

Abstract
Severe acute respiratory syndrome-associated coronavirus (SARS-CoV) emerged, in November 2002, as a novel agent causing severe respiratory illness. To study sequence variation in the SARS-CoV genome, we determined the nucleic acid sequence of the S and N genes directly from clinical specimens from 10 patients—1 specimen with no matched SARS-CoV isolate, from 2 patients; multiple specimens from 3 patients; and matched clinical-specimen/ cell-culture-isolate pairs from 6 patients. We identified 3 nucleotide substitutions that were most likely due to natural variation and 2 substitutions that arose after cell-culture passage of the virus. These data demonstrate the overall stability of the S and N genes of SARS-CoV over 3 months during which a minimum of 4 generations for transmission events occurred. These findings are a part of the expanding investigation of the evolution of how this virus adapts to a new host.
Severe acute respiratory syndrome (SARS) likely emerged in Guangdong Province, China, late in 2002 and subsequently spread to many other countries. Shortly after SARS and its potential for global transmission were recognized by the World Health Organization (WHO) in March 2003, the etiology of SARS was identified as a previously unknown coronavirus (CoV), SARS-CoV. The complete genome sequences of 2 viral isolates were soon determined, and, since then, many isolates have been sequenced completely or partially. The sequences show a genomic organization typical of a coronavirus. The 5' two-thirds of the genome encodes a series of proteases and the replicase proteins, and the 3' one-third encodes the spike (S), envelope (E), membrane (M), and nucleocapsid (N) structural proteins, plus several other open reading frames (ORFs) of unknown function. The S and N proteins are considered to be the most important targets for the humoral and cellular immune responses to SARS-CoV [1, 2].
The available sequence information shows limited genetic change in this virus but does document the specific changes that may be useful for understanding the source and the transmission patterns of SARS-CoV [3, 4]. One uncertainty about interpretation of these data is the possibility that virus isolation in cell culture selects for some of the observed changes [5]. In the present report, we describe (1) a strategy for direct sequencing of the S and N genes from clinical specimens and (2) the sequence data on primary clinical specimens and/or matched cell-culture isolates from 10 patients. Our data provide both additional information on the genetic stability of SARS-CoV and evidence for the existence of nucleotide mutations associated with cell-culture passage of the virus.
Patients, Materials, and Methods
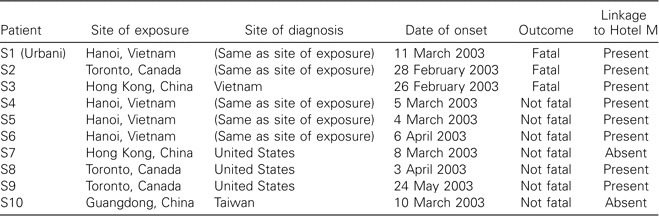
Case descriptions. Clinical specimens and epidemiologic data used in the present study were obtained as part of the emergency public health response to the outbreak of SARS; therefore, Federal regulations for human-subject research did not apply, and approval by the Institutional Review Board was not required. The clinical specimens used in this study (table 1) were from 10 case patients meeting the case definition for probable SARS [6] and having laboratory-confirmed SARS-CoV infection [7, 8]. Most of the cases occurred outside mainland China, but many were linked directly or indirectly, through multiple chains of transmission, to a single ill traveler from Guangdong Province, China, who exposed multiple guests at Hotel M in Hong Kong in late February 2003 [9]. The resulting cluster of infections initiated the global spread of SARS. Patient S3 was the Hotel M contact who transmitted the infection to Vietnam. Patient S1 was likely infected through close contact with patient S3 or other early cases of SARS in Vietnam [10]. All other patients in Vietnam (S4–S6) [11], the patient from Canada (S2) [12], and 2 patients from the United States (S8 [13] and S9 [14]) were infected through chains of transmission originating from the Hotel M cluster. Two cases in the present study are not known to be directly linked to the Hotel M cluster: patient S7 developed symptoms after returning home to the United States after a trip to Hong Kong but without known exposure to Hotel M [15], and patient S10 was a secondary contact, in Taiwan, of a traveler to Guangdong, China, who was diagnosed with SARS and who transmitted the illness after returning to Taiwan [16].
Table 1.
Epidemiologic data and outcomes for 10 patients with severe acute respiratory syndrome.
RNA extraction. Clinical specimens included nasal- and throat-swab, sputum, stool, and tissue specimens. Stool specimens and organ tissues were homogenized and clarified as described elsewhere [17, 18]. A 100-εL aliquot of each specimen was added to vials containing 900 εL of NucliSens lysis buffer (BioMérieux). Stool samples were prepared as 10% clarified suspensions in Tris-HCl buffer before extraction of total nucleic acid for reverse transcriptase-polymerase chain reaction (RTPCR) testing. The extraction procedures were performed by the automated NucliSens extraction system (BioMérieux), according to the manufacturer's instructions, and nucleic acid was recovered in a final volume of 50 εL.
One-step RT-PCR and sequence analysis. To determine the S and N gene sequences, overlapping RT-PCR products were generated by use of a SuperScript One Step RT-PCR kit (Invitrogen). Eight overlapping RT-PCR amplicons with an average size of ∼500 bp covered the S gene ORF (3768 bp), whereas 2 RT-PCR products with an average size of ∼700 bp covered the N gene ORF (1269 bp). The RT-PCR reactions were set up according to the manufacturer's instructions, with each 50 εL of RT-PCR reaction mixture containing 5 εL of extracted nucleic acid sample, 1 × Reaction Mix, 2 mmol of MgSO4/L, 0.2 εg of sense primer, 0.2 εg of antisense primer, 20 U of RNase inhibitor, and 1 εL of SSII/Platinum Taq Mix. Reverse transcription was performed at 50°C for 30 min, followed by a 5-min denaturation at 94°C. This was followed by 40 cycles of amplification, each consisting of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min. Amplification was concluded with a single final extension at 72°C for 7 min. Amplification products were visualized on 2% agarose gels containing 0.5 εg of ethidium bromide/mL and were purified by use of a QIAquickPCR purification kit (Qiagen). The 8 RT-PCR products for the S gene were sequenced, in both directions, by use of the same 16 RTPCR primers, and the 2 RT-PCR products for the N gene were sequenced, in both directions, by use of 7 different sequencing primers. Automated sequencing was performed by use of a BigDye Terminator v3.1 Cycle Sequencing Kit, on an ABI PRISM 3100 automated sequencer (Applied Biosystems). Sequences were assembled and analyzed by Sequencher software (Gene Code). The sequence of the Urbani strain (GenBank accession number AY278741) of SARS-CoV was used as a reference for all sequence comparisons.
Viral culture. Clinical specimens from each patient were inoculated and cultured on Vero E6 cells, as described elsewhere [18]. The SARS-CoV was identified by cytopathic effect and was confirmed by electronic microscopy (EM) and SARS-CoV-specific RT-PCR [18]. To prepare a virus stock for use as an inactivated vaccine, SARS-CoV was isolated from a clinical specimen from patient S7 and was passed 2 times on “Good Manufacture Procedure”-grade Vero cells (batch FA139414, passage 141; courtesy of Aventis Pasteur). The sequence of the entire genome of this viral isolate was determined by the approach described elsewhere by Rota et al. (GenBank accession number AY714217) [2].
Results
Sequence variations. The clinical specimens from the 10 patients included 1 specimen with no matched SARS-CoV isolate, from 2 patients (S9 and S10); multiple specimens from 3 patients (S2, S3, and S8), and matched specimen/isolate pairs from 6 patients (S1 and S3–S7). The S and N gene sequences of clinical specimens from these 10 patients were compared with those of the original isolate from patient S1 (Urbani), whose sequence had been generated previously and was used as the reference sequence for the present study (table 2). Specimens from all 10 patients showed the same 3768-nt ORF encoding the 1255-aa S protein, as well as the 1269-nt ORF encoding the 422-aa N protein, indicating an absence of frameshifts, insertions, and deletions in these ORFs. There were only 5 nucleotide positions that differed relative to the previously published Urbani sequence; 3 of these sequence variations arose independently, as true mutations, in 3 patients (mutation 29347-G in patient S4, mutation 23445-G in patient S7, and mutation 22570-C in patient S8), and 2 were sequence variations that were present in 2 cultured isolates (mutation 21938-C in the isolate from patient S3 and mutation 24872-C in the isolate from patient S1 [Urbani]) but not in the matched clinical specimens. Of the 3 true substitutions, 2 were observed in specimen/isolate pairs (patients S4 and S7) and 1 was observed in multiple clinical specimens (patient S8). The 2 cell culture-related changes were noted in isolates from patients S1 (Urbani) and S3. The sequence derived from the isolate from patient S1 had a C at nucleotide position 24872, whereas the matched clinical specimen had a T at this position; all other published SARS-CoV sequences had a T at this position, suggesting that a T→C transition was selected during isolation or cell-culture passage of the virus. The sequence derived from the isolate from patient S3 had a C at nucleotide position 21938, and multiple clinical specimens from patient S3 had a T at that position; again, because all other published sequences have a T at this position, it is likely that the C at nucleotide position 21938 in the isolate from patient S3 was selected during isolation or cell-culture passage of the virus.
Table 2.
Coding-sequence variations in severe acute respiratory syndrome (SARS) co-ronavirus S and N genes, in 10 patients.

The 3 nucleotide substitutions found in patients S4, S7, and S8 are all transitions and are nonsynonymous. The A→G change at nucleotide position 29347 in the N gene from patient S4 leads to a predicted nonconserved amino acid change from asparagine (N) to aspartic acid (D). The T→C change at nucleotide position 22570 in the S gene from patient S8 leads to a predicted amino acid change, from phenylalanine (F) to serine (S). The A→G change at nucleotide position 23445 in the S gene from patient S7 also leads to a predicted amino acid change, from isoleucine (I) to valine (V). Because all 3 changes were present either in 2 or more clinical specimens or in matched specimen/isolate pairs, these changes probably reflect the genetic characteristic of the infecting virus. The 2 cell culture-related changes were synonymous.
Sequence comparisons of SARS-CoV in multiple samples from a single patient. In patient S8, serial stool samples were collected on days 14, 18, 21, and 26 after the onset of SARS. Identical S and N gene sequences were observed in these temporally distinct stool specimens, and, compared with the reference Urbani strain, all showed the same nonsynonymous nucleotide substitution, T22570C (mentioned above).
We also compared S and N gene sequences of different tissue specimens collected, at autopsy, fromthe same individual. Specimens for this evaluation were from the right middle and right upper lung of patient S2 and from the left upper and right middle lung, kidney, and lymph node of patient S3. The S and N gene sequences at these different anatomical sites were identical, in both patients.
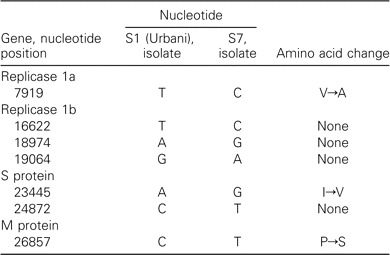
Sequence of the complete genome of the isolate frompatient S7. Analysis of the complete genomic sequence of the isolate from patient S7 underscores the genetic stability of SARS-CoV. In addition to the 2 nucleotide variations already described in the S gene, only 5 additional nucleotide substitutions, including 2 nonsynonymous substitutions, were detected in the remainder of the genome; 1 of them occurred in the predicted M protein, and the other 4 occurred in the predicted polymerase gene (table 3).
Table 3.
Genome-sequence variations in severe acute respiratory syndrome-coronavirus in patient S7.
Discussion
We compared S and N gene sequences of SARS-CoV-positive specimens obtained from 10 patients during 3 months at the height of the 2002–2003 global SARS outbreak and found that the SARS-CoV S and N genes were very stable during this period. Comparison of patients S3 and S9 illustrates this most clearly. Patient S3 represents the starting point for multiple transmission chains emanating from the Hotel M cluster [9]. Although many details of these transmission events remain uncertain, ∼3 months and at least 4 generations of transmission separate the SARS-CoV infection of patient S3 from that of patient S9, an American visitor to Canada [14]. Despite the time interval and these discrete transmission events, we detected no variation between the SARS-CoV S and N gene sequences of clinical specimens from these 2 patients. We also evaluated SARS-CoV S and N gene genetic stability during the course of infection in a single person, by comparing viral genomes from 4 serial stool specimens obtained, between days 14 and 26 after the onset of SARS, from patient S8, and we found the sequences to be invariant. Finally, we compared sequences amplified from lung, lymph node, and kidney tissues obtained at autopsy of patient S3, the index patient of the Hotel M cluster, and again found no variation in the S and N gene sequences of the SARSCoV present in these different tissues.
In all the SARS-CoV S and N genes evaluated, we identified only 3 mutations (2 in the S gene and 1 in the N gene) likely to be present in the viruses infecting 3 different patients. The viruses present in the other 6 patients had S and N gene sequences identical to those of the earliest specimen—that is, that from patient S3. We also identified 2 substitutions in the S gene that were likely to have arisen as a result of cell-culture passage of the virus. The cell culture-related changes underscore the need, when one is analyzing sequence data from isolates, to consider the potential effect of cell-culture changes; for example, 14 SARS-CoV genome sequences based solely on cell culture isolates have been reported to include a total of 127 nucleotide-sequence variations; variations at 4 loci appeared to be phylogenetically significant, and some of them were cell culture-related changes [3].
There are several limitations in the present study. First, as is the case with respect to other RNA viruses, it is likely that SARSCoV in humans is represented by a quasi species and that the sequences that we obtained reflect the average sequence in a specimen and may not represent the full spectrum of variants replicating in an individual; however, our data suggest that individual variants are not prominent, because we did not see, at any loci, sequence ambiguity that would suggest the presence of significant heterogeneity. The second limitation is that the specimens studied were from only 10 patients and were only from a period of 3 months. Furthermore, because we limited our analysis to the S and N gene sequences (i.e., only 5037 nt), it is possible that different patterns of variation may be observed in other regions of the SARS-CoV genome [3]; however, both the conservation in the complete genomic sequence of the isolate from S7 and other published data argue against this. Last, the 3 apparent mutations that we identified were not seen in the other patients in the present study or in sequences published elsewhere, and therefore their significance remains unknown.
Genes that code for proteins that are likely to interact with the host immune response (i.e., S and N proteins) presumably show changes resulting from immunologic selective pressure; however, the data from the present study suggest that the SARSCoV S and N genes were relatively stable during a 3-month period of the outbreak. These data are consistent with the conclusion by The Chinese SARS Molecular Epidemiology Consortium—that is, that the SARS-CoV genome was stable during the late epidemic phase, with emergence of a predominant genotype [4]. Low rates of variation have also been reported for other human coronaviruses (e.g., 229E) [19], whereas still other coronaviruses have been found to have greater variation and higher rates of mutation. If SARS-CoV recurs, it will be important to continue to monitor its clinical, epidemiologic, and genetic patterns, for changes that might be associated with improvements in its ability to infect and replicate in humans and for changes that might present new challenges to the control of it.
Acknowledgements
We acknowledge the support of the following people from the collaborating sites who helped with the collection of specimens: Allison McGeer and Donald Low (Department of Microbiology, Mt. Sinai Hospital, Toronto, Canada); Wilina Lim (Department of Health, Hong Kong, China); Su, Ih-Jen (CDC Taiwan, Taipei); Nguyen Thi Hong Hanh, Hoang Thuy Long, NgHiem Kim Ha, and Nguyen Le Khang Hang (Department of Virology, National Institute of Hygiene and Epidemiology, Hanoi, Vietnam) and staff of the French Hospital (Hanoi, Vietnam); D. Weber and K. Dail (North Carolina Department of Health, Raleigh); F. Alvarez (Utah Department of Health, Salt Lake City); and personnel from the Pennsylvania Department of Health, The Bethlehem Bureau of Health, and Lehigh Valley Hospital, Harrisburg. In addition, personnel at the US Centers for Disease Control and Prevention (Atlanta, Georgia) who assisted with the collection, processing, and sequencing of specimens included Scott Dowell, Daniel R. Feikin, Kathryn Felton, Mike Frace, Elmira T. Isakbaeva, Olen Kew, Nino Khetsuriani, Sara A. Lowther, Yumi Matsuoka, L. Clifford McDonald, Joel M. Mongomery, E. Claire Newbern, Angela Peck, Sheen Scott, Tim M. Uyeki, and Marc-Alain Widdowson.
Footnotes
Presented in part: International Conference on SARS—One Year after the (First) Outbreak, Lübeck, Germany, 8–11 May 2004 (abstract 6.07).
References
- 1.Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 2.Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–9. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Ruan YJ, Wei CL, Ee AL, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361:1779–85. doi: 10.1016/S0140-6736(03)13414-9. [erratum: Lancet 2003;361:1832]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinese SARS Molecular Epidemiology Consortium Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–9. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 5.Chim SS, Tsui SK, Chan KC, et al. Genomic characterisation of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong. Lancet. 2003;362:1807–8. doi: 10.1016/S0140-6736(03)14901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization . Geneva, Switzerland: World Health Organization; 2003. Case definitions for surveillance of severe acute respiratory syndrome (SARS) [Google Scholar]
- 7.Updated interim surveillance case definition for severe acute respiratory syndrome (SARS)—United States, April 29, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:391–3. [PubMed] [Google Scholar]
- 8.Update: severe acute respiratory syndrome—United States. MMWR Morb Mortal Wkly Rep. 2003;52:664–665. [PubMed] [Google Scholar]
- 9.Tsang KW, Ho PL, Ooi GC, et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1977–85. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 10.Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 11.Update: outbreak of severe acute respiratory syndrome—worldwide. MMWR Morb Mortal Wkly Rep. 2003;52:241–6. 248. [PubMed] [Google Scholar]
- 12.Poutanen SM, Low DE, Henry B, et al. Identification of severe acute respiratory syndrome in Canada. N Engl J Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 13.Update: severe acute respiratory syndrome—United States, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:357–60. [PubMed] [Google Scholar]
- 14.Update: severe acute respiratory syndrome—United States, June 11, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:550. [PubMed] [Google Scholar]
- 15.Severe acute respiratory syndrome (SARS) and coronavirus testing—United States, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:297–302. [PubMed] [Google Scholar]
- 16.Severe acute respiratory syndrome—Taiwan, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:461–6. [PubMed] [Google Scholar]
- 17.Tai JH, Ewert MS, Belliot G, Glass RI, Monroe SS. Development of a rapid method using nucleic acid sequence-based amplification for the detection of astrovirus. J Virol Methods. 2003;110:119–27. doi: 10.1016/s0166-0934(03)00108-3. [DOI] [PubMed] [Google Scholar]
- 18.Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 19.Hays JP, Myint SH. PCR sequencing of the spike genes of geographically and chronologically distinct human coronaviruses 229E. J Virol Methods. 1998;75:179–93. doi: 10.1016/S0166-0934(98)00116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]