

Summary
Analysis of endogenous protein localization, function, and dynamics is fundamental to the study of all cells, including the diversity of cell types in the brain. However, current approaches are often low-throughput and resource-intensive. Here we describe a CRISPR/Cas9-based Homology-independent Universal Genome Engineering (HiUGE) method for endogenous protein manipulation that is straightforward, scalable, and highly flexible in terms of genomic target and application. HiUGE employs AAV vectors of autonomous insertional sequences (payloads) encoding diverse functional modifications, that can integrate into any genomic target loci specified by easily assembled gene-specific guide-RNA (GS-gRNA) vectors. We demonstrate that universal HiUGE donors enable rapid alterations of proteins in vitro or in vivo for protein labeling and dynamic visualization, neural circuit-specific protein modification, subcellular rerouting and sequestration, as well as truncation-based structure-function analysis. Thus, the “plug and play” nature of HiUGE enables high-throughput and modular analysis of mechanisms driving protein functions in cellular neurobiology.
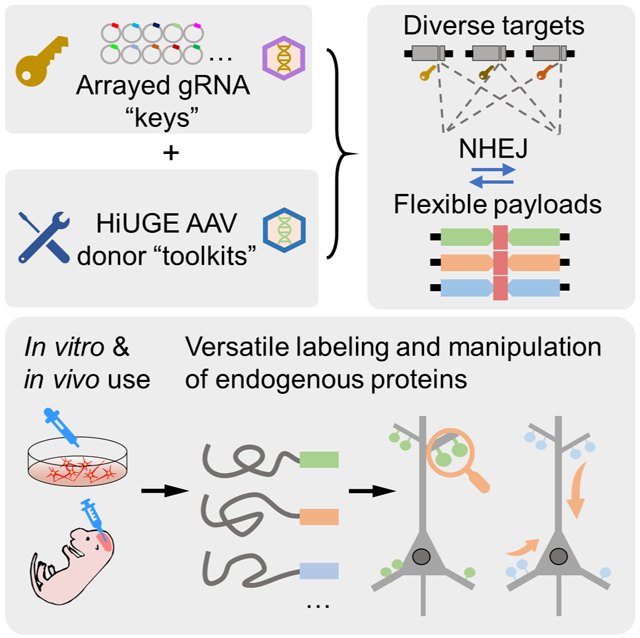
Graphical Abstract
In Brief (eTOC blurb)
Gao et al. developed an AAV-based CRISPR-mediated Homology-independent Universal Genome Engineering (HiUGE) method for high-throughput modification of endogenous proteins, enabling scalable, flexible, and diverse applications such as localization mapping and functional interrogation of proteins in vitro and in vivo.
Introduction
Selective labeling and manipulation of endogenous proteins are essential to delineating the molecular mechanisms of cell and organismal biology. Recent advances in exploratory proteomics and gene expression analysis generate sizable datasets that urgently require high-throughput and reliable methods for protein visualization and functional manipulation purposes. However, current techniques to enable these strategies are often inefficient or resource-intensive. For example, due to the limited availability and the cost of antibodies, it is often not a viable option for single labs to immunolabel tens to hundreds of proteomic candidates. Further, a large fraction of available antibodies may have limited utility due to unsuspected cross-reactivity to other proteins, lot-to-lot variability in quality, and insufficient application-specific validation (Berglund et al., 2008; Bradbury and Pluckthun, 2015; Egelhofer et al., 2011; Michel et al., 2009). Additionally, overexpression of recombinant constructs to map protein localization or conduct functional analyses is also resource-intensive, and can be highly sensitive to available cellular docking sites or unforeseen artifactual cellular effects associated with protein overexpression.
Clustered regularly interspaced short palindromic repeats (CRISPR)-associated endonuclease Cas9-based strategies have great promise to enable highly precise genome editing of mammalian cells to address many of the above limitations (Cong et al., 2013; Doudna and Charpentier, 2014; Hsu et al., 2014; Jinek et al., 2012; Mali et al., 2013). CRISPR-Cas9 introduces double-strand breaks (DSBs) at guide RNA (gRNA) specified genomic sites. These genomic DSBs are repaired via two main pathways in cells; non-homologous end joining (NHEJ) is the prevailing pathway, while homology-directed repair (HDR) is reportedly less frequent, especially in non-dividing cells outside of the S- and G2 phase of cell cycle (Heyer et al., 2010; Hsu et al., 2014; Mao et al., 2008; Saleh-Gohari and Helleday, 2004). Both HDR and NHEJ pathways are currently utilized to insert foreign DNA sequences into genes of interest (GOIs), for applications such as labeling of translated endogenous protein products.
Examples of these methods include single cell labeling of endogenous proteins (SLENDR and viral (v)SLENDR), and homology-independent targeted integration (HITI), both of which have been shown to function in post-mitotic cells, including neurons. SLENDR is based on HDR, using oligonucleotides or Adeno-Associated Virus (AAV) donors containing gene-specific homology sequences to facilitate insertion into GOIs (Mikuni et al., 2016; Nishiyama et al., 2017). Alternatively, HITI leverages NHEJ to insert foreign sequences into a GOI. It utilizes donor vectors containing gene-specific gRNA recognition sequences, which simultaneously direct DSB cuts to the gene and donor vector, facilitating directional insertion during NHEJ (Suzuki et al., 2016). Although SLENDR and HITI are flexible in their ability to modify proteins both in vitro and in vivo for neuronal applications, they both necessitate the cloning of gRNA vectors for the GOIs, as well as customized gene-specific donor vectors for each insertion. By requiring the generation of tailored donor vectors for each DSB cut site in each GOI, the scalability and throughput of testing multiple insertional sequences using either SLENDR or HITI are limited. Alternatively, CRISPaint pairs NHEJ with modular donor vectors that are linearized and integrated into GOIs, and demonstrates improved throughput for insertional editing in cell lines (Schmid-Burgk et al., 2016). However, it requires specially prepared mini-circular vectors that are not compatible with viral delivery methods important for many in vivo applications, or the bacterial vector backbones are necessarily co-inserted into genomes, which can interfere long-term transgene expression (Chen et al., 2003; Chen et al., 2001b). These issues may limit its widespread utility, especially for applications where viral transduction is preferred in vitro or in vivo.
Here we describe Homology-independent Universal Genome Engineering (HiUGE), a new generalized method which obviates many of the constraints to the current state-of-art. HiUGE is an AAV-mediated method that uses a two-vector modular approach for higher-throughput genomic knock-in (KI) applications. The donor vector contains an insertional DNA fragment (payload) that is flanked on both ends by an artificial DNA sequence non-homologous to the target genome. This sequence is recognized by a donor-specific gRNA (DS-gRNA) that directs the Cas9-mediated autonomous excision and release of the payload. The payload can then be integrated across diverse GOIs, both in vitro and in vivo, at genomic loci specified by a separate panel of high-throughput and low-cost gene-specific gRNA (GS-gRNA) vectors. This design frees the donor vectors of any gene-specific sequences, rendering them universally compatible for virtually any CRISPR/Cas9-accessible genomic loci. Importantly, these AAV donors can be premade for ubiquitous applications, greatly simplifying strategies for insertional genome editing. We have tested payloads for a variety of applications and demonstrate their interchangeability within and between GOIs in both cells and tissues. These applications include antibody epitope and fluorescent protein labeling for localization mapping and dynamic visualization of endogenous proteins, protein subcellular re-routing and sequestration, protein truncation for structure-function relationship analysis, and neural circuit-specific genome engineering. Because this method is highly modular, scalable, and suitable for in vitro as well as in vivo applications, it opens new avenues to pair higher-throughput proteomic and genomic applications with experimental validation and phenotypic screening to address molecular mechanisms of cellular neurobiology.
Results
HiUGE Concept and its Specificity to Modify Endogenous Proteins
Recent advances in proteomics and gene expression studies generate sizable protein/gene network datasets, which urgently require novel methods to analyze them on larger scales with greater precision. Higher-throughput genome engineering techniques targeting candidate proteins/genes could satisfy these needs. We thus designed a method utilizing the delivery of universal DNA inserts (payloads) that can ubiquitously integrate across genes to achieve this. In the HiUGE system (Figure 1A), a two-vector approach was used to deliver gene-specific gRNA (GS-gRNA) and universal payloads, with adeno-associated virus (AAV) as the delivery vehicle for its flexible use in vitro and in vivo. HiUGE GS-gRNA vectors are prepared by ligations of 23-24mer oligonucleotides to the backbone vector, which is scalable to generate panels of gRNA expressing vectors targeting diverse GOIs. HiUGE donor vectors are autonomous, expressing a synthetic donor-specific gRNA (DS-gRNA) that is non-homologous to the targeted genome, but directs Cas9-mediated self-cleavage and release of the universal payloads for genomic insertion. This design allows the separation of the donor vector construction from any specific sequence of genomic targets, thus enabling universal applications of the donor “toolkits”. Such utilities include localization mapping and functional manipulation of the endogenous proteins, by introducing tags at their carboxy- or amino-terminus (C- or N-term) within diverse CRISPR/Cas9-accessible genomic loci unlocked by the GS-gRNA “keys”.
Figure 1. Illustration of the HiUGE system.

(A) Schematic of HiUGE method. The HiUGE donor vector expresses a donor-specific gRNA (DS-gRNA), which specifically recognizes the donor recognition sequence (DRS) and directs the cleavage and release of the donor payload for insertion into gene-specific gRNA (GS-gRNA) targeted loci for modification of proteins. (B) HiUGE donor vectors harboring short epitope tags employ a dual-orientation design for efficient expression following either forward or reverse genomic integration. A cassette of stop codons for all six ORFs (denoted by an “X” boxed in red, sequence listed in Table S1) is used to terminate translation. (C) Workflow using a small-scale, low-cost, and higher-throughput AAV supernatant production method for in vitro applications. (D-F) Proof-of-principle data showing HA-epitope KI to mouse Tubb3 gene in primary neuron culture of Cas9 mice after transduction with a combination of small-scale GS-gRNA AAV and HA-epitope donor AAV. (D) Representative (i) confocal and (ii) stimulated emission depletion (STED) images of immunocytochemistry staining for HA-epitope, showing the characteristic microtubule expression pattern of the HA-epitope labeled ßIII-tubulin. GFP fluorescence of the Cas9-2A-GFP, nuclei labeling with DAPI (4',6-diamidino-2-phenylindole), and synaptic marker Synapsin I staining are also shown. (E) Western blot for HA-epitope in a comparison against a negative control (transduced with empty GS-gRNA backbone and HiUGE donor), showing a single band of labeled protein at the expected molecular mass (~51kD) for ßIII-tubulin. (F) Representative sequencing results showing correct integration in forward and reverse orientation. Regions of DNA sequence containing genomic Tubb3, DRS (blue shade), restriction enzymes (yellow shade), HA-epitope (green shade) and stop codon cassette (red box) are indicated. Truncated terminal amino acids are noted. Translated amino acid sequences encoded by the payload and the in-frame stop codon (red asterisk) are indicated as well.
