 years of age.
FTD has a strong genetic component, with up to 40% of patients reporting family a history of dementia, psychiatric, and/or motor symptoms, although only about 10% show a clear autosomal dominant transmission.1 The most common causative FTD genes are chromosome 9 open reading frame 72 (C9orf72),2,3 microtubule-associated protein tau (MAPT),4 and progranulin (GRN),5,6 whereas other rare pathogenic variants have been identified in TAR DNA binding protein (TARDBP),7 FUS RNA binding protein (FUS),8 charged multivesicular body protein 2B (CHMP2B),9 valosin containing protein (VCP),10 sequestosome 1 (SQSTM1),11 and TANK binding kinase 1 (TBK1).12
The Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) consortium is dedicated to conduct clinical research in sporadic (project 1) and familial (project 2) North American FTD spectrum patients to better characterize and understand these disorders and to develop new clinical outcome measures. A subset of the ARTFL participants are also enrolled in a closely connected study aimed at understanding familial FTD, the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) study. In contrast to project 2 of ARTFL, which enrolls participants with a strong FTD family history regardless of whether a causative variant has been identified, LEFFTDS only enrolls families in which a known pathogenic variant in one of the three main FTD genes (C9orf72, MAPT, and GRN) has been identified. Herein, we aim to characterize the genetic variation within the main genes associated with dementia in the sporadic and familial participants enrolled in the ARTFL and LEFFTDS studies.
2 |. METHODS
2.1 |. Series
The ARTFL series comprises to date >1000 participants referred from 18 clinical research sites in North America. Familial cohort participants are enrolled in ARTFL project 2, based on a strong family history or a known FTD-associated mutation in the family, whereas sporadic participants are enrolled in ARTFL project 1. All participants are evaluated as baseline assessment with a standard battery of neuropsychological tests, clinical evaluation, and biomarker collection, and participants with a family history of FTD (symptomatic or at-risk) return after 1 year. A subset of these ARTFL participants are also enrolled in the closely connected study LEFFTDS. The LEFFTDS series comprises asymptomatic or symptomatic individuals in kindreds with known mutations in MAPT, GRN, or C9orf72 genes who are evaluated at approximately yearly intervals using a standardized battery of measures. Investigators are blinded to individuals’ genotypes. Herein, we report the demographic, clinical, and genetic characteristics of 736 of these participants, who were screened for pathogenic and risk variants in the main dementia genes through June 2018. All DNA specimens are available at the National Centralized Repository for Alzheimer’s Disease and Related Dementia (https://ncrad.iu.edu/). Data and biospecimens may be requested via links on the ARTFL website (https://www.rarediseasesnetwork.org/cms/artfl/).
2.2 |. Targeted sequencing
This series was screened using a panel of ≈300 RefSeq genes previously implicated in neurodegenerative disorders. Exonic regions were captured using a custom designed library (SeqCap EZ Choice Library, NimbleGen) and sequenced on an Illumina HiSeq4000 at the UCLA Neuroscience Genomics Core (http://www.semel.ucla.edu/ungc). Sequence reads were mapped to the GRCh37/hg19 reference genome and variants were joint-called with the Genome Analysis Toolkit (GATK).13 The joint variant calling file was annotated using ANNOVAR and the Ensembl Variant Effect Predictor tool.14,15
2.3 |. Screening of the main dementia genes
The coding and exon–intron boundary regions of the seven most common FTD and AD genes (GRN, MAPT, TARDBP, FUS, and amyloid beta precursor protein [APP], presenilin 1 [PSEN1], presenilin 1 [PSEN2]) were screened for known (listed in the AD&FTD Mutation Database: http://www.molgen.ua.ac.be/ADMutations) or novel (likely) pathogenic variants (classified according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guidelines16). Known and novel (likely) pathogenic variants were confirmed by Sanger sequencing. Summarized coverage and variant frequency data for these seven genes is available on the ARTFL/LEFFTDS browser (https://coppolalab.ucla.edu/exac_browser/artfl), a website generated using the ExAC database interface.
2.4 |. Screening of other rare familial frontotemporal dementia genes
Coding and exon–intron boundaries of CHMP2B, SQSTM1, TBK1, and VCP were also examined. Variants within these genes were filtered for (1) protein truncating (nonsense, frameshift, canonical splice sites) and missense variants that were (2) novel or rare (minor allele frequency [MAF] <0.0001 in the non-Finnish European [NFE] population from the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), as it corresponds to the highest MAF of known (likely) pathogenic variants in the GRN and MAPT genes) and (3) predicted to be damaging by at least one of the following in silico software algorithms: SIFT, PolyPhen-2, and CADD (Phred score ≥20 in v1.4, corresponding to the 1% most deleterious variants in the genome). The filtered candidate variants were confirmed by Sanger sequencing and classified according to ACMG-AMP guidelines.16
2.5 |. C9orf72 repeat screening
The presence of a pathological hexanucleotide repeat expansion in C9orf72 was detected using both fluorescent and repeat-primed polymerase chain reaction (PCR), as described previously.2 Fragment length analysis was performed on an ABI 3730 genetic analyzer, and data were analyzed using the Peak Scanner Software, including a positive control sample for reference. To confirm the presence of a homozygous C9orf72 repeat expansion, Southern blotting was performed, as described previously.17 In brief, a digoxigenin-labeled probe (Roche) was utilized to detect an expanded C9orf72 repeat using ≈10 μg of genomic DNA as input material. The expansion was visualized with CDP-star substrate (Roche) on autoradiography film.
2.6 |. Dementia risk alleles
Genotypes for the rare risk alleles MAPT p.A152T (rs143624519) and triggering receptor expressed on myeloid cells 2 (TREM2) p.R47H (rs75932628), and the common apolipoprotein E (APOE) (rs429358 and rs7412), and 17q haplotype (rs1560310), were obtained using TaqMan SNP assays and/or extracted from the targeted sequencing data.
3 |. RESULTS
3.1 |. Series overview
We have screened, through June 2018, a total of 736 individuals enrolled in the ARTFL and/or LEFFTDS studies, with mean age of 57.8 years at baseline (range 18 to 87 years), of which 364 (49.5%) were female. Principal component analysis on our targeted genetic data was performed to evaluate population substructure (see Supplementary Material) and confirmed European descent for most of these samples (Fig. S1). Among these participants, 346 were enrolled in ARTFL project 1 (hereinafter referred as sporadic). As expected, based on the inclusion criteria for enrollment in ARTFL project 1, these sporadic cases were not related to any of the other participants, based on identity-by-descent estimates (see Supplementary Material), with the exception of two predicted sets of twins. The remaining 390 individuals (hereinafter referred as familial) were enrolled in ARTFL project 2 and/or LEFFTDS and had a strong family history of FTD. Based on identity-by-descent analysis (see Supplementary Material), we estimated that 240 of these individuals had at least one first-degree relative also enrolled, which resulted in 88 predicted kindreds. Due to our limited genetic data, we were not able to call more distant relatives, so we cannot exclude the possibility that some of these predicted kindreds, as well as the remaining 150 unrelated individuals, might be more distantly related.
3.2 |. Sporadic familial frontotemporal dementia series
Among the sporadic participants screened from ARTFL project 1, 43 were found to have non-FTD diagnoses (such as AD, Parkinson’s disease, psychiatric disorders), and one was clinically normal at the time of the analysis. After excluding these samples, we had a total of 302 sporadic cases diagnosed with FTD syndromes (mean age ±SD at baseline 65.87 ± 8.29 years, range 34 to 87). The most frequent phenotypes were bvFTD (37%) and PSP (26%), followed by svPPA (14%), nfvPPA (11%), CBS (9%), and FTD/ALS (3%) (Fig. 1a, left).
FIGURE 1.
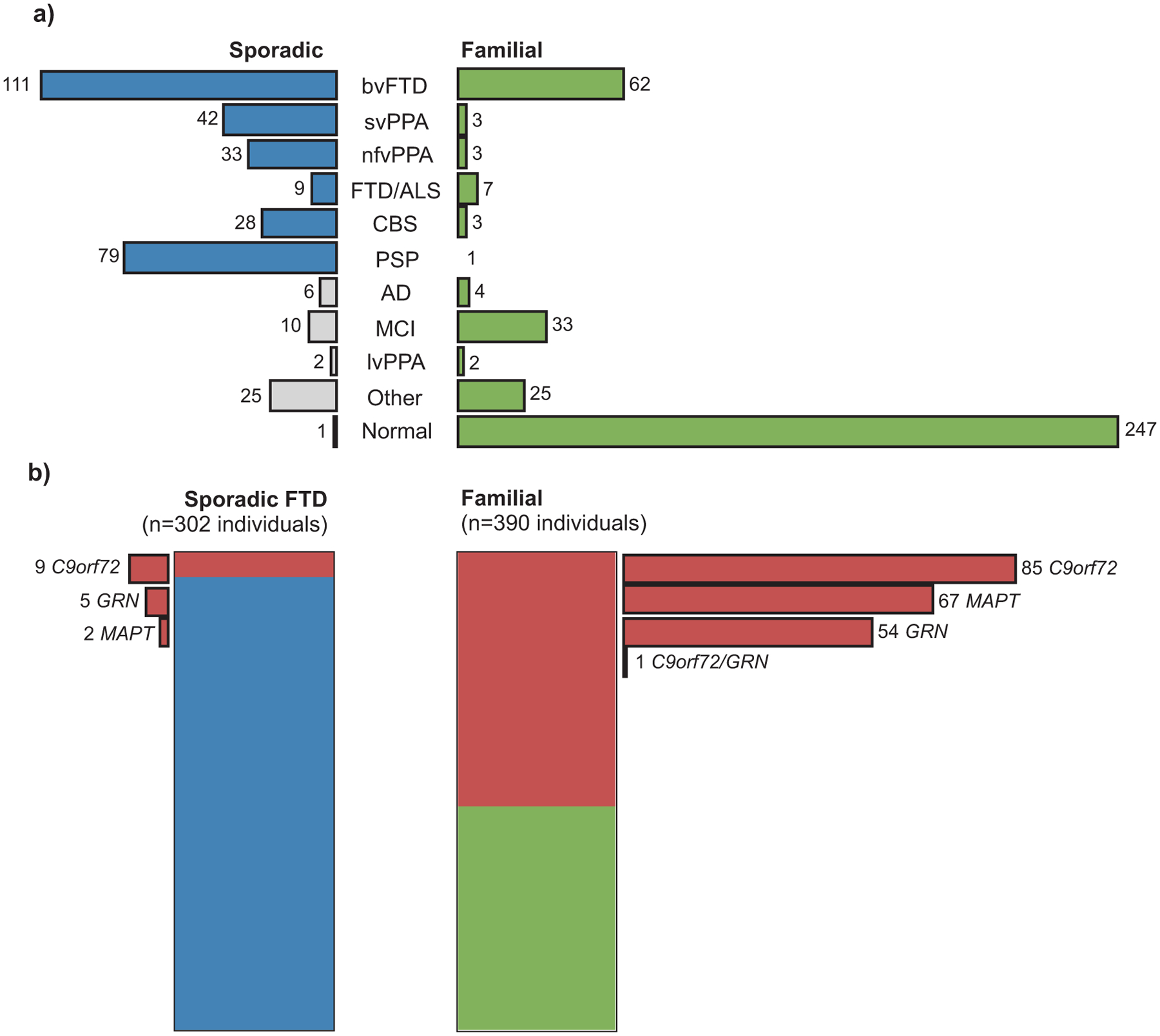
Clinical and genetic characteristics of individuals enrolled in the ARTFL/LEFFTDS studies. (a) Clinical diagnoses among 346 sporadic patients with dementia (left) and 390 individuals with a family history of FTD (right). (b) Carriers of (likely) pathogenic variants in the main FTD genes (red) among 302 sporadic cases diagnosed with FTD-spectrum phenotypes (blue, left), and in 390 individuals with family history of FTD (green, right). One individual carried both an expanded C9orf72 repeat and a known pathogenic GRN variant. (AD, Alzheimer’s disease dementia; ALS, amyotrophic lateral sclerosis; bvFTD, behavioral variant FTD; CBS, corticobasal syndrome; FTD, frontotemporal dementia; lvPPA, logopenic variant PPA; MCI, mild cognitive impairment; nfvPPA, non-fluent variant PPA; PPA, primary progressive aphasia; PSP, progressive supranuclear palsy; svPPA, semantic variant PPA)
Sixteen of the 302 sporadic FTD cases harbored a known pathogenic variant - nine carriers of an expanded C9orf72 repeat, including one homozygous carrier (Fig. S2), five of GRN and two of MAPT variants (Fig. 1b, left, and Table 1). These correspond to a total frequency of 5.3% (16 of 302) carriers of pathogenic variants in this sporadic FTD series. Due to inclusion criteria for ARTFL project 1, parents of sporadic cases were not originally enrolled in ARTFL, and therefore it was not possible to assess if any of these were possible de novo pathogenic variants.
TABLE 1.
