

Abstract
This review focuses on the association between vascular calcification and arterial stiffness, highlighting the important genetic factors, systemic and local microenvironmental signals, and underlying signaling pathways and molecular regulators of vascular calcification. Elevated oxidative stress appears to be a common pro-calcification factor that induces osteogenic differentiation and calcification of vascular cells in a variety of disease conditions such as atherosclerosis, diabetes mellitus and chronic kidney disease (CKD). Thus, the role of oxidative stress and oxidative stress-regulated signals in vascular smooth muscle cells (VSMC) and their contributions to vascular calcification are highlighted. In relation to diabetes mellitus, the regulation of both hyperglycemia and increased protein glycosylation, by advanced glycation end products (AGEs) and O-GlcNAcylation, and its role in enhancing intracellular pathophysiological signaling that promotes osteogenic differentiation and calcification of VSMC are discussed. In the context of CKD, this review details the role of calcium and phosphate homeostasis, parathyroid hormone, and specific calcification inhibitors in regulating vascular calcification. In addition, the impact of these systemic and microenvironmental factors on respective intrinsic signaling pathways that promote osteogenic differentiation and calcification of VSMC and osteoblasts are compared and contrasted, aiming to dissect the commonalities and distinctions that underlie the paradoxical vascular-bone mineralization disorders in aging and diseases.
Keywords: Vascular calcification, arterial stiffness, atherosclerosis, diabetes mellitus, chronic kidney disease
Graphical Abstract
INTRODUCTION
Arterial stiffening is an important determinant of vascular aging and independently predicts cardiovascular events, including left ventricular hypertrophy, myocardial infarction, hypertension, atherosclerosis and stroke, as well as cardiovascular complications in diabetic mellitus and chronic kidney disease (CKD)1–9. As stiffening of the large elastic arteries progresses with aging, it accelerates the development of cardiovascular diseases. Epidemiological studies have demonstrated an independent prognostic link of aortic pulse wave velocity (PWV), the indicator for arterial stiffness, with cardiovascular outcomes in older adults and patients with diabetes mellitus and hypertension7, 8, 10. On the other hand, vascular stiffening is further exacerbated by metabolic syndromes, diabetes mellitus, obesity, CKD, and cardiovascular diseases including atherosclerosis and hypertension11. Accordingly, pharmacological interventions that target these conditions, such as advanced glycation end-product (AGE) crosslink breaker, angiotensin-converting enzyme (ACE) inhibitors, and statins, are emerging as potential candidates to reduce arterial stiffness, although no selective and highly effective treatment for arterial stiffness has been identified10, 12–14.
This is partly due to the lack of comprehensive and precise understanding of the mechanisms responsible for arterial stiffening. Nonetheless, it is well documented that arterial stiffening is linked to the structural and functional changes of the large elastic arteries with aging and during the development of cardiovascular diseases. As summarized by previous reviews, remodeling of extracellular matrix (ECM), including elastin degradation and excessive collagen deposition and glycosylation, is critical in regulating arterial stiffness and the development of cardiovascular diseases15–19. At the cellular level, while endothelial cells, fibroblasts and inflammatory cells all contribute to arterial stiffening, vascular smooth muscle cells (VSMC) are essential to maintain vascular homeostasis in the arterial system, including the elastic arteries such as aorta, muscular arteries such as coronary and femoral arteries, as well as arterioles. Increased VSMC proliferation, migration, senescence and mineralization play predominant roles in age-related medial and intimal thickening and arterial stiffening as well as pathogenesis of cardiovascular diseases4. In particular, VSMC in the tunic media of the elastic arteries regulate vascular tone, thus VSMC dysfunction leads to impaired vascular homeostasis with aging and/or the development of major cardiovascular diseases.
VASCULAR CALCIFICATION AND ARTERIAL STIFFNESS
Intrinsic stiffness of VSMC is aggravated with aging20. In addition, VSMC regulate arterial stiffness by overproducing various ECM components, such as collagen and elastin, which provide biomechanical, structural integrity and signaling regulation of the ECM to maintain vascular homeostasis under normal healthy conditions16, 21. Furthermore, VSMC undergo osteogenic differentiation and mineralization in response to a variety of stimuli, which drives vascular calcification. Both vascular calcification and remodeling of ECM directly mediate arterial stiffening4. The remodeling of ECM in turn promotes VSMC calcification, which further accelerates vascular stiffening. For example, increased expression of matrix proteases, such as calpain-1 and matrix metalloproteinase 2 (MMP2), has been identified in aged human aortic wall and atherosclerotic lesions22. Studies with cultured VSMC in vitro and carotid artery rings ex vivo have shown that increased calpain-1 and MMP2 promote VSMC calcification, elastin degradation, alkaline phosphatase (ALP) activation, collagen production; and reduce expression of calcification inhibitors, such as osteopontin and osteonectin22. Emerging studies have linked vascular calcification to reduced arterial elasticity, decreased vessel walls compliance, increased arterial stiffening and accelerated development of cardiovascular diseases23–25.
By definition, there are two major types of vascular calcification: medial calcification and intimal calcification. Medial arterial calcification, also called Mönckeberg sclerosis, is localized mainly in the tunica media that contains VSMC and elastic tissues. Medial arterial calcification exists in aging arteries in patients with diabetes mellitus and CKD, which often occurs independent of atherosclerosis26, 27. Intimal arterial calcification, on the other hand, is present in atherosclerotic lesions in the tunica intima, which is often associated with hyperlipidemia, macrophage infiltration and vascular inflammation28, 29. Intimal arterial calcification occurs mostly in patients with atherosclerosis, but also is found in patients with diabetes mellitus and CKD26. An early study examining over 540 aorta samples from human autopsies has revealed an increased incidence of medial and intimal calcification with aging30. In patients with diabetes mellitus and CKD, increased calcification in the media is associated with arterial stiffening as well as an increased risk of cardiovascular morbidity and mortality26, 31. In addition, recent studies have linked vascular calcification to formation, progression and rupture of aortic aneurysm32, 33. Consistently, combing coronary artery calcium score with conventional risk factors for cardiovascular diseases significantly improves the clinical risk reclassification of cardiovascular outcomes, in cohorts in the Multi-Ethnic Study of Atherosclerosis, Dallas Heart Study, Prospective Army Coronary Calcium Project, and the Framingham Heart Study34–37. These clinical observations have supported a strong link between vascular calcification and arterial stiffening, with aging and multiple cardiovascular diseases. On the other hand, arterial stiffening in atherosclerosis, diabetes and CKD may further accelerate vascular calcification, creating a vicious cycle. Therefore, better understanding of molecular mechanisms underlying vascular calcification should lead to identifying potential targets and strategies to selectively block vascular calcification and calcification-associated adverse cardiovascular outcomes.
REGULATION OF VASCULAR CALCIFICATION
Vascular calcification is now recognized as an active osteogenic process of vascular cells, mainly VSMC, resembling osteoblast formation that is defined by the osteogenic differentiation, matrix maturation and matrix mineralization stages38. It is regulated by multifaceted mechanisms underlying intracellular osteogenic differentiation and remodeling of extracellular matrix, involving genetic, (micro)environmental and cellular signaling modulators39. Osteogenic differentiation and calcification of VSMC is a common feature existing in both medial and intimal arterial calcification. VSMC possesses phenotypic plasticity. In response to environmental changes with aging and cardiovascular diseases, VSMC undergo phenotypic transition from the contractile phenotype, characterized by the expression of SMC-specific contractile proteins such as smooth muscle α-actin (SMA), SM22α and smooth muscle cell myosin heavy chain (SMMHC), to a synthetic phenotype as well as trans-differentiation into other cells of mesenchymal lineages, such as osteoblasts, chondrocytes and adipocytes. Osteogenic differentiation of VSMC features increased expression of bone markers and concurrently decreased expression of VSMC markers40–42. Similar to the osteogenic differentiation of bone cells, increased expression of alkaline phosphatase (ALP) and matrix proteins, such as type 1 collagen (Col1) that marks the matrix maturation38 was evident in vascular calcification. Extracellular matrix mineralization involves the formation and secretion of matrix vesicles (MVs), membrane-bound nanoparticles that carry and transport calcium/phosphate and matrix proteins, followed by nucleation of calcium phosphate crystals and aggregation on collagen fibers in the extracellular matrix29. The ALP catalyzes the degradation of inorganic pyrophosphate (PPi), a major inhibitor for amorphous calcium phosphate nucleation, hydroxyapatite aggregation and crystal growth43, thus promoting hydroxyapatite deposition and matrix mineralization (Figure 1).
Figure 1.

Vascular calcification resembles the process of osteoblast differentiation and mineralization, involving osteogenic differentiation, matrix maturation and mineralization. Systemic and local microenvironmental conditions associated with atherosclerosis, diabetes mellitus, CKD and aging induce osteogenic differentiation of vascular cells and promote matrix remodeling and mineralization, leading to vascular calcification.
The importance of MVs in regulating cardiovascular calcification in atherosclerosis, diabetes mellitus and CKD has been documented44, 45. MVs are evident in atherosclerotic lesions as well as in medial calcification46, 47. In vitro studies with human VSMC have further illustrated the critical role of MVs in promoting vascular calcification in response to extracellular calcium and phosphate stress48. In addition, many bone matrix proteins and key osteogenic regulators have been identified in calcified arteries and vascular cells, including Col I, matrix Gla protein (MGP), osteopontin (OPN), matrix metalloproteinases (MMP), bone morphogenetic protein-2 (BMP-2) and the master osteogenic transcription factor, Runt-related transcription factor-2 (Runx2)29, 49. In vitro studies with VSMC have provided direct evidence for osteogenic differentiation and calcification of VSMC in response to extracellular stimuli, which features decreased expression of SMC marker genes and concurrent upregulation of bone markers29. These multiple lines of evidence have supported the concept that vascular calcification represents a regulated osteogenic differentiation and calcification of vascular cells. This review discusses the integrated regulation of vascular calcification that promotes arterial stiffening, focusing on genetic inhibitors as well as pro-osteogenic regulators associated with atherosclerosis, diabetes mellitus and CKD, including oxidative stress, hyperglycemia and dysregulation of calcium/phosphate.
Genetic determinants of vascular calcification
Multiple genetic regulators of vascular calcification have been identified using genome-wide association studies (GWAS) and single nucleotide polymorphism (SNP)-based analyses in human, as well as genetically modified animal models. These diverse regulators include matrix proteins such as MGP and OPN, circulating calcium binding protein, Fetuin-A, as well as regulators for phosphate and pyrophosphate metabolism, including ectonucleototide pyrophosphatase/phosphodiesterase-1 (ENPP1), pyrophosphate extracellular transporter progressive ankylosis protein (ANK), ecto-5′-nucleotidase CD73 and the ATP-binding cassette transporter subtype 6 (ABCC6). Increased vascular calcification and corresponding arterial stiffness are observed in each monogenic mutation mapped to these genetic factors. The identification of these genetic factors supports the concept that “loss-of-inhibitors” promotes the pathogenesis of vascular calcification (Table 1).
Table 1.
Genetic regulators of vascular calcification in mouse and human
| Mechanisms | Gene Symbol | Mouse Models | Human Disease Correlation (SNPs / Mutations) | ||
|---|---|---|---|---|---|
| Genotype | Vascular Phenotype | Bone Phenotype | |||
| Calcium binding, inhibiting hydroxyapatite crystal formation | MGP | Mgp−/− | Arterial calcification | Cartilage calcification, Short stature, Osteopenia, fractures | Increased arterial calcification in men, Keutel syndrome |
| SPP1 | Opn−/− | Enhancing calcification of the mgp−/− mice, Aortic fibrosis and vascular stiffening in LDLR−/− mice; | Hypermineralized, Resistant to ovariectomy-induced bone resorption | ||
| AHSG | Ahsg−/− | Diffused arterial calcification, Calcified intimal plaques upon vascular injury | Alopecia-mental retardation syndrome 1, Increased arterial calcification and stiffness in patients with atherosclerosis, diabetes mellitus and CKD | ||
| Impaired phosphate and pyrophosphate homeostasis (ATP metabolism) | ENPP1 | ttw/ttw (tiptoe-walking mouse) | Aortic calcification in early life | Articular cartilage calcification, hyperostosis | Generalized arterial calcification of infancy 1, Increased aortic arch calcification in patients with type II diabetes, Cole disease, Autosomal recessive hypophosphatemic rickets 2 |
| enpp1−/− | Arterial calcification | Spine and peripheral joint fusion | |||
| ANKH | ank/ank | Arterial calcification | Arthritis, bone outgrowths, joint destruction | Craniometaphysial dysplasia, Chondrocalcinosis-2 | |
| NT5E | e-5’NT/ CD73−/− | Prothrombotic and proinflammatory phenotype of the vasculature | Arterial calcification due to deficiency of CD73 (ACDC); Calcification of joints and arteries | ||
| ABCC6 | abcc6−/− | Myocardial calcification, Arterial and retinal calcification | Generalized Arterial Calcification of Infancy 2, Pseudoxanthoma Elasticum | ||
MGP is a small secreted matrix protein, a member of the vitamin K-dependent protein family containing γ-carboxylated glutamate residues, and binds to calcium and calcified matrices50, 51. In humans, MGP SNPs with defective MGP function are linked to arterial calcification and mutations in the gene encoding the human MGP cause Keutel syndrome52–54. Consistently, genetic ablation of the mgp gene in mice induces severe ectopic calcification in the arteries and cartilage55. Additionally, inhibition of vitamin K metabolism leads to under carboxylation of MGP and subsequent medial calcification of the arterial vessel wall56, further indicating the importance of MGP production and activity in inhibiting vascular calcification. Another matrix protein, OPN, has been shown to inhibit deposition of hydroxyapatite crystals and calcification of VSMC similarly to PPi57,58. Opn deficiency does not cause calcification in mice, but increases arterial medial calcification in the Mgp−/− mice59 and vascular stiffening in LDLR−/− mice 60, suggesting an inducible role of OPN in inhibiting vascular calcification and stiffening.
The circulating calcium binding protein alpha2-Heremans-Schmid glycoprotein (AHSG, Fetuin-A) has been reported to be a potent circulating calcification inhibitor. Fetuin-A is a serum glycoprotein produced in the liver, and is capable of binding ~100 Ca2+ ions per molecule. It binds to clusters of amorphous calcium phosphate to form primary calciprotein particles, which maintain calcium phosphate in solution and prevent it from precipitating61, 62. Fetuin-A polymorphismswith decreased serum fetuin-A levels in humans63 are associated with increased arterial calcification and stiffness in patients with atherosclerosis, diabetes mellitus and CKD64–66. Consistently, mice deficient in fetuin-A develop heavy and diffused soft tissue calcifications throughout the whole body, as well as in calcified intimal plaques upon vascular injury67, supporting the critical role of fetuin-A as a major systemic inhibitor of vascular calcification.
The importance of phosphate and pyrophosphate metabolism in regulating vascular calcification is bolstered by the identification of several key regulators in the ATP metabolism network (Figure 2). ENPP1 is the ectoenzyme that hydrolyzes ATP at the cell membrane into AMP and pyrophosphate (PPi), and the resulting PPi is subsequently transported to the extracellular matrix via the membrane transporter, ANK68, 69. As PPi is the major inhibitor for hydroxyapatite formation and matrix mineralization, the ENPP1 deficiency is associated with generalized arterial calcification of infancy (GACI) in humans70, a genetic defect with increased arterial calcification, particularly in patients with atherosclerosis, diabetes mellitus and CKD70–72. Mice deficient in enpp1 have reduced pyrophosphate level and increased aortic calcification73. Similarly, mice with an ank mutation exhibit some defect in extracellular pyrophosphate and develop medial arterial calcification69. Treatment of pyrophosphate analogs, bisphosphonates, have decreased arterial calcification and improved the overall survival of patients with GACI due to ENPP1 mutations74. Accordingly, deficiency of either ENPP1 or ANK results in decreased PPi in the extracellular matrix and increased formation of hydroxyapatite crystals and matrix calcification, supporting the inhibitory role of both ENPP1 and ANK in vascular calcification.
Figure 2.

Genetic and cellular determinants that regulate pyrophosphate and phosphate homeostasis and the formation of hydroxyapatite crystals in the ECM. The key enzymes (ENPP1, CD73 and ALP) and transporters (ANK and ABCC6) that mediate the ATP metabolic pathway and pyrophosphate catalysis are detailed. The functions of matrix vesicle (MV) that carry and transport calcium/phosphate (Ca++/Pi) and matrix proteins to ECM for the formation of hydroxyapatite crystals and the secretion factors that are important for calcium binding and inhibition of the formation of hydroxyapatite crystals (MGP, OPN and Fetuin-A) are highlighted.
Another enzyme in the ATP metabolism pathway, CD73 ectoenzyme encoded by the ecto-5′-nucleotidase gene NT5E, has also been linked to vascular calcification75. CD73 is a cell membrane protein that catalyzes the generation of extracellular adenosine and inorganic phosphate (Pi) from AMP, which acts directly downstream of extracellular ATP degradation by ENPP1. CD73 deficiency in mice promotes thrombosis and inflammatory response in the vasculature76. NT5E mutations in human cause arterial calcification due to deficiency of CD73 (ACDC). NT5E mutations increase accumulation of calcium phosphate crystals. This is likely due to decreased adenosine-enhanced activity of tissue non-specific alkaline phosphatase (TNAP), since adenosine supplementation reverses the increased TNAP activity in CD73-deficient cells75.
In addition, deficiency in ABCC6, a gene encoding a member of the transmembrane ATP-binding cassette transport proteins, is associated with arterial calcification and pseudoxanthoma elasticum (PXE) 77,78. The Abcc6 deficiency in mice causes increased arterial calcification79. Because of the similarity in arterial lesions found in the PXE patients and ACDC patients, it was suggested that PXE and ACDC share common pathogenic pathways, and ABCC6 may serve as an adenosine transporter80. Overexpression of fetuin-A rescues vascular calcification in Abcc6−/− mice81, supporting the notion that lack of calcification inhibitors may contribute to ABCC6 deficiency-increased vascular calcification. Jansen et al identified that plasma PPi levels in the Abcc6−/− mice were significantly reduced compared to those in wild-type mice, and demonstrated the important role of ABCC6-mediated ATP secretion and generation of extracellular PPi82. By crossing Abcc6−/− mice with the Enpp1−/− and Nt5e−/− mice, Ziegler et al validated the metabolic crosstalk among ENPP1, CD73, and ABCC6 as well as the important role of ABCC6 in mediating extracellular ATP metabolism83. A bisphosphonate and an inhibitor for TNAP activity attenuates the development and progression of calcification in Abcc6−/− mice83, further validating the role of ABCC6-mediated phosphate metabolism in vascular calcification..
Altogether, monogenic deficiencies of ENPP1, CD73 or ABCC6 cause human diseases (GACI, ACDC, PXE) that share a common pathology of spontaneous, and premature onset of arterial calcification, in which the homeostasis of the phosphate/pyrophosphate metabolism is critical. In addition to pyrophosphate, matrix proteins that bind to calcium phosphate, such as MGP and OPN, as well as circulating calcium binding protein, fetuin-A, have also been recognized as potent inhibitors for vascular calcification. These genetic determinants highlight the importance of inhibition of matrix mineralization under physiological conditions in the vascular cells, as they act predominantly at the matrix mineralization stage by blocking accumulation of calcium phosphate, and formation and growth of hydroxyapatite crystals. The mechanistic insights gained from these rare genetic disorders have not only improved our understanding of the fundamental regulation of matrix mineralization in the development of vascular calcification; but also shed new lights on developing potential strategies to prevent and inhibit vascular calcification associated with specific genetic defects, and beyond.
Regulatory signals for vascular calcification
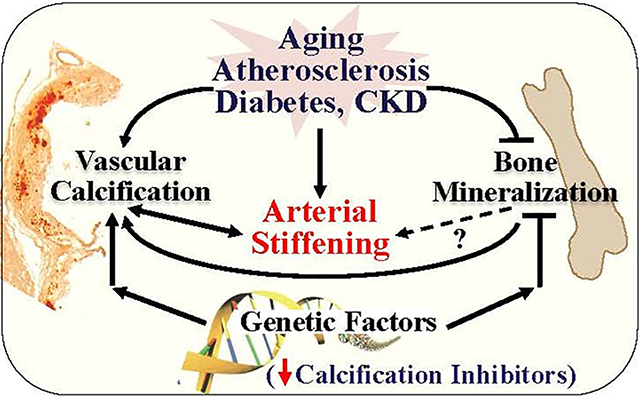
Despite the common osteogenic and mineralization process shared by bone and vascular cells, decreased bone mineralization (osteoporosis) occurs concurrently with increased vascular mineralization (vascular calcification) in human and mice associated with aging, atherosclerosis and CKD. These paradoxically opposite outcomes in bone and vascular niche indicate that distinct intrinsic signals driven by the tissue-specific microenvironmental cues govern the mineralization differently84. Mounting clinical and experimental findings in the past two decades have uncovered various important microenvironmental conditions as well as major signaling pathways and molecular regulators for both vascular calcification and bone mineralization. Elevation of oxidative stress, hyperglycemia and dysregulated calcium and phosphate homeostasis promote vascular calcification and inhibit bone mineralization. The regulation of vascular calcification by these pro-calcification conditions, and their activation of the major signaling pathways as well as their impacts on bone mineralization (Figure 3) are discussed below.