To test this concept, a dual-orientation payload design was used to facilitate the expression of an HA- epitope tag following either forward or reverse integration into the C-term of proteins of interest (Figure 1B). We adopted a protocol to produce small-scale transduction-ready AAVs for higher-throughput use in vitro, complimenting traditional AAV purifications for use in vivo (Figure 1C and Methods). As a proof-of-concept, small-scale AAVs of a GS-gRNA targeting the mouse Tubb3 gene and a dual-orientation HA-epitope HiUGE donor were used to co-transduce primary neurons prepared from neonatal pups of conditional Cas9 mice. The GS-gRNA AAV also expresses Cre-recombinase to induce Cas9-2A-GFP expression. Approximately 1 week following infection, immunofluorescent detection of HA-epitope showed successful labeling of endogenous βIII-tubulin, with localization characteristics specific to microtubules (Figure 1D & Figure S1A). Western blot detection of the HA-epitope showed a single band (~51kD) consistent with the predicted molecular mass of βIII-tubulin (Figure 1E). Genomic insertion of the payload was verified by sequencing the Tubb3 locus, confirming the successful HA-epitope integration (Figure 1F). In control experiments, the relative selectivity of frame-dependent labeling was tested by comparing the correct ORF donor (ORF+1, in reference to the Cas9 cleavage locus specified by the Tubb3 gRNA), to out-of-frame donors (ORF+0 or ORF+2) (Figure S1B i-iii). HA-labeling in the correct ORF was substantially more efficient (Figure S1C), with occasional HA-positive cells observed from out-of-frame ORFs, indicating comparatively less frequent frame-shifting insertion or deletion (indel) events during payload integration. Negative controls demonstrated the absolute requirement for both GS-gRNA and HiUGE donor vectors for HA-epitope KI (Figure S1B iv-vi). To demonstrate that donor vectors for all 3 ORFs are equally capable of facilitating HiUGE, three GS-gRNAs, one for each ORF, were designed to target the mouse Map2 gene. Pairing these GS-gRNA AAVs with HA-epitope donor AAVs in their corresponding ORFs resulted in comparable and efficient labeling (Figure S1D & E).
An important consideration is that the HiUGE donor vectors should be suitable to target diverse coding sequences without introducing premature stop codons that prevent proper expression of the insert. This is achieved by using donor recognition sequences (DRS) specifically designed such that premature stop codons cannot be introduced during genomic integration. We compared HA-epitope donor vectors with different DRS sequences and found similar cellular labeling efficiencies amongst them (Figure S2A-C), demonstrating flexibility in DRS usage. Also, we found that the cellular labeling efficiency for HiUGE was comparable to HITI (Figure S2D-I).
Higher-Throughput Modification of Diverse Protein Targets by Universal HiUGE Payloads
We next tested whether a single HiUGE payload can be integrated across diverse genomic loci and label multiple target proteins for subcellular localization mapping. We used oligonucleotide ligation to rapidly construct an arrayed panel of GS-gRNA AAV vectors targeting the C-term of twelve proteins, which exhibit previously described and highly patterned subcellular localizations in neurons and glia. These included proteins of the microtubule network (βIII-tubulin, Tubb3; Microtubule associated protein 2, Map2), the nucleus (Methyl-CpG-binding protein 2, Mecp2, (Chen et al., 2001a)), the synaptic actin cytoskeleton (Actin-related protein 2, Actr2, (Kim et al., 2013; Spence and Soderling, 2015)), clathrin-coated vesicles (Clathrin light chain A, Clta), the axonal initial segment (AIS) (Neuronal adhesion molecule, Nrcam; Ankyrin-G, Ank3; βIV-spectrin, Sptbn4; Sodium channel subunit Nav1.2, Scn2a, (Jones and Svitkina, 2016)), intermediate filaments (Glial fibrillary acidic protein, Gfap), mitochondria (Pyruvate dehydrogenase E1 component subunit alpha, Pdha1), and the distal tips of neurites (Doublecortin, Dcx, (Francis et al., 1999; Gdalyahu et al., 2004)). Colony screening demonstrated that the cloning reactions were highly efficient (Figure S1F-H). Primary neuron / glia cultures of Cas9 mice were co-transduced with one of these GS-gRNA AAVs, in combination with the same HA-epitope donor AAV (Figure 2A). Approximately seven days following infection, we observed the predicted localization of the twelve protein targets by HA immunostaining (Figure 2B-M). Further, genomic PCRs were performed for five targets to amplify the region around the edited junctions. HA-epitope integrations were confirmed (Figure S3A & B), with deep sequencing of the amplicons revealing the positional indel occurrences, the proportions of forward and reverse payload insertion without indels, and the proportions of allelic mutations without donor integration (Figure S3C). We also tested for the integration of donor payloads in top-ranked predicted off-target sites and found no evidence for payload insertion (Figure S3D-E). In addition, experimental genome-wide analysis was performed to detect potential off-target payload integrations, including those at non-predicted sites. We found that while genomic off-target integrations can occur, their estimated frequencies are low (Figure S3F-H). In each instance the observed off-target integration was into non-coding regions that would not result in unintended labeling of non-target proteins. Further, the cellular labeling efficiencies across various conditions were quantified for several AIS-specific targets, as each neuron typically has only one AIS, and their distinct expression pattern is suitable for easy visual identification and quantification. The ratios of HiUGE-labeled AIS structures compared to all AIS (stained with an antibody against the AIS-marker Ankyrin-G) were quantified (Figure S4A-C). Estimated efficiencies were shown across multiple viral doses (Sptbn4: 34%; Scn2a: 29% at the high dose), and across various ratios between the GS-gRNA and the donor (ratios around 1:1 are optimal). Further, we tested HiUGE-mediated labeling to visualize several members of the recently uncovered inhibitory postsynaptic density (iPSD) proteome (Uezu et al., 2016). Spaghetti-monster HA (smFP-HA), a larger insert that exhibits enhanced antigenicity suitable for detecting low-expression proteins (Viswanathan et al., 2015), was used as the payload (Figure 2A). These iPSD proteomic candidates included Inhibitory Synaptic protein 1 (Insyn1), Inhibitory Synaptic protein 2 (Insyn2), and Rho GTPase Activating Protein 32 (Arhgap32). Immunocytochemistry demonstrated that they were localized juxtaposed to the Vesicular GABA Transporter (VGAT) (Figure 2N-P), confirming their presence at inhibitory synapses. In addition, three different GS-gRNAs (Table S1) were tested for Insyn1, and all of them yield comparable inhibitory synaptic labeling (Figure S4D-G), demonstrating the flexibility of GS-gRNA selection and the robustness of the labeling method. Finally, we performed mixed labeling of diverse protein targets by applying a single C-term HA-epitope donor with a mixture of GS-gRNA AAVs (Figure S4H-I). Immunocytochemistry revealed simultaneous labeling of the targeted proteins (βIV-spectrin, GFAP, and MeCP2) in a single experiment, and provided a proof-of-concept for other applications that require simultaneous modification of multiple proteins.
Figure 2. Rapid Protein Modification Across Diverse Genomic and Protein Targets with HiUGE In Vitro.

(A) Schematic of HiUGE KI application for C- or N-term protein labeling in vitro. Primary hippocampal cells from Cas9 mice were transduced with a combination of GS-gRNA and HiUGE donor AAVs and immunostained to detect epitope or smFP-HA labeling, with representative images displayed in panels B-S. (B-M) Examples of C-term HA-epitope KI to diverse targets (mouse Tubb3, Map2, Mecp2, Actr2, Clta, Nrcam, Ank3, Sptbn4, Scn2a, Gfap, Pdha1, and Dcx), showing the expected expression patterns of the translated proteins respectively. (N-P) C-term smFP-HA KI to mouse Insyn1, Insyn2, and Arhgap32, which encode the inhibitory postsynaptic density (iPSD) proteomic candidates. Colocalization of the HA-immunoreactivity with the juxtaposed inhibitory presynaptic marker vesicular GABA transporter (VGAT) immunosignal is shown in the insets. (Q-S) N-term Myc-epitope KI to mouse Actb, Lmnb1, and Nefm, showing the expected expression patterns of the translated proteins respectively. Scale bar is indicated in each panel, or within insets (2μm). GFP fluorescence of the Cas9-2A-GFP and nuclei labeling with DAPI are also shown. Arrowheads represent the subcellular features associated with the targeted genes, such as the dendritic spines, AIS, mitochondria, distal end of neurites, inhibitory synapses, and neurofilaments.
Next, we tested N-term modifications using a single Myc-epitope payload with an upstream stop codon cassette to constrain its expression to the N-term (Figure 2A). GS-gRNAs targeting the N-term of three proteins were tested, including proteins of the actin cytoskeleton (ß-Actin, Actb), nuclear envelope (Lamin B1, Lmnb1), and neurofilaments (Neurofilament medium, Nefm). Myc-epitope immunolabeling, consistent with the expected localization pattern, was observed for each target (Figure 2Q-S). Further, like previous studies (Mikuni et al., 2016), we tested selective dual-labeling of different proteins with different tags. This is achieved by combining the N-term selective Myc-epitope payload with the HA-epitope HiUGE payload described above, which contains a stop codon cassette following the epitope tag that enforces its expression at the C-term (Figure S4J). These donors were co-infected with GS-gRNA AAVs targeting the N-term of dendritic MAP2 (Map2) and the C-term of AIS-enriched bIV-spectrin (Sptbn4). Co-staining for HA and Myc-epitopes revealed selective labeling for each protein, with instances of co-staining in single neurons (Figure S4K). Thus, HiUGE N- and C-term selective payloads can be used to co-label proteins simultaneously to reveal the spatial relationships of two endogenous proteins, which may be more useful than the mixed labeling of multiple proteins with identical tags described above.