MAPT and GRN pathogenic variants identified in the ARTFL/LEFFTDS series Abbreviation: NFE, non-Finnish Europeans.
| Variant | cDNA change | Aminoacid change | Historical name | gnomAD NFE frequency | Sporadic | Familial (kindreds) | Total number of carriers |
|---|---|---|---|---|---|---|---|
| MAPT (NM_001123066) | |||||||
| 1 | c. 1828–10G>T | - | IVS9–10G>T | - | - | 4 (2) | |
| 2 | c.1842T>G | P.N614K | Asn279Lys | - | - | 9 (6) | |
| 3 | C.1907C>T | P.P636L | Pra301Leu | 1.3E-05 | - | 21 (15) | |
| 4 | c.1919G>A | P.S640N | Ser305Asn | - | - | 2 (1) |  |
| 5 | c.1919G>T | P.S640I | Ser305lle | - | - | 2 (1) |  |
| 6 | c.1920T>C | p S640= | Ser305 | - | - | 1 (1) |  |
| 7 | c.1920+16C>T | - | IVS10+16C>T | - | 1 | 5 (3) | |
| 8 | c.2014G>A | P.V672M | Val337Met | - | - | 9 (2) | |
| 9 | c.2170G>A | p G724R | Gly389Arg | 1.8E-05a | - | 3 (1) | |
| 10 | c.2221C>T | P.R741W | Arg406Trp | 2.7E-05b | 1 | 11 (6) | |
| GRN (NM_002087) | |||||||
| 11 | c.26C>A | p.A9D | Ala9Asp | - | - | 7 (5) | |
| 12 | c.87_90dup CTGC | p.C31Lfs*35 | Cys31fs | - | - | 8 (4) | |
| 13 | c.154delA | p.T52Hfs*2 | Thr52fs | - | - | 7 (4) | |
| 14 | c.264+2T>C | - | IVS3+2T>C | - | - | 4 (2) | |
| 15 | c.328C>T | p.R110* | Arg110X | 9.0E-06 | - | 7 (4) | |
| 16 | c.349+1G>A | - | - | - | - | 1 (1) |  |
| 17 | c.592_593delAG | p.R198Gfs*19 | Arg198fs | - | - | 3 (1) |  |
| 18 | c.598+1G>A | - | - | 9.0E-06 | 1 | - |  |
| 19 | c.675_676delCA | p.S226Wfs*28 | Ser226fs | - | - | 1 (1) |  |
| 20 | C.708+1G>A | - | IVS7+1G>A | .c | 1 | 1 (1) |  |
| 21 | c.708+6_708+9delTGAG | - | c.708+6_9delTGAG | 1.8E-05 | 1 | - |  |
| 22 | c.709–2A>G | - | IVS7–2A>G | - | - | 1 (1) |  |
| 23 | c.836–1G>C | - | IVS8–1G>C | - | - | 1 (1) |  |
| 24 | c.898C>T | p.Q300* | Gln300X | - | - | 1 (1) |  |
| 25 | c.910_911dupTG | p.W304Cfs*58 | Trp304fs | 9.0E-06 | - | 1 (1) |  |
| 26 | c.911G>A | p.W304* | Trp304X | - | 1 | - |  |
| 27 | c.1252C>T | p.R418* | Arg418X | - | - | 3 (3) | |
| 28 | c. 1256_1263dup GAAGCGAG | p.l422Efs*72 | Ile422fs | - | - | 1 (1) |  |
| 29 | c.1477C>T | p.R493* | Arg493X | .d | - | 6 (5) | |
| 30 | c.1414–2A>G | - | c.1414–2A>G | - | 1 | - |  |
| 31 | c.1535delC | p.P512Lfs*5 | Pro512fs | - | - | 2 (1) |  |
Also found in one East Asian individual (MAF = 5.8E-05) and
in one Finnish individual (MAF = 4.5E-05). Found in two Finnish individuals (MAF = 9.1E-05) and
in one Finnish individual (MAF = 4.5E-05).
When stratified by phenotype, we identified pathogenic variants within the three main genes in 10 (9.0%) of the 111 bvFTD cases, with the majority carrying C9orf72 repeat expansions. In contrast, 3 (10.7%) of 28 CBS cases carried pathogenic variants, all within the GRN gene, whereas 3 (33.3%) of 9 FTD/ALS cases carried repeat expansions in the C9orf72 gene. Among 33 nfvPPA cases, only one carried a pathogenic variant (in the GRN gene), whereas none was found within 79 PSP and 42 svPPA cases. In addition to MAPT, GRN, and C9orf72, we also identified variants in less common FTD genes. Two bvFTD cases carried likely pathogenic variants in the VCP gene, whereas five other sporadic cases carried variants of unknown significance (VUS) in TBK1, CHMP2B, and SQSTM1 (Table 2).
TABLE 2.
Other variants found in the ARTFL/LEFFTDS series and their classification according to the ACMG-AMP guidelines
| Variant | cDNA change | Aminoacid change | Historical name | gnomAD NFE frequency | SIFT | PolyPhen | CADD | ACMG-AMP classification | Sporadic | Familial (kindreds) | Total number of carriers |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAPT (NM_001123066) | |||||||||||
| 32 | c.2194C>T | p.P732S | - | - | D | D | 23.6 | VUS | 1 | - |  |
| TARDBP (NM_007375) | |||||||||||
| 33 | c.1147A>G | p.I383V | Ile383Val | 2.5E-05 | T | B | 18.6 | Likely Pathogenic | - | 4 (2) | |
| PSEN1 (NM_000021) | |||||||||||
| 34 | c.488A>G | p.H163R | His163Arg | - | D | P | 23.9 | Likely Pathogenic | - | 2 (1) |  |
| VCP (NM_007126.3) | |||||||||||
| 35 | c.383G>C | p.G128A | Gly128Ala | - | T | D | 25.1 | Likely Pathogenic | - | 1 (1) |  |
| 36 | c.460G>T | p.V154F | - | - | D | D | 32.0 | Likely Pathogenic | 1 | - |  |
| 37 | c.464G>A | p.R155H | Arg155His | - | T | B | 24.6 | Pathogenic | - | 1 (1) |  |
| 38 | c.811+2T>C | - | - | - | - | - | 32.0 | Likely Pathogenic | 1 | - |  |
| 39 | c.1184A>G | p.D395G | - | - | D | P | 29.7 | Likely Pathogenic | - | 2 (1) |  |
| TBK1 (NM_013254) | |||||||||||
| 40 | c.1717C>T | p.R573C | - | 1.1E-05 | T | D | 25.8 | VUS | 1 | - |  |
| 41 | c.2048T>C | p.L683S | - | 9.1E-06 | T | B | 22.8 | VUS | - | 1 (1)a |  |
| CHMP2B (NM_014043 | |||||||||||
| 42 | c.64C>T | p.R22* | - | 6.3E-05 | - | - | 37.0 | VUS | 1 | - |  |
| 43 | c.95G>A | p.R32Q | - | 3.6E-05 | D | B | 24.3 | VUS | 1 | - |  |
| 44 | c.292A>G | p.M98V | - | - | T | B | 22.2 | VUS | 1 | - |  |
| SQSTM1 (NM_003900) | |||||||||||
| 45 | c.416G>A | p.R139H | - | 6.7E-05 | D | D | 23.9 | VUS | 1 | - |  |
Carrier of an expanded C9orf72 repeat.
Abbreviations: B, benign; D, deleterious (SIFT) or probably damaging (PolyPhen); NFE: non-Finnish Europeans; P, possibly damaging; T, tolerated; VUS, variant of unknown significance.
We also screened this sporadic series for the rare risk-associated variants MAPT p.A152T (associated with FTD and PSP18) and TREM2 p.R47H (associated with AD19,20), and identified two (0.66%) MAPT p.A152T and five (1.66%) TREM2 p.R47H carriers. Finally, we screened for two common risk variants: the haplotype at the 17q21.31 locus (commonly referred to as 17q or tau haplotype), which has been linked to many neurological diseases, with the H1 haplotype being consistently associated with PSP; and the APOE ε4 allele, the strongest genetic risk factor for sporadic AD. The majority of the sporadic FTD samples carried the H1 haplotype (225 H1/H1 and 69 H1/H2), with 74 of the 79 PSP cases (94%) carrying an H1/H1 tau haplotype (the other five PSP cases had an H1/H2 haplotype). On the other hand, we identified 69 carriers (22.8%) of at least one APOE ε4 allele (3 ε2/ε4, 64 ε3/ε4, and 2 ε4/ε4).
3.3 |. Familial familial frontotemporal dementia series
Among the individuals with a family history of FTD (Fig. 1a, right), 247 (63%) were family members with no symptoms at baseline assessment (mean age 46.72 ± 13.86 years, range 18 to 80). The subset of 143 family members who were symptomatic at baseline (mean age 57.60 ± 11.13 years, range 28 to 85) spanned a range of different FTD, AD dementia, and other related phenotypes, with the majority being diagnosed with bvFTD (43% of all symptomatic) and mild cognitive impairment (MCI; 23%).
Among these participants, we identified a total of 85 carriers of an expanded C9orf72 repeat (from 70 kindreds), 67 (from 38 kindreds) of MAPT, and 54 (from 37 kindreds) of GRN pathogenic variants, and one participant who carried both an expanded C9orf72 repeat and a GRN pathogenic variant (p.C31Lfs*35) (Fig. 1b, right, and Table 1). These correspond to a total frequency of 61.3% (146 of 238) unrelated carriers of pathogenic variants in the three main FTD genes within our familial series. We also identified four (from two kindreds) carriers of TARDBP, and two (from one kindred) carriers of PSEN1 likely pathogenic variants, whereas four participants (from three kindreds) carried (likely) pathogenic variants in the VCP gene (Table 2). There was one participant, with a C9orf72 repeat expansion, that was found to also carry a VUS in the TBK1 gene (Table 2).
3.4 |. Overall genetic findings
In addition to the expanded C9orf72 repeat (identified in 95 individuals), we identified a total of 31 different pathogenic variants among the other two most common FTD genes, MAPT and GRN. Within the MAPT gene, we identified a total of 69 individuals carrying 10 distinct pathogenic variants (Table 1, Fig. 2a), all of which were reported previously in the literature as associated with dementia. The most frequent MAPT variant in this series was p.P636L (historically known as P301L), observed in 21 participants (from 15 predicted kindreds), followed by p.R741W (historically known as R406W) found in 12 participants (6 kindreds). These two variants, as well as the p.G724R (historically known as G389R) variant, were found in one to four individuals in the Genome Aggregation Database (gnomAD: http://gnomad.broadinstitute.org/),21 whereas the other seven MAPT variants were absent from this population database. We also found a novel MAPT missense variant, c.2194C>T:p.P732S (P397S according to the historical nomenclature), in a sporadic bvFTD case (Table 2). Although this variant is absent from the gnomAD database and predicted deleterious by both PolyPhen and SIFT in silico tools, and it affects a residue adjacent to known pathogenic variants, it was classified as a VUS due to the lack of sufficient evidence for pathogenicity.
FIGURE 2.

Schematic representation of GRN and MAPT variants found in the ARTFL/LEFFTDS series. Pathogenic variants (red) are represented in the topologic protein models of (a) tau (Uniprot accession P10636–9) and (b) progranulin (Uniprot accession P28799). (a) The canonical MAPT transcript (GenBank accession NM_001123066) contains 15 exons (including non-coding exon 1), with exons 6, 8, and 10 (white) not expressed in human brain, whereas exons 3, 4, and 12 (yellow) are alternatively spliced and adult brain-specific. The tubulin-binding region (blue) in the C-terminal can include either three (R1, R3, and R4) or four (R1, R2, R3, and R4) repeat regions, with the fourth microtubule-binding domain being encoded by the alternatively spliced exon 10 (bottom panel). Together, these give rise to six different possible tau isoforms in the human brain. Historical names for the MAPT mutations are shown in brackets. (b) Progranulin, encoded by the GRN gene (GenBank accession NM_002087), is a secreted precursor protein comprising a signal peptide (SP, dark gray), paragranulin (GrnP), and granulins (Grn) A to G. Cleavage of the signal peptide produces mature granulin, which can undergo further proteolytic cleavage resulting in a variety of active granulins
We identified a total of 60 carriers of 21 different GRN variants (Table 1, Fig. 2b). To the best of our knowledge, two of these were novel (not yet reported in the literature) splice-donor site variants c.349+1G>A and c.598+1G>A (Table 1). Although the c.598+1G>A variant was found in one individual in the gnomAD database, c.349+1G>A was absent from this population database. Altogether, there was sufficient evidence to classify these novel variants as pathogenic.
In addition to variants in the three most common FTD genes, we also identified 13 different variants in the other genes screened. Four participants (from two kindreds) carried the same missense variant (p.I383V) in TARDBP (Table 2). Although this variant is predicted to be tolerated by both PolyPhen and SIFT and it was observed in three non-Finnish Europeans in the gnomAD database, it has also been reported in at least four familial ALS cases,22,23 with biochemical analysis revealing a substantial increase in TDP-43 caspase cleaved fragments.22,24 Based on the available evidence, we classified this variant as likely pathogenic. Pathogenic variants in PSEN1 are the main genetic causes of AD; however, they have occasionally been linked in the literature to FTD phenotypes.25–28 We identified two related individuals with a missense variant (p.H163R) in the PSEN1 gene (Table 2). This variant is absent from gnomAD, is predicted to be damaging, and has been associated with multiple cases of AD, warranting classification as likely pathogenic. We also found six participants, including two seemingly sporadic cases, with four predicted deleterious missense and one splice site variant in the VCP gene-all of which were absent from gnomAD (Table 2). Three of the missense variants (p.G128A, p.V154F, and p.R155H) are located in the N-terminal domain of the VCP protein, which is associated with ubiquitin and recruiting co-factor binding, and where most symptomatic VCP reported mutations occur, including the p.G128A and p.R155H variants found in our series.10,29 Furthermore, VCP is essential for clearance of ubiquitinated protein by autophagy, and p.R155H and other disease-causing VCP variants have been shown to impair autophagy maturation.30 The other missense variant (p.D395G) is located within the ATPase domain D1, and was found in two asymptomatic relatives. Based on the different levels of evidence, p.R155H was classified as pathogenic while the other variants were considered likely pathogenic. An additional six variants were identified in TBK1 (2), CHMP2B (3), and SQSTM1 (1) (Table 2). Although these were absent or very rare in the gnomAD database and predicted deleterious by in silico tools, they were classified as VUS due to the current lack of sufficient evidence for pathogenicity.
3.5 |. Clinical presentation of (likely) pathogenic carriers at baseline visit
Most carriers of pathogenic variants within the three most common FTD genes were asymptomatic at baseline assessment (44.09 ± 14.31 years) (Fig. 3), including the individual carrying both a C9orf72 repeat expansion and a GRN pathogenic variant. For C9orf72 symptomatic carriers (59.33 ± 10.28 years at baseline), the most common phenotypes were bvFTD, diagnosed in 28 of the 55 symptomatic carriers (50.9%), followed by MCI (10 carriers, 18.2%) and FTD/ALS (eight carriers, 14.5%, including one homozygous carrier). Similarly, pathogenic variants in the MAPT gene were found mostly in symptomatic individuals (52.94 ± 9.75 years at baseline) diagnosed with bvFTD (23 of 33 symptomatic carriers, 69.7%) and MCI (5 symptomatic carriers, 15.2%). In contrast, GRN pathogenic variants were found in a more diverse range of phenotypes (62.50 ± 8.16 years at baseline), including CBS and MCI (each with 6 out of 26 symptomatic carriers), bvFTD (5 carriers), nfvPPA (3 carriers), AD dementia (2 carriers), and lvPPA (1 carrier). Of interest, one of the two related individuals carrying a likely pathogenic PSEN1 variant was diagnosed with bvFTD, whereas the TARDBP carriers were diagnosed with either bvFTD (3) or svPPA (1). Although most of the familial VCP carriers were asymptomatic at baseline, the two sporadic carriers were diagnosed with bvFTD.
FIGURE 3.
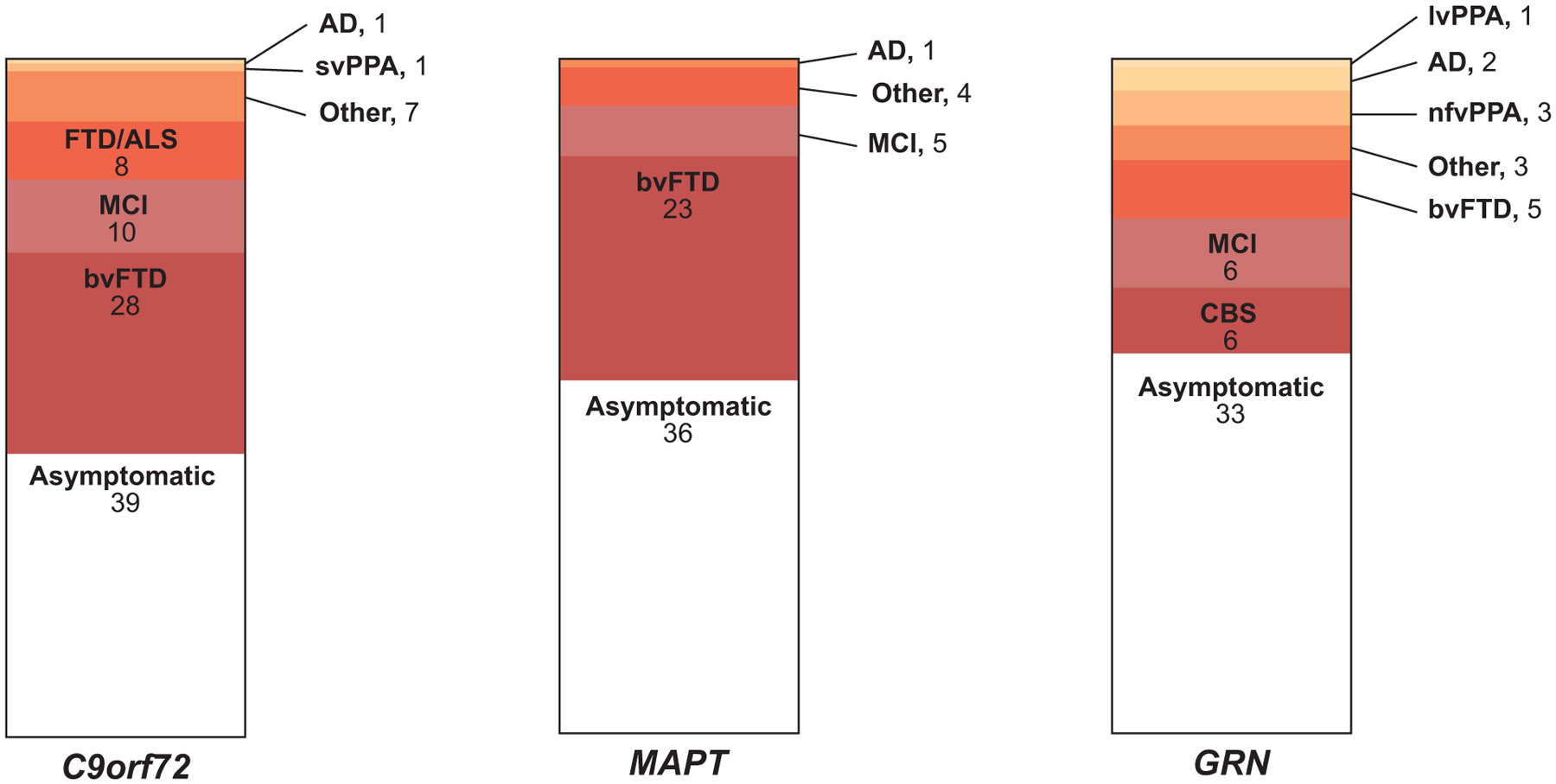
Clinical presentation among C9orf72, MAPT, and GRN mutation carriers. Although the majority of carriers were asymptomatic at baseline assessment, bvFTD was the most common phenotypic presentation among C9orf72 and MAPT mutation carriers, whereas the clinical spectrum was more heterogeneous among GRN carriers. Numbers of individuals with gene mutations are listed under each diagnostic category. (AD, Alzheimer’s disease dementia; ALS, amyotrophic lateral sclerosis; bvFTD, behavioral variant frontotemporal dementia (FTD); CBS, corticobasal syndrome; lvPPA, logopenic variant PPA; MCI, mild cognitive impairment; nfvPPA, non-fluent variant PPA; PPA, primary progressive aphasia; svPPA, semantic variant PPA)
4 |. DISCUSSION
We describe the genetic screen of nine FTD (C9orf72, MAPT, GRN, TARDBP, FUS, VCP, TBK1, CHMP2B, and SQSTM1) and three AD (APP, PSEN1, PSEN2) genes in 302 sporadic and 390 familial (symptomatic or at-risk) individuals enrolled to date in the ARTFL/LEFFTDS studies. We identified 31 different pathogenic variants within the main FTD genes (MAPT and GRN), in addition to the expanded C9orf72 repeat, in a total 223 participants, including 16 seemingly sporadic cases. In addition, we found 12 participants, mostly with positive family history, with seven distinct (likely) pathogenic variants in the TARDBP, PSEN1, and VCP genes. There were also six sporadic cases with VUS in the MAPT, TBK1, CHMP2B, and SQSTM1 genes, and one participant with a positive family history who carried both a C9orf72 repeat expansion and a VUS in the TBK1 gene.
Several studies have reported C9orf72 as the major cause of both familial (~25%) and sporadic (~5%) FTD,31 followed by GRN, which is estimated to be responsible for 5% to 20% of familial and 1% to 5% of sporadic cases, and MAPT, with a frequency ranging between 5% and 20% of familial FTD cases.32 In our series, the major FTD genes accounted for 5.3% of sporadic cases, consistent with other FTD genetic screens, with C9orf72 being the most frequent (3.0%), followed by GRN (1.7%) and MAPT (0.7%). However, due to the inclusion criteria for both ARTFL project 2 and LEFFTDS studies, we cannot speculate about the frequency of these genes in our familial series, as these are expected to be overrepresented by design. Of interest, in our series we found one asymptomatic familial participant with both an expanded C9orf72 repeat and a GRN pathogenic variant. A small number of patients with behavioral phenotypes have been found to carry both C9orf72 repeat expansions and a GRN or MAPT variant.33 In order to understand the contribution of each mutation to clinical presentation (i.e., whether they are both pathogenic, or one of them is merely affecting risk), further studies involving multiple patients from families carrying double mutations are necessary. In addition, we identified one homozygous carrier of a C9orf72 repeat expansion. Although this participant was originally enrolled in ARTFL project 1 as a sporadic case, family history information later revealed consanguinity in the family, and several relatives had developed disease symptoms. Similar to what was observed in the first homozygous C9orf72 patient reported in the literature,34 disease symptoms were not more severe in this homozygous patient, diagnosed with FTD/ALS, compared to the presumed heterozygous carrier relatives.
In terms of clinical presentation, symptomatic C9orf72 repeat expansion carriers were, as reported in other studies, mostly diagnosed with bvFTD and FTD/ALS. Because LEFFTDS is focused on asymptomatic and early familial FTD, unsurprisingly a number of C9orf72 carriers in our series were diagnosed with behavioral (n = 3) or cognitive (n = 6) MCI variants. One participant had an amnestic clinical syndrome (AD-like). Several earlier studies have reported C9orf72 expansions in clinically diagnosed or pathologically confirmed AD (accounting for <1% of all the AD population, as reviewed in35), suggesting that C9orf72 expansions might contribute to AD pathology. In contrast, some clinically diagnosed AD cases have been shown to be pathologically consistent with FTLD-TDP,36–38 suggesting that early onset of memory impairment in some C9orf72 carriers might mislead to a clinical diagnosis of AD. In our series, symptomatic GRN carriers showed a range of both FTD and AD dementia-spectrum phenotypes, with bvFTD, but also CBS and MCI behavior (n = 3) being among the most frequent. Although rarely observed at onset, parkinsonian features often manifesting as CBS have been reported in up to 40% of patients with GRN mutations.39 Symptomatic MAPT carriers showed high frequency of bvFTD, followed by MCI behavior. Overall, it should be noted that the majority of the C9orf72, GRN, and MAPT carriers were asymptomatic at baseline assessment, since both ARTFL project 2 and LEFFTDS were designed as longitudinal studies aimed at tracking disease progression in familial FTD, and therefore asymptomatic carriers are expected to be overrepresented in our series.
Although the majority of causative variants were identified in the major FTD-causative genes, we identified in our series two kindreds that carried the same likely pathogenic variant in TARDBP (p.I383V). Mutations in TARDBP have been associated with 2% to 3% of ALS patients and more rarely with FTD.40 Among the few FTD cases reported so far in the literature, most present a heterogeneous phenotype, with significant motor neuron involvement, whereas the most common pure FTD presentations are bvFTD followed by PPA, in particular svPPA,40 which is consistent with our findings. We also identified a PSEN1 variant (p.H163R) that has, to the best of our knowledge, only been associated with AD in a patient with clinical bvFTD. However, it should be noted that a frontal variant of AD characterized by predominant behavioral or dysexecutive deficits might mimic bvFTD in up to 40% of clinically diagnosed bvFTD cases.41 Of interest, we found six participants with (likely) pathogenic variants in the VCP gene, making this the most common disease-associated gene in our series after the three main FTD genes (C9orf72, GRN, and MAPT). More than 45 different VCP variants have been reported to be associated with heterogeneous clinical presentations, including inclusion body myopathy, Paget disease of the bone, and FTD and ALS, even in patients carrying the same variant, suggesting that additional genetic (or other) factors may determine the clinical phenotype. In our series, the symptomatic VCP carriers were mainly apparently sporadic bvFTD.
Even though most familial cases enrolled in these series were recruited from known C9orf72, MAPT, and GRN families, there was a small subset (about 30 unrelated familial participants) that had strong family history, but no known causative variant was found. It should be noted that although targeted sequencing has facilitated the concurrent testing of multiple causative dementia genes, it is unable to detect large insertions/deletions and repeat expansions, while other types of mutations such as intronic or copy number variations can go undetected. In addition, a dementia-specific panel, such as the one used herein, may fail to reveal an unexpected or novel causative gene.
Although genetic testing for dementia is complex, and normally only recommended in familial cases, it can have a significant role in the disease management.42,43 Genetic screening may enable an accurate and earlier diagnosis. For example, early onset familial AD and FTD may present with overlapping clinical features that may be distinguished by genetic findings. It may also clarify the risk for dementia in asymptomatic relatives of affected individuals. Furthermore, the presence of a specific genetic mutation (or risk factor) is, at the moment, still the most specific predictor of the underlying pathophysiology of a patient with a clinical FTD diagnosis, leading to improved selection of patients for clinical trials designed to target a specific type of pathology. And, as more clinical trials of drugs targeting specific genes become available and demonstrate success regarding primary/secondary outcomes, more at-risk people may elect to get tested to determine their trial eligibility. Finally, the discovery of novel causative/risk genes will contribute to clarifying the pathogenic mechanism of dementias and lead to effective treatment(s).
With next-generation sequencing and the concurrent genetic testing of many genes, generating large amounts of data, comes an increase difficulty in interpreting genetic findings. Therefore, it is of great interest to generate a comprehensive catalog of not only causative and risk variants, but also benign variants, in the known dementia genes, to aid physicians and genetic counselors in the interpretation of genetic test results and refinement of clinical diagnoses for patients with FTD. The ARTFL/LEFFTDS genetics database (https://coppolalab.ucla.edu/exac_browser/artfl) currently lists all variants within the main AD and FTD genes found in this series, along with associated annotations (including pathogenicity), and frequency.
In conclusion, we present an in-depth genetic characterization of both symptomatic and asymptomatic FTD individuals enrolled in the ARTFL and LEFFTDS studies. This is a critical resource necessary to interpret all the clinical, neuropsychological, and biomarker data generated by these consortia, and will contribute to more standardized diagnostic criteria and, importantly, facilitate the identification of homogeneous series for upcoming disease-modifying therapeutic trils in familial frontotemporal lobar degeneration. Our study confirms the importance of unbiased sequencing approaches, either targeted to dementia-associated genes or whole-exome sequencing, in screening dementia cohorts, independent of their clinical presentation.
Supplementary Material
HIGHLIGHTS.
In-depth genetic screen of participants enrolled in the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) studies.
Identified a total of 223 carriers of chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT) and progranulin (GRN) pathogenic variants.
Found 31 different pathogenic variants within the MAPT and GRN genes.
Twelve additional participants with likely pathogenic variants in TAR DNA binding protein (TARDBP), presenilin 1 (PSEN1), and valosin containing protein (VCP).
RESEARCH IN CONTEXT.
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources, meeting abstracts, and presentations. Disease-causing variants in frontotemporal dementia (FTD) have been widely reported in the literature and all relevant citations are appropriately cited.
Interpretation: Our in-depth genetic characterization of both symptomatic and asymptomatic FTD individuals enrolled in the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) studies is a critical resource necessary to interpret all the clinical, neuropsychological, and biomarker data generated by these consortia.
Future directions: This article provides a framework for the generation of new hypotheses and additional studies. The data and samples described herein, which are available to interested investigators worldwide, may contribute to more standardized diagnostic criteria and facilitate the identification of homogeneous series for upcoming disease-modifying therapeutic trials in familial FTD.
ACKNOWLEDGMENTS
Samples from the National Centralized Repository for Alzheimer’s Disease and Related Dementia (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. We thank the UCLA Neuroscience Genomics Core (www.semel.ucla.edu/ungc) for assistance with sequencing data generation and the support of the National Institute of Neurological Disorders and Stroke (NINDS) Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691). We also thank the MacArthur lab at the Broad Institute for assistance in implementing the ARTFL/LEFFTDS browser, generated using their ExAC database interface. We extend our appreciation to Dr. John Hsiao from NIA and Dr. Margaret Sutherland from NINDS.
FUNDING/SUPPORT
This work was supported by The Frontotemporal Lobar Degeneration Clinical Research Consortium (U54 NS092089) and the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS, U01 AG045390), as well as by R35 NS097261, P01 NS084974, R21 NS099631, U01 AG016976, the ALS Association, and the Robert Packard Center for ALS.
ROLE OF THE FUNDER/SPONSOR
The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
DECLARATION OF INTEREST
Eliana Marisa Ramos has nothing to disclose. Deepika Reedy Dokuru has nothing to disclose. Victoria Van Berlo has nothing to disclose. Kevin Wojta has nothing to disclose. Qing Wang has nothing to disclose. Alden Y. Huang has nothing to disclose. Sandeep Deverasetty has nothing to disclose. Yue Qin has nothing to disclose. Marka van Blitterswijk receives research support from the National Institutes of Health (NIH), the ALS Association, and the Robert Packard Center for ALS Research. Jazmyne Jackson has nothing to disclose. Brian Appleby receives research support from the Centers for Disease Control and Prevention (CDC) and NIH. Yvette Bordelon has served as an investigator for clinical trials sponsored by AbbVie, Biogen, Bristol-Myers Squibb, and C2N Diagnostics. Patrick Brannelly is employed by the Rainwater Charitable Foundation. Danielle E. Brushaber has nothing to disclose. Bradford Dickerson receives research support from NIH. Susan Dickinson is on staff at the Association for Frontotemporal Degeneration and a member of the National Institute for Neurological Disorders and Stroke Advisory Council. Kimiko Domoto-Reilly has served as an investigator for clinical trials sponsored by Avid Radiopharmaceuticals, Biogen, and Janssen Pharmaceuticals; he has served as an advisory board consultant for Biogen and receives research support from NIH. Kelley Faber receives research support from NIH. Julie Fields receives research support from NIH. Jamie Fong has nothing to disclose. Tatiana Foroud receives research support from NIH. Leah Forsberg has nothing to disclose. Ralitza Gavrilova receives research support from NIH. Nupur Ghoshal has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by the following companies: Bristol Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen Immunotherapy, Novartis, Pfizer, Wyeth, SNIFF (The Study of Nasal Insulin to Fight Forgetfulness) study, and A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial. She receives research support from Tau Consortium and Association for Frontotemporal Dementia and is funded by the NIH. Jill Goldman is serving as a consultant to the Novartis Alzheimer’s Prevention Advisory Board. She receives research support from NIH, HDSA, and New York State Department of Health (RFA #1510130358). Jonathan Graff-Radford receives research support from NIH. Neill Graff-Radford receives royalties from UpToDate, and has participated in multicenter therapy studies sponsored by Biogen, TauRx, AbbVie, Novartis, and Lilly; he receives research support from NIH. Ian Grant has nothing to disclose. Murray Grossman receives grant support from NIH, Avid, and Piramal; participates in clinical trials sponsored by Biogen, TauRx, and Alector; serves as a consultant to Bracco and UCB; and serves on the Editorial Board of Neurology. Hilary W. Heuer has nothing to disclose. Ging-Yuek R. Hsiung has served as an investigator for clinical trials sponsored by AstraZeneca, Eli Lilly, and Roche/Genentech. He receives research support from Canadian Institutes of Health Research and the Alzheimer Society of British Columbia. Edward Huey receives grant support from NIH, the Association for Frontotemporal Degeneration, and the Alzheimer’s Drug Discovery Foundation. He participates in clinical trials sponsored by NIH and the Lawson Health Research Institute. David Irwin receives support from NIH, BrightFocus Foundation, and Penn Institute on Aging. Kejal Kantarci served on the Data Safety Monitoring Board for Takeda Global Research & Development Center, Inc.; data monitoring boards of Pfizer and Janssen Alzheimer Immunotherapy; research support from the Avid Radiopharmaceuticals, Eli Lilly, the Alzheimer’s Drug Discovery Foundation and NIH. Anna Karydas has nothing to disclose. Daniel Kaufer has served as an investigator for clinical trials sponsored by Abbvie, Axovant, Janssen Research & Development, Navidea Biopharmaceuticals, and TauRx. He has consulted for Abbvie, Axovant, Janssen Research & Development, Takeda/Zinfandel. He serves on the Scientific Advisory Board of the Lewy Body Dementia Association. He receives research funding from the NIH, HRSA, and Bryan Family Foundation. Diana Kerwin has served on an Advisory Board for AbbVie and as site PI for studies funded by Roche/Genentech, AbbVie, Avid, Novartis, Eisai, Eli Lilly, and UCSF. David Knopman serves on the DSMB of the DIANTU study; is a site PI for clinical trials sponsored by Biogen, Lilly, and the University of Southern California; and is funded by NIH. John Kornak has provided expert witness testimony for Teva Pharmaceuticals in Forest Laboratories Inc. et al. v. Teva Pharmaceuticals USA, Inc., Case Nos. 1:14-cv-00121 and 1:14-cv-00686 (D. Del. filed Jan. 31, 2014 and May 30, 2014) regarding the drug Memantine; for Apotex/HEC/Ezra in Novartis AG et al. v. Apotex Inc., No. 1:15-cv-975 (D. Del. filed Oct. 26, 2015), regarding the drug Fingolimod. He has also given testimony on behalf of Puma Biotechnology in Hsingching Hsu et al., v. Puma Biotechnology, INC., et al. 2018 regarding the drug Neratinib. He receives research support from the NIH. Joel H. Kramer receives research support from NIH and serves on an advisory board for Biogen. Walter Kremers receives research funding from AstraZeneca, Biogen, Roche, DOD, and NIH. Walter Kukull receives research support from NIH. Irene Litvan receives research support from NIH, Parkinson Study Group, Parkinson Foundation, Michael J Fox Foundation, AVID Pharmaceuticals, C2N Diagnostics/Abbvie, and Bristol-Myers Squibb. She was a member of the Biogen and Bristol-Myers Squibb Advisory Boards, Biotie/Parkinson Study Group Medical Advisory Board, and a consultant for Toyama Pharmaceuticals. She receives her salary from the University of California San Diego and as Editor of Frontiers in Neurology. Peter Ljubenkov has nothing to disclose. Codrin Lungu received honoraria for editorial work from Elsevier, Inc. Ian Mackenzie receives research funding from Canadian Institutes of Health Research. Mario Mendez supported by NIH (NIA) research grants and has received research support from Biogen. Bruce Miller receives research support from NIH. Chiadi Onyike receives research funding from the NIH, the CIHR, and Biogen, Inc. He is also supported by the Jane Tanger Black Fund for Young-Onset Dementias, the Nancy H. Hall Fund for Geriatric Psychiatry, and the gift from Joseph Trovato. Alexander Pantelyat receives research support from the NIH and AbbVie, Inc., participates in a research trial sponsored by Biogen, Inc., and has served as consultant for AbbVie, Inc. Rodney Pearlman is employed by The Bluefield Project. Len Petrucelli receives research support from NIH. Madeline Potter receives research support from NIH. Katherine P. Rankin receives research support from NIH, Quest Diagnostics, and the Rainwater Charitable Foundation. Katya Rascovsky receives research support from NIH. Erik D. Roberson receives research support from NIH, Bluefield Project to Cure Frontotemporal Dementia, Alzheimer’s Association, BrightFocus Foundation, Biogen, Alector, and owns intellectual property related to tau. Emily Rogalski receives research support from NIH and the Association for Frontotemporal Dementia. Leslie Shaw receives research support from NIH. Jeremy Syrjanen has nothing to disclose. Maria Carmela Tartaglia receives research funding from CIHR and NIH, and is an investigator on pharmaceutical studies with Biogen, Roche, Eli Lilly, and Boehringer. Nadine Tatton is employed by the Association for Frontotemporal Degeneration. Joanne Taylor has nothing to disclose. Arthur Toga receives research support from the NIH and the Alzheimer’s Association. John Trojanowski may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is coinventor, and he received revenue from the sale of Avid to Eli Lily as coinventor on Aβ amyloid imaging-related patents submitted by the University of Pennsylvania. He receives research support from the NIH and several nonprofits. Sandra Weintraub receives research support from NIH. Bonnie Wong receives research support from NIH. Zbigniew Wszolek is supported by the NIH, Mayo Clinic Center for Regenerative Medicine, the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa. He has also received grant funding support from Allergan, Inc. (educational grant), and Abbvie (medication trials). Rosa Rademakers receives research funding from NIH and the Bluefield Project to Cure Frontotemporal Dementia. Brad Boeve has served as an investigator for clinical trials sponsored by GE Healthcare and Axovant. He receives royalties from the publication of a book entitled Behavioral Neurology of Dementia (Cambridge Medicine, 2009, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from NIH, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, and the Little Family Foundation. Howard J. Rosen has received research support from Biogen Pharmaceuticals, has consulting agreements with Wave Neuroscience and Ionis Pharmaceuticals, and receives research support from NIH. Adam L. Boxer receives research support from NIH, the Tau Research Consortium, the Association for Frontotemporal Degeneration, Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, the Alzheimer’s Drug Discovery Foundation, and the Alzheimer’s Association. He has served as a consultant for Aeton, Abbvie, Alector, Amgen, Arkuda, Ionis, Iperian, Janssen, Merck, Novartis, Samumed, Toyama, and UCB; and received research support from Avid, Biogen, BMS, C2N, Cortice, Eli Lilly, Forum, Genentech, Janssen, Novartis, Pfizer, Roche, and TauRx. Giovanni Coppola receives research support from the NIH, the Tau Consortium, the Adelson Medical Research Foundation, Takeda Pharmaceutical Company Ltd., the John Douglas French Alzheimer’s Foundation, the Friedreich’s Ataxia Research Alliance, the CHDI foundation, the Hillblom Foundation, the Eleanor Leslie Chair in Innovative Brain Research from the Brain Research Institute, and the Semel Institute for Neuroscience and Human Behavior at the University of California Los Angeles.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 5.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 6.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. [DOI] [PubMed] [Google Scholar]
- 7.Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. [DOI] [PubMed] [Google Scholar]
- 8.Van Langenhove T, van der Zee J, Sleegers K, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–371. [DOI] [PubMed] [Google Scholar]
- 9.Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. [DOI] [PubMed] [Google Scholar]
- 10.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. [DOI] [PubMed] [Google Scholar]
- 11.Rubino E, Rainero I, Chio A, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. [DOI] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Blitterswijk M, Dejesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12(10):978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez A, Lee SE, Wojta K, et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain. 2017;140:1128–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ticozzi N, LeClerc AL, van Blitterswijk M, et al. Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol Aging. 2011;32:2096–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendron TF, Rademakers R, Petrucelli L. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis. 2013;33(suppl 1):S35–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernardi L, Tomaino C, Anfossi M, et al. Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol Aging. 2009;30:1825–1833. [DOI] [PubMed] [Google Scholar]
- 26.Robles A, Sobrido MJ, Garcia-Murias M, et al. Clinical picture of a patient with a novel PSEN1 mutation (L424V). Am J Alzheimers Dis Other Demen. 2009;24:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahoney CJ, Downey LE, Beck J, et al. The presenilin 1 P264L mutation presenting as non-fluent/agrammatic primary progressive aphasia. J Alzheimers Dis. 2013;36:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riudavets MA, Bartoloni L, Troncoso JC, et al. Familial dementia with frontotemporal features associated with M146V presenilin-1 mutation. Brain Pathol. 2013;23:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Tahan S, Al-Obeidi E, Yoshioka H, et al. Novel valosin-containing protein mutations associated with multisystem proteinopathy. Neuromuscul Disord. 2018;28:491–501. [DOI] [PubMed] [Google Scholar]
- 30.Tresse E, Salomons FA, Vesa J, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Blitterswijk M, Baker MC, DeJesus-Hernandez M, et al. C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology. 2013;81:1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fratta P, Poulter M, Lashley T, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chi S, Jiang T, Tan L, Yu JT. Distinct neurological disorders with C9orf72 mutations: genetics, pathogenesis, and therapy. Neurosci Biobehav Rev. 2016;66:127–142. [DOI] [PubMed] [Google Scholar]
- 36.Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobson-Stone C, Hallupp M, Bartley L, et al. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology. 2012;79:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majounie E, Abramzon Y, Renton AE, et al. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med. 2012;366:283–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131:732–746. [DOI] [PubMed] [Google Scholar]
- 40.Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138:2732–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldman JS, Deerlin VM. Alzheimer’s disease and frontotemporal dementia: the current state of genetics and genetic testing since the advent of next-generation sequencing. Mol Diagn Ther. 2018;22(5):505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldman JS. New approaches to genetic counseling and testing for Alzheimer’s disease and frontotemporal degeneration. Curr Neurol Neurosci Rep. 2012;12(5):502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.