Figure 3. Differential responses to pathogenic inducers in vasculature and bone environment leads to opposite outcomes in mineralization of bone and vascular cells.

Aging and pathogenic conditions including atherosclerosis, diabetes and CKD are associated with systemic and tissue microenvironmental changes, such as increased oxidative stress, hyperglycemia, and impaired Ca++/Pi homeostasis. The difference in basal levels of TNAP, high in bone and low in VSMC, and their different responses to the pathologic conditions activate various intrinsic signaling cascades that eventually lead to opposite effects on the key osteogenic transcription factor Runx2, upregulating Runx2 in VSMC that promotes vascular calcification whereas inhibiting Runx2 in osteoblasts that decrease bone mineralization.
Increased oxidative stress
Increased oxidative stress and oxidative stress-induced signaling are a common feature in aging, cardiovascular and metabolic diseases, representing a major driving factor for vascular calcification and stiffness4, 49, 85, 86. The mechanisms underlying increased oxidative stress-induced vascular calcification in the atherosclerotic lesions are linked to oxidized lipids, vascular inflammation and matrix protein secretion49, 86–88. Elevated production of hydrogen peroxide (H2O2) production by residential vascular cells and infiltrated cells activates signaling pathways that lead to increased expression of inflammatory cytokines and adhesion molecules, which in turn promote the generation of H2O2 and increase oxidative stress in atherosclerotic lesions89. In vitro studies have further demonstrated a direct effect of H2O2 on promoting osteogenic differentiation and calcification of VSMC42 and calcifying bovine vascular cells85.
Increased oxidative stress is also manifested in patients with diabetes mellitus and animal models of diabetes90. High levels of blood sugar (hyperglycemia) in diabetes increases the accumulation of advanced glycation endproducts (AGEs), which bind to the receptor for advanced glycation (RAGE) that activates inflammatory signals and increases oxidative stress60, 91, 92. Increased AGEs/RAGE signaling promotes vascular calcification in vitro and in vivo92, 93. In CKD patients and animals, aortic and systemic increase in oxidative stress is a key feature of the uremic state94, which is highly associated with elevated vascular calcification95. High phosphate, a characteristic of CKD, induces production of mitochondria-mediated ROS and promotes VSMC calcification, which can be attenuated by antioxidants that limit high phosphate-induced ROS96. Inhibition of oxidative stress by antioxidants also slows downs the progression of vascular calcification in uremic CKD animals, demonstrating a critical role of uremia-induced oxidative stress in regulating vascular calcification in CKD94.
Of note, oxidative stress modulates osteogenic differentiation of vascular cells and bone cells in an opposite manner, inducing VSMC calcification while inhibiting differentiation and mineralization of osteoblasts 42, 85, which may explain the inverse correlation of bone and vascular mineralization mediated by the systemic oxidative stress that is often found with aging, cadioavascualr diseases and CKD conditions. Inhibition of oxidative stress via enhancing antioxidant systems and suppressing NADPH oxidase activity by statins promoted bone formation thus inhibiting osteoporosis in aged and ovariectomized rats97. In contrast, statins block vascular calcification in the LDLR−/− mice98. Consistently, in vitro studies have demonstrated that elevation of intracellular oxidative stress, by oxidized lipids and H2O2, inhibits osteogenic differentiation of bone cells but induces osteogenic differentiation and calcification of VSMC. These paradoxical mineralization outcomes are related to the opposite effects of oxidative stress on the expression of Runx2, a key osteogenic transcription factor42, 86, 99. As Runx2 is the essential regulator for osteogenic differentiation and mineralization in both bone cells and VSMC42, 88, 100, oxidative stress may distinctly activate or inhibit intrinsic signaling pathways in two different cell types that ultimately converge to Runx2 for differential regulation of osteogenic transition. Uncovering oxidative stress-induced intrinsic signals in both bone and vascular cells may help to understand the inverse pathological outcomes in skeletal and vascular mineralization, and develop precision therapy targeting vascular calcification.
Hyperglycemia and protein glycosylation
Similar to the effects of oxidative stress, hyperglycemia and protein glycosylation signals are also associated with opposite mineralization outcomes in the bone and vasculature. Increased oxidized lipids and glucose promotes non-enzymatic glycation and accumulation of AGEs. The AGEs and their receptor RAGE-regulated signaling axis contribute significantly to age-associated cardiovascular diseases, including increased vascular calcification in diabetes, atherosclerosis and CKD92, 101, 102. In addition to promoting oxidative stress, increased vascular accumulation of AGEs facilitates the binding of AGEs to RAGE and thus activates vascular inflammatory and multiple pro-osteogenic signaling pathways103. In cultured VSMC, AGEs induce the expression of RAGE, thus accelerating VSMC osteogenic differentiation and calcification104. AGEs form irreversible cross-links of matrix collagen and elastin, which results in stiffer ECM and arterial stiffening105. Non-enzymatic glycation of collagen renders their resistance to enzymatic degradation and their accumulation in the arterial wall106, 107. AGEs-modified elastin has also been linked to increased calcium binding and aortic media calcification105. RAGE mediates Pi-induced VSMC calcification in vitro 108 and vascular calcification in the enpp1–/– mice109. On the other hand, the AGEs/RAGE signaling pathways mediate diabetic osteopathy via regulating bone remodeling110. In cell cultures, AGEs inhibit osteogenic differentiation of osteoblasts by downregulating Runx2111. Consistently, RAGE knockout mice exhibit increased bone mass112. Overall, increased AGEs/RAGE signaling axis promotes vascular calcification but inhibits bone mineralization.
Chronic elevation of protein O-linked β-N-acetylglucosamine modification (O-GlcNAcylation), by increased glucose metabolism via the hexosamine biosynthesis pathway, has emerged as an important regulator for the pathogenesis of diabetes mellitus and diabetic cardiovascular complications90. O-GlcNAcylation is a dynamic posttranslational modification on serine or threonine residues of proteins by a single sugar moiety GlcNAc, which is different from the classic N-glycosylation and O-glycosylation90. O-GlcNAcylation is reversibly catalyzed by the sugar-adding O-GlcNAc transferase (OGT) and the sugar-removing O-GlcNAcase (OGA). A SNP in the MGEA5 gene encoding OGA is associated with type 2 diabetes in Mexican Americans113, supporting a potential role of upregulation of O-GlcNAcylation in the pathogenesis of diabetes mellitus. Diabetes-associated chronic hyperglycemia and oxidative stress both can increase protein O-GlcNAcylation114, which accelerates the development of cardiovascular complications in diabetes114, 115. We have recently demonstrated a novel role of increased protein O-GlcNAcylation in promoting VSMC calcification in vitro and vascular calcification in diabetes mice in vivo, via upregulation of Runx2116. The in vivo function of O-GlcNAcylation in regulating the formation of bone cells, including osteoblasts and osteoclasts, awaits further investigation. Increased O-GlcNAcylation inhibits BMP2-induced osteogenic differentiation of C2C12 cells, due to the suppressed Runx2 transcription activity117. However, another study has shown that increasing O-GlcNAcylation by OGA inhibition enhances osteoblastic differentiation of MC3T3-E1118. These in vitro studies need to be validated in vivo in animal models. The global OGT and OGA deletion mice are lethal119, 120, which precludes in vivo characterization of the role of O-GlcNAcylation in regulating bone and vascular mineralization. Therefore, developing animal models with tissue-specific alteration of O-GlcNAcylation levels is necessary to dissect and define a causative role of O-GlcNAcylation in regulating bone and vascular mineralization in vivo.
Dysregulation of calcium and phosphate homeostasis
Genetic studies in humans and animal models discussed above have clearly linked impaired calcium, phosphate and pyrophosphate homeostasis to vascular calcification. In patients with CKD, particularly dialysis patients, decreased fetuin-A and pyrophosphates as well as increased serum phosphate (hyperphosphatemia), parathyroid hormone (PTH, hyperparathyroidism) and fibroblast growth factor 23 (FGF23) all compromise homeostasis of calcium and phosphate and promote vascular calcification, leading to increased arterial stiffness and cardiovascular mortality in these impacted patients95, 121, 122.
Reduced concentrations of plasma pyrophosphate and fetuin-A, inhibitors of calcium phosphate, are documented in hemodialysis patients121, 122. Both pyrophosphate and fetuin-A directly inhibit inorganic phosphate-induced VSMC calcification, by blocking osteogenic differentiation of VSMC and formation of hydroxyapatite crystals84. Studies in CKD patients and human VSMC have demonstrated the uptake of the serum fetuin-A by VSMC and its secretion into ECM via matrix vesicles, thus inhibiting matrix vesicles-mediated nucleation of calcium phosphate123. Despite the similar action of pyrophosphate and fetuin-A on bone and VSMC calcification, the vascular-bone mineralization paradox in CKD patients and mouse models remains. As noted in the pyrophosphate deficient enpp1−/− mice, decreased bone density coexists with increased vascular calcification124. On the other hand, the pyrophosphate analogs bisphosphonates inhibit vascular calcification in CKD patients and animal models of CKD125, 126, suggesting that bone and VSMC may respond differently to the changes of extracellular PPi. A recent study investigating phosphate-induced calcification has revealed that cultured VSMC exhibit much higher total NPP activity (generating PPi) than osteoblasts. In contrast, osteoblasts possess over 100-fold higher TNAP activity (hydrolyzing PPi and generating Pi) than calcifying VSMC127. These differences in the basal PPi/Pi metabolism in the two cell types may account for different levels of reactions between VSMC and bone cells in response to changes of PPi/Pi ratios in their respective local microenvironment. Consistently, a TNAP inhibitor markedly inhibits bone formation in a concentration-dependent manner, but has no effect on VSMC calcification at the highest concentration used127. In addition, ENPP1 appears to be necessary for early osteoblast differentiation via a mechanism independent of its enzymatic activity and generation of extracellular inorganic PPi128, which may contribute to the bone defects observed in the enpp1−/− mice.
Increased serum phosphate and PTH concentrations in CKD are associated with increased arterial calcification as well as decreased bone mineralization in hemodialysis patients and animal models of CKD129, 130. Hyperparathyroidism induces release of serum phosphate and calcium from bone, the primary reservoir for calcium and phosphate that maintains systemic mineral homeostasis, thus resulting in bone mineral defects in CKD. Additionally, PTH has also been shown to induce the expression of RANKL, the master regulator for osteoclast formation, in osteoblasts; thus promoting osteoclast-mediated bone resorption that accelerates bone remodeling and high turnover of bone, the most common form of metabolic bone disorders in CKD131. Hyperparathyroidism-induced impaired phosphate excretion in CKD patients leads to hyperphosphatemia, which further stimulates PTH secretion132. Vitamin D analogues are used to reduce hyperparathyroidism, but the treatment has a side effect, often resulting in increased serum calcium and phosphate, and concurrently increased vascular calcification133. In animals, vitamin D treatment induces medial arterial calcification134.
Hyperphosphatemia is one of the major factors that promotes the development of vascular calcification in patients with CKD, and increases the risk of cardiovascular events and mortality132. Inorganic pyrophosphate, transported via sodium-dependent Pi cotransporter, Pit-1, upregulates Runx2 and induces osteogenic differentiation and calcification of VSMC135. Treatment of CKD patients with non-calcium-containing phosphate blocker delays progression of vascular calcification136; whereas calcium-containing phosphate blockers accelerate the development of vascular calcification in CKD patients137. Animal studies also demonstrate that both calcium containing and non-calcium-containing phosphate blockers exhibit similar effects on reducing phosphate levels in CKD mice, but produce different results. Only non-calcium-containing phosphate blocker reduces calcium-phosphate and PTH levels, and inhibits progression of vascular calcification138, suggesting an added role for calcium in promoting vascular calcification independent of phosphate levels.
The bone cells-derived FGF23 is an important regulator of PTH secretion, vitamin D metabolism and serum phosphate139. Progressive increase in FGF23 is considered as a biomarker for CKD. Animals with deficiency in either fgf23 or its co-receptor klotho exhibit increased vascular calcification and decreased bone mineral density concurrently, supporting an opposite regulatory role of FGF23/KLOTHO axis in bone and vascular mineralization140, 141. Hyperphosphatemia and increased serum level of vitamin D are demonstrated in both fgf23 and klotho deficient mice140, 141, indicating that impaired calcium phosphate metabolism may contribute to FGF23/KLOTHO-mediated vascular calcification. Direct effects of high phosphate, and high calcium with or without high phosphate, on promoting VSMC calcification have been linked to multiple integrated cellular events, including increases in secretion of calcium phosphate-loaded matrix vesicles, apoptosis, and augmentation of osteogenic differentiation of VSMC135, 142–144. Better understanding of how the different underlying mechanisms converge to modulate osteogenic differentiation and mineralization in bone and VSMC should advance our understanding of the bone-vascular paradox.
Pro-osteogenic signaling for VSMC calcification
In addition to regulating the essential matrix mineralization process in the ECM, systemic and local environmental changes differentially induce and activate intrinsic signaling pathways in vascular cells that promote osteogenic differentiation and expression of matrix proteins, leading to pathogenic calcification in the vasculature. Similar to osteoblast cells, upregulation of the osteogenic transcription factor, Runx2, is the hallmark for osteogenic differentiation of VSMC145. Over the past two decades, numerous important signaling pathways and integrated regulatory mechanisms have been identified that define the transition of VSMC into “bone-like” cells, featuring decreased expression of SMC markers and increased expression of bone markers, including the master osteogenic transcription factor Runx2146. A few key regulatory pathways are briefly highlighted below.
Upregulation of Runx2 is associated with calcification of multiple vascular cells, including calcifying vascular cells (CVC), VSMC, endothelial cells (via mesenchymal transition) and vascular progenitor cells42, 143, 147, 148. A wide variety of stimuli, such as oxidative-stress, high phosphate, calcium and uremia, increase expression and activation of Runx2 and downregulate VSMC markers, inducing VSMC osteogenic differentiation and calcification42, 44, 60, 143, 149. Using gain- and loss-of-function genetic approaches in VSMC and SMC-specific Runx2 ablation mice, we and others have determined an essential role of Runx2 in promoting vascular calcification and increasing production of matrix proteins. In contrast to the Runx2 null mice, which exhibit impaired early bone formation and neonatal lethality150, SMC-specific Runx2 deficiency does not affect normal mouse development and arterial function88, 151. However, the Runx2 deficiency attenuates VSMC calcification in vitro as well as intimal and medial arterial calcification in mouse models of atherosclerosis and CKD42, 88, 151. Accordingly, upregulation of Runx2 is a key determinant for VSMC calcification in vitro and vascular calcification in vivo.
Multiple pro-osteogenic signaling pathways are known to induce Runx2 upregulation and Runx2-mediated osteogenic function in VSMC, including the bone morphogenetic proteins (BMP), the msh homeobox 2 (Msx-2) as well as protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) signaling pathways. We have recently demonstrated that activation of p38 MAPK promotes VSMC calcification in vitro and vascular calcification in vivo152. The MAPK pathway increases phosphorylation of Runx2 on residues within the C-terminal proline-serine-threonine-rich domain, which may enhance binding of Runx2 to other co-factors, such as the coregulatory Smad proteins, thus enhancing osteoblast differentiation153, 154. In addition, phosphorylation of Runx2 may also initiate epigenetic changes in the chromatin structure that are necessary for chromatin decondensation and increased transcription155. In VSMC, p38 knockout inhibits Runx2 transactivity and calcification, supporting the role p38 MAPK in regulating Runx2 osteogenic function in VSMC152. Activation of the p38 MAPK signaling pathway by local stimuli, such as oxidative stress, extracellular matrix, mechanical loading, and BMP, may also interface with other signaling pathways such as Wnt 155, which further promotes upregulation of Runx2 and osteogenic differentiation of VSMC.
Activation of BMP signalling by lipid, glucose metabolism and inflammatory cytokines induces upregulation of Runx2 and osteogenic differentiation of VSMC156, 157. The important regulation of the BMP-Msx2-Wnt signaling axis in promoting vascular calcification has been demonstrated, particularly in the context of diabetes. Msx2 promotes vascular calcification via Wnt signaling158. SMC-specific ablation of Msx1 and Msx2 attenuates atherosclerotic calcification and aortic stiffness in diabetic mice60, 159. Similarly, mice deficient in the Msx2 gene manifest defects in skull ossification and a marked reduction in bone formation associated with decreases in osteoblast numbers160. Furthermore, both Runx2 and Msx2 repress myocardin-mediated VSMC differentiation and enhance osteogenic differentiation, via direct binding to the SMC-specific transcription factors, serum response factor and myocardin161, 162. Accordingly, upregulation of Runx2 and Msx2 in VSMC not only directly promotes osteogenic differentiation, but also inhibits the expression of VSMC markers genes that facilitates osteogenic differentiation and calcification.
Using cultured VSMC and animal models of diabetes and atherosclerosis, we have demonstrated that activation of AKT signaling pathway plays a major role in upregulating Runx2 and promoting VSMC calcification in vitro and vascular calcification in vivo42, 88, 95, 116, 163. Oxidative stress activates multiple signaling pathways, including the phosphatidyl inositol 3-kinase (PI3K)/AKT164. Inhibition of AKT activation blocks oxidative stress-induced upregulation of Runx2 and osteogenic differentiation of VSMC42. Furthermore, activation of AKT/FoxO1/3 signaling axis inhabits ubiquitination-mediated Runx2 degradation, leading to Runx2 upregulation and VSMC calcification42, 163. In osteoblasts, AKT signaling is also critical for osteoblast proliferation and Runx2-dependent differentiation165. Activation of the PI3K/AKT/FOXO1 signaling mediates 1α,25-Dihydroxyvitamin D3-induced osteoblast differentiation and bone formation166. However, in contrast to activating PI3K/AKT signaling in VSMC, increased oxidative stress inhibits AKT and Wnt signaling-mediated proliferation and differentiation of osteoblasts167; and increased activation of AKT blocks oxidative stress-induced osteoblast apoptosis168. Therefore, although activation of PI3K/AKT/FOXO1 signaling axis promotes proliferation, survival and osteogenic differentiation signals similarly in bone and VSMC cells; its opposite outcomes in in osteoblasts and VSMC may partially explain the differential mineralization seen in bone and VSMC.
In an animal model of diabetes, direct modification by O-GlcNAcylation is important for AKT activation, AKT-induced Runx2 upregulation as well as osteogenic differentiation and calcification of VSMC116. Chronic hyperglycemia increased O-GlcNAcylation in the vasculature of diabetic mice; and increased O-GlcNAcylation-mediated activation of AKT and upregulation of Runx2 promote vascular calcification and vascular stiffness in the diabetes mice116. O-GlcNAc modification of Runx2 has been reported in bone marrow mesenchymal stem cells169, but the functional consequence of such modification remains unclear. Further investigations to determine whether Runx2 is directly modified by O-GlcNAcylation in VSMC and how O-GlcNAcylation of Runx2 may contribute to its osteogenic function will provide new molecular insights into Runx2-regulated osteogenic differentiation and mineralization of bone and vascular cells.
CONCLUSIONS AND PROSPECTIVE
Arterial stiffening and vascular calcification are emerging as markers of vascular aging and cardiovascular disease risk. Studies in humans and animal models of atherosclerosis, diabetic mellitus and CKD, in which vascular calcification is highly prevalent, have revealed a few important genetic determinants, systemic and local microenvironmental conditions, and intrinsic molecular regulators for vascular calcification. We have begun to appreciate the similar osteogenic differentiation and mineralization process shared by bone and vascular cells; and their distinct, often completely opposite responses and outcomes to the changes in their microenvironment. As a result, “soft tissues become hard while hard tissue become soft” are common pathologies associated with aging and metabolic disorders.
This review covers the contribution of the major disease-promoting factors, including oxidative stress, hyperglycemia and impaired calcium phosphate homeostasis, and their regulation of bone and vascular cells that account for the vascular-bone mineralization paradox. As bone is the major reservoir for calcium and phosphate minerals, increased release of serum phosphate and calcium from bone, by FGF23 deficiency and hyperparathyroidism, is responsible for decreased bone mineral density and subsequently induced vascular calcification in CKD. On the other hand, the loss of calcium and phosphate inhibitors, such as fetuin-A and pyrophosphate, promotes matrix mineralization of the vascular cells while reducing bone mineralization. The differences in the basal PPi and Pi metabolism in bone and vascular cells and their response threshold to the changes of local PPi and Pi is also responsible for the inverse mineralization seen in bone and vascular cells. Therefore, managing local and systemic PPi and Pi levels may offer an effective strategy to mitigate the adverse mineral outcomes in both bone and vasculature.
It is encouraging that inhibition of oxidative stress and improving glucose and calcium phosphate metabolism are beneficial, at least partially, in reducing vascular calcification and increasing bone mineralization, notably in animal models. The cellular impact of these environmental stresses on VSMC is highlighted in this review, as VSMC are key to maintaining vascular homeostasis in all arterial system. The intrinsic stiffness and extracellular matrix remodeling of VSMC contribute to the osteogenic differentiation and calcification of VSMC. At the molecular level, upregulation of Runx2 is a major determinant for VSMC osteogenic differentiation and calcification. In atherosclerosis, diabetes mellitus and CKD, increased oxidized lipids, hyperglycemia and protein glycosylation, as well as high calcium and phosphate conditions all induce upregulation of Runx2, which is mediated via the activation of multiple signaling pathways, including BMP, Msx2/Wnt, MAPK and AKT signals. Of note, some of these signaling pathways are operating differently in bone cells. Many are inhibited by oxidative stress, hyperglycemia and high phosphate in osteoblasts, which leads to reduced bone mineralization.
To date, there is no effective and selective treatment for vascular calcification and arterial stiffness. Lessons learned from human and animal models have provided strong mechanistic explanations as to why some treatments, such as statins, ACE inhibitors, AGEs crosslink breaker, phosphate binders or pyrophosphate analogs, exhibit beneficial effects on vascular calcification and arterial stiffening10, 12–14. Some of these treatments have also demonstrated positive impact on bone mineral density. For instance, in vivo animal studies showed that statins increased bone formation and inhibited osteoporosis via inhibiting oxidative stress97. On the other hand, statins blocked vascular calcification98. Recent studies found that treatment of CKD rats with the AGE crosslink breaker alagebrium decreased total AGEs amounts in bone but not aortas. However, alagebrium reduced aorta expression of RAGE and inhibited oxidative stress and aorta calcification, whereas did not improve bone mechanics170, suggesting that specific matrix AGEs may regulate bone and arterial mineralization differently. The bone-resorbing bisphosphonates have generally shown beneficial effects on aortic and coronary calcification125, 126, 171, while the effects may differ based on the type, potency, dosage and administration route of bisphosphonates. In addition, combination therapy such as statin plus bisphosphonate is significantly more effective in reducing atherosclerotic aortic plaques in hypercholesterolemic patients than either monotherapy172, supporting the use of mechanism-driven combination therapies for arterial stiffness in different underlying conditions. We have recently found that dietary potassium supplements effectively inhibit vascular calcification and arterial stiffness in the ApoE−/− mice, which is associated with inhibition of Runx2 and VSMC calcification173. Accordingly, introducing dietary approaches to lifestyle may help to prevent the development of vascular calcification and stiffening, thus improving vascular health.
Evidence from human, animal and cellular studies has advanced our knowledge on the development of vascular calcification and its links to arterial stiffening. A more comprehensive and precise understanding of the regulation of VSMC osteogenic differentiation, eg. identifying VSMC subpopulations and progenitor cells, and the key cellular events and regulators that mediate VSMC de-differentiation, will help to define useful biomarkers and structural changes for early detection and prevention of vascular calcification. Furthermore, since bone and vascular cells share similar mineralization process, treatment strategies for vascular calcification should not ignore the potential impact on the bone, and vice versa. Further investigation to systemically compare vascular and bone cells in aging and vascular calcification-prevalent diseases may reveal cell type-specific regulatory network and/or disease-specific regulators. Uncovering the intrinsic cellular regulation of bone and vascular cells in responses to the systemic and local environmental changes should lead to identifying precise targets that are amendable for therapeutic interventions for vascular calcification, without affecting the skeletal system.
HIGHLIGHTS.
This review focuses on the contribution of vascular calcification to arterial stiffness. The important genetic factors, systemic and local microenvironmental signals, and underlying signaling pathways and regulatory molecules that promote vascular calcification-driven arterial stiffness are highlighted:
Monogenic genetic mutations in humans, featuring dysregulation of PPi/Pi metabolism and loss of inhibitors of calcium and phosphate, display vascular calcification and arterial stiffening,
Elevation of oxidative stress, hyperglycemia and dysregulated calcium and phosphate homeostasis enhance vascular calcification and arterial stiffening,
Activation of the major signaling pathways and their regulation on the master osteogenic transcription factor Runx2 in promotes osteogenic differentiation and calcification of VSMC,
Differential regulation of intrinsic signaling pathways by local and systemic factors in bone and vasculature contributes to the vascular-bone mineral disorder paradox,
Potential therapeutic strategies targeting molecular events that determines vascular calcification under different microenvironments.
ACKNOWLEDGEMENTS
Due to the scope and limitation, we apologize for not being able to include all the important work in the field.
SOURCES OF FUNDING
The original research of the authors has been supported by grants from the National Institutes of Health HL092215, HL136165, HL146103 and DK100847 as well as United States Department of Veterans Affairs BX003617 and BX004426 to YC.
ABBREVIATIONS
- ABCC6
ATP-binding cassette transporter subtype 6
- ACDC
arterial calcification due to deficiency of CD73
- AGEs
advanced glycation end products
- AKT
protein kinase B
- ALP
alkaline phosphatase
- ANK
ankylosis protein
- ApoE
apolipoprotein E
- ATP
adenosine triphosphate
- BMP-2
bone morphogenetic protein-2
- CKD
chronic kidney disease
- Col 1
type 1 collagen
- ECM
extracellular matrix
- ENPP1
ectonucleototide pyrophosphatase/phosphodiesterase-1
- FGF23
fibroblast growth factor 23
- GACI
generalized arterial calcification of infancy
- GWAS
genome-wide association studies
- H2O2
hydrogen peroxide
- JNK
c-Jun N-terminal kinases
- LDL
low-density lipoprotein
- MAPK
mitogen-activated protein kinases
- MGP
matrix Gla protein
- MMP
matrix metalloproteinases
- MVs
matrix vesicles
- NADPH
nicotinamide adenine dinucleotide phosphate
- NPP
nucleototide pyrophosphatase/phosphodiesterase-1
- O-GlcNAcylation
O-linked β-N-acetylglucosamine modification
- OPN
osteopontin
- OxLDL
oxidized low-density lipoprotein
- Pi
inorganic phosphate
- PI3K
phosphatidyl inositol 3-kinase
- PTH
parathyroid hormone
- PWV
pulse wave velocity
- PXE
pseudoxanthoma elasticum
- RAGE
Receptor for advanced glycation end products
- RANKL
receptor activator of nuclear factor kappa-B ligand
- ROS
reactive oxygen species
- Runx2
Runt-related transcription factor 2
- SMA
smooth muscle α-actin
- SMMHC
smooth muscle cell myosin heavy chain
- SNP
single nucleotide polymorphism
- TNAP
tissue non-specific alkaline phosphatase
- VSMC
vascular smooth muscle cells
Footnotes
DISCLOSURES
The authors have no potential conflicts of interests to disclose.
REFERENCES
- 1.Benetos A, Waeber B, Izzo J, Mitchell G, Resnick L, Asmar R, Safar M. Influence of age, risk factors, and cardiovascular and renal disease on arterial stiffness: Clinical applications. American journal of hypertension. 2002;15:1101–1108 [DOI] [PubMed] [Google Scholar]
- 2.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA, Seta F. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62:1105–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Popele NM, Grobbee DE, Bots ML, Asmar R, Topouchian J, Reneman RS, Hoeks AP, van der Kuip DA, Hofman A, Witteman JC. Association between arterial stiffness and atherosclerosis: The rotterdam study. Stroke. 2001;32:454–460 [DOI] [PubMed] [Google Scholar]
- 4.Lakatta EG. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises. 2003;107:490–497 [DOI] [PubMed] [Google Scholar]
- 5.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: Cause and logic of therapy. Hypertension. 2005;46:200–204 [DOI] [PubMed] [Google Scholar]
- 6.Fesler P, Safar ME, du Cailar G, Ribstein J, Mimran A. Pulse pressure is an independent determinant of renal function decline during treatment of essential hypertension. J Hypertens. 2007;25:1915–1920 [DOI] [PubMed] [Google Scholar]
- 7.Ben-Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen CH, Cruickshank JK, Hwang SJ, Lakatta EG, Laurent S, Maldonado J, Mitchell GF, Najjar SS, Newman AB, Ohishi M, Pannier B, Pereira T, Vasan RS, Shokawa T, Sutton-Tyrell K, Verbeke F, Wang KL, Webb DJ, Willum Hansen T, Zoungas S, McEniery CM, Cockcroft JR, Wilkinson IB. Aortic pulse wave velocity improves cardiovascular event prediction: An individual participant meta-analysis of prospective observational data from 17,635 subjects. Journal of the American College of Cardiology. 2014;63:636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–670 [DOI] [PubMed] [Google Scholar]
- 9.Ford ML, Tomlinson LA, Smith ER, Rajkumar C, Holt SG. Fetuin-a is an independent determinant of change of aortic stiffness over 1 year in non-diabetic patients with ckd stages 3 and 4. Nephrol Dial Transplant. 2010;25:1853–1858 [DOI] [PubMed] [Google Scholar]
- 10.Van Bortel LM, Struijker-Boudier HA, Safar ME. Pulse pressure, arterial stiffness, and drug treatment of hypertension. Hypertension. 2001;38:914–921 [DOI] [PubMed] [Google Scholar]
- 11.Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: Jacc state-of-the-art review. Journal of the American College of Cardiology. 2019;74:1237–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, Lakatta EG. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104:1464–1470 [DOI] [PubMed] [Google Scholar]
- 13.Dudenbostel T, Glasser SP. Effects of antihypertensive drugs on arterial stiffness. Cardiol Rev. 2012;20:259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrier KE, Muhlmann MH, Baguet JP, Cameron JD, Jennings GL, Dart AM, Kingwell BA. Intensive cholesterol reduction lowers blood pressure and large artery stiffness in isolated systolic hypertension. Journal of the American College of Cardiology. 2002;39:1020–1025 [DOI] [PubMed] [Google Scholar]
- 15.Tsamis A, Krawiec JT, Vorp DA. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: A review. Journal of the Royal Society, Interface. 2013;10:20121004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cocciolone AJ, Hawes JZ, Staiculescu MC, Johnson EO, Murshed M, Wagenseil JE. Elastin, arterial mechanics, and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018;315:H189–h205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duca L, Blaise S, Romier B, Laffargue M, Gayral S, El Btaouri H, Kawecki C, Guillot A, Martiny L, Debelle L, Maurice P. Matrix ageing and vascular impacts: Focus on elastin fragmentation. Cardiovascular research. 2016;110:298–308 [DOI] [PubMed] [Google Scholar]
- 18.Kwon GP, Schroeder JL, Amar MJ, Remaley AT, Balaban RS. Contribution of macromolecular structure to the retention of low-density lipoprotein at arterial branch points. Circulation. 2008;117:2919–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, Pure E, Phillips MC, Lund-Katz S, Janmey PA, Assoian RK. Cardiovascular protection by apoe and apoe-hdl linked to suppression of ecm gene expression and arterial stiffening. Cell reports. 2012;2:1259–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: Vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Back M, Gasser TC, Michel JB, Caligiuri G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovascular research. 2013;99:232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L, Zhang J, Monticone RE, Telljohann R, Wu J, Wang M, Lakatta EG. Calpain-1 regulation of matrix metalloproteinase 2 activity in vascular smooth muscle cells facilitates age-associated aortic wall calcification and fibrosis. Hypertension. 2012;60:1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payne RA, Wilkinson IB, Webb DJ. Arterial stiffness and hypertension: Emerging concepts. Hypertension. 2010;55:9–14 [DOI] [PubMed] [Google Scholar]
- 24.Lacolley P, Regnault V, Avolio AP. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovascular research. 2018;114:513–528 [DOI] [PubMed] [Google Scholar]
- 25.Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, St Hilaire C, Shanahan C. Medial vascular calcification revisited: Review and perspectives. European heart journal. 2014;35:1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740 [DOI] [PubMed] [Google Scholar]
- 27.Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with monckeberg’s sclerosis: Evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176 [DOI] [PubMed] [Google Scholar]
- 28.Libby P, Sukhova G, Lee RT, Galis ZS. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J. Cardiovasc. Pharmacol 1995;25 Suppl 2:S9–12.:S9-12 [DOI] [PubMed] [Google Scholar]
- 29.Demer LL, Tintut Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation. 2008;117:2938–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blumenthal HT, Lansing AI, Wheeler PA. Calcification of the media of the human aorta and its relation to intimal arteriosclerosis, ageing and disease. Am J Pathol. 1944;20:665–687 [PMC free article] [PubMed] [Google Scholar]
- 31.Raffield LM, Cox AJ, Criqui MH, Hsu FC, Terry JG, Xu J, Freedman BI, Carr JJ, Bowden DW. Associations of coronary artery calcified plaque density with mortality in type 2 diabetes: The diabetes heart study. Cardiovasc Diabetol. 2018;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees B, Schurgers L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2019;39:1351–1368 [DOI] [PubMed] [Google Scholar]
- 33.Buijs RV, Willems TP, Tio RA, Boersma HH, Tielliu IF, Slart RH, Zeebregts CJ. Calcification as a risk factor for rupture of abdominal aortic aneurysm. European journal of vascular and endovascular surgery : the official journal of the European Society for Vascular Surgery. 2013;46:542–548 [DOI] [PubMed] [Google Scholar]
- 34.Paixao AR, Ayers CR, El Sabbagh A, Sanghavi M, Berry JD, Rohatgi A, Kumbhani DJ, McGuire DK, Das SR, de Lemos JA, Khera A. Coronary artery calcium improves risk classification in younger populations. JACC. Cardiovascular imaging. 2015;8:1285–1293 [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann U, Massaro JM, D’Agostino RB, Kathiresan S, Fox CS, O’Donnell CJ. Cardiovascular event prediction and risk reclassification by coronary, aortic, and valvular calcification in the framingham heart study. Journal of the American Heart Association. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeboah J, Young R, McClelland RL, Delaney JC, Polonsky TS, Dawood FZ, Blaha MJ, Miedema MD, Sibley CT, Carr JJ, Burke GL, Goff DC Jr., Psaty BM, Greenland P, Herrington DM. Utility of nontraditional risk markers in atherosclerotic cardiovascular disease risk assessment. Journal of the American College of Cardiology. 2016;67:139–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khera A, Budoff MJ, O’Donnell CJ, Ayers CA, Locke J, de Lemos JA, Massaro JM, McClelland RL, Taylor A, Levine BD. Astronaut cardiovascular health and risk modification (astro-charm) coronary calcium atherosclerotic cardiovascular disease risk calculator. Circulation. 2018;138:1819–1827 [DOI] [PubMed] [Google Scholar]
- 38.Lian JB, Stein GS. Development of the osteoblast phenotype: Molecular mechanisms mediating osteoblast growth and differentiation. The Iowa orthopaedic journal. 1995;15:118–140 [PMC free article] [PubMed] [Google Scholar]
- 39.Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109:578–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154 [DOI] [PubMed] [Google Scholar]
- 41.Tintut Y, Parhami F, Bostrom K, Jackson SM, Demer LL. Camp stimulates osteoblast-like differentiation of calcifying vascular cells. Potential signaling pathway for vascular calcification. J Biol Chem. 1998;273:7547–7553 [DOI] [PubMed] [Google Scholar]
- 42.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor runx2 by akt signaling. J Biol Chem. 2008;283:15319–15327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleisch H, Bisaz S. Mechanism of calcification: Inhibitory role of pyrophosphate. Nature. 1962;195:911. [DOI] [PubMed] [Google Scholar]
- 44.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12 [DOI] [PubMed] [Google Scholar]
- 45.Goettsch C, Hutcheson JD, Aikawa E. Microrna in cardiovascular calcification: Focus on targets and extracellular vesicle delivery mechanisms. Circ Res. 2013;112:1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanimura A, McGregor DH, Anderson HC. Matrix vesicles in atherosclerotic calcification. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine (New York, N.Y.). 1983;172:173–177 [DOI] [PubMed] [Google Scholar]
- 47.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nature materials. 2016;15:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–1323 [DOI] [PubMed] [Google Scholar]
- 49.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-upregulated receptor activator of nuclear factor kappab ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cancela ML, Laize V, Conceicao N. Matrix gla protein and osteocalcin: From gene duplication to neofunctionalization. Archives of biochemistry and biophysics. 2014;561:56–63 [DOI] [PubMed] [Google Scholar]
- 51.Cranenburg EC, Vermeer C, Koos R, Boumans ML, Hackeng TM, Bouwman FG, Kwaijtaal M, Brandenburg VM, Ketteler M, Schurgers LJ. The circulating inactive form of matrix gla protein (ucmgp) as a biomarker for cardiovascular calcification. Journal of vascular research. 2008;45:427–436 [DOI] [PubMed] [Google Scholar]
- 52.Herrmann SM, Whatling C, Brand E, Nicaud V, Gariepy J, Simon A, Evans A, Ruidavets JB, Arveiler D, Luc G, Tiret L, Henney A, Cambien F. Polymorphisms of the human matrix gla protein (mgp) gene, vascular calcification, and myocardial infarction. Arterioscler. Thromb. Vasc. Biol 2000;20:2386–2393 [DOI] [PubMed] [Google Scholar]
- 53.Crosier MD, Booth SL, Peter I, Dawson-Hughes B, Price PA, O’Donnell CJ, Hoffmann U, Williamson MK, Ordovas JM. Matrix gla protein polymorphisms are associated with coronary artery calcification in men. Journal of nutritional science and vitaminology. 2009;55:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munroe PB, Olgunturk RO, Fryns JP, Van Maldergem L, Ziereisen F, Yuksel B, Gardiner RM, Chung E. Mutations in the gene encoding the human matrix gla protein cause keutel syndrome. Nature genetics. 1999;21:142–144 [DOI] [PubMed] [Google Scholar]
- 55.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix gla protein. Nature. 1997;386:78–81 [DOI] [PubMed] [Google Scholar]
- 56.Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. 1998;18:1400–1407 [DOI] [PubMed] [Google Scholar]
- 57.Scatena M, Liaw L, Giachelli CM. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–2309 [DOI] [PubMed] [Google Scholar]
- 58.Wada T, McKee MD, Steitz S, Giachelli CM. Calcification of vascular smooth muscle cell cultures: Inhibition by osteopontin. Circ. Res 1999;84:166–178 [DOI] [PubMed] [Google Scholar]
- 59.Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, Giachelli CM. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. 2002;161:2035–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao JS, Sierra OL, Cohen R, Mecham RP, Kovacs A, Wang J, Distelhorst K, Behrmann A, Halstead LR, Towler DA. Vascular calcification and aortic fibrosis: A bifunctional role for osteopontin in diabetic arteriosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:1821–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heiss A, DuChesne A, Denecke B, Grotzinger J, Yamamoto K, Renne T, Jahnen-Dechent W. Structural basis of calcification inhibition by alpha 2-hs glycoprotein/fetuin-a. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278:13333–13341 [DOI] [PubMed] [Google Scholar]
- 62.Schinke T, Amendt C, Trindl A, Poschke O, Muller-Esterl W, Jahnen-Dechent W. The serum protein alpha2-hs glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J Biol Chem. 1996;271:20789–20796 [DOI] [PubMed] [Google Scholar]
- 63.Jung JY, Hwang YH, Lee H, Ro H, Lee H, Chung W, Chae DW, Joo KW, Ahn C, Oh KH. Association of ahsg gene polymorphisms and aortic stiffness in peritoneal dialysis patients. American journal of nephrology. 2010;31:510–517 [DOI] [PubMed] [Google Scholar]
- 64.Andersen G, Burgdorf KS, Sparso T, Borch-Johnsen K, Jorgensen T, Hansen T, Pedersen O. Ahsg tag single nucleotide polymorphisms associate with type 2 diabetes and dyslipidemia: Studies of metabolic traits in 7,683 white danish subjects. Diabetes. 2008;57:1427–1432 [DOI] [PubMed] [Google Scholar]
- 65.Lehtinen AB, Burdon KP, Lewis JP, Langefeld CD, Ziegler JT, Rich SS, Register TC, Carr JJ, Freedman BI, Bowden DW. Association of alpha2-heremans-schmid glycoprotein polymorphisms with subclinical atherosclerosis. The Journal of clinical endocrinology and metabolism. 2007;92:345–352 [DOI] [PubMed] [Google Scholar]
- 66.Stenvinkel P, Wang K, Qureshi AR, Axelsson J, Pecoits-Filho R, Gao P, Barany P, Lindholm B, Jogestrand T, Heimburger O, Holmes C, Schalling M, Nordfors L. Low fetuin-a levels are associated with cardiovascular death: Impact of variations in the gene encoding fetuin. Kidney Int. 2005;67:2383–2392 [DOI] [PubMed] [Google Scholar]
- 67.Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, Muller-Esterl W, Schinke T, Jahnen-Dechent W. The serum protein alpha 2-heremans-schmid glycoprotein/fetuin-a is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–c11 [DOI] [PubMed] [Google Scholar]
- 69.Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270 [DOI] [PubMed] [Google Scholar]
- 70.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P. Mutations in enpp1 are associated with ‘idiopathic’ infantile arterial calcification. Nature genetics. 2003;34:379–381 [DOI] [PubMed] [Google Scholar]
- 71.Lee JE, Choi YK, Seo HA, Jeon JH, Jeong JY, Moon SS, Kim JG, Kim BW, Kim SW, Min Y, Kim JY, Lee IK. Impact of enpp1 and mmp3 gene polymorphisms on aortic calcification in patients with type 2 diabetes in a korean population. Diabetes research and clinical practice. 2010;88:87–96 [DOI] [PubMed] [Google Scholar]
- 72.Eller P, Hochegger K, Feuchtner GM, Zitt E, Tancevski I, Ritsch A, Kronenberg F, Rosenkranz AR, Patsch JR, Mayer G. Impact of enpp1 genotype on arterial calcification in patients with end-stage renal failure. Nephrol Dial Transplant. 2008;23:321–327 [DOI] [PubMed] [Google Scholar]
- 73.Johnson K, Polewski M, van Etten D, Terkeltaub R. Chondrogenesis mediated by ppi depletion promotes spontaneous aortic calcification in npp1−/− mice. Arterioscler Thromb Vasc Biol. 2005;25:686–691 [DOI] [PubMed] [Google Scholar]
- 74.Rutsch F, Boyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW, Davies JH, Loirat C, Strom TM, Schnabel D, Nurnberg P, Terkeltaub R, Group GS. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. Nt5e mutations and arterial calcifications. N Engl J Med. 2011;364:432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sevigny J, Gear A, Weber AA, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H, Godecke A, Schrader J. Targeted disruption of cd73/ecto-5’-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821 [DOI] [PubMed] [Google Scholar]
- 77.Meng H, Vera I, Che N, Wang X, Wang SS, Ingram-Drake L, Schadt EE, Drake TA, Lusis AJ. Identification of abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci U S A. 2007;104:4530–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier MF, Chassaing N, Roche O, Feenstra I, Loechner K, Deshpande C, Garber SJ, Chikarmane R, Steinmann B, Shahinyan T, Martorell L, Davies J, Smith WE, Kahler SG, McCulloch M, Wraige E, Loidi L, Hohne W, Martin L, Hadj-Rabia S, Terkeltaub R, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either enpp1 or abcc6. American journal of human genetics. 2012;90:25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, van Kuppevelt TH, Levelt CN, de Wolf A, Loves WJ, Scheper RJ, Peek R, Bergen AA. Disruption of abcc6 in the mouse: Novel insight in the pathogenesis of pseudoxanthoma elasticum. Human molecular genetics. 2005;14:1763–1773 [DOI] [PubMed] [Google Scholar]
- 80.Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in nt5e deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Molecular genetics and metabolism. 2011;103:44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang Q, Dibra F, Lee MD, Oldenburg R, Uitto J. Overexpression of fetuin-a counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (abcc6(−/−)). The Journal of investigative dermatology. 2010;130:1288–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jansen RS, Kucukosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, Bergen AA, Gorgels TG, Borst P, van de Wetering K. Abcc6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110:20206–20211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ziegler SG, Ferreira CR, MacFarlane EG, Riddle RC, Tomlinson RE, Chew EY, Martin L, Ma CT, Sergienko E, Pinkerton AB, Millan JL, Gahl WA, Dietz HC. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free radical biology & medicine. 2001;31:509–519 [DOI] [PubMed] [Google Scholar]
- 86.Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016;9:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sutra T, Morena M, Bargnoux AS, Caporiccio B, Canaud B, Cristol JP. Superoxide production: A procalcifying cell signalling event in osteoblastic differentiation of vascular smooth muscle cells exposed to calcification media. Free radical research. 2008;42:789–797 [DOI] [PubMed] [Google Scholar]
- 88.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park JG, Yoo JY, Jeong SJ, Choi JH, Lee MR, Lee MN, Hwa Lee J, Kim HC, Jo H, Yu DY, Kang SW, Rhee SG, Lee MH, Oh GT. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein e-deficient mice. Circ Res. 2011;109:739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Zhao X, Wu H. Metabolic stress and cardiovascular disease in diabetes mellitusthe role of protein o-glcnac modification. Arterioscler Thromb Vasc Biol. 2019:ATVBAHA119312192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brodeur MR, Bouvet C, Bouchard S, Moreau S, Leblond J, Deblois D, Moreau P. Reduction of advanced-glycation end products levels and inhibition of rage signaling decreases rat vascular calcification induced by diabetes. PloS one. 2014;9:e85922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tada Y, Yano S, Yamaguchi T, Okazaki K, Ogawa N, Morita M, Sugimoto T. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: Functional roles of nad(p)h-oxidase. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2013;45:267–272 [DOI] [PubMed] [Google Scholar]
- 94.Yamada S, Taniguchi M. [kidney and bone update : The 5-year history and future of ckd-mbd. Disorders of musculoskeletal system in ckd; bone fracture and periarticular calcification]. Clinical calcium. 2012;22:1000–1007 [PubMed] [Google Scholar]
- 95.Byon CH, Chen Y. Molecular mechanisms of vascular calcification in chronic kidney disease: The link between bone and the vasculature. Curr Osteoporos Rep. 2015;13:206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, Tang C, Wang X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79:1071–1079 [DOI] [PubMed] [Google Scholar]
- 97.Yin H, Shi ZG, Yu YS, Hu J, Wang R, Luan ZP, Guo DH. Protection against osteoporosis by statins is linked to a reduction of oxidative stress and restoration of nitric oxide formation in aged and ovariectomized rats. Eur J Pharmacol. 2012;674:200–206 [DOI] [PubMed] [Google Scholar]
- 98.Lin CP, Huang PH, Lai CF, Chen JW, Lin SJ, Chen JS. Correction: Simvastatin attenuates oxidative stress, nf-kappab activation, and artery calcification in ldlr−/− mice fed with high fat diet via down-regulation of tumor necrosis factor-alpha and tnf receptor 1. PloS one. 2016;11:e0148590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mody N, Tintut Y, Radcliff K, Demer LL. Vascular calcification and its relation to bone calcification: Possible underlying mechanisms. J. Nucl. Cardiol 2003;10:177–183 [DOI] [PubMed] [Google Scholar]
- 100.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754 [DOI] [PubMed] [Google Scholar]
- 101.Yamagishi SI. Role of advanced glycation endproduct (age)-receptor for advanced glycation endproduct (rage) axis in cardiovascular disease and its therapeutic intervention. Circulation journal : official journal of the Japanese Circulation Society. 2019;83:1822–1828 [DOI] [PubMed] [Google Scholar]
- 102.Senatus LM, Schmidt AM. The age-rage axis: Implications for age-associated arterial diseases. Frontiers in genetics. 2017;8:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wei Q, Ren X, Jiang Y, Jin H, Liu N, Li J. Advanced glycation end products accelerate rat vascular calcification through rage/oxidative stress. BMC cardiovascular disorders. 2013;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ren X, Shao H, Wei Q, Sun Z, Liu N. Advanced glycation end-products enhance calcification in vascular smooth muscle cells. The Journal of international medical research. 2009;37:847–854 [DOI] [PubMed] [Google Scholar]
- 105.Sell DR, Monnier VM. Molecular basis of arterial stiffening: Role of glycation - a mini-review. Gerontology. 2012;58:227–237 [DOI] [PubMed] [Google Scholar]
- 106.Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031 [DOI] [PubMed] [Google Scholar]
- 107.Konova E, Baydanoff S, Atanasova M, Velkova A. Age-related changes in the glycation of human aortic elastin. Experimental gerontology. 2004;39:249–254 [DOI] [PubMed] [Google Scholar]
- 108.Belmokhtar K, Ortillon J, Jaisson S, Massy ZA, Boulagnon Rombi C, Doué M, Maurice P, Fritz G, Gillery P, Schmidt AM, Rieu P, Touré F. Receptor for advanced glycation end products: A key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter pit-1 expression. Nephrology Dialysis Transplantation. 2019;34:2018–2030 [DOI] [PubMed] [Google Scholar]
- 109.Cecil DL, Terkeltaub RA. Arterial calcification is driven by rage in enpp1−/− mice. Journal of vascular research. 2011;48:227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Asadipooya K, Uy EM. Advanced glycation end products (ages), receptor for ages, diabetes, and bone: Review of the literature. Journal of the Endocrine Society. 2019;3:1799–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Franke S, Siggelkow H, Wolf G, Hein G. Advanced glycation endproducts influence the mrna expression of rage, rankl and various osteoblastic genes in human osteoblasts. Archives of physiology and biochemistry. 2007;113:154–161 [DOI] [PubMed] [Google Scholar]
- 112.Ding KH, Wang ZZ, Hamrick MW, Deng ZB, Zhou L, Kang B, Yan SL, She JX, Stern DM, Isales CM, Mi QS. Disordered osteoclast formation in rage-deficient mouse establishes an essential role for rage in diabetes related bone loss. Biochem Biophys Res Commun. 2006;340:1091–1097 [DOI] [PubMed] [Google Scholar]
- 113.Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, Hamlington J, Dyer TD, Arya R, Abboud H, Goring HH, Duggirala R, Blangero J, Konrad RJ, Stern MP. A single nucleotide polymorphism in mgea5 encoding o-glcnac-selective n-acetyl-beta-d glucosaminidase is associated with type 2 diabetes in mexican americans. Diabetes. 2005;54:1214–1221 [DOI] [PubMed] [Google Scholar]
- 114.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem. 2019;294:2211–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell’Italia LJ, Chatham JC, Wu H, Chen Y. Activation of akt by o-linked n-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. 2014;114:1094–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gu H, Song M, Boonanantanasarn K, Baek K, Woo KM, Ryoo HM, Baek JH. Conditions inducing excessive o-glcnacylation inhibit bmp2-induced osteogenic differentiation of c2c12 cells. International journal of molecular sciences. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Koyama T, Kamemura K. Global increase in o-linked n-acetylglucosamine modification promotes osteoblast differentiation. Experimental cell research. 2015;338:194–202 [DOI] [PubMed] [Google Scholar]
- 119.Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional knock-out reveals a requirement for o-linked n-acetylglucosaminase (o-glcnacase) in metabolic homeostasis. The Journal of Biological Chemistry. 2015;290:7097–7113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The o-glcnac transferase gene resides on the x chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97:5735–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lomashvili KA, Garg P, Narisawa S, Millan JL, O’Neill WC. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008;73:1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ketteler M, Westenfeld R, Schlieper G, Brandenburg V. Pathogenesis of vascular calcification in dialysis patients. Clin. Exp. Nephrol 2005;9:265–270 [DOI] [PubMed] [Google Scholar]
- 123.Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, Jahnen-Dechent W, Shanahan CM. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J. Am. Soc. Nephrol 2005;16:2920–2930 [DOI] [PubMed] [Google Scholar]
- 124.Johnson K, Goding J, Van Etten D, Sali A, Hu SI, Farley D, Krug H, Hessle L, Millan JL, Terkeltaub R. Linked deficiencies in extracellular pp(i) and osteopontin mediate pathologic calcification associated with defective pc-1 and ank expression. J Bone Miner Res. 2003;18:994–1004 [DOI] [PubMed] [Google Scholar]
- 125.Nitta K, Akiba T, Suzuki K, Uchida K, Watanabe R, Majima K, Aoki T, Nihei H. Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am J Kidney Dis. 2004;44:680–688 [PubMed] [Google Scholar]
- 126.Price PA, Roublick AM, Williamson MK. Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int. 2006;70:1577–1583 [DOI] [PubMed] [Google Scholar]
- 127.Patel JJ, Bourne LE, Davies BK, Arnett TR, MacRae VE, Wheeler-Jones CP, Orriss IR. Differing calcification processes in cultured vascular smooth muscle cells and osteoblasts. Experimental cell research. 2019;380:100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nam HK, Liu J, Li Y, Kragor A, Hatch NE. Ectonucleotide pyrophosphatase/phosphodiesterase-1 (enpp1) protein regulates osteoblast differentiation. J Biol Chem. 2011;286:39059–39071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roman-Garcia P, Carrillo-Lopez N, Fernandez-Martin JL, Naves-Diaz M, Ruiz-Torres MP, Cannata-Andia JB. High phosphorus diet induces vascular calcification, a related decrease in bone mass and changes in the aortic gene expression. Bone. 2010;46:121–128 [DOI] [PubMed] [Google Scholar]
- 130.Liu F, Fu P, Fan W, Gou R, Huang Y, Qiu H, Zhong H, Huang S. Involvement of parathyroid hormone-related protein in vascular calcification of chronic haemodialysis patients. Nephrology (Carlton). 2012;17:552–560 [DOI] [PubMed] [Google Scholar]
- 131.Huang JC, Sakata T, Pfleger LL, Bencsik M, Halloran BP, Bikle DD, Nissenson RA. Pth differentially regulates expression of rankl and opg. J. Bone Miner. Res 2004;19:235–244 [DOI] [PubMed] [Google Scholar]
- 132.Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ. Res 2004;95:560–567 [DOI] [PubMed] [Google Scholar]
- 133.Jono S, Nishizawa Y, Shioi A, Morii H. 1,25-dihydroxyvitamin d3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation. 1998;98:1302–1306 [DOI] [PubMed] [Google Scholar]
- 134.Norman PE, Powell JT. Vitamin d, shedding light on the development of disease in peripheral arteries. Arterioscler Thromb Vasc Biol. 2005;25:39–46 [DOI] [PubMed] [Google Scholar]
- 135.Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, pit-1, in vascular smooth muscle cell calcification. Circ. Res 2006;98:905–912 [DOI] [PubMed] [Google Scholar]
- 136.Block GA, Spiegel DM, Ehrlich J, Mehta R, Lindbergh J, Dreisbach A, Raggi P. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005;68:1815–1824 [DOI] [PubMed] [Google Scholar]
- 137.West SL, Swan VJ, Jamal SA. Effects of calcium on cardiovascular events in patients with kidney disease and in a healthy population. Clinical journal of the American Society of Nephrology : CJASN. 2010;5 Suppl 1:S41–47 [DOI] [PubMed] [Google Scholar]
- 138.Cozzolino M, Staniforth ME, Liapis H, Finch J, Burke SK, Dusso AS, Slatopolsky E. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003;64:1653–1661 [DOI] [PubMed] [Google Scholar]
- 139.Memon F, El-Abbadi M, Nakatani T, Taguchi T, Lanske B, Razzaque MS. Does fgf23-klotho activity influence vascular and soft tissue calcification through regulating mineral ion metabolism? Kidney Int. 2008;74:566–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin d-mediated process. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:720–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51 [DOI] [PubMed] [Google Scholar]
- 142.Chen NX, Duan D, O’Neill KD, Wolisi GO, Koczman JJ, Laclair R, Moe SM. The mechanisms of uremic serum-induced expression of bone matrix proteins in bovine vascular smooth muscle cells. Kidney Int. 2006;70:1046–1053 [DOI] [PubMed] [Google Scholar]
- 143.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–17 [DOI] [PubMed] [Google Scholar]
- 144.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in esrd. J. Am. Soc. Nephrol 2004;15:2857–2867 [DOI] [PubMed] [Google Scholar]
- 145.Karsenty G Minireview: Transcriptional control of osteoblast differentiation. Endocrinology. 2001;142:2731–2733 [DOI] [PubMed] [Google Scholar]
- 146.Towler DA, Shao JS, Cheng SL, Pingsterhaus JM, Loewy AP. Osteogenic regulation of vascular calcification. Ann. N. Y. Acad. Sci 2006;1068:327–333 [DOI] [PubMed] [Google Scholar]
- 147.Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687 [DOI] [PubMed] [Google Scholar]
- 148.Bostrom K, Demer LL. Regulatory mechanisms in vascular calcification. Crit Rev. Eukaryot. Gene Expr 2000;10:151–158 [PubMed] [Google Scholar]
- 149.Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol 2004;15:2959–2964 [DOI] [PubMed] [Google Scholar]
- 150.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764 [DOI] [PubMed] [Google Scholar]
- 151.Lin ME, Chen T, Leaf EM, Speer MY, Giachelli CM. Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. Am J Pathol. 2015;185:1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang Y, Sun Y, Chen J, Bradley WE, Dell’Italia LJ, Wu H, Chen Y. Akt-independent activation of p38 map kinase promotes vascular calcification. Redox Biol. 2018;16:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Afzal F, Pratap J, Ito K, Ito Y, Stein JL, van Wijnen AJ, Stein GS, Lian JB, Javed A. Smad function and intranuclear targeting share a runx2 motif required for osteogenic lineage induction and bmp2 responsive transcription. J. Cell Physiol 2005;204:63–72 [DOI] [PubMed] [Google Scholar]
- 154.Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E. Multiple signaling pathways converge on the cbfa1/runx2 transcription factor to regulate osteoblast differentiation. Connect. Tissue Res 2003;44 Suppl 1:109–16.:109–116 [PMC free article] [PubMed] [Google Scholar]
- 155.Franceschi RT, Ge C. Control of the osteoblast lineage by mitogen-activated protein kinase signaling. Curr Mol Biol Rep. 2017;3:122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hruska KA. Vascular smooth muscle cells in the pathogenesis of vascular calcification. Circ Res. 2009;104:710–711 [DOI] [PubMed] [Google Scholar]
- 157.Bostrom KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108:446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine wnt signals. J. Clin. Invest 2005;115:1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cheng SL, Behrmann A, Shao JS, Ramachandran B, Krchma K, Bello Arredondo Y, Kovacs A, Mead M, Maxson R, Towler DA. Targeted reduction of vascular msx1 and msx2 mitigates arteriosclerotic calcification and aortic stiffness in ldlr-deficient mice fed diabetogenic diets. Diabetes. 2014;63:4326–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Satokata I, Ma L, Ohshima H, Bei M, Woo I, Nishizawa K, Maeda T, Takano Y, Uchiyama M, Heaney S, Peters H, Tang Z, Maxson R, Maas R. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet 2000;24:391–395 [DOI] [PubMed] [Google Scholar]
- 161.Tanaka T, Sato H, Doi H, Yoshida CA, Shimizu T, Matsui H, Yamazaki M, Akiyama H, Kawai-Kowase K, Iso T, Komori T, Arai M, Kurabayashi M. Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells. Mol. Cell Biol 2008;28:1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hayashi K, Nakamura S, Nishida W, Sobue K. Bone morphogenetic protein-induced msx1 and msx2 inhibit myocardin-dependent smooth muscle gene transcription. Mol. Cell Biol 2006;26:9456–9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deng L, Huang L, Sun Y, Heath JM, Wu H, Chen Y. Inhibition of foxo1/3 promotes vascular calcification. Arterioscler Thromb Vasc Biol. 2015;35:175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ardanaz N, Pagano PJ. Hydrogen peroxide as a paracrine vascular mediator: Regulation and signaling leading to dysfunction. Exp. Biol. Med. (Maywood. ) 2006;231:237–251 [DOI] [PubMed] [Google Scholar]
- 165.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking akt1 and akt2. Genes Dev. 2003;17:1352–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xiong Y, Zhang Y, Xin N, Yuan Y, Zhang Q, Gong P, Wu Y. 1alpha,25-dihydroxyvitamin d3 promotes bone formation by promoting nuclear exclusion of the foxo1 transcription factor in diabetic mice. J Biol Chem. 2017;292:20270–20280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor alpha increase oxidative stress and suppress wnt protein signaling in osteoblasts. J Biol Chem. 2011;286:44326–44335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dai P, Mao Y, Sun X, Li X, Muhammad I, Gu W, Zhang D, Zhou Y, Ni Z, Ma J, Huang S. Attenuation of oxidative stress-induced osteoblast apoptosis by curcumin is associated with preservation of mitochondrial functions and increased akt-gsk3beta signaling. Cell Physiol Biochem. 2017;41:661–677 [DOI] [PubMed] [Google Scholar]
- 169.Nagel AK, Ball LE. O-glcnac modification of the runt-related transcription factor 2 (runx2) links osteogenesis and nutrient metabolism in bone marrow mesenchymal stem cells. Molecular & cellular proteomics : MCP. 2014;13:3381–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen NX, Srinivasan S, O’Neill K, Nickolas TL, Wallace JM, Allen MR, Metzger CE, Creecy A, Avin KG, Moe SM. Effect of advanced glycation end-products (age) lowering drug alt-711 on biochemical, vascular, and bone parameters in a rat model of ckd-mbd. J Bone Miner Res. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Caffarelli C, Montagnani A, Nuti R, Gonnelli S. Bisphosphonates, atherosclerosis and vascular calcification: Update and systematic review of clinical studies. Clin Interv Aging. 2017;12:1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kawahara T, Nishikawa M, Kawahara C, Inazu T, Sakai K, Suzuki G. Atorvastatin, etidronate, or both in patients at high risk for atherosclerotic aortic plaques: A randomized, controlled trial. Circulation. 2013;127:2327–2335 [DOI] [PubMed] [Google Scholar]
- 173.Sun Y, Byon CH, Yang Y, Bradley WE, Dell’Italia LJ, Sanders PW, Agarwal A, Wu H, Chen Y. Dietary potassium regulates vascular calcification and arterial stiffness. JCI Insight. 2017;2(19). e 94920. [DOI] [PMC free article] [PubMed] [Google Scholar]