Together, using rapidly constructed GS-gRNA libraries, HiUGE integrates universal payloads into diverse protein coding regions in a highly specific and higher-throughput manner for mapping endogenous protein localizations. HiUGE is thus suitable for rapid and robust labeling of proteins, including post hoc localization analysis following large-scale proteomic studies.
In Vivo HiUGE Labeling of Proteins via Viral Delivery
We next tested in vivo labeling by intracranial injections of HiUGE AAVs (Figure 3A). Approximately 2 weeks following infection, immunohistochemistry was performed. Images of sections from each injection revealed the expected HA immunoreactivity localized to the AIS (Sptbn4 and Scn2a; Figure 3B & C), microtubules (Tubb3; Figure 3D), and nuclei (Mecp2; Figure 3E). Of note, the detection for each of four targets was consistent across independent cells with no visible mis-localization, suggesting off-target protein coding integration was undetectable or rare. In addition, local injections of HiUGE AAVs into the motor cortex of adult Cas9 mice were performed, and the cellular labeling efficiencies for the AIS-localized targets at the injection sites were quantified (Sptbn4: 30%; Scn2a: 23%. Figure 3F-J). These data demonstrated that a single HiUGE donor can be paired with multiple GS-gRNAs to modify diverse endogenous proteins in vivo.
Figure 3. Endogenous Protein Labeling with HiUGE In Vivo.

(A) Schematic of C-term HA-epitope KI in vivo. Neonatal Cas9 pups were intracranially injected with a combination of purified GS-gRNA AAV and HiUGE donor AAV, and euthanized after P15 for immunohistochemistry to detect HA-epitope KI. (B-E) Representative images of HA-epitope KI to mouse Sptbn4, Scn2a, Tubb3, and Mecp2 gene, showing the expected expression patterns of the translated proteins respectively. Cortical regions of coronal sections are shown. Image on the right within each panel shows a magnified view of the boxed region from the left image. (F) Adult Cas9 mice were locally injected with a combination of purified GS-gRNA and HA-epitope donor AAVs (titer of 3 × 1010 GC / μL) into the motor cortex and euthanized 2 weeks later for immunohistochemistry. (G-H) Representative immunofluorescent images of HA-epitope (green) following HiUGE-mediated KI to mouse Sptbn4 and Scn2a. Antibody labeling of the AIS-marker (ßIV-spectrin) is also shown (red). (I-J) Quantification of cellular labeling efficiency showing the fractions of the ßIV-spectrin-positive AIS-structures labeled with HiUGE at the injection sites (Sptbn4: 30.2 ± 3.4 %; Scn2a: 22.8 ± 2.9 %, n=3 mice), in comparison to negative controls (uninjected, 0%). Error bars represent standard error of the mean (SEM). Scale bar is indicated in each panel.
Multiplex Labeling Demonstrates HiUGE Donor Payloads are Interchangeable at Single Genomic Locus.
While the above data demonstrated that a single HiUGE payload could integrate across diverse gene targets in vitro and in vivo, an additional feature of the HiUGE method is that the donor payloads should be interchangeable for multiplexing and flexible selection of protein modifications. The ability to mix-and-match premade HiUGE donors would simplify the experimental selection of optimal epitopes, diverse fusion proteins, or fusion proteins with variable linkers. To demonstrate the interchangeability of HiUGE payloads, we modified proteins either in vitro or in vivo with a combination of epitope tags (HA, Myc, and/or V5) to create a mosaic labeling of infected cells (Figure 4A). Immunostaining of cultured neurons infected with a mixture of these HiUGE epitope payload AAVs paired with GS-gRNA AAV targeting the Tubb3 locus revealed a visual montage of multi-colored βIII-tubulin labeling, with occasional double-labeled cells indicating the two alleles of Tubb3 were modified by different epitopes (Figure 4B, magenta neurons). Thus, after genomic cleavage directed by the GS-gRNA, different HiUGE payloads can be inserted interchangeably. Further, multiplexed labeling was also tested in vivo, where a mixture of HiUGE epitope payload AAVs were co-injected with the Tubb3 GS-gRNA AAV into neonatal pups intracranially. Two weeks following infections, immunohistochemistry revealed extensive multiplexed labeling of neurons throughout multiple brain regions of the cerebrum (Figure 4C). Higher magnification images (Figure 4D) demonstrated heterogeneous epitope labeling of ßIII-tubulin in neighboring neurons of the cortex, hippocampus, and thalamus, as well as projections representing thalamo-cortical connectivity, globus pallidus bundles, and corpus callosum fibers. Together, these data demonstrated the interchangeability of HiUGE donor payloads within a single gene locus, thus enabling flexible selection of diverse protein modifications.
Figure 4. HiUGE Donor Payload Interchangeability for Multiplexed Protein Modification.

(A) Schematic of C-term KI of epitope mixture in vitro and in vivo. (B) Labeling of HA, Myc, and V5-epitope following mosaic KI to mouse Tubb3, showing stochastically integrated epitopes in neighboring neurons in vitro. Occasionally, cells positive for two epitopes can be seen (e.g. the magenta-colored cell in this image, showing both HA and V5 immunoreactivity). (C) Coronal section demonstrating labeling of HA and Myc-epitope following mosaic KI to mouse Tubb3 in vivo. (D) Zoomed images showing the cortex, hippocampus, thalamus, thalamo-cortical projections, globus pallidus, and corpus collosum of panel (C). Scale bar is indicated in each panel.
Neural Circuit-based HiUGE Payload Delivery
We further reasoned that it should be possible to limit the genome editing activity to specific neural circuits by utilizing a recently reported retrograde-transported AAV2-retro serotype (Tervo et al., 2016). Injection of AAV2-retro GS-gRNA into a brain area containing axon terminals of a circuit could be paired with injection of AAV2/9 HiUGE donor into a specific projection region, allowing retrograde access of projection neurons from a target brain region for neural circuit-selective protein manipulations. We tested this possibility in the well-defined cortico-striatal circuit and the thalamo-cortical circuit. For the cortico-striatal circuit, Tubb3 GS-gRNA AAV2-retro was injected to the striatum, whereas individual AAV2/9 HiUGE donor AAVs were injected laterally into either the primary motor cortex (MOp, HA-epitope) or secondary motor cortex (MOs, Myc-epitope) of adult conditional Cas9 mice (Figure 5A). Retrograde access to the projection neurons in the motor cortex was confirmed by positive GFP labeling, indicating Cre-dependent activation of Cas9-2A-GFP (Figure 5B). HiUGE-edited projection neurons within the motor cortex were clearly delineated by HA and Myc-epitope labeled βIII-tubulin, corresponding to the injection sites (Figure 5C & D). Bundles of GFP-positive fibers were also observed in the striatum representing cortico-striatal projections (Figure 5E & F). Individual axons immuno-positive for either the HA or Myc-epitope, which presumably originated from neurons of different motor cortical sub-regions, were present within neighboring cortico-striatal axon bundles (Figure 5G & H). Similar circuit-selective HA-epitope tagging of ßIII-tubulin within thalamic projection neurons was also observed in the thalamo-cortical circuit (Figure 5I-K). Thus, retrograde infection of projection neurons with AAV2-retro enabled HiUGE-mediated protein modification specific to neural circuit connectivity.
Figure 5. Neural Circuit-based HiUGE Labeling.

(A) Illustration of cortico-striatal circuit-selective C-term labeling of ßIII-tubulin by injection of AAV2-retro mouse Tubb3 GS-gRNA into the striatum and 2 lateral injections of AAV2/9 HA and Myc-epitope donors in the motor cortex. (B) Representative image showing GFP labeling in the motor cortex, indicating retrogradely accessed Cre-dependent Cas9– 2A-GFP expression in projection neurons. (C) Immunolabeling of HA (arrows) and Myc-epitope (arrowheads) tagged ßIII-tubulin, imaged from the boxed area in (B). (D) Enlarged images from the boxed areas in (C), showing cells positive for (i) HA or (ii) Myc-epitope. (E) GFP signal from the AAV2-retro injected striatum. (F) Zoomed image of the boxed area in (E), showing GFP-positive axon bundles that contain fibers positive for HA or Myc-epitope. (G, H) Enlarged images showing fibers positive for (i) HA or (ii) Myc-epitope within GFP-positive axon bundles from boxed areas in (F). (I) Illustration of thalamo-cortical circuit-selective C-term labeling of ßIII-tubulin by injection of AAV2- retro mouse Tubb3 GS-gRNA in the somatosensory cortex and injection of AAV2/9 HA-epitope donor in the thalamus. (J) Representative image showing retrogradely activated Cas9-2A-GFP expression within the thalamus (boxed area) and local cortical networks (mostly cells within layer II/III and layer VI). (K) Zoomed image of the boxed area in (J), showing retrogradely accessed and HiUGE edited thalamic neurons positive for HA-epitope (arrows) and Cas9-2A-GFP. Scale bar is indicated in each panel.
Diverse HiUGE Payloads Enable Approaches to Study Endogenous Protein Dynamics and Functions.
Modification of endogenous proteins in mammalian cells in order to delineate their physiological functions is a highly promising area of CRISPR-genome editing. Leveraging the “plug and play” interchangeability of HiUGE payloads, we tested various payload sequences for studying the dynamics and functions of endogenous proteins.
Fluorescent proteins (FPs) are commonly used for imaging protein localization and dynamics. However, conventional experiments usually rely on exogenic overexpression of fusions that are susceptible to artifacts associated with the elevated expression levels. To test HiUGE-mediated KI of FPs to label endogenously expressed proteins, the coding sequence for mCherry (Shaner et al., 2004) containing a flexible linker was used as the payload to tag several genes (Figure 6A), including mouse Tubb3 (Figure 6B), Gfap (Figure 6C), and Pdha1 (Figure 6D). HiUGE-mediated KI of mCherry was readily visualized by either immunostaining (Figure 6B), or by direct fluorescence (Figure 6C & D). Similar labeling pattern was also observed using a mCherry donor without the flexible linker (Figure S4L & M). If desired, donor vectors with or without linkers can be paired with panels of GS-gRNA vectors to rapidly optimize the labeling strategy. Further, HiUGE-mediated mCherry labeling was also sufficient for live imaging (Movie S1), showing the dynamics of mitochondria following KI of mCherry into mouse Pdha1. Together, HiUGE-mediated KI of FPs can facilitate dynamic visualization of endogenous proteins.
Figure 6. HiUGE Payloads Enable Flexible Selection Across Multiple Methods to Interrogate Endogenous Protein Functions.

(A) Schematic of fluorescent protein (FP) KI with flexible (GGGGS)4 linker. (B-D) Detection of mCherry (mCh) KI to different genomic targets (mouse Tubb3, Gfap, and Pdha1) with (B) antibody against mCh, or (C, D) direct visualization of mCh fluorescence. (E) Schematic of HiUGE subcellular re-routing constructs using cellular trafficking tags as the payloads. (F) Example using a HiUGE payload with HA-epitope and nuclear localization signal (HA-NLS) to sequester an actin cytoskeletal protein Arp2 to the nucleus. (G) Representative image of immunostaining following C-term HA-NLS KI to mouse Actr2 gene, showing the HA-NLS-tagged Arp2 (red) redirected to the nucleus. Simultaneously, the Myc-epitope (no NLS) tagged Arp2 (green) was enriched at the dendritic spines, consistent with the normal localization of Arp2. (H) Schematic illustration of HiUGE HA-3’ untranslated region (3’-UTR) KI to truncate endogenous proteins for conducting structure-function relationship studies. (I) Example using HA-3’UTR KI strategy to truncate ßIV-spectrin, composed of calponin-homology (CH) domains, spectrin repeats, and a pleckstrin homology (PH) domain. (J) C-term KI using a GS-gRNA targeting near the stop codon of the last coding exon 36 (e.36) of ßIV-spectrin is compared to (K) truncation of PH domain by targeting exon 31 (e.31), which still retains AIS enrichment, or (L) disruption of spectrin repeat 14 and truncation of downstream sequences by targeting exon 26 (e.26), which completely disrupts the AIS localization. (M) Western blot of HA-epitope showing stepwise reduction of molecular mass consistent with the serial truncation conditions (arrowheads). Arrowheads indicate the Σ1 isoform of ßIV-spectrin, while arrows indicate the Σ6 isoform. The Σ6 isoform of the truncation at exon 26 (e.26) appeared undetectable. Molecular mass is marked in reference to the ladder. Scale bar is indicated in each panel.
As proper localization is key to the physiological actions of proteins, one mechanism to alter protein function is to manipulate their subcellular residency with exogenous localization tags (Bear et al., 2000; Niopek et al., 2014). A classic example is the nuclear localization signal (NLS) from SV40 T-antigen that targets proteins into the nucleus via importins (Kalderon et al., 1984; Kosugi et al., 2009; Lange et al., 2007). Here we chose Arp2 (Actr2), an actin-regulating protein that is normally enriched in dendritic spines to exemplify subcellular rerouting and sequestration using HiUGE. A dual-orientation HA-NLS HiUGE payload (Figure 6E) was targeted to the C-term of mouse Arp2 to determine if this cytoskeletal protein could be relocated to the nucleus (Figure 6F). Neurons were transduced with mouse Actr2 GS-gRNA AAV and a mixture of HA-NLS and Myc-epitope (no NLS) HiUGE payloads. HA-NLS-tagged Arp2 (red) was completely redirected to the nuclei, whereas the Myc-tagged Arp2 (green) was mostly found in the dendritic spines, the normal localization of Arp2 (Figure 6G). Thus, HiUGE payloads of subcellular trafficking tags, such as an NLS, can be harnessed to manipulate the subcellular residency of endogenous proteins, for purposes such as loss-of-function or gain-of-function experiments.
Protein truncation experiments are crucial for identifying functional domains, unveiling protein interactions, and delineating structure-function relationships. Once again, conventional methods typically rely on over-expression of exogenic constructs. To enable truncation studies of endogenously expressed proteins, an HA-Stop-3’UTR HiUGE payload was designed (Figure 6H). This construct enables the labeling of truncated endogenous proteins with an epitope, while facilitating escape from non-sense mediated decay by the addition of the 3’UTR containing a poly-adenylation sequence. This application is exemplified by truncation of ßIV-spectrin, as it is enriched at the AIS and contains modular domain structures (Figure 6I). Pairing the GS-gRNA targeting the last coding exon near the stop codon (exon 36 of the canonical Σ1 isoform) resulted in strong HA-epitope immunoreactivity restricted to the AIS (Figure 6J). Targeting exon 31 to delete the pleckstrin homology (PH) domain from ßIV-spectrin did not disrupt its AIS localization (Figure 6K), consistent with previous work suggesting the PH domain is not required for its localization to the AIS (Yang et al., 2007). Remarkably, targeting the more upstream exon 26 to truncate ßIV-spectrin within the 14th spectrin repeat completely abrogated its AIS-enriched immunoreactivity and resulted in a diffuse cytosolic staining (Figure 6L). These results demonstrated that the region between the 14th spectrin repeat and the PH domain of ßIV-spectrin is required for its enrichment within the AIS. This is consistent with previous studies that indicate ßIV-spectrin interacts with the AIS-scaffold protein Ankyrin within the 14th-15th spectrin repeats (Ipsaro et al., 2008; Kennedy et al., 1991; Yang et al., 2007), suggesting this interaction is important for its proper AIS localization. Further, Western blot analysis confirmed a stepwise reduction of the protein molecular mass that was consistent with the serial truncation (Figure 6M). Thus, HiUGE enables rapid serial truncation to interrogate structure-function relationships associated with underlying cellular mechanisms.
Together, libraries of HiUGE payloads with different functional moieties facilitate independent approaches to study endogenous proteins. Due to the plug and play nature of HiUGE, other DNA sequences, such as an inducible degron tag (Natsume et al., 2016; Nishimura et al., 2009), can also be incorporated as payloads to be delivered to diverse genomic loci, opening a myriad of new possibilities for gene/protein modification and manipulation.
All-in-One HiUGE Vectors Utilizing Intein-Based Cas9 Protein Trans-splicing.
Thus far, the HiUGE system relies on additional Cas9 expression strategies. To simplify the application in diverse wild-type (WT) cells and animals, we incorporated the sequences for Cas9 expression into the vectors. To overcome the constraint of AAV packaging size, spCas9 was split between the donor and GS-gRNA vectors, with each half incorporating Npu split-intein sequences (Cheriyan et al., 2013; Truong et al., 2015). Intein-mediated protein trans-splicing would thus enable the reconstitution of full-length spCas9, facilitating HiUGE applications in WT targets (Figure 7A-B). This design was tested in WT primary neurons and common cell lines using HA-epitope or GFP payloads paired with GS-gRNAs targeting either mouse Tubb3, Map2, Pdha1, Tubb5, or human TUBB genes. Immunostaining of AAV-transduced neurons showed successful HA-epitope and GFP KI (Figure 7C-H). Likewise, HiUGE vectors delivered by plasmid co-transfection into common cell lines such as HeLa, HEK293T, and NIH3T3 also showed successful HA-epitope and GFP KI (Figure 7I-N). Specificity control experiments showed that the incorporation of the HA-epitope or GFP payloads requires both the GS-gRNA and the donor vectors (Figure S5A). Like the HiUGE system without intein-split-Cas9, we found that around 1: 1 ratio between the GS-gRNA and donor is optimal, although a higher overall viral concentration was needed (Figure S5B-D). Further, intein-split-Cas9 mediated HiUGE was tested in vivo. Purified AAVs targeting mouse Map2 were unilaterally co-injected into the dorsal hippocampus of adult WT mice. Two weeks after infection, HA-epitope immunostaining revealed robust MAP2 labeling in dendrites and cell bodies within the injected hippocampus, but not on the contralateral uninfected side (Figure 7O & P). Together, HiUGE with intein-split-Cas9 simplifies its application in WT cells and animals.
Figure 7. HiUGE Vectors with Intein-mediated Split-Cas9 Trans-splicing.

(A) Schematic of the HiUGE system that harbors intein-mediated split-Cas9 expression for use in WT cells and animals. (B) Illustration of delivering interchangeable payloads to diverse genomic loci using these vectors. (C-H) Wild-type (WT) primary mouse neurons were AAV transduced and (I-N) common human or mouse cell lines were plasmid transfected with a combination of GS-gRNAs and HiUGE donors, followed by immunostaining for HA-epitope or GFP to detect payload KI. (C, E, G) Representative images of HA-epitope KI to the mouse Tubb3, Map2 and Pdha1 genes, showing the expected (C, E) microtubule and (G) mitochondrial localization of the tagged proteins. (D, F, H) Representative images of GFP KI to the mouse Tubb3, Map2 and Pdha1 genes, showing the expected (D, F) microtubule and (H) mitochondrial localization of the tagged proteins. (I, K, M) Representative images of HA-epitope KI to the human TUBB or mouse Tubb5 gene, showing the expected microtubule localization of the tagged proteins in (I) HeLa cell, (K) HEK293T cell and (M) NIH3T3 cell. (J, L, N) Representative images of GFP KI to the human TUBB or mouse Tubb5 gene, showing the expected microtubule localization of the tagged proteins in (J) HeLa cell, (L) HEK293T cell and (N) NIH3T3 cell. (O) Representative image of HA-epitope KI to mouse Map2 gene following local AAV injection in the dorsal hippocampus of adult WT mice, showing efficient labeling of the neurons at the injection site compared to negative labeling on the contralateral side. Zoomed view of the boxed area is shown in (P). Scale bar is indicated in each panel.
Discussion
Here we report the development of HiUGE, a simplified approach that allows the flexible targeting of multiple insertional payloads across GOIs to rapidly manipulate and engineer endogenous proteins in vitro and in vivo. We demonstrate that single HiUGE donor vectors can be used to modify multiple proteins at their C- or N-terminus, and that multiple HiUGE payloads of different functional moieties can be inserted into a single protein. Thus, HiUGE payloads are universal with respect to gene identity, provided that the modifications (insertion of the tag and linker / truncation of the terminal amino acids) are functionally tolerated, and retained during post-translational maturation of the protein products (some proteins may be post-translationally processed at both their C- and N-termini, thus inherently difficult to target). Overall, because custom donor vectors to deliver foreign sequences into GOIs are no longer needed, HiUGE dramatically increases the throughput and scalability of localization mapping and functional interrogation of endogenous proteins. To utilize HiUGE, a simple ligation of 23-24mer duplexed oligos into the GS-gRNA backbone vector is sufficient to unlock multiple applications to study gene/protein functions based on homology-independent insertions of diverse HiUGE payloads. Thus, testing different antibody epitopes, fluorescent protein fusions, linker amino acid sequences, and functional modifiers, which are common steps for optimizing protein labeling and manipulation, is easily achievable. Prior methods such as HITI or SLENDR would require additional tailored donor vectors for each optimization strategy. Further, due to the molar excess of oligos, the cloning reaction of GS-gRNA vectors is highly efficient, and easily scalable to tens to hundreds of protein targets by single labs. Moreover, we also note a high success-rate for protein labeling during the development of HiUGE (24 genes successful tagged out of 25 tested). We anticipate that HiUGE will enable higher-throughput mapping and functional interrogation of proteomes, which is urgently needed for analyzing larger-scale proteomic and genomic datasets.
The HiUGE method we outline relies on NHEJ, which has advantages to approaches dependent on HDR. First, while HDR is less frequently observed outside of the S- and G2 phases (Heyer et al., 2010; Hsu et al., 2014; Saleh-Gohari and Helleday, 2004), NHEJ repair is operational at all phases of the cell cycle (Mao et al., 2008; Suzuki et al., 2016). Furthermore, quantitative analysis of HDR versus NHEJ suggests the latter is the prevailing repair pathway following DSBs (Mao et al., 2008). Thus, NHEJ-based applications for genome engineering may be more broadly applicable than HDR-dependent approaches. Moreover, dual-orientation design is used for C-term epitope-tag payloads, which allows translation and expression of the tag following both forward and reverse NHEJ-mediated integration. This design maximizes the potential of efficient protein modification and is especially desirable for analyses that require large populations of edited cells. Optimization by testing several GS-gRNAs, which is easy to accomplish with HiUGE, can also be done to identify the most efficient ones.
An important consideration of any CRISPR-based application is the fidelity of genome modification and availability of genomic sites for Cas9-mediated cleavage. One potential drawback to HiUGE, as well as all CRISPR-based approaches, is the formation of indels at the targeted loci. For this reason, phenotypic analysis following these approaches should rely on cells without indels or multiple independent cells to confirm the observation. Rates of indels are likely reduced by the availability of experimental insertional fragments, such as the HiUGE payloads we describe, that disrupt genomic GS-gRNA recognition sites after integration, thus abrogating repetitive rounds of cleavage and repair. Besides, for C-term HiUGE payloads, frame-shifting indel events at the integration site abrogate the detectability or reactivity of the translated inserts, thus effectively removing them from the analysis. For N-term fusions, the consistency of labeled proteins across cells is especially important as frame-shifting indel events at the integration site could potentially lead to protein depletion or the expression of payloads tagged with wrongly encoded peptides in some cells. However, detectable expressions of frame-shifted N-term fusions are likely to be very limited due to nonsense-mediated decay triggered by premature stop codons (Baker and Parker, 2004; Hug et al., 2016). Further, off-target insertion of HiUGE payloads can be reduced by computational methods to minimize the selection of gRNAs with potentially high off-target liability (Bae et al., 2014; Haeussler et al., 2016). By genome-wide analysis we found rare off-target integrations of the payload (Figure S3F-H). In each instance the integration was into non-coding regions and did not result in unintended labeling of non-target proteins. Additionally, the consistency of protein labeling across cells within an experimental condition demonstrates that the cell-to-cell variability due to off-target integration was minimal enough to prevent detectable expression from non-target coding regions. High-fidelity spCas9 variants, including eSpCas9, SpCas9-HF1, and HypaCas9, are reported to have less off-target tolerance (Chen et al., 2017; Kleinstiver et al., 2016; Slaymaker et al., 2016), and can be incorporated with the HiUGE method to further improve specificity. Likewise, additional variants of spCas9 with less stringent PAM sequence usage, such xCas9 and SpCas9-NG (Hu et al., 2018; Nishimasu et al., 2018), could further increase the flexibility of selecting genomic targets, placing them immediately adjacent to either the natural start or stop sites and minimizing the undesired truncation of terminal amino acids.
We have demonstrated that HiUGE is easily adopted for a variety of applications, exemplified here by: (1) localization mapping and dynamic visualization of endogenous proteins; (2) simultaneous N-term and C-term labeling of multiple proteins; (3) targeted re-routing and sequestration of protein subcellular residency; and (4) protein truncation for disrupting its function or performing structure-function relationship analysis. The donor payloads can be easily interchanged with other custom DNA sequences for additional applications. Further, neighboring neurons within the brain can be heterogeneously modified with diverse payloads, enabling multiplexed labeling that might facilitate lineage tracing or cellular connectomics studies. Moreover, by using AAV2-retro, projection neurons can be retrogradely accessed and edited by HiUGE based on circuit connectivity. This novel approach opens future possibilities for interrogating molecular mechanisms of behaviorally relevant neural circuits with rapid insertional genome engineering.
To generalize HiUGE for applications in WT cells and organisms without relying on Cas9 transgenics, we have shown that the intein-split-Cas9 mediated HiUGE is functional in diverse targets, including multiple cell-types, species of origin, and delivery route. We envision that HiUGE can be adapted to broader range of organisms, such as models of avian or aquatic species, by modularly optimizing the following components: (1) the promoter driving gRNA expression; (2) the promoter driving Cas9 expression; (3) Cas9 codon optimization, and (4) viral serotype / delivery route. We also anticipate that cell-type selectivity can be achieved by using selective promoters to restrict Cas9 expression within the cell-type of interest, facilitating differential genome modification across diverse subpopulations of cells. Together, the higher-throughput and modular features of HiUGE to modify endogenous proteins are especially desirable downstream of genomic and proteomic screens that generate hypotheses requiring further analysis. Using HiUGE, we successfully labeled the recently uncovered iPSD proteins Insyn1 and Insyn2, and confirmed their endogenous localization at inhibitory synapses. We anticipate the “plug and play” nature of HiUGE we describe here will facilitate future studies that leverage multiple functionalities to delineate the molecular mechanisms underlying cellular neurobiology.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. Scott Soderling (scott.soderling@duke.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh (Cas9) and C57BL/6J mice
For in vivo studies, Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh mice (Cas9 mice, Jackson Laboratory) or wild-type (WT) C57BL/6J mice (Jackson Laboratory) were used. For intracranial injections, neonatal pups (P0-P2, undetermined sex) were used. Sex determination at this development stage is both visually difficult and unnecessary for the experiment. For stereotaxic local injections, adult male mice (> 8 weeks of age) were used. Mice were group-housed in the Duke University's Division of Laboratory Animal Resources facility. All procedures were performed with a protocol approved by the Duke University Institutional Animal Care and Use Committee in accordance with US National Institutes of Health guidelines.
Primary mouse hippocampal and cortical neuron / glia culture
Primary neuron / glia cultures were derived from Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh or C57BL/6J (Jackson Laboratory) neonatal pups (P0-P2). Primary culture was performed following a previously described method (Uezu et al., 2016). Briefly, after euthanasia, brains were rapidly collected, and hippocampi or cortices were isolated. Cells were gently dissociated with fire polished glass pipettes following papain digestion (Worthington), and plated at a density of ~100,000 cells per cm2 (for imaging, hippocampal cells), or ~300,000 cells per cm2 (for DNA / protein preparation, cortical cells) on poly-L-lysine (Sigma P2636) coated surfaces. Cells were then maintained in Neurobasal A medium (ThermoFisher 10888022) supplemented with 2% B27 (ThermoFisher 17504044) and 1% GlutaMAX (ThermoFisher 35050061) at 37 °C, 5% CO2. Glia growth was inhibited by adding cytosine arabinoside (Sigma C1768, 5uM final concentration) together with media change between DIV 4-6, when necessary.
Cell culture of HEK293T, HeLa, and NIH3T3 cell lines.
Cell lines used in this study include HEK293T (ATCC CRL-11268), HeLa (ATCC CCL-2), and NIH3T3 (ATCC CRL-1658). All cell lines were maintained in DMEM (Gibco 11965-092) supplemented with 10% fetal bovine serum (Sigma F4135) and 1% Penicillin-Streptomycin (ThermoFisher 15140122) at 37 °C, 5% CO2, and passaged by trypsin/EDTA digestion (ThermoFisher 25200056) upon reaching ~95% confluency.
METHOD DETAILS
Construction of HiUGE Vectors.
Both the gene-specific vector and the HiUGE donor vector were derived from a backbone vector AAV:ITR-U6-sgRNA-hSyn-Cre-2A-EGFP-KASH-WPRE-shortPA-ITR (a gift from Feng Zhang (Platt et al., 2014), Addgene plasmid # 60231).
Gene-specific gRNA (GS-gRNA) vector backbone were prepared by removing the HA-2A-EGFP-KASH fragment from the backbone plasmid. These vectors express GS-gRNAs driven by a U6 promoter, as well as Cre-recombinase driven by a neuronal specific hSyn promoter. For gRNA targeting non-neuronal cells (glia, etc.), the hSyn promoter was swapped with the ubiquitous Ef1α promoter to drive Cre expression. These vectors were used in conjunction with Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh mice (Cas9 mice) for Cre-dependent Cas9 expression. For each gene of interest (GOI), GS-gRNA sequences targeting either the N-term or the C-term of the GOI were designed using an online gRNA evaluation tool “CRISPOR” (Haeussler et al., 2016), and cloned into the SapI site using a highly efficient (typically > 90% positive rate of colony picking, Figure S1F-H) restriction digestion / ligation cloning method. In brief, 10ng (~2fmol) of the GS-gRNA backbone plasmid was digested with SapI enzyme (NEB R0569, 1uL) and simultaneously ligated with 50fmol of annealed 23-24mer (including SapI sticky ends) GS-gRNA oligos with T4 DNA ligase (NEB M0202, 1uL) in a 10uL reaction, by 10 repeats of thermocycling between 37°C (5 min) and 21°C (5 min). Colony PCR was performed to detect GS-gRNA integration into the backbone plasmid using a forward primer in the upstream U6 promoter region, paired with the reverse GS-gRNA oligo as the reverse primer to amplify an ~100bp amplicon. GS-gRNA target sequences used in this study were listed in Table S1.
For HiUGE donor vectors, a donor-specific gRNA (DS-gRNA) was cloned into the SapI site behind the U6 promoter. The fragment between the XbaI and PmlI restriction sites was replaced with payload sequences to be inserted to the targeted genomic loci, such as epitope tags (HA, Myc, and V5), fluorescent proteins (Shaner et al., 2004; Zacharias et al., 2002), “spaghetti monster” fluorescent protein- HA (a gift from Loren Looger (Viswanathan et al., 2015), Addgene plasmid # 59759), and cellular trafficking tag (SV40 nuclear localization signal, NLS). The payload sequences were flanked on both ends by a donor recognition sequence (DRS), that is an artificial sequence not present in the human or mouse genome and is specifically recognized by the DS-gRNA with low genomic off-target liability (exemplary DS-gRNA has a CRISPOR specificity score of 98 or higher against human or mouse genome, with no genomic targets within 3-basepair mismatches), as predicted by CRISPOR and Cas-OFFinder (Bae et al., 2014; Haeussler et al., 2016; Hsu et al., 2013). The DS-gRNA directs Cas9-ediated DRS cleavage and release of the payload sequence to be inserted into the targeted genomic locus. Exemplary DRS and payload sequences used in this study can be found in Table S1.
In addition, to facilitate genome editing in wild-type cells and animals without extra source of Cas9 expression, an intein-split-Cas9 system (Truong et al., 2015) was used to distribute Cas9 coding sequences to both the GS-gRNA vector and the HiUGE donor vector. In this case, the GS-gRNA vectors were constructed by replacing hSyn-Cre with the EF1α/HTLV (nEF) promoter (nEF-Cas9, a gift from Juan Belmonte (Suzuki et al., 2016), Addgene plasmid # 87115) followed by C-intein-C-Cas9 coding sequences between XbaI and PmlI sites. The donor vectors were constructed by the addition of N-Cas9- N-intein coding sequences, also driven by the nEF promoter, behind the payload sequences of the donor vectors.
AAV Production.
Purified AAV was produced following a previously described method (Uezu et al., 2016). Briefly, HEK293T cells were plated on six 15cm dishes and transfected with 15¼g HiUGE AAV vector (either GS-gRNA or donor payload), 30μg helper plasmid pAd-DeltaF6, and 15μg AAV serotype 2/9 plasmid per six dishes with PEI Max (Polysciences 24765) when reaching 60-80% confluency. Cells were then incubated in DMEM with glutamine and pyruvate (Corning 10-013-CV), supplemented with 10% FBS (Sigma F4135). Cells were harvested 3 days after transfection and lysed with 3 repeats of freeze-thaw cycles, followed by treatment with Benzonase (Novagen 70664) at 37°C for 30 minutes. Cell lysate was then applied to an Optiprep density gradient (Sigma D1556, 15%, 25%, 40% and 60%) for ultracentrifugation purification. AAV-enriched fraction was collected, then further concentrated by repeated washes with sterile PBS in an Amicon Ultra-15 filtration unit (NMWL: 100kDa, Millipore UFC910008) to a final volume of ~100μL and aliquoted for storage at −80°C. For preparing AAV2-retro, 15μg rAAV2-retro plasmid (a gift from Alla Karpova & David Schaffer (Tervo et al., 2016), Addgene plasmid # 81070) was used as the serotype plasmid. Similar transfection and viral purification procedure described above was used. Real-time PCR reactions were performed with PowerUp SYBR Green Master Mix (Thermo A25741) using primers targeting the ITR of AAV2 to measure viral titers against copy number standards (Aurnhammer et al., 2012). Primer sequences are listed in Table S1. Titers of purified AAVs used in this study were: Sptbn4 C-term GS-gRNA (AAV2/9 serotype. Batch 1: 4.7×1011 GC / μL, Figure 3B; Batch 2: 2.9ϗ1011 GC / μL, Figure S3A-C; Batch 3: 3.9ϗ1011 GC / μL, Figure 3F-J & S4A-C; Batch 4: 1.8×1011 GC / μL, Figure S2D-F); Scn2a C-term GS-gRNA (AAV2/9 serotype. Batch 1: 4.7×1011 GC / μL, Figure 3C & S3F-H; Batch 2: 9.7×1011 GC / μL, Figure S3A-E; Batch 3: 3.6×1011 GC / μL, Figure 3F-J, S2A-C & G-I, S4A-B; Batch 4: 9.6×1010 GC / μL, Figure S2G-I); Tubb3 C-term GS-gRNA (AAV2/9 serotype. Batch 1: 6.7×1011 GC / μL, Figure 3D & 4C-D; Batch 2: 2.7×1011 GC / μL, Figure S1B-C); Mecp2 C-term GS-gRNA (AAV2/9 serotype. 6.4×1011 GC / μL, Figure 3E); Map2 C-term GS-gRNA (AAV2/9 serotype. 4.4×1011 GC / μL, Figure S3A-C); Actr2 C-term GS-gRNA (AAV2/9 serotype. 1.3×1011 GC / μL, Figure S3A-C); Clta C-term GS-gRNA (AAV2/9 serotype. 2.3×1011 GC / μL, Figure S3A-C); empty GS-gRNA (AAV2/9 serotype. Batch 1: 9.1×1011 GC / μL, Figure S1B-C; Batch 2: 2.7×1011 GC / μL, Figure S2D-I); HA-epitope donor (AAV2/9 serotype, ORF+1. Batch 1: 1.4×1011 GC / μL, Figure 3A-E, 4C-D & S3F-H; Batch 2: 1.1×1011 GC / μL, Figure 5 & S1B-C; Batch 3: 1.8×1010 GC / μL, Figure S3A-E; Batch 4: 3.8×1010 GC / μL, Figure S3A-C; Batch 5: 1.4×1011 GC / μL, Figure S4A-C; Batch 6: 3.4×1010 GC / μL, Figure 3F-J & S2A-C; Batch 7: 1.2×1010 GC / μL, Figure S2D-I; Batch 8: 2.2×1010 GC / μL, Figure S2D-I); HA-epitope donor (AAV2/9 serotype, ORF+0. 1.3×1010 GC / μL, Figure S1B-C); HA-epitope donor (AAV2/9 serotype, ORF+2. 5.9×1010 GC / μL, Figure S1B-C); HA-epitope donor (AAV2/9 serotype, ORF+1 with DRS-2. 2.2×1010 GC / μL, Figure S2A-C); HA-epitope donor (AAV2/9 serotype, ORF+1 with DRS-3. 1.6×1010 GC / μL, Figure S2A-C); Myc-epitope donor (AAV2/9 serotype, ORF+1. 2.4×1011 GC / μL, Figure 4C-D & 5A-H); Tubb3 C-term GS-gRNA (AAV2-retro serotype. 2.7×109 GC / μL, Figure 5); Map2 C-term GS-gRNA with intein-split-Cas9 (AAV2/9 serotype. 7.6×1010 GC / μL, Figure 7O-P); Sptbn4 C-term GS-gRNA with intein-split-Cas9 (AAV2/9 serotype. 8.5×1010 GC / μL, Figure S5B-D); HA-epitope donor with intein-split-Cas9 (AAV2/9 serotype, ORF+1. Batch 1: 3.0×1010 GC / μL, Figure 7O-P, Batch 2: 2.2×1011 GC / μL, Figure S5B-D); Sptbn4-HITI HA-epitope donor (AAV2/9 serotype, 1.6×1011 GC / μL, Figure S2D-F); Scn2a-HITI HA-epitope donor (AAV2/9 serotype, 8.1×1010 GC / μL, Figure S2G- I). For quantitative experiments, AAVs were added to primary cultures at a final concentration of 5×1010 GC / mL per virus in the culture medium, unless otherwise stated.
For small-scale AAV supernatant, HEK293T cells were plated on a 12 well plate, then transfected with 0.4μ AAV plasmid, 0.8μ helper plasmid pAd-DeltaF6, and 0.4μ serotype 2/1 plasmid per well with PEI Max (Polysciences 24765) when cell density reached 60-80% confluency. Cells were then incubated in glutamine-free DMEM (ThermoFisher 11960044) supplemented with 1% Glutamax (ThermoFisher 35050061) and 10% FBS (Sigma F4135) for 3 days. The AAV-containing supernatant medium was collected and filtered through a 0.45 μm cellulose acetate Spin-X centrifuge tube filter (Costar 8162), and temporarily stored at 4°C for no more than 2 weeks. Titers of small-scale AAVs were typically 108 - 109 GC / μL.
Stereotaxic Injections
For stereotaxic injections, adult mice were anesthetized through inhalation of 1.5% isofluorane gas and placed in a stereotaxic frame (Kopf Instruments). Mice were administered meloxicam (~10 μL / 25 g) subcutaneously before the beginning of surgery to reduce inflammation. After confirming that Lambda and Bregma were on the same dorsal-ventral plane, craniotomies were made with a high speed drill (Foredom MH-170) over either motor cortex (0.5 A/P, 0.6-1.0 L, 1.2 V), striatum (0.5 A/P, 2.0 L, 3.2 V), thalamus (−1.7 A/P, 1.2 L, 3.4 V), posterior somatosensory cortex (−1.7 A/P, 3.5 L, 1.2 V), or dorsal hippocampus (−1.6 A/P, 2.2 L, 1.3 V), in reference to the Allen Mouse Brain Atlas (Lein et al., 2007). Using a precision pressure injection system (Drummond Nanoject), a glass pipette filled with virus was lowered to the desired depth, slightly retracted (~0.2mm), and small amounts of virus were injected over a period of ~10 minutes (30 injections of 18-32 nL every 20 seconds). After waiting for an additional 5-10 minutes to prevent efflux of virus during pipette retraction, the glass pipette was retracted from the brain and the skin over the craniotomy was sutured shut. After applying several drops of topical anesthetic to the incision (bupivacaine) and administering an analgesic subcutaneously (buprenorphine, ~25 μL / 25 g), mice were allowed to recover under a heat lamp for 20-30 minutes and then placed in their home cage.
In Vitro and In Vivo Protein Modification by HiUGE.
For in vitro qualitative experiments, primary cultures were prepared using neonatal pups (P0-P2) of Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh mice (Cas9 mice), and transduced with combinations of small-scale AAV supernatants (GS-gRNA and donor payload). The filtered supernatants were added directly to primary cultures together with media change on DIV 4-6 for transduction (100-200 μL per well on a 24 well plate). NMDA receptor antagonist D-APV (Tocris 0106, ~50μM final concentration) was added to protect neurons against toxicity associated with media change (Hogins et al., 2011). Alternatively, combinations of small-scale AAV supernatants were added to primary cultures at DIV 1 (in this case D-APV is not necessary); and incubated together with the neurons until the day of fixation to extend the accumulation period of the nascently modified proteins. For quantitative in vitro experiments, purified AAVs were added to primary cultures with the final concentration of 5×1010 GC / mL per virus in the culture medium, unless otherwise stated. On the day of fixation (DIV 11-14), cells were treated with 4% PFA + 4% sucrose in PBS for 15 minutes at 4°C and used for immunocytochemistry to detect HiUGE-mediated KI. For qualitative in vivo applications, purified high-titer AAVs were combined (GS-gRNA and donor payload, 1:1 v/v) and intracranially injected (~2μL per hemisphere) to neonatal pups of Cas9 mice (P0-P2). For quantitative in vivo experiments, purified AAVs were combined with a final titer of 3×1010 GC/μL for each virus, and locally injected to the motor cortex of adult Cas9 mice. For neural circuit-selective applications, adult Cas9 mice received a stereotaxic injection of purified AAV2-retro GS-gRNA virus into the brain area containing axon terminals of a specific neural circuit, whereas separate injections of purified AAV2/9 HiUGE donor virus were delivered into the brain area containing projection neuron cell bodies. 2-3 weeks after injection, mice were euthanized, and brains were isolated either fresh frozen with dry ice, or after transcardial perfusion, for immunohistochemistry to detect HiUGE-mediated KI.
For HiUGE vectors that have built-in intein-split-Cas9 expression, primary culture derived from WT C57BL/6J mice (P0-P2) or other common cell lines (HeLa, HEK293T, NIH3T3) were used. Small-scale AAVs were used to transduce primary neurons as described above in similar applications. Further, to demonstrate that these HiUGE components were also suitable to be delivered via plasmid transfection rather than AAV transduction in common cell lines of human or mouse origin, HiUGE GS-gRNA and donor plasmids were co-transfected into HeLa, HEK293T, or NIH3T3 cells with PEI MAX (Polysciences 24765) or Lipofectamine 3000 (ThermoFisher L3000008) following manufacturer's protocol. Cells were fixed 2-4 days after transfection for immunocytochemistry to detect HiUGE-mediated KI. For in vivo testing, purified high-titer AAVs (GS-gRNA and donor payload) were combined, then stereotaxically injected to specific brain areas of adult WT mice. 2-3 weeks after injection, mice were euthanized, and transcardially perfused. Brains were isolated for immunohistochemistry to detect HiUGE-mediated KI.
Immunohistochemistry and Immunocytochemistry.
For immunohistochemistry (IHC), both fresh frozen specimens and perfused specimens were used. For fresh frozen specimens (when working with AIS proteins), the animal was euthanized, and its brain was quickly isolated, frozen on crushed dry ice, coronally sectioned at 20μm thickness and mounted on glass slides (VWR). The specimen was fixed with 4% PFA + 4% sucrose in PBS for 15 minutes at 4°C. IHC was then performed. The specimen was blocked and permeabilized with blocking buffer containing 0.3% Triton-X, 5% normal goat serum (NGS, Sigma) in PBS for 30-60 min at room temperature, and primary antibodies were applied for 2 hr at room temperature or at 4°C overnight. Fluorescent secondary antibodies were applied for 30-60 min at room temperature, and counterstained with DAPI. For perfused specimens (when working with non-AIS proteins), the animal was euthanized and transcardially perfused with ice cold heparinized (25U / mL) PBS, followed by 4% PFA in PBS. Brains were harvested and post-fixed in 4% PFA overnight at 4°C and cryoprotected in 30% sucrose + 10% glycerol in PBS at 4°C. Brains were coronally sectioned at 40-80μm thickness for free-float antibody incubation. Similar IHC incubation conditions as described above were used for free-float sections 50μm or thinner. For thicker sections (>50μm), incubation durations were doubled to allow sufficient penetration of antibody. The specimens were then mounted to glass slides and coverslipped with FluorSave reagent (Millipore 345789).
For immunocytochemistry (ICC), cells grown on glass coverslips were fixed with 4% PFA + 4% sucrose in PBS for 15 minutes at 4°C, then blocked and permeabilized with blocking buffer containing 0.3% Triton-X, 5% NGS in PBS for 30-60 min at room temperature. Primary antibodies were applied for 2 hr at room temperature or at 4°C overnight. Fluorescent secondary antibodies were applied for 30-60 min at room temperature, and counterstained with DAPI. The coverslips were then mounted to glass slides with FluorSave reagent (Millipore 345789).
Primary antibodies used for IHC/ICC were listed as following with dilutions indicated in parentheses: rat anti-HA (Roche/Sigma 11867423001, 1:1000), mouse anti-HA (BioLegend MMS-101P, 1:250), rabbit anti-Myc (Santa Cruz sc-789, 1:250), mouse anti-V5 (ThermoFisher R960-25, 1:500), chicken anti-GFP (Abcam ab13970, 1:2000), rabbit anti-mCherry (Abcam ab167453, 1:1000), rabbit anti-MAP2 (Synaptic Systems 188002, 1:5000), mouse anti-Ankyrin G (Santa Cruz sc-12719, 1:250), rabbit anti-βIV-spectrin (ThermoFisher PA5-62972, 1:1000), guinea pig anti-VGAT (Synaptic Systems 131004, 1:2000), guinea pig anti-Synapsin I (Synaptic Systems 106104, 1:250), mouse anti-βIII-tubulin (Sigma T8660, 1:1000). Fluorophore-conjugated secondary antibodies or reagents used for IHC/ICC were listed as following: goat anti-mouse Alexa 405 (ThermoFisher A31553 1:1000), donkey anti-rat Alexa 488 (ThermoFisher A21208, 1:1000), goat anti-chicken Alexa 488 (ThermoFisher A11039 1:1000), goat anti-rabbit Alexa 555 (ThermoFisher A21428, 1:1000), goat anti-rat Alexa 568 (ThermoFisher A11077 1:1000), goat anti-rabbit Alexa 568 (ThermoFisher A11036, 1:1000), goat anti-guinea pig Alexa 594 (ThermoFisher A11076, 1:100 for STED), goat anti-rat Alexa 594 (Jackson ImmunoResearch 112-585-167, 1:1000), donkey anti-rabbit Alexa 647 (ThermoFisher A31573, 1:1000), donkey anti-mouse Alexa 647 (Jackson ImmunoResearch 715-605-150, 1:1000), goat anti-mouse Alexa 647 (Jackson ImmunoResearch 115-605-166, 1:1000), donkey anti-guinea pig Alexa 647 (Jackson ImmunoResearch 706-605-148, 1:1000), goat anti-mouse Atto 647N (Sigma 50185, 1:100 for STED).
Microscopic Fluorescence Imaging
Microscopic images were acquired with Zeiss 710 or Zeiss 880 inverted confocal microscopes, or with Zeiss Imager.M2 upright microscope equipped with an Apotome module. Stimulated emission depletion (STED) images were acquired on a Leica STED system. Exposures were manually adjusted for optimal dynamic range. Tiling and z-stack were performed when necessary. Tiled images were stitched in Zeiss Zen for composite presentation (Figures 3, 4, 5, and 7). Brightness, contrast, and gamma correction were adjusted for each channel using Zeiss Zen or FIJI (Schindelin et al., 2012; Schneider et al., 2012). When comparisons against control conditions were necessary, identical exposure and adjustment parameters were used across the cohort. Live imaging was performed on Zeiss 710 microscope at 37°C, with 5% CO2 incubation. Time series of z-stacked images were acquired every 100s, then processed and exported at 6 fps in FIJI as videoclip followed by annotation in Adobe After Effects. All images were pseudocolored for presentation with color assignments indicated in each panel.
Western Blot
Cells were collected in ice cold RIPA buffer supplemented with proteinase inhibitor cocktail and sonicated. Protein concentrations were determined by BCA method and equal amounts of protein were heated in sample buffer at 95°C for 5 min, loaded for SDS/PAGE electrophoresis, then transferred to a nitrocellulose membrane. Following blocking in blocking buffer (Rockland MB-070), the membrane was sequentially probed for HA-epitope (primary: rat anti-HA, Roche/Sigma 11867423001, 1:2000; secondary: IRDye 800CW goat anti-rat, 1:10,000) and house-keeping gene GAPDH (primary: rabbit anti-GAPDH, Abcam ab9485, 1:5000; secondary: IRDye 680RD goat anti-rabbit, 1:10,000). Both primary and secondary antibodies were incubated for 1hr at room temperature. Immunofluorescence signal was detected on Odyssey FC imager (LI-COR). For immunoprecipitation (IP)-enriched Western blot to detect the serial truncation of βIV-spectrin, equal amount of inputs (300μg) were mixed with 30μL mouse anti-HA beads (ThermoFisher 26181) and incubated at 4°C overnight on a nutator. After washes with RIPA buffer, proteins were eluted by boiling in 80μL sample buffer at 95°C for 5 min. Equal volume of the eluted IP-samples and equal amount of the input samples were loaded for SDS/PAGE and then transferred to nitrocellulose membranes. The IP-membrane was sequentially probed for HA-epitope (primary: Sigma H6908, 1:2000; secondary: IRDye 800CW goat anti-rabbit, 1:10,000) and the input membrane was probed for house-keeping gene GAPDH (primary: Abcam ab9485, 1:5000; secondary: IRDye 680RD goat anti-rabbit, 1:10,000). Both primary and secondary antibodies were incubated for 1 hr at room temperature in CanGetSignal solutions (TOYOBO NKB-101T). Immunofluorescence signal was detected and processed on an Odyssey FC imager (LI-COR).
Genomic PCR, TOPO Cloning and DNA Sequencing.
Genomic DNA was extracted by MyTaq Extraction-PCR kit (Bioline BIO-21127) or PureLink Genomic DNA Mini Kit (ThermoFisher K182001) from primary neurons after HiUGE editing. Genomic polymerase chain reaction (PCR) using MyTaq HS polymerase (Bioline BIO-25045 or BIO-25047) was performed to detect the insertion of the dual-orientation HA-epitope payload using primers indicated in Table S1. A primer specific for the endogenous genomic sequence upstream of the edited locus was paired with an orientation-selective HA-epitope payload primer to amplify the edited genomic locus and differentiate forward versus reverse payload integration. PCR products were isolated by gel purification (NucleoSpin Gel and PCR Clean-up kit, Macherey Nagel 740609) and TOPO-cloned (ThermoFisher K457501) for sequencing using a common M13F(−21) primer (Eton Bioscience). For indel estimation, genomic DNA were extracted from triplicate neuronal samples following HiUGE-mediated HA-epitope KI. Genomic PCRs were performed using primer pairs that were either insert-specific (described above), or across the edited junction. The PCR products were cleaned up (NucleoSpin Gel and PCR Clean-up kit, Macherey Nagel 740609), combined into three independent pools, and deep sequenced on a MiSeq system (Illumina) by the Duke Sequencing and Genomic Technologies Shared Resource.
Assessment of genomic off-target integration.
The GS-gRNA targeting mouse Scn2a, which has a mediocre CRISPOR specificity score of 66 out of 100 (best), was used as an example to assess the off-target effects of HiUGE-mediated genome editing. First, genomic primers surrounding the top CRISPOR predicted off-target loci (3 for DS-gRNA, 5 for Scn2a GS-gRNA) were designed using NCBI Primer-BLAST (Ye et al., 2012) (sequences listed in Table S1). Genomic DNA was prepared from HiUGE-edited primary neurons incubated with a combination of Scn2a GS-gRNA and HA-epitope donor, using PureLink Genomic DNA Mini Kit (ThermoFisher K182001). PCR reactions were performed by pairing the genomic primer with the insert-specific primer to detect genomic integration of the HA-epitope payload in either forward or reverse orientation. Meanwhile, PCR using pairs of forward and reverse genomic primers spanning the off-target loci were performed as controls. PCR products were electrophoresed on agarose gel and imaged under UV light. Further, genome-wide identification of potential off-target integration of the payload was performed using Genome Walker kit (Clontech 636406). Genomic DNA was prepared from in vivo edited mouse brain injected with a combination of Scn2a GS-gRNA and HA-epitope donor, using DNeasy Blood & Tissue Kit (Qiagen 69504). Restriction enzyme digestion, adapter ligation and nested PCR was performed according to the manufacturer’s protocol (primer sequences listed in Table S1), followed by band purification and sequencing. Three potential genomic off-target integrations of the payload were identified. Primers surrounding these sites were designed (sequences listed in Table S1). PCR reactions were performed by pairing the genomic primer with the insert-specific primer to detect genomic integration of the HA-epitope payload in either forward or reverse orientation. In addition, real-time PCR was performed in duplicates using SensiFAST Real-Time PCR Kit (Bioline BIO-86005).
QUANTIFICATION AND STATISTICAL ANALYSIS
For immunofluorescence quantification of C-term HA-epitope KI to mouse Tubb3, microscopic images were taken by an experimenter blinded to the experimental groups, using identical exposure parameters across the cohort. Three coverslips were used per experimental group, with three microscopic images taken per coverslip at random locations. The HA immunofluorescence intensity of an image was quantified by FIJI after automatic background subtraction. The total cell count of an image was estimated by particle analysis of DAPI fluorescence in FIJI. A ratio of mean HA immunoreactivity over cell count was computed, then normalized on a scale of 0-100 arbitrary unit (a.u.), against the negative group (no virus added) and the positive group (Tubb3 GS-gRNA plus HiUGE donor ORF+1). The normalized ratio was defined as the “mean fluorescence intensity”. Also, cells positive for HA immunoreactivity were tallied and their percentages over total cell counts were calculated. The quantification results were averaged for each coverslip and reported (n=3). For immunofluorescence quantification of C-term HA-epitope KI to AIS protein targets in vitro, microscopic images were taken by an experimenter blinded to the experimental groups, using identical exposure parameter across the cohort. Three coverslips were used per experimental group, with three microscopic images taken per coverslip at random locations. The number of AIS structures positive for HA-epitope immunofluorescence and the total number of Ank-G-positive AIS were manually counted by a blinded experimenter, then converted into percentages for analysis. The quantification results were averaged for each coverslip and reported (n=3). For quantification of HiUGE labeling efficiency in vivo, three adult Cas9 mice were stereotaxically injected with HiUGE AAVs into the motor cortex per group and analyzed by immunohistochemistry 2 weeks following injection. Four microscopic images were taken for each mouse at the injection site from separate coronal sections. The number of AIS structures positive for HA-epitope immunofluorescence and the total number of βIV-Spectrin-positive AIS structures were manually counted by a blinded experimenter, then converted into percentages for analysis. The quantification results were averaged for each mouse and reported (n=3). For the comparison experiment with HITI, the Cre-expressing empty GS-gRNA AAV was used to induce Cas9 expression in primary neuronal cultures from Cas9 mice, together with the HA-epitope HITI donor vectors to facilitate genome editing. Two targets (mouse Sptbn4 and Scn2a) were edited by either HiUGE or HITI with 7 coverslips used for each group. Three microscopic images were taken per coverslip at random locations by a blinded experimenter. The number of AIS structures positive for HA-epitope immunofluorescence and the total number of Ank-G-positive AIS were manually counted by a blinded experimenter, converted into percentages, averaged for each coverslip, and normalized to the HITI group of each gene to assess the comparative efficiencies between the two methods (n=7). To detect pairwise differences, two-tailed t-test or one-way ANOVA followed by post-hoc Tukey-Kramer HSD test was performed in JMP Pro (v13, SAS), with significance level set at 0.05.
For quantitative estimation of the indel rate at the edited junctions upstream of the payload, raw singleend read sequences of the insert-specific PCR products were trimmed of adapter and poor-quality sequences using the Trim Galore Toolkit (v 0.5.0, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), which employs Cutadapt (Martin, 2011) (v 1.18). The quality of the raw reads was verified by examining the FastQC (v 0.11.5, https://www.bioinformatics.babraham.ac.uk/projects/fastqc) output from Trim Galore. A reference FASTA file containing the amplicon sequence for each of the genes by integration (forward and reverse) combinations was prepared, which included ten sequences in total. Quality-trimmed reads were aligned to the reference FASTA file with bwa-mem (Li and Durbin, 2009) (v0.7.17) with default parameters and post-processed with SAMtools (Li et al., 2009) (v1.9). A custom Python script was used to filter the SAM file for aligned reads with the supplementary alignment (0×800) flag, and to filter for reads with overlap of the integration site. Specifically, the start alignment position of the read was used to verify that the read started at least 30bp downstream of the integration site and the cigar string was parsed to verify that the read extended past 30bp upstream of the integration site; the integration site position varied with each gene. For cumulative read counts, the SAM file was then parsed with a custom Python script and each read that contained an indel was tallied. For positional read counts, Picard (http://broadinstitute.github.io/picard) was used to add read group information and index the bam file. Indels were then called with GATK HaplotypeCaller (McKenna et al., 2010) (v3.8-1-0-gf15c1c3ef). In order to call indels on all reads, some parameters were modified from default (min_mapping_quality_score=0; min_base_quality_score=0, max_alternate_alleles=50, output_mode=EMIT_ALL_SITES, emitRefConfidence=BP_RESOLUTION). The resulting VCF file was then filtered to exclude any sites with single nucleotide polymorphisms and to count the number of reads at each site containing an insertion or deletion. For all analyses, each of the three pools were treated independently until calculation of mean and standard error of the mean for both positional and cumulative data.
For quantitative estimation of allelic mutation rate without donor insertion, raw paired-end read sequences of the across-junction PCR products were trimmed of adapter and poor-quality sequences using Trimmomatic (Bolger et al., 2014) and demultiplexed using Cutadapt (Martin, 2011). The quality of the raw reads was verified by examining the FastQC (v 0.11.8). The paired-end reads were merged with FLASH (Magoc and Salzberg, 2011). The merged reads were then aligned to reference amplicon sequences using EMBOSS Needle (Chojnacki et al., 2017). Finally, the aligned reads were analyzed using CRISPResso for quantification of position and type of mutations within the amplicon sequences (Pinello et al., 2016). Rates of allelic mutations without donor insertion were estimated by the fractions of reads with mutations within the population that lack donor integration. For all analyses, each of the three pools were treated independently until calculation of mean and standard error of the mean.
Supplementary Material
Supplemental Table S1 DNA Sequences of gRNA Targets, Donor Payloads, and Primers, Related to the STAR Methods.
Supplemental Video S1 Live Imaging of Neuron with HiUGE-mediated mCherry Labeling of Pyruvate Dehydrogenase E1 Component Subunit Alpha (Pdha1), Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-HA | Roche/Sigma | Cat#11867423001 |
| Rabbit polyclonal anti-HA | Sigma | Cat#H6908 |
| Mouse monoclonal anti-HA | BioLegend | Cat#MMS-101P |
| Mouse monoclonal anti-HA Agarose | ThermoFisher (Pierce) | Cat#26181 |
| Rabbit polyclonal anti-Myc | Santa Cruz | Cat#sc-789 |
| Mouse monoclonal anti-V5 | ThermoFisher | Cat#R960-25 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970 |
| Rabbit polyclonal anti-mCherry | Abcam | Cat#ab167453 |
| Rabbit polyclonal anti-MAP2 | Synaptic Systems | Cat#188002 |
| Mouse monoclonal anti-Ankyrin G | Santa Cruz | Cat#sc-12719 |
| Rabbit polyclonal anti-βIV-Spectrin | ThermoFisher | Cat#PA5-62972 |
| Guinea pig polyclonal anti-VGAT | Synaptic Systems | Cat#131004 |
| Guinea pig polyclonal anti-Synapsin I | Synaptic Systems | Cat#106104 |
| Mouse monoclonal anti-βIII-tubulin | Sigma | Cat#T8660 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat#ab9485 |
| Goat anti-mouse Alexa 405 | ThermoFisher | Cