

Dynein action requires a complex of the dynein motor, the dynactin activator, and an adaptor that confers cargo specificity. The functions of different dynein adaptors are poorly understood. Spriggs et al. find that BICD adaptors can remodel a viral cargo to promote infection, revealing an unexpected function for these cellular motor adaptors in virus membrane penetration and disassembly.
Abstract
During entry, viruses must navigate through the host endomembrane system, penetrate cellular membranes, and undergo capsid disassembly to reach an intracellular destination that supports infection. How these events are coordinated is unclear. Here, we reveal an unexpected function of a cellular motor adaptor that coordinates virus membrane penetration and disassembly. Polyomavirus SV40 traffics to the endoplasmic reticulum (ER) and penetrates a virus-induced structure in the ER membrane called “focus” to reach the cytosol, where it disassembles before nuclear entry to promote infection. We now demonstrate that the ER focus is constructed proximal to the Golgi-associated BICD2 and BICDR1 dynein motor adaptors; this juxtaposition enables the adaptors to directly bind to and disassemble SV40 upon arrival to the cytosol. Our findings demonstrate that positioning of the virus membrane penetration site couples two decisive infection events, cytosol arrival and disassembly, and suggest cargo remodeling as a novel function of dynein adaptors.
Introduction
During entry, viruses must undergo a series of decisive events such as trafficking along the complex endomembrane network, penetration of a host membrane, and capsid disassembly in order to reach the appropriate subcellular destination to cause infection (Helenius, 2018; Spriggs et al., 2019). How viruses exploit host proteins to coordinately accomplish these tasks is not fully understood. This study identifies cellular factors that couple membrane penetration and disassembly during entry of the nonenveloped polyomavirus (PyV) to promote infection.
PyVs are responsible for causing debilitating human diseases, especially in immunocompromised individuals (DeCaprio and Garcea, 2013). Prominent human PyVs include BK PyV, which induces hemorrhagic cystitis and nephropathy; JC PyV, which triggers progressive multifocal leukoencephalopathy; and Merkel cell PyV, which causes the often-fatal Merkel cell carcinoma. Simian virus 40 (SV40) is the archetype PyV, not only possessing structural and genetic similarities to human PyVs but also sharing the same infection pathway (Howley and Livingston, 2009). Not surprisingly, studies on SV40 entry have illuminated much of the molecular basis of human PyV infection. Structurally, SV40 consists of 72 pentamers of the VP1 major capsid protein that encases its DNA genome, with each pentamer harboring an internal hydrophobic protein VP2 or VP3 (VP2/3; Liddington et al., 1991; Chen et al., 1998). When properly assembled, the viral particle displays a diameter of ∼45 nm.
To infect cells, SV40 binds to the ganglioside GM1 receptor on the plasma membrane, is endocytosed, and is targeted to the endosome (Tsai et al., 2003; Anderson et al., 1996). The virus then traffics in a retrograde manner to reach the ER, where it penetrates the ER membrane to reach the cytosol (Kartenbeck et al., 1989; Chen et al., 2019). From the cytosol, SV40 mobilizes to the nucleus, where transcription and replication of the viral genome lead to lytic infection or cellular transformation (Clever et al., 1991; Nakanishi et al., 1996). How SV40 penetrates the ER membrane to reach the cytosol and then the nucleus remains largely enigmatic, although aspects of these processes are slowly being revealed.
According to the current model, upon reaching the ER lumen from the cell surface, disulfide bonds present in the SV40 capsid are reduced and isomerized by the ER-resident protein disulfide isomerase (PDI) family proteins (Schelhaas et al., 2007; Walczak and Tsai, 2011). These reactions induce viral conformational changes that expose the internal VP2/3, generating a hydrophobic particle that binds to and inserts into the ER membrane (Magnuson et al., 2005; Norkin et al., 2002). Despite these structural changes, SV40 remains relatively intact as it penetrates the ER membrane (Inoue and Tsai, 2011). Importantly, insertion of SV40 into the ER membrane triggers reorganization of select ER membrane proteins into discrete puncta called “foci” where the viral particle enters the cytosol (Dupzyk and Tsai, 2016). For instance, SV40 triggers transmembrane J proteins (B12, B14, and C18), as well as BAP31, to accumulate in the focus, where the cytosolic chaperone complex (composed of Hsc70, Hsp105, SGTA, and Bag2) is thought to be recruited (Bagchi et al., 2015; Geiger et al., 2011; Goodwin et al., 2011; Walczak et al., 2014). This cytosolic complex in turn extracts membrane-embedded SV40 into the cytosol to complete the membrane penetration event (Dupzyk et al., 2017; Dupzyk and Tsai, 2018; Ravindran et al., 2015; Walczak et al., 2014). Only upon reaching the cytosol does the virus experience pronounced disassembly, a step that enables a subviral particle harboring the genome to enter the nucleus to cause infection (Inoue and Tsai, 2011). Although we recently reported that the host cytoplasmic dynein-1 (hereafter referred as dynein) plays an important role in promoting SV40 disassembly (Ravindran et al., 2018), the precise mechanism by which SV40 is disassembled and whether this event is coupled to ER-to-cytosol membrane penetration are still unknown.
Processive movement by dynein requires the formation of a three-member protein complex composed of the dynein motor, dynactin activator, and a cargo adaptor that confers cargo specificity (Schlager et al., 2014a; McKenney et al., 2014; Zhang et al., 2017). In this study, we clarified the role of the dynein motor complex in virus disassembly and the specific contribution of cargo adaptors to this process. Specifically, we found that the bicaudal-D (BICD) dynein cargo adaptors BICD2 and BICDR1 interact directly with the virus and are required for virus disassembly in the cytosol. Unexpectedly, in vitro reconstitution of virus disassembly revealed that these adaptors are sufficient to disassemble the virus, independent of dynein and dynactin. These findings identify a novel function of the BICD adaptors during virus infection. Moreover, we show that the SV40-induced focus is constructed adjacent to a high concentration of Golgi-associated BICD2/BICDR1. This spatial localization allows these adaptors to directly disassemble the virus upon cytosol arrival and provides the first evidence of Golgi involvement in PyV infection. Our data thus demonstrate that positioning of the virus membrane penetration site couples ER-to-cytosol arrival and disassembly, two decisive steps required for successful infection, and raise the possibility that cargo remodeling is a novel activity of dynein cargo adaptors.
Results
The dynein cargo BICD adaptors support SV40 infection
We previously reported that a dynein-dependent activity promotes SV40 disassembly to support infection (Ravindran et al., 2018), prompting us to examine which dynein cargo adaptor might play a role in this step. Accordingly, we used an siRNA approach to individually knock down six established dynein adaptors, including BICD2, BICDR1, HOOK3, Ninein, Rab11-FIP3, and SPDL1, in simian CV-1 cells, which are used classically to study SV40 entry. Cells transfected with a scrambled siRNA served as the negative control. RT-PCR analysis confirmed that, in each case, the corresponding mRNA was depleted (Fig. 1 A).
Figure 1.

The dynein cargo BICD adaptors support SV40 infection. (A) CV-1 cells were transfected with 5 nM of the indicated siRNA for 48 h. RNA was isolated and RT-PCR used to assess transcript levels. GAPDH was used as a loading control. (B) CV-1 cells were transfected with 5 nM of the indicated siRNA and infected with SV40 (MOI ∼1). At 24 hours post infection (hpi), cells were fixed and stained for large T antigen (TAg). Data were normalized to the scrambled control. (C) CV-1 cells were transfected with either a scrambled control siRNA or siRNA against BICD2 or BICDR1, and BICD2 and BICDR1 protein levels were assessed by immunoblotting. Hsp90 was used as a loading control. (D) CV-1 cells were transfected with the scrambled control siRNA or siRNA against BICD2 for 24 h. Cells were then either transfected with the FLAG-HA-GFP control construct or mouse HA-BICD2 for an additional 24 h before infection with SV40 (MOI ∼1). At 24 hpi, cells were fixed and stained for TAg. Data were normalized to the scrambled control with FLAG-HA-GFP. (E) CV-1 cells were transfected with the scrambled control siRNA or siRNA against BICDR1 for 24 h. Cells were then either transfected with the FLAG-HA-GFP control construct or mouse HA-BICDR1 for an additional 24 h before infection with SV40 (MOI ∼1). At 24 hpi, cells were fixed and stained for TAg. Data were normalized to the scrambled control with FLAG-HA-GFP. (F) CV-1 cells were transfected with 5 nM of the indicated siRNA and infected with BK PyV (MOI ∼0.5). At 48 hpi, BK TAg levels were assessed by immunoblotting. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. *, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005; ****, P ≤ 0.0001. See also Fig. S1.
To monitor infection, we measured expression of the virally encoded large T antigen, which occurs only upon successful arrival of SV40 to the host nucleus. Strikingly, under the knockdown conditions, only depletion of BICD2 and BICDR1 markedly blocked SV40 nuclear arrival (81% and 70%, respectively), while their concurrent knockdown led to an even stronger block in virus infection (90%; Fig. 1 B). We also tested the two other mammalian BICD proteins, BICD1 and BICDR2, using the same knockdown approach (Fig. S1 A). We found that BICD1 (67% block), but not BICDR2 (26% block), is important for SV40 infection (Fig. S1 B). Note that in triple-knockdown experiments (BICD2, BICD1, and BICDR1), we were unable to achieve sufficient knockdown of all of the BICD proteins to see a further drop in infection, likely due to the cell’s inability to simultaneously uptake all three siRNAs.
Figure S1.

BICD1, but not BICDR2, supports SV40 infection (related to Fig. 1). (A) CV-1 cells were transfected with 5 nM of the indicated siRNA for 48 h. RNA was isolated and RT-PCR used to assess the transcript levels. GAPDH was used as a loading control. (B) CV-1 cells were transfected with 5 nM of the indicated siRNA and infected with SV40 (MOI ∼1). At 24 hpi, cells were fixed and stained for TAg. Data were normalized to the scrambled control. Values are the averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. **, P ≤ 0.005; ***, P ≤ 0.0005.
Although the BICD2 and BICDR1 siRNAs robustly decreased the protein level of their intended targets (Fig. 1 C), we nonetheless considered the possibility that the effect of these siRNAs on SV40 infection might be due to unintended off-target effects. To unambiguously establish the integrity of these siRNAs, cells transfected with scrambled or BICD2 siRNA were cotransfected with either a control FLAG-HA-GFP construct or mouse HA-BICD2 (whose sequence is sufficiently divergent from simian BICD2 to render it resistant to the siRNA). Only HA-expressing cells were scored for large T antigen expression. Using this approach, we found that expression of HA-BICD2 in cells depleted of BICD2 fully restored the block in SV40 infection caused by the BICD2 siRNA (Fig. 1 D). Likewise, expression of mouse HA-BICDR1 in BICDR1-depleted cells completely rescued the block in infection due to the BICDR1 siRNA (Fig. 1 E). These results demonstrate that the BICD2 and BICDR1 dynein cargo adaptors execute important functions during SV40 infection. Moreover, we found that knockdown of BICD2 and BICDR1 also impaired infection by the human BK PyV (Fig. 1 F), indicating that these adaptors are broadly important for PyV infection.
BICD2 and BICDR1 promote neither SV40 ER arrival from the cell surface nor cytosol arrival from the ER
To establish when BICD2 and BICDR1 execute their roles during PyV entry, we probed the effect of siRNA knockdown on ER and cytosol arrival of SV40. Upon reaching the ER lumen from the cell surface, PDI family proteins reduce and isomerize SV40 disulfide bonds to generate VP1 monomers, as well as disulfide-bonded dimers and trimers, which prepare the virus for subsequent ER-to-cytosol membrane penetration (Schelhaas et al., 2007; Walczak and Tsai, 2011). Hence, a convenient assay to examine arrival of SV40 to the ER is to assess the formation of VP1 monomers, dimers, and trimers using a nonreducing SDS-PAGE system (Walczak and Tsai, 2011; Inoue and Tsai, 2011). This analysis revealed that knockdown of BICD2 or BICDR1 had no effect on VP1 monomer, dimer, or trimer formation, compared with cells treated with brefeldin A (BFA), which impairs SV40 trafficking to the ER from the cell surface (Fig. 2 A, nonreducing; Norkin et al., 2002; Damm et al., 2005); the total levels of VP1 also were unchanged under knockdown conditions (Fig. 2 A, reducing). These findings suggest that BICD2 and BICDR1 are not acting to promote SV40 arrival to the ER from the plasma membrane.
Figure 2.

BICD2 and BICDR1 do not promote SV40 ER arrival from the cell surface or cytosol arrival from the ER. (A) CV-1 cells were transfected with 5 nM of the indicated siRNA, or treated with BFA, and infected with SV40 (MOI ∼2). At 13 hpi, cells were lysed in the presence of 10 mM NEM and VP1 levels assessed by either nonreducing or reducing SDS-PAGE followed by immunoblotting. Hsp90 was used as a loading control. (B) CV-1 cells were transfected with 5 nM of the indicated siRNA or treated with BFA and infected with SV40 (MOI ∼5). At 16 hpi, cells were subjected to the ER-to-cytosol membrane transport assay (see Materials and methods). VP1 levels were assessed by immunoblotting. Cytosolic Hsp90 and ER-resident PDI were used as markers for the cytosol and membrane fractions, respectively. (C) Relative VP1 band intensities from the cytosol fraction of B were determined using FIJI. Data were normalized to the scrambled control. (D) CV-1 cells were transfected with 5 nM of the indicated siRNA and stained for β-tubulin (green). Cells were counterstained with DAPI (blue). Scale bars, 10 µm. Graph represents the number of cells with radial MT structure normalized to the scrambled control. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. ****, P ≤ 0.0001.
We then assessed whether these two cargo adaptors support cytosol arrival of SV40 from the ER instead. To test this, we used a cell-based, semipermeabilization assay in which SV40-infected CV-1 cells transfected with the indicated siRNA (or treated with BFA) were harvested and incubated with a low concentration of digitonin to permeabilize the plasma membrane without damaging internal membranes. Cells were then subjected to centrifugation to produce two fractions: a supernatant fraction that harbors cytosolic proteins and virus that reaches the cytosol (“cytosol” fraction) and a pellet fraction that contains membranes, including the ER, as well as membrane-associated virus (“membrane” fraction). The integrity of the fractionation protocol can be verified by the release of the cytosolic marker Hsp90 to the cytosolic fraction and the pelleting of ER-resident PDI with the membrane fraction (Fig. 2 B). Using this approach, we found that knockdown of BICD2 or BICDR1 did not affect arrival of SV40 into the cytosol, in contrast to the BFA control (Fig. 2 B, top panel; quantified in Fig. 2 C). This indicates that BICD2 and BICDR1 do not promote SV40 cytosol arrival from the ER and are, therefore, necessary for a subsequent entry step. It is also important to note that SV40’s ability to successfully traffic from the plasma membrane to the cytosol under BICD2 or BICDR1 knockdown conditions indicates that general cellular integrity is largely maintained in the absence of these dynein cargo adaptors. Consistent with this, the overall microtubule (MT) organization, as assessed by β-tubulin staining, was unperturbed in cells depleted of BICD2 or BICDR1 (Fig. 2 D).
BICD2 and BICDR1 bind to and promote SV40 disassembly in the cytosol during entry
As BICD2 and BICDR1 support SV40 infection (Fig. 1) without impacting virus arrival to either the ER or the cytosol (Fig. 2), we reasoned that these cargo adaptors must execute a critical function in promoting cytosol-to-nuclear entry of SV40. Because a dynein-dependent activity disassembles SV40 in the cytosol (Ravindran et al., 2018), we hypothesized that BICD2 and BICDR1 might mediate the interaction between SV40 and the dynein motor. Immunoprecipitation of endogenous BICD2 from virus-infected cells pulled down both VP1 and the viral genome (Fig. 3 A), indicating that BICD2 associates with the infectious viral particle. Intriguingly, BICD2 preferentially binds to the VP3 minor capsid protein when compared with VP2 (Fig. 3 B). Similar to BICD2, BICDR1 pulled down VP1 as well as the viral genome (Fig. 3 C). Consistent with these findings, in vitro pull-down experiments showed that both purified BICD2 and BICDR1 (see Fig. 4 A) interact with native SV40 pretreated with the reductant DTT and the calcium chelator EGTA (Fig. 3 D), suggesting that these adaptors can directly bind to the virus during infection. (DTT serves to mimic disulfide bond reduction experienced by the virus in the ER, while EGTA removes the stabilizing interpentameric calcium ions that are released from the capsid when SV40 enters the cytosol from the ER.)
Figure 3.

BICD2 and BICDR1 bind to and promote SV40 disassembly in the cytosol during entry. (A) CV-1 cells were infected with SV40 (MOI ∼25) for 16 h. Endogenous BICD2 was immunoprecipitated (IP) from whole-cell extracts and the eluted samples subjected to SDS-PAGE followed by immunoblotting for VP1. DNA was isolated from the eluted sample and PCR performed to identify SV40 genomic DNA. (B) As in A, except immunoprecipitation by BICD2 was followed by immunoblotting for VP2/3. (C) As in A, except endogenous BICDR1 was immunoprecipitated. (D) In vitro binding assay of SV40 and full-length BICD2 and BICDR1. Purified His-tagged BICD2 and BICDR1 were separately linked to Nickel- Nitrilotriacetic acid (NTA) agarose resin. After incubation of these adaptors with SV40 (treated with DTT and EGTA), the immobilized proteins were eluted with imidazole and blotted with anti-VP1 antibody. The line indicates that lanes from the same immunoblot have been spliced together. (E) CV-1 cells were transfected with either a scrambled control siRNA, siRNA against BICD2, BICDR1 or RAB11-FIP3, and infected with SV40 (MOI∼5). At 16 hpi, the cytosolic fraction was isolated, layered on top of a 20% sucrose cushion, and centrifuged. Top and bottom fractions were collected and the presence of SV40 (VP1) assessed by immunoblotting. The levels of VP1 in the top fraction, corresponding to disassembled virus, and the bottom fraction, representing the intact virus, are quantified. (F) As in E, except the cytosolic fraction was layered over a discontinuous sucrose gradient (20–40% sucrose). Fractions were collected from the top of the gradient and the presence of SV40 (VP1) assessed by immunoblotting. The levels of VP1 in fractions 1–7, corresponding to disassembled virus, and fraction 8, representing the intact virus, are quantified. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. *, P ≤ 0.05; **, P ≤ 0.005.
Figure 4.

Reconstitution of BICD2-dependent disassembly of SV40 in vitro. (A) SDS-PAGE of purified dynein, dynactin, LIS1, NDEL, BICD2, and BICDR1. (B) Native SV40 (±DTT/EGTA) and SV40 isolated from the cytosol or ER of infected CV-1 cells were layered over a discontinuous sucrose gradient (20–40% sucrose) and centrifuged. Fractions were collected from the top of the gradient and the presence of SV40 (VP1) assessed by immunoblotting. The levels of VP1 in fractions 1–7, corresponding to disassembled virus, and fraction 8, representing the intact virus, are quantified. (C) Native SV40 was incubated with DTT/EGTA, the indicated purified proteins, ATP, and polymerized MTs where indicated. Samples were then layered over a discontinuous sucrose gradient and disassembly analyzed as in B. (D) Native SV40 was incubated with DTT/EGTA and BICD2 (without ATP or MTs). Disassembly was analyzed as in B. (E) Native SV40 was incubated with DTT/EGTA and increasing molar ratios of BICD2. Disassembly was assessed as in B. (F) △VP3 SV40 was incubated with DTT/EGTA and BICD2 as in D. Disassembly was analyzed as in B. (G) Native SV40 (without DTT/EGTA) was incubated with BICD2 and disassembly analyzed as in B. (H) Negative-stained images of DTT/EGTA-treated SV40 incubated with BICD2 at different molar ratios. (I) Quantification of negative staining. Graph represents the number of intact or partially disassembled viruses and formed VP1 pentamers per field of view. Intact and disassembled viruses were averaged from 3 × 3 µM fields of view, while pentamers are averaged from 0.3 × 0.3 µM views. (J) Native SV40, treated with or without DTT/EGTA, was incubated with BSA or BICD2 or denatured at 65°C. EtBr was added to each sample and EtBr incorporation measured by fluorimetry. Data were normalized to the BSA control. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. *, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005; ****, P ≤ 0.0001. See also Figs. S2 and S3.
To test whether BICD2 and BICDR1 are involved in dynein-dependent disassembly of SV40 during entry, we first used a previously established sucrose “cushion” sedimentation assay to measure viral disassembly (Inoue and Tsai, 2011). In this assay, cytosol-localized SV40 was isolated and layered over a 20% sucrose cushion. Following centrifugation, a top (disassembled virus) and bottom (intact virus) fraction was collected. In control cells, a significant portion of cytosol-localized SV40 (VP1) appeared in the top fraction (Fig. 3 E, first panel), indicating that a pool of the virus is disassembled, in agreement with our previous reports (Inoue and Tsai, 2011; Ravindran et al., 2018). Strikingly, depletion of either BICD2 or BICDR1 decreased virus disassembly when compared with control cells (Fig. 3 E, compare second and third panels to first panel) or RAB11-FIP3–depleted cells (fourth panel), which we previously found had no effect on virus infection (Fig. 1 B). To evaluate virus disassembly under these knockdown conditions with greater resolution, we also used a 20–40% discontinuous sucrose gradient approach to assess disassembly (Inoue and Tsai, 2011). In this case, following centrifugation, large intact virus will sediment to the bottom fraction (fraction 8), while smaller disassembled viral particles and pentamers appear in the lighter fractions (fractions 1–7). We found that while cytosol-localized SV40 appeared throughout the entire gradient in control and RAB11-FIP3 knockdown cells, loss of either BICD2 or BICDR1 again impaired virus disassembly (Fig. 3 F, compare first and fourth panels to second and third panels), consistent with the sucrose cushion sedimentation data. Together, these results indicate that SV40 disassembly during virus entry is dependent on BICD2 and BICDR1.
Reconstitution of BICD2-dependent disassembly of SV40 in vitro
We initially posited that BICD-dependent virus disassembly likely operates in the context of the entire dynein machinery, including dynein, dynactin, a cargo adaptor, and potentially additional cofactors such as LIS1 and Nuclear distribution protein nudE-like 1 (NDEL; Toropova et al., 2014; Cianfrocco et al., 2015). This machinery could, in principle, generate a force that drives the disassembly reaction via an energy-dependent, MT-based mechanism. To test this possibility, we developed an in vitro reconstitution strategy to assess whether these factors are sufficient to stimulate virus disassembly. Accordingly, we used an affinity purification approach to purify dynein, dynactin, LIS1, and NDEL, as well as BICD2 and BICDR1 (Fig. 4 A). These dynein components were then incubated with native SV40 pretreated with DTT/EGTA, and the reaction supplemented with polymerized MTs and ATP. Importantly, DTT/EGTA treatments did not artificially induce virus disassembly (Fig. 4 B).
When the control protein (BSA) was incubated with DTT/EGTA–treated SV40 and the samples subjected to sucrose gradient centrifugation, the virus sedimented to the heaviest fraction (Fig. 4 C, first panel), indicating that it remained intact. However, when we incubated the virus with dynein, dynactin, LIS1, NDEL, and BICD2, a pool of virus floated to the lighter sucrose fractions (Fig. 4 C, second panel), demonstrating that a portion of this virus was disassembled. Surprisingly, when MTs were removed from the reaction, virus disassembly still occurred (Fig. 4 C, third panel), suggesting that conventional MT-based dynein activity is not responsible for driving SV40 disassembly. As our cell-based assays indicate that the BICD adaptors are essential for SV40 disassembly, these findings led us to suspect that BICD2 might directly induce virus disassembly. Strikingly, we found that compared with the BSA control, BICD2 was sufficient to disassemble SV40 in vitro using a MT- and ATP-independent mechanism (Fig. 4 D). When treated with each of the other dynein components individually, no disassembly was observed (Fig. S2 A). BICD2 appears mainly in the lower fraction of the gradient, although a small portion can also be observed in the heaviest fraction (Fig. 4 D). Similar to BICD2, BICDR1 also induced virus disassembly (Fig. S2 B), suggesting that these adaptors can directly disassemble the virus during infection.
Figure S2.

Additional characterization of BICD-dependent binding and disassembly of SV40 in vitro (related to Fig. 4). (A) Native SV40 was incubated with DTT/EGTA and the indicated purified proteins. Samples were then layered over a discontinuous sucrose gradient (20–40% sucrose) and centrifuged. Fractions were collected from the top of the gradient and the presence of SV40 (VP1) assessed by immunoblotting. The levels of VP1 in fractions 1–7, corresponding to disassembled virus, and fraction 8, representing the intact virus, are quantified. (B) Native SV40 was incubated with DTT/EGTA and BICDR1 (without ATP or MTs). Disassembly was analyzed as in A. (C) Native SV40 was incubated with DTT/EGTA and increasing molar ratios of BICDR1. Disassembly was assessed as in A. (D) In vitro pull-down of BICD2 with VP1 in the presence or absence of DTT/EGTA. Values are the averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. **, P ≤ 0.005.
We next focused on clarifying the molecular mechanism of BICD2-dependent disassembly. To determine the minimum amount of BICD2 required to disassemble the virus, we performed the in vitro disassembly assay using varying concentrations of BICD2. Structurally, SV40 is composed of 360 monomers of VP1 arranged as 72 VP1 pentamers (Liddington et al., 1991). At a molar ratio of one viral particle per BICD2 molecule (1:1), we observed no virus disassembly compared with the BSA control (Fig. 4 E, second panel). Increasing the stoichiometry to one viral particle with 72 BICD2 molecules (1:72) triggered a low level of virus disassembly (Fig. 4 E, third panel). Only when a high molar ratio of viral particle to BICD2 (1:500) was used, as in Fig. 4 D, was efficient disassembly observed (Fig. 4 E, fourth panel), suggesting that at least one molecule of BICD2 must engage a VP1 monomer to stimulate virus disassembly. Similar results were seen for BICDR1, although some modest virus disassembly was observed at the lower molar ratio (Fig. S2 C).
Our data suggest that the BICD adaptors directly bind to SV40 during infection and, in fact, might preferentially associate with the VP3 minor capsid protein (Fig. 3). To determine whether VP3 is required for BICD2-dependent disassembly, we used a mutant SV40 that lacks VP3, but still contains VP2 (△VP3; Inoue and Tsai, 2011; Bagchi et al., 2015). Strikingly, BICD2 was unable to stimulate disassembly of △VP3 in vitro (Fig. 4 F). Because the internal VP3 is exposed through conformational changes in the ER, we performed the in vitro disassembly assay on native SV40 that had not been treated with DTT/EGTA. Similar to △VP3, BICD2 cannot induce efficient disassembly of native SV40 (Fig. 4 G), indicating that VP3 is indeed required for virus disassembly. In vitro pull-down experiments suggest that this is, at least in part, due to a greater affinity for BICD2 binding to SV40 in the presence of DTT/EGTA (Fig. S2 D).
To support these results, we used EM to independently assess BICD2-dependent disassembly of SV40. For this, DTT/EGTA–treated SV40 was incubated with BICD2; and the samples were subjected to negative-stain EM analysis. In agreement with our sucrose gradient centrifugation data, DTT/EGTA–treated virus (1:0) showed little disassembly and remained spherical and largely intact at ∼45 nm in diameter in the absence of BICD2 (Fig. 4 H, top left; quantified in Fig. 4 I), as did virus incubated with a low molar ratio of SV40 to BICD2 (1:10; Fig. 4 H, top right). However, when BICD2 was increased to a 1:50 ratio, partially disassembled and distorted virus can begin to be observed (Fig. 4 H, bottom left). With the highest molar ratio used (1:100), the number of observable intact virus decreased dramatically with a concurrent increase in partially disassembled particles and individual pentamers (Fig. 4 H, bottom right). A 1:100 ratio was used for this assay because higher concentrations of BICD2 obscured the appearance of viral particles and pentamers. Additional representative images are shown in Fig. S3. These findings support our in vitro sucrose gradient disassembly data, suggesting that the presence of BICD2 leads to concentration-dependent conformational changes in the viral capsid that results in the release of VP1 pentamers from the core particle.
Figure S3.

Supporting images for negative staining of SV40 in the presence of FL BICD2 at different molar ratios (related to Fig. 4). Negative staining of SV40 incubated with FL BICD2 at different molar ratios. The purified SV40 concentration was kept constant in all samples. All samples were treated at 37°C for 10 min in presence of DTT at 0.1 mM and EGTA at 0.05 mM (in Hepes 50 mM, NaCl 150 mM).
We previously reported that the SV40 genome is exposed in the cytosol during entry (Ravindran et al., 2018). We therefore asked whether virus disassembly by BICD2 is sufficient to expose the viral DNA. After incubating the DTT/EGTA–treated virus with BICD2 in vitro, we used ethidium bromide (EtBr) fluorescence as a probe for genome exposure. Compared with both native SV40 and DTT/EGTA–treated SV40 incubated with the control BSA protein, BICD2 disassembly of DTT/EGTA–treated SV40 significantly enhanced incorporation of EtBr, as measured by an increase in fluorescent emission (Fig. 4 J). This increase was comparable to genome exposure following heat-induced unfolding of SV40 (Fig. 4 J), indicating that BICD2-dependent disassembly of the SV40 VP1 coat protein is sufficient to unmask the virus genome.
Domain analysis of BICD2-dependent SV40 disassembly
Structurally, BICD2 harbors three coiled-coil (CC) domains, including the N-terminal CC1, which interacts with dynein and dynactin; the central CC2, which associates with kinesin-1; and the C-terminal CC3 region, which associates with the nuclear pore component RanBP2 or the small GTPase Rab6 (Splinter et al., 2010; Hoogenraad et al., 2001). We therefore generated and purified proteins corresponding to BICD2 CC1, CC2, and CC3, as well as truncated proteins containing CC1-2 or CC2-3 (Fig. 5 A). When CC1-CC2 was coincubated with CC3, or CC1 with CC2-3, so that together the full-length BICD2 was present, virus disassembly was readily observed (Fig. 5 B, second and third panels; quantified in bottom graph). Importantly, CC2-3 also was able to stimulate virus disassembly with almost the same efficiency as CC1 plus CC2-3 (Fig. 5 B, compare fourth to third panels). Although the classic cargo-binding domain CC3 alone was not sufficient to trigger virus disassembly (Fig. 5 B, fifth panel), CC2 did partially trigger disassembly (Fig. 5 B, sixth panel). Not surprisingly, CC1 was unable to disassemble the virus (Fig. 5 B, seventh panel). These findings indicate that CC2 and CC3 comprise the minimum domains within BICD2 essential to stimulate disassembly of SV40. Binding studies revealed that although CC3 has a greater binding affinity for the virus than CC2 and displays an affinity for the virus more similar to CC2-3 (Fig. 5 C), its canonical cargo-binding domain is not sufficient to trigger SV40 disassembly.
Figure 5.

Domain analysis of BICD2-dependent SV40 disassembly. (A) Coomassie stain of full-length (FL) and truncated BICD2 variants. (B) Native SV40 was incubated with DTT/EGTA and the indicated purified proteins before being layered over a discontinuous sucrose gradient (20–40% sucrose), and centrifuged. Fractions were collected from the top of the gradient and the presence of SV40 (VP1) assessed by immunoblotting. A schematic of the BICD2 domain structure used in the different experimental conditions is shown. The levels of VP1 in fractions 1–7, corresponding to disassembled virus, and fraction 8, representing the intact virus, are quantified in the graph below. (C) In vitro binding of SV40 and truncated BICD2 proteins. ZZ-tagged BICD2 proteins were linked to IgG Sepharose beads. After incubation of these truncated adaptors with SV40 (treated with DTT and EGTA), the immobilized proteins were eluted with TEV protease and blotted with anti-VP1 antibody. Coomassie staining was used to detect released BICD2 proteins. The levels of VP1 bound to the truncated adaptors are quantified. NC, negative control. Values are averages of the means (n = 3) ± SD.
The SV40-induced ER focus is constructed proximal to Golgi-associated BICD2
Our results thus far revealed that cytosol-localized SV40 is disassembled directly by a high concentration of the BICD dynein adaptors. We therefore asked if SV40 might be localized close to BICD2 in virus-infected cells. By confocal microscopy, we observed a concentrated region of BICD2 in a ring-like structure around VP2/3-exposed virus (Fig. 6 A, see inset). This phenomenon could be seen in the majority (84%) of cells with exposed VP2/3, and VP1 could also be detected within this BICD2 ring structure (Fig. S4 A, see inset). These findings indicate that VP2/3-exposed SV40 is proximal to a high concentration of BICD2 in infected cells.
Figure 6.

The SV40-induced ER focus is constructed proximal to Golgi-associated BICD2. (A) Confocal analysis of CV-1 cells that were infected with SV40 (MOI ∼5) for 16 h and stained with anti-VP2/3 (green) and anti-BICD2 (red) antibodies. Cells were counterstained with DAPI (blue). Percentage represents the number of VP2/3 foci surrounded by BICD2 out of total cells with VP2/3 foci. (B) Confocal analysis of uninfected or SV40-infected cells (MOI ∼5) stained with anti-BAP31 (green) and anti-BICD2 (red) antibodies. Cells were counterstained with DAPI (blue). Percentage represents the number of BAP31 foci surrounded by BICD2 out of total cells with virus-induced BAP31 foci. (C) Confocal analysis of control and BICD2-depleted CV-1 cells infected with SV40 (MOI ∼5). At 16 hpi, cells were stained with anti-VP2/3 (green) and anti-BAP31 (red) and counterstained with DAPI (blue). The graph represents the percentage of cells with BAP31+ foci normalized to the scrambled control. (D) As in B, except cells were stained with anti-γ-tubulin (white, arrows), anti-BICD2 (red), and anti-VP2/3 (green) antibodies. Cells were counterstained with DAPI (blue). (E) As in B, except cells were stained with anti-GM130 (green) and anti-BICD2 (red) antibodies. Cells were counterstained with DAPI (blue). Scale bars, 10 µm. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. See also Fig. S4.
Figure S4.

The SV40-induced ER focus is surrounded by BICD2 and BICDR1 (related to Fig. 6). (A) Confocal analysis of CV-1 cells that were infected with SV40 (MOI ∼5) for 16 h and stained with anti-VP1 (green) and anti-BICD2 (red) antibodies. Cells were counterstained with DAPI (blue). (B) Confocal analysis of uninfected and SV40-infected cells (MOI ∼5) stained with anti-VP2/3 (pink), anti-BAP31 (green), and anti-BICD2 (red) antibodies. Cells were counterstained with DAPI (blue). (C) Confocal analysis of control and BICDR1-depleted CV-1 cells infected with SV40 (MOI ∼5). At 16 hpi, cells were stained with anti-VP2/3 (green), anti-BAP31 (red), and counterstained with DAPI (blue). The graph represents the percentage of cells with BAP31+ foci normalized to the scrambled control. (D) As in A, except stained with anti-GM130 (green) and anti-BICDR1 (red) antibodies. Cells were counterstained with DAPI (blue). Scale bars, 10 µm. Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance.
The hydrophobic VP2 and VP3 proteins are exposed only upon arrival of the virus to the ER lumen, where this newly generated hydrophobic virus is targeted to a virus-induced structure at the ER membrane called “focus” that serves as the cytosol entry site for the virus (Gilbert et al., 2006; Inoue and Tsai, 2011; Geiger et al., 2011). This focus structure harbors select ER membrane proteins such as BAP31 that are recruited by the virus to facilitate SV40 ER-to-cytosol membrane transport (Geiger et al., 2011). Hence, we reasoned that the virus-induced BAP31 focus may also be proximal to the BICD2 ring structure. Indeed, confocal analysis revealed that the majority of BAP31 foci (76%) are formed near the concentrated BICD2 ring structure (Fig. 6 B, see inset, compare second to first row). Moreover, by performing four-color confocal microscopy, we confirmed that the virus-triggered BAP31 focus colocalizes with the VP2/3 focus, as expected (Geiger et al., 2011), and that this focus structure is also at the BICD2 ring (Fig. S4 B, see inset, second row). Because BICD2 also displays a concentrated ring-like pattern in uninfected cells (Fig. 6 B, see inset, first row), this localization pattern is not a virus-induced phenomenon.
Two unresolved questions in the field are how and why does the viral focus form at this particular site in the ER membrane. Given the proximity of BICD2 to the ER focus, we asked if BICD2 might act as a signal for viral focus formation in cells. Knockdown of BICD2 (Fig. 6 C) or BICDR1 (Fig. S4 C), however, did not affect foci formation in cells, again indicating that the role of these adaptors in SV40 infection occurs only after the virus has reached the cytosol. Additional characterization unveiled that the VP2/3 focus is constructed near, but not at, the MT organizing center (MTOC), because staining for γ-tubulin, a common MTOC marker, showed that the MTOC is localized adjacent to the VP2/3 focus (Fig. 6 D, see inset, second row). As the Golgi has been proposed to serve as an MTOC (Chabin-Brion et al., 2001), as well as to recruit BICD2 (Matanis et al., 2002; Hoogenraad et al., 2001), we asked if BICD2 colocalizes with the Golgi in these cells. In both uninfected and SV40-infected cells, we found colocalization of BICD2 with the Golgi marker GM130 (Fig. 6 E, see inset, first and second rows). Similarly, GM130 colocalized with BICDR1 in infected cells (Fig. S4 D, see inset). Together, these results indicate that the SV40-induced ER focus is constructed proximal to a concentrated region of the Golgi-associated BICD cargo adaptors.
BICD2 localization to the Golgi is crucial to promote SV40 disassembly and infection
The surprising finding that Golgi-associated BICD2 surrounds the virus-induced focus raises the possibility that SV40 penetrates the ER membrane proximal to the BICD disassembly machinery for efficient uncoating upon entering the cytosol. To test this, we asked if mislocalizing BICD2 away from the membrane penetration site would impair virus infection. To disrupt BICD2 localization, we used a previously established Golgi dispersion assay (Engelke et al., 2016), which we anticipated would concomitantly disperse BICD2.
In this assay, CV-1 cells are transfected with an active kinesin-1 motor construct that is targeted to the Golgi via the Golgi targeting sequence (GTS) of GMAP210. Thus, [RnKif5C(1–560)-mCit-GTS] specifically binds to and “pulls apart” the tightly packed Golgi complex to generate small Golgi-derived vesicles that are dispersed throughout the cytosol (Engelke et al., 2016). Compared with the mCitrine control (mCit), the characteristic Golgi-associated BICD2 signal was dramatically dispersed in cells expressing RnKif5C(1–560)-mCit-GTS (Fig. 7 A). Importantly, although expression of RnKif5C(1–560)-mCit-GTS did not reduce the endogenous level of BICD2 in cells (Fig. 7 B), it markedly blocked SV40 infection (Fig. 7 C). Dispersing BICD2 away from the ER membrane penetration site had no effect on either virus-induced BAP31 foci formation (Fig. 7 D) or cytosol arrival of the virus (Fig. 7 E). However, when BICD2 was dispersed by RnKif5C(1–560)-mCit-GTS, we observed a dramatic disruption in SV40 disassembly in the cytosol (Fig. 7 F). This disassembly assay was performed in CV-1–derived COS-7 cells, as they afford the high transfection efficiency required for the experiment; expression of RnKif5C(1–560)-mCit-GTS in these cells also dispersed BICD2 (Fig. S5).
Figure 7.

BICD2 localization to the Golgi is crucial to promote SV40 disassembly and infection. (A) CV-1 cells were transfected with either the mCitN1 control or RnKIF5C(1–560)-mCit-GTS construct for 24 h and stained for BICD2 (red/white). Cells were counterstained with DAPI (blue). (B) COS-7 cells were transfected with either the mCitN1 control or RnKIF5C(1–560)-mCit-GTS construct for 24 h. Cells were harvested and the BICD2 levels assessed by immunoblotting. Hsp90 was used as a loading control. Anti-GFP was used to detect mCit levels. The BICD2 band intensities in cells expressing mCitN1 or RnKIF5C(1–560)-mCit-GTS are quantified in graph below. (C) CV-1 cells were transfected with either the mCitN1 control or RnKIF5C(1–560)-mCit-GTS construct for 24 h and infected with SV40 (MOI∼1). At 24 hpi, cells were fixed and stained for TAg. Only mCit expressing cells were counted (green). (D) As in C, except cells were infected for 16 h and stained with anti-VP2/3 (red), anti-BAP31 (pink) and counterstained with DAPI (blue). Data on graph was normalized to the mCitN1 control. (E) COS-7 cells were transfected with either the mCitN1 control or RnKIF5C(1–560)-mCit-GTS construct for 24 h and then infected with SV40 (MOI∼5). At 12 hpi, the cytosolic fraction was isolated and VP1 levels assessed by immunoblotting. Cytosolic Hsp90 was used as a loading control. The VP1 band intensities are quantified in the graph below. (F) As in E, except at 12 hpi, the cytosolic fraction was layered on top of a 20% sucrose cushion and centrifuged. Top and bottom fractions were collected and the presence of SV40 (VP1) assessed by immunoblotting. The levels of VP1 in the top fraction, corresponding to disassembled virus, and the bottom fraction, representing the intact virus, are quantified. (G) CV-1 cells were transfected with either EGFP-Rab6A or EGFP-Rab6A T27N. Cells were stained with anti-GM130 (green), anti-BICD2, and counterstained with DAPI (blue). (H) CV-1 cells were transfected with either EGFP-Rab6A or EGFP-Rab6A T27N for 24 h and infected with SV40 (MOI∼1). At 24 hpi, cells were fixed and stained for TAg. Only EGFP expressing cells were counted (white). Values are averages of the means (n = 3) ± SD. A standard Student’s t test was used to determine statistical significance. **, P ≤ 0.005; ***, P ≤ 0.0005; ****, P ≤ 0.0001. Scale bars, 10 µm. See also Fig. S5.
Figure S5.
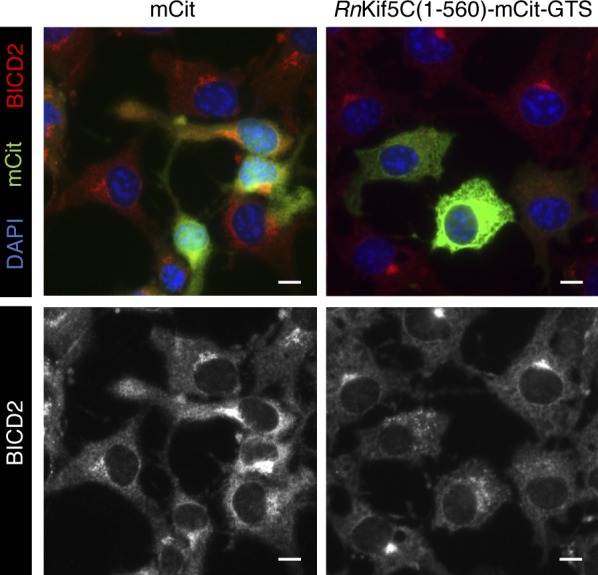
Golgi dispersion mislocalizes BICD2 in COS-7 cells (related to Fig. 7). COS-7 cells were transfected with either the mCitN1 control or RnKIF5C(1–560)-mCit-GTS construct for 24 h and stained for BICD2 (red/white). Cells were counterstained with DAPI (blue). Scale bars, 10 µm.
To confirm that the effect of Golgi dispersion on virus infection is due to the selective loss of BICD2 recruitment to this site and not a global disruption of other Golgi constituents, we used an independent method to disrupt BICD2 localization. To this end, we employed a dominant-negative approach to inactivate Rab6A, which is required for BICD2 targeting to the Golgi (Matanis et al., 2002). As previously reported, overexpression of EGFP-Rab6A T27N (but not wild-type EGFP-Rab6A) impaired BICD2 recruitment to the Golgi without perturbing the overall distribution of the Golgi, as evidenced by GM130 staining (Fig. 7 G). Under this condition, SV40 infection was blocked to a similar level as seen with RnKif5C(1–560)-mCit-GTS expression (Fig. 7 H). Collectively, these data indicate that localization of the Golgi-associated BICD2 proximal to the ER focus, and not simply its presence in cells, is critical for supporting SV40 disassembly required for successful infection.
Discussion
Dynein is a major molecular motor that transports different cargos along MTs within the cell. As part of a three-member protein complex composed of the dynein motor, dynactin activator, and a cargo adaptor, the activity and regulation of the latter is by far the most enigmatic. In addition to physically linking cargos to the dynein motor, cargo adaptors have been proposed to regulate dynein motility and control the motility switch between the use of dynein and kinesin motors (McKenney et al., 2014; Grigoriev et al., 2007; Larsen et al., 2008).
In this paper, we report an unexpected function of the BICD adaptors, cargo remodeling, that can be exploited to promote virus infection. Strikingly, our analysis also revealed that the select Golgi localization of these cargo adaptors can couple two decisive virus entry steps essential for infection. Specifically, our findings revealed a model in which a penetration site in the ER membrane (called focus) that enables SV40 to escape into the cytosol from the ER, a critical infection step, is constructed proximal to the Golgi-associated BICD cargo adaptors. This juxtaposition enables Golgi-localized BICD2/BICDR1 to bind to and disassemble SV40 when the virus enters the cytosol. Virus disassembly represents yet another important infection step, as it generates a subviral particle that can successfully enter the nucleus to cause infection.
Our results have provided significant insights into the molecular mechanism by which BICD2 disassembles SV40. First, we found that this dynein adaptor can efficiently disassemble DTT/EGTA–treated (but not native) SV40. DTT/EGTA–treated virus mimics native SV40 that has trafficked from the cell surface to the ER, where it penetrates the ER membrane to escape into the cytosol, en route to infection. Thus, BICD2 can disassemble SV40 only if the virus trafficked along its infectious route to reach the cytosol so that it is properly “primed” for BICD2 engagement.
Next, our binding studies revealed that BICD2 interacts with VP1 and the VP3 minor capsid protein, suggesting that BICD2 recognizes VP1 pentamers with exposed VP3. Because a SV40 particle contains 72 VP1 pentamers, with each pentamer harboring either VP2 or VP3, this finding suggests that BICD2 recognizes VP1 pentamers with VP3. These data are consistent with the notion that BICD2 disassembles the DTT/EGTA–treated virus, because DTT treatment reduces the viral disulfide bonds essential to expose VP3.
Guided by this binding study, we used a mutant SV40 devoid of VP3 and found that BICD2 cannot disassemble this mutant virus, further suggesting that BICD2 acts on a distinct form of the viral cargo: VP1 pentamers containing VP3. Why BICD2 might prefer VP3 over VP2 is unclear, especially given that VP2 contains all the sequences of VP3 and only harbors a unique N-terminal region. One possibility is that this VP2 N-terminal region occludes a BICD2 recognition site that is unmasked in VP3.
Using an in vitro reconstitution strategy with purified components, we then found that the central CC2 and C-terminal CC3 domains of BICD2 are necessary and sufficient to drive SV40 disassembly. Because the CC3 domain is known to bind to cargos (Matanis et al., 2002; Hoogenraad et al., 2001), its ability to support SV40 binding and disassembly is perhaps not surprising. However, whereas the CC2 domain can associate with the kinesin motor (Grigoriev et al., 2007), it has not been shown to serve as a cargo-binding domain. A role of the CC2 domain in virus disassembly raises the possibility that kinesin may also play a role in the disassembly of SV40, as is the case of adenovirus (Strunze et al., 2011). Because we previously reported that kinesin-1 activity is essential for the virus to construct the ER foci in order to reach the cytosol (Ravindran et al., 2017), assessing a role of this motor during SV40 disassembly in the cytosol is not trivial, unless an inducible system that inactivates kinesin-1 only after the virus arrives in the cytosol can be developed. Regardless, the observation that these two BICD2 domains are essential to drive efficient viral disassembly suggest a bivalent mechanism of engaging the SV40 VP1 monomer. Whether BICDR1 (and BICD1) use a similar binding mechanism to uncoat SV40 remains unknown.
A critical question is how the BICD2-SV40 interaction functionally leads to disassembly of the VP1 coat protein. In addition to the covalent disulfide bonds in SV40 that provide architectural support of the viral particle (Stehle et al., 1996; Liddington et al., 1991), noncovalent interactions between the C-terminal tail of an individual VP1 monomer, which extends into an adjacent pentamer, stabilizes interpentamer interactions (Stehle et al., 1996). Individual pentamers can be released by disruption of these noncovalent bonds. Hence, one scenario is that the BICD-SV40 engagement destabilizes this interpentamer interaction and, as a consequence, releases the VP1 pentamers (or oligomers of the pentamers). This idea would be consistent with the observed formation of numerous VP1 species across the sucrose gradient during disassembly, as well as the EM data, which show the appearance of VP1 pentamers in the presence of high concentrations of BICD2.
Finally, our data suggest that the hidden DNA genome of SV40 is exposed upon BICD2-mediated disassembly. Because SV40 VP1 is known to bind to the viral genome (Moreland et al., 1991), uncoating of VP1 pentamers likely results in some degree of exposure of its genetic material. The extent of genome exposure due to BICD-dependent disassembly is unknown but has clear implication in virus detection by the host immune system. To evade immune detection, it is possible that the BICD adaptors remain bound to the partially disassembled virus, masking the exposed genome until the cargo adaptors reach the nuclear membrane, where they release the viral particle immediately before nuclear entry. In this scenario, it is worth noting that a pool of BICD2 has indeed been reported to localize to the nuclear membrane (Splinter et al., 2010).
Another prominent cellular location of the BICD cargo adaptors is the Golgi. In fact, BICD2, BICD1, and BICDR1 have been shown to bind to the Golgi membrane via the small GTPase Rab6 (Matanis et al., 2002; Schlager et al., 2010). Surprisingly, we discovered that the ER focus structure, representing SV40’s site of cytosol entry from the ER, is constructed proximal to the Golgi-associated BICD cargo adaptors. This juxtaposition creates an optimal situation during SV40 entry in which cytosol arrival of the viral particle is efficiently coupled to its subsequent disassembly.
In this context, we found that only BICD2, BICD1, and BICDR1, but not BICDR2, promote SV40 infection. Why BICDR2 does not support virus infection is unclear. One intriguing possibility is that while BICD2, BICD1, and BICDR1 all bind to Rab6 and localize to the Golgi, BICDR2 was shown to not interact with Rab6 but associate with Rab13 instead (Schlager et al., 2010). Because Rab13 is primarily associated with endosomal compartments (Goud et al., 2018), lack of BICDR2 localization at the Golgi may explain why it does not support virus infection. The other possibility is that the domain within BICDR2 that corresponds with the CC2 and CC3 region of BICD2 is vastly different from the counterparts in BICD2, BICD1, and BICDR1. However, the CC2 and CC3 domains of BICDR2, as defined by Schlager et al. (2014b), display 52% sequence conservation to BICD2, which is similar to that of BICDR1 (55%). Hence, it is unlikely that structural differences in the CC2 and CC3 domains of BICDR2 account for this adaptor’s inability to participate in SV40 infection.
Our data revealed that the loss of a single Rab6-associated BICD adaptor significantly impaired virus disassembly and that restoring expression rescued this phenotype. It is surprising that knockdown of any one of these adaptors results in such a strong level of inhibition in the presence of the remaining adaptors. Knockdown of BICD2 and BICDR1 disrupted SV40 infection by 81% and 70%, respectively, while simultaneously blocking both decreased infection by 90%. Given the high concentration of the BICD adaptors required to disassemble the virus, as indicated by our in vitro reconstitution assays, it appears that any disruption in the total level of these adaptors surrounding the site of ER membrane penetration is sufficient to impair disassembly.
We previously reported that virus disassembly requires a dynein-dependent action (Ravindran et al., 2018), but here, we report that the BICD adaptors disassemble SV40 in a dynein- and MT-independent manner. Depletion of the dynein motor leads to a relocalization of BICD2 toward the cell periphery (Splinter et al., 2010) and reduced pericentrosomal localization of BICDR1 (Schlager et al., 2010); therefore, one possible explanation is that inactivation of the dynein motor displaces the BICD adaptors away from the virus membrane penetration site to block virus disassembly indirectly. Alternatively, despite physical proximity between the ER focus and Golgi-associated BICD adaptors, SV40 that has just entered the cytosol might nonetheless need to be conducted to the Golgi by an MT-based, dynein-dependent transport mechanism. An obvious question in this model is the identity of the cargo adaptor that mediates SV40 transport to the Golgi.
In sum, by revealing how Golgi-associated dynein cargo adaptors couple ER membrane penetration and disassembly of a viral cargo, this study raises many intriguing questions. For instance, beyond a viral cargo, can a cellular cargo also be remodeled by a cargo adaptor? If so, what functional advantage does cargo remodeling achieve? If a cargo is to be transported by dynein, does cargo remodeling affect cargo loading or release from the dynein motor? The juxtaposition of the ER focus and Golgi-localized BICD adaptors also raises the question of whether cellular cargos exiting the ER might communicate with these Golgi-localized BICD adaptors for cargo delivery to other cellular destinations. For example, ER-coupled autophagy, in which ER-resident protein aggregates exit the ER and are delivered to the lysosome for degradation, might require the action of the Golgi-localized BICD adaptors for transport. If true, positioning the ER exit site of these aggregates adjacent to the Golgi should have distinct advantages. No doubt, additional experiments are needed to address all of these outstanding questions.
Materials and methods
Cell lines and reagents
CV-1 and COS-7 cells were obtained from the ATCC. Cells were grown in complete DMEM containing 10% fetal bovine serum, 10 U/ml penicillin, and 10 µg/ml streptomycin (Gibco). DMEM, Opti-MEM, and 0.25% trypsin-EDTA were purchased from Invitrogen. BSA, N-ethylmaleimide (NEM), DTT, and BFA were purchased from MilliporeSigma. PMSF was purchased from Thermo Fisher Scientific.
Antibodies
SV40 large T antigen (Santa Cruz; sc-147), monoclonal VP1 was provided by Dr. Walter Scott (University of Miami, Miami, FL), SV40 VP2/3 (Abcam; ab53983), BICD2 (Abcam; ab117818), BICDR1 (Thermo Fisher Scientific; PA5-66367), Hsp90 (Santa Cruz; sc-13119), PDI (Abcam; ab2792), BAP31 (Pierce; MA3-002), γ-tubulin (MilliporeSigma; T5326), GM130 (Abcam), GFP (Proteintech; 66002), and 6xHis tag (MilliporeSigma; SAB1306084).
Preparation of SV40
SV40 was prepared using the OptiPrep gradient system (MilliporeSigma) as previously described (Inoue and Tsai, 2011). Briefly, cells transfected with the SV40 viral genome were lysed in HN buffer (50 mM Hepes, pH 7.5, and 150 mM NaCl) with 0.5% Brij58 for 30 min on ice. Following centrifugation, the supernatant was layered on top of a discontinuous 20% and 40% OptiPrep gradient. Tubes were centrifuged at 49,500 rpm for 2 h at 4°C in a SW55Ti rotor (Beckman Coulter). Purified virus was collected from the white interface that forms between the OptiPrep layers and the aliquots stored at −80°C for future use. BK PyV and antibody against BK TAg were kindly provided by Dr. Michael Imperiale (University of Michigan, Ann Arbor, MI).
Plasmids
HA-BICD2 and HA-BICDR1 were generous gifts from Dr. Casper Hoogenraad (Utrecht University, Utrecht, Netherlands). FLAG-HA-GFP was a gift from Dr. Wade Harper (Harvard Medical School, Boston, MA; AddGene; plasmid #22612; http://n2t.net/addgene:22612; RRID: Addgene_22612). RnKIF5C(1–560)-mCit-GTS has been described (Engelke et al., 2016). EGFP-Rab6 and EGFP-Rab6T27N were gifts from Dr. William Britt (UAB). Human dynein-1 intermediate chain2 (isoform 2C, 612 aa) and human dynactin p62 subunit (isoform 1, 460 aa) were received as a gift from the Reck-Peterson laboratory (University of California, San Diego, San Diego, CA) in pcDNA vectors, pcDNA5/FRT-IC2-5×GA-BioID-fSNAP-3×FLAG, and pcDNA5/FRT-p62-5×GA-BioID-HALO-3×FLAG, respectively (Redwine et al., 2017). Full-length human NDEL with N-terminal GST tag and C-terminal 6xHis tag in pKL backbone vector for insect cell expression was received from the Reck-Peterson laboratory. Full-length BICDR1 (Mus musculus) was designed for expression in insect cells with 8xHis-ZZ N-terminal tag, and Tobacco Etch Virus (TEV) site to cleave 8x-His-ZZ in pOmniBac vector was a gift from Dr. Andrew Carter (MRC Laboratory of Molecular Biology, Cambridge, UK; Addgene plasmid #111859; http://n2t.net/addgene:111859; RRID:Addgene_111859; Urnavicius et al., 2018). 8xHis-zz-TEV-BICD2 (full-length M. musculus BICD2) was codon optimized for expression in SF9 insect cell, synthesized by Genscript, and subsequently cloned into pFastBac vector. The truncated versions of full-length BICD2 in pFastBac vector were subcloned using NEBuilder HiFi DNA assembly (NEB E2621S) as listed in Table 1.
Table 1. BICD2 constructs used for protein expression.
| BICD2 construct | Segment of FL BICD2 (aa) |
|---|---|
| 8xHis-zz-TEV-BicD2 (full length) | 1-820 |
| 8xHis-zz-TEV-BicD2 (CC1) | 1-286 |
| 8xHis-zz-TEV-BicD2 (CC2) | 326-560 |
| 8xHis-zz-TEV-BicD2 (CC3) | 643-820 |
| 8xHis-zz-TEV-BicD2 (CC1+CC2) | 1-560 |
| 8xHis-zz-TEV-BicD2 (CC2+CC3) | 326-820 |
| 8xHis-zz-TEV-BicD2 (CC1+CC2+CC3) | 1-804 |
siRNA transfection
All Star Negative (Qiagen) was used as a scrambled control siRNA. The following siRNAs were all purchased from Dharmacon: siBICD2, 5′-GCAAGGAGCUGUCACACUA-3′; siBICDR1, 5′-GCACUUAGAGCAAGAGAAA-3′; siHOOK3, 5′-GAAGAAACAUUCAGACUAG-3′; siNinein, 5′-GAACUUAAUCAACGUCUAA-3′; siRAB11-FIP3, 5′-GCAAGAAGGUGGCAAGGUA-3′; and siSPDL1, 5′-GGGAGAAGUUUAUCGAUUA-3′. The remaining were purchased from MilliporeSigma (9siBICD1, 5′-GAGGATGGGAGTGAACCAA-3′; and siBICDR2, 5′-AGATGCTTCTGGAGCGAAA-3′). For knockdown experiments, CV-1 cells were reverse transfected with 5 nM of the indicated siRNA using Lipofectamine RNAiMAX (Invitrogen). Infections and biochemical assays were all performed at 24–48 h after transfection.
DNA transfection
For CV-1 cells, plasmids were transfected into 50% confluent cells using the FuGENE HD (Promega) transfection reagent. For COS-7 cells, polyethylenimine (PEI; Polysciences) was used. DNA was allowed to express for at least 24 h before experimentation.
ER arrival and ER-to-cytosol membrane transport assays
These assays were performed as previously described (Inoue and Tsai, 2011). For ER arrival, following siRNA treatment, cells were washed with PBS containing 10 mM NEM and harvested by scrapping. Cells were then centrifuged and the pellet treated with either nonreducing or reducing (+BME) SDS sample buffer before being run on an SDS-PAGE gel. For cytosol arrival, cells were lysed in HN buffer with 0.05–0.1% digitonin and 1 mM PMSF for 10 min on ice. Cells were then centrifuged at 13,000 rpm for 10 min at 4°C. The supernatant (cytosol fraction) was removed and stored in a fresh tube. SDS sample buffer was added to the resulting pellet (membrane fraction) and the samples boiled at 95°C for 10 min.
Immunoprecipitation
Cells were lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.25% sodium deoxycholate, 1% NP-40, and 1 mM EDTA) with 1 mM PMSF for 15 min on ice. Cells were then centrifuged at 13,000 rpm for 10 min at 4°C. A 5% input was collected from the resulting supernatant before 2 µg of the indicated antibody was added to the lysate and rotated overnight at 4°C. The next day, Protein A/G agarose beads (Pierce) were washed three times with RIPA buffer and rotated with the lysate for 30 min at 4°C. The tubes were then centrifuged at 7,000 rpm for 1 min, and the beads washed three times with RIPA buffer. For genome extraction, 40 µl of RIPA with 1% SDS was added to the beads and vortexed at room temperature for 5 min. Of this, 20% (8 µl) was removed, 40 µl of PB buffer (Qiagen) was added, and samples were run through a Qiagen column. DNA was extracted according to the miniprep protocol. SDS sample buffer was added to the remaining immunoprecipitation lysate and the beads boiled for 10 min at 95°C. Samples were then run on an SDS-PAGE gel and immunoblotted for VP1.
SV40 disassembly assay
For the sucrose cushion, cytosol fractions were obtained as described above and layered over 20% sucrose and centrifuged at 50,000 rpm for 30 min at 4°C. A top (80 µl) and bottom (20 µl) fraction were collected and analyzed by SDS-PAGE with immunoblotting. For sucrose gradients, the cytosolic fraction was layered over a 20–30–40% or 30–40–60% discontinuous sucrose gradient. Tubes were centrifuged for 30 min at 4°C for either 50,000 or 30,000 rpm, respectively. From the top, 25-µl aliquots were collected and subjected to SDS-PAGE followed by immunoblotting.
Protein purification
Dynein and dynactin
The human dynein and dynactin complexes were purified from FreeStyle HEK293 cells transiently expressing IC2-SNAPf-3xFLAG or p62-HALO-3xFLAG, respectively. In brief, 35 µg plasmid DNA diluted in Opti-MEM (Gibco) was added to the same volume of Opti-MEM solution of PEI (MilliporeSigma; 408727) at 35 µg/ml. After 30-min incubation at room temperature, the solution of DNA–PEI complexes was added to a low passage of HEK293 cell suspension with final cell density of ∼1 × 106 viable cells/ml in fresh FreeStyle 293 Expression Medium for each 30-ml transfection. The cells were incubated in a 37°C incubator with a humidified atmosphere of 8% CO2 in air at 125 rpm and harvested ∼48 h posttransfection. Subsequent purification steps for dynein and dynactin using M2-FLAG antibody resin were described previously (Redwine et al., 2017).
NDEL and LIS1
Baculovirus were prepared from pKL donor vectors containing the ORFs of these human genes. SF9 cells at the density of 2 × 106 viable cells/ml were inoculated with the passage 2 of virus at 1:100 ratio. The infected cells were harvested after 60 h incubation at 27°C. The cell pellets were resuspended in 1 ml lysis buffer (Hepes 50 mM, pH 7.5, NaCl 150 mM, 10% glycerol, DTT 1 mM, 0.2% Triton X-100, imidazole 10 mM, Pefabloc 0.5 mM, and 1X SigmaFast protease inhibitor) for each 10-ml culture. The cells were lysed using Dounce homogenizer and centrifuged at 200,000 g. The C-terminally histidine-tagged proteins were initially purified using Ni-NTA column (Thermo Fisher Scientific; His-trap) with 350 mM imidazole in elution buffer. For LIS1, purification continued to the next step and eluted LIS1 (in elution buffer, without further dilution) was incubated with IgG Sepharose 6 Fast Flow beads (Amersham Pharmacia) for 2–16 h at 4°C. The protein treated beads were washed with 10 × 1 ml lysis buffer and 10 × 1 ml with TEV buffer (10 mM Tris-HCl, pH 8.0, 150 mM KCl, 10% glycerol, 0.2% Triton X-100, 0.5 mM EGTA, and 1 mM DTT). LIS1 was released from beads via incubation with TEV protease at 25 µg/ml for 1 h at 16°C, resulting in cleavage from the 8xHis-ZZ tag.
BICD2 and BICDR1
Baculovirus were prepared from pFastBac (BICD2 and truncated versions) and pOmniBac (BICDR1) donor vector using standard methods. SF9 cells at 2 × 106/ml density were infected with passage 2 of virus at 1:100 ratio and harvested 48 h later. We followed the method explained previously (Huynh and Vale, 2017) for lysing the cells and purification of His-tagged BICD2s and BICDR1 by His-trap column (GE; 17524801) with some modifications. The eluted proteins from Ni-NTA purification step were diluted three times in diluting buffer (10% glycerol, 2 mM BME, 2 mM EGTA, 10 mM Hepes, and 0.15% Tween) and incubated with IgG Sepharose Fast Flow beads to be further purified via their ZZ tag. Washing steps and release of proteins with TEV protease were the same as explained for LIS1.
In vitro reconstitution of disassembly
Native SV40 was treated with DTT and EGTA (400 µM) and incubated in HN buffer with purified dynein components for either 30 min at room temperature or 10 min at 37°C. The reaction was supplemented with 1 mM MgCl2 and, where indicated, 2 mM ATP and polymerized MTs. The reaction was then layered over a 20–40% discontinuous sucrose gradient and disassembly assessed as described above. To measure genome exposure, native SV40 was incubated with BSA or BICD2 at 37°C on ice or with 1 mM DTT at 65°C shaking. Following a 10-min incubation, 0.02% EtBr was added to the disassembled material and mixed thoroughly. EtBr fluorescence intensity was then measured on a SpectraMax M5 spectrofluorimeter (Molecular Devices), where the samples were excited at 520 nm and the emission measured from 550 to 700 nm.
In vitro binding assay
8xHis-ZZ fused BICD2 (full-length and truncated versions) and BICDR1 purified by His-Trap column were mixed separately with SV40 intact viruses (CsCl purified) in at least 72 times excess in molar ratio. Incubation/binding buffer included 50 mM Hepes, pH 7.5, 150 mM NaCl, 0.4 mM DTT, and 0.4 mM EGTA with BSA 0.1% and Triton-X100 0.02%. IgG Sepharose resin preequilibration was done per the manufacturer’s protocol in the presence of 0.1–0.2% BSA. 100 µl packed resin was treated with 200 µl of each protein sample mixture at 4°C for 2 h on tube roller. Unbound proteins were washed out with 5 × 3 ml binding buffer, 2 × 3 ml binding buffer + high salt (400 mM NaCl), and 4 × 3 ml TEV buffer (10 mM Tris, pH 8.0, 150 mM KCl, 2 mM MgCl2, 0.5 mM EGTA, and 10% glycerol). The proteins were eluted from the IgG Sepharose in 200 µl TEV buffer containing 60 µg/ml TEV protease. Following SDS-PAGE with immunoblotting, eluted SV40 was detected by anti-VP1 antibody, and eluted BICD2s and BICDR1 were visualized by Coomassie blue staining. For Fig. S2 D, native SV40 was treated with or without DTT and EGTA and incubated with BICD2 in PBS with 1 mM MgCl2. Samples were then rotated with 1 µg BICD2 antibody overnight at 4°C. The next day, Protein A/G agarose beads (Pierce) were washed three times with PBS and rotated with the lysate for 20 min at 4°C. The tubes were then centrifuged at 7,000 rpm for 1 min and the beads washed three times with PBS. SDS sample buffer was added to the beads and boiled for 10 min at 95°C. Samples were then run on an SDS-PAGE gel and assessed by Western blot. Three replicates of each single assay were used to quantify the results using ImageJ.
Negative-stain EM
CsCl-purified SV40 at constant concentration (0.2 mg/ml) was incubated with full-length BICD2 at 1:0, 1:10, 1:50, or 1:100 SV40:BicD2 molar ratio at 37°C for 10 min in presence of 50 mM Hepes, pH 7.5, 150 mM NaCl, 0.1 mM DTT, and 0.05 mM EGTA. Grid samples for negative-stain EM were prepared from these samples without further dilution using conventional negative staining procedures (0.075% uranyl formate on glow-discharged Formwar Carbon Film 400-mesh copper). Imaging was performed at room temperature with Tecnai T12 transmission electron microscope (FEI) equipped with a tungsten filament operated at an acceleration voltage of 120 kV and a mounted K2 Rio camera (Gatan) with leginon for automated image collection. Intact and partially disassembled viral particles were averaged from 3 × 3 µM views (magnification 6,500 and pixel size 20.40 Å), and released VP1 pentamers were averaged from 0.3 × 0.3 µM views (magnification 67,000 and pixel size 0.93 Å). At least 10–30 views out of different squares on two grids were counted for the purpose of quantification.
Immunofluorescence and confocal microscopy
Cells were grown on no. 1 glass coverslips and fixed with either 1% formaldehyde for 15 min or ice-cold 100% methanol for 10 min followed by 1% formaldehyde for 15 min. Cells were then permeabilized in PBS with 0.2% Triton X-100 for 5 min and blocked with 5% milk containing 0.02% Tween-20. Primary antibodies were incubated in milk for 1 h at room temperature or overnight at 4°C. Coverslips were then washed three times in milk and incubated with Alexa Fluor secondary antibodies (Invitrogen) for 30 min at room temperature. Coverslips were again washed and mounted using ProLong Gold with DAPI (Invitrogen). Images were taken on either a Nikon Eclipse TE2000-E inverted epifluorescence microscope or a Zeiss LSM 800 confocal laser scanning microscope. FIJI software was used for image processing and analysis.
Quantification of Western blots
All Western blots were developed on film and quantified using FIJI software. At least three independent replicates were quantified for each experiment.
Protein sequence analysis
Protein sequences were analyzed using homology extension by the T-Coffee Server (Di Tommaso et al., 2011).
Online supplemental material
Fig. S1 shows that BICD1, but not BICDR2, supports SV40 infection. Fig. S2 provides additional characterization of BICD-dependent binding and disassembly of SV40 in vitro. Fig. S3 shows supporting images for negative staining of SV40 in the presence of FL BICD2 at different molar ratios. Fig. S4 shows the SV40-induced ER focus is surrounded by BICD2 and BICDR1. Fig. S5 shows that Golgi dispersion mislocalizes BICD2 in COS-7 cells.
Supplementary Material
Acknowledgments
We thank members of the Tsai laboratory for thoughtful discussions throughout this work and the Coulombe laboratory at the University of Michigan for sharing its microscope facility. We also thank Megan Procario (University of Michigan, Ann Arbor, MI) from the Imperiale laboratory at the University of Michigan for providing BK PyV.
This work was supported by the National Institutes of Health (grant RO1AI064296 to B. Tsai and grants T32 AI007528 and F32 GM133099 to C.C. Spriggs).
The authors declare no competing financial interests.
Author contributions: Conceptualization, C.C. Spriggs, K.J. Verhey, M.A. Cianfrocco, and B. Tsai; Methodology, C.C. Spriggs and B. Tsai; Investigation, C.C. Spriggs and S. Badieyan; Validation, C.C. Spriggs and B. Tsai; Writing – Original Draft, C.C. Spriggs and B. Tsai; Writing – Review & Editing, C.C. Spriggs, S. Badieyan, K.J. Verhey, M.A. Cianfrocco, and B. Tsai; Funding Acquisition, C.C. Spriggs and B. Tsai; Supervision, B. Tsai.
References
- Anderson H.A., Chen Y., and Norkin L.C.. 1996. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 7:1825–1834. 10.1091/mbc.7.11.1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi P., Walczak C.P., and Tsai B.. 2015. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J. Virol. 89:4058–4068. 10.1128/JVI.03574-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabin-Brion K., Marceiller J., Perez F., Settegrana C., Drechou A., Durand G., and Poüs C.. 2001. The Golgi complex is a microtubule-organizing organelle. Mol. Biol. Cell. 12:2047–2060. 10.1091/mbc.12.7.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.S., Stehle T., and Harrison S.C.. 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 17:3233–3240. 10.1093/emboj/17.12.3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.J., Liu X., and Tsai B.. 2019. SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection. Viruses. 11:E917 10.3390/v11100917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocco M.A., DeSantis M.E., Leschziner A.E., and Reck-Peterson S.L.. 2015. Mechanism and regulation of cytoplasmic dynein. Annu. Rev. Cell Dev. Biol. 31:83–108. 10.1146/annurev-cellbio-100814-125438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clever J., Yamada M., and Kasamatsu H.. 1991. Import of simian virus 40 virions through nuclear pore complexes. Proc. Natl. Acad. Sci. USA. 88:7333–7337. 10.1073/pnas.88.16.7333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm E.M., Pelkmans L., Kartenbeck J., Mezzacasa A., Kurzchalia T., and Helenius A.. 2005. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 168:477–488. 10.1083/jcb.200407113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio J.A., and Garcea R.L.. 2013. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 11:264–276. 10.1038/nrmicro2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.M., Taly J.F., and Notredame C.. 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39(suppl):W13-7 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupzyk A., and Tsai B.. 2016. How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses. 8:242 10.3390/v8090242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupzyk A., and Tsai B.. 2018. Bag2 Is a Component of a Cytosolic Extraction Machinery That Promotes Membrane Penetration of a Nonenveloped Virus. J. Virol. 92:e00607-18 10.1128/JVI.00607-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupzyk A., Williams J.M., Bagchi P., Inoue T., and Tsai B.. 2017. SGTA-Dependent Regulation of Hsc70 Promotes Cytosol Entry of Simian Virus 40 from the Endoplasmic Reticulum. J. Virol. 91:e00232-17 10.1128/JVI.00232-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke M.F., Winding M., Yue Y., Shastry S., Teloni F., Reddy S., Blasius T.L., Soppina P., Hancock W.O., Gelfand V.I., and Verhey K.J.. 2016. Engineered kinesin motor proteins amenable to small-molecule inhibition. Nat. Commun. 7:11159 10.1038/ncomms11159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R., Andritschke D., Friebe S., Herzog F., Luisoni S., Heger T., and Helenius A.. 2011. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 13:1305–1314. 10.1038/ncb2339 [DOI] [PubMed] [Google Scholar]
- Gilbert J., Ou W., Silver J., and Benjamin T.. 2006. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 80:10868–10870. 10.1128/JVI.01117-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin E.C., Lipovsky A., Inoue T., Magaldi T.G., Edwards A.P., Van Goor K.E., Paton A.W., Paton J.C., Atwood W.J., Tsai B., and DiMaio D.. 2011. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio. 2:e00101–e00111. 10.1128/mBio.00101-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goud B., Liu S., and Storrie B.. 2018. Rab proteins as major determinants of the Golgi complex structure. Small GTPases. 9:66–75. 10.1080/21541248.2017.1384087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I., Splinter D., Keijzer N., Wulf P.S., Demmers J., Ohtsuka T., Modesti M., Maly I.V., Grosveld F., Hoogenraad C.C., and Akhmanova A.. 2007. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell. 13:305–314. 10.1016/j.devcel.2007.06.010 [DOI] [PubMed] [Google Scholar]
- Helenius A. 2018. Virus Entry: Looking Back and Moving Forward. J. Mol. Biol. 430:1853–1862. 10.1016/j.jmb.2018.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad C.C., Akhmanova A., Howell S.A., Dortland B.R., De Zeeuw C.I., Willemsen R., Visser P., Grosveld F., and Galjart N.. 2001. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 20:4041–4054. 10.1093/emboj/20.15.4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley P.M., and Livingston D.M.. 2009. Small DNA tumor viruses: large contributors to biomedical sciences. Virology. 384:256–259. 10.1016/j.virol.2008.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh W., and Vale R.D.. 2017. Disease-associated mutations in human BICD2 hyperactivate motility of dynein-dynactin. J. Cell Biol. 216:3051–3060. 10.1083/jcb.201703201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T., and Tsai B.. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 7:e1002037 10.1371/journal.ppat.1002037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartenbeck J., Stukenbrok H., and Helenius A.. 1989. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 109:2721–2729. 10.1083/jcb.109.6.2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen K.S., Xu J., Cermelli S., Shu Z., and Gross S.P.. 2008. BicaudalD actively regulates microtubule motor activity in lipid droplet transport. PLoS One. 3:e3763 10.1371/journal.pone.0003763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddington R.C., Yan Y., Moulai J., Sahli R., Benjamin T.L., and Harrison S.C.. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature. 354:278–284. 10.1038/354278a0 [DOI] [PubMed] [Google Scholar]
- Magnuson B., Rainey E.K., Benjamin T., Baryshev M., Mkrtchian S., and Tsai B.. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell. 20:289–300. 10.1016/j.molcel.2005.08.034 [DOI] [PubMed] [Google Scholar]
- Matanis T., Akhmanova A., Wulf P., Del Nery E., Weide T., Stepanova T., Galjart N., Grosveld F., Goud B., De Zeeuw C.I., et al. 2002. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 4:986–992. 10.1038/ncb891 [DOI] [PubMed] [Google Scholar]
- McKenney R.J., Huynh W., Tanenbaum M.E., Bhabha G., and Vale R.D.. 2014. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science. 345:337–341. 10.1126/science.1254198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland R.B., Montross L., and Garcea R.L.. 1991. Characterization of the DNA-binding properties of the polyomavirus capsid protein VP1. J. Virol. 65:1168–1176. 10.1128/JVI.65.3.1168-1176.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi A., Clever J., Yamada M., Li P.P., and Kasamatsu H.. 1996. Association with capsid proteins promotes nuclear targeting of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA. 93:96–100. 10.1073/pnas.93.1.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norkin L.C., Anderson H.A., Wolfrom S.A., and Oppenheim A.. 2002. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 76:5156–5166. 10.1128/JVI.76.10.5156-5166.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran M.S., Bagchi P., Inoue T., and Tsai B.. 2015. A Non-enveloped Virus Hijacks Host Disaggregation Machinery to Translocate across the Endoplasmic Reticulum Membrane. PLoS Pathog. 11:e1005086 10.1371/journal.ppat.1005086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran M.S., Engelke M.F., Verhey K.J., and Tsai B.. 2017. Exploiting the kinesin-1 molecular motor to generate a virus membrane penetration site. Nat Commun. 8:15496. 10.1038/ncomms15496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran M.S., Spriggs C.C., Verhey K.J., and Tsai B.. 2018. Dynein engages and disassembles cytosol-localized SV40 to promote infection. J. Virol. 92:e00353-18 10.1128/JVI.00353-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redwine W.B., DeSantis M.E., Hollyer I., Htet Z.M., Tran P.T., Swanson S.K., Florens L., Washburn M.P., and Reck-Peterson S.L.. 2017. The human cytoplasmic dynein interactome reveals novel activators of motility. eLife. 6:e28257 10.7554/eLife.28257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelhaas M., Malmström J., Pelkmans L., Haugstetter J., Ellgaard L., Grünewald K., and Helenius A.. 2007. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 131:516–529. 10.1016/j.cell.2007.09.038 [DOI] [PubMed] [Google Scholar]
- Schlager M.A., Kapitein L.C., Grigoriev I., Burzynski G.M., Wulf P.S., Keijzer N., de Graaff E., Fukuda M., Shepherd I.T., Akhmanova A., and Hoogenraad C.C.. 2010. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. EMBO J. 29:1637–1651. 10.1038/emboj.2010.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager M.A., Hoang H.T., Urnavicius L., Bullock S.L., and Carter A.P.. 2014a In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J. 33:1855–1868. 10.15252/embj.201488792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager M.A., Serra-Marques A., Grigoriev I., Gumy L.F., Esteves da Silva M., Wulf P.S., Akhmanova A., and Hoogenraad C.C.. 2014b Bicaudal d family adaptor proteins control the velocity of Dynein-based movements. Cell Rep. 8:1248–1256. 10.1016/j.celrep.2014.07.052 [DOI] [PubMed] [Google Scholar]
- Splinter D., Tanenbaum M.E., Lindqvist A., Jaarsma D., Flotho A., Yu K.L., Grigoriev I., Engelsma D., Haasdijk E.D., Keijzer N., et al. 2010. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 8:e1000350 10.1371/journal.pbio.1000350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs C.C., Harwood M.C., and Tsai B.. 2019. How non-enveloped viruses hijack host machineries to cause infection. Adv. Virus Res. 104:97–122. 10.1016/bs.aivir.2019.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle T., Gamblin S.J., Yan Y., and Harrison S.C.. 1996. The structure of simian virus 40 refined at 3.1 A resolution. Structure. 4:165–182. 10.1016/S0969-2126(96)00020-2 [DOI] [PubMed] [Google Scholar]
- Strunze S., Engelke M.F., Wang I.H., Puntener D., Boucke K., Schleich S., Way M., Schoenenberger P., Burckhardt C.J., and Greber U.F.. 2011. Kinesin-1-mediated capsid disassembly and disruption of the nuclear pore complex promote virus infection. Cell Host Microbe. 10:210–223. 10.1016/j.chom.2011.08.010 [DOI] [PubMed] [Google Scholar]
- Toropova K., Zou S., Roberts A.J., Redwine W.B., Goodman B.S., Reck-Peterson S.L., and Leschziner A.E.. 2014. Lis1 regulates dynein by sterically blocking its mechanochemical cycle. eLife. 3:e03372 10.7554/eLife.03372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B., Gilbert J.M., Stehle T., Lencer W., Benjamin T.L., and Rapoport T.A.. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 22:4346–4355. 10.1093/emboj/cdg439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnavicius L., Lau C.K., Elshenawy M.M., Morales-Rios E., Motz C., Yildiz A., and Carter A.P.. 2018. Cryo-EM shows how dynactin recruits two dyneins for faster movement. Nature. 554:202–206. 10.1038/nature25462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak C.P., and Tsai B.. 2011. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J. Virol. 85:2386–2396. 10.1128/JVI.01855-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak C.P., Ravindran M.S., Inoue T., and Tsai B.. 2014. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog. 10:e1004007 10.1371/journal.ppat.1004007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Foster H.E., Rondelet A., Lacey S.E., Bahi-Buisson N., Bird A.W., and Carter A.P.. 2017. Cryo-EM Reveals How Human Cytoplasmic Dynein Is Auto-inhibited and Activated. Cell. 169:1303–1314.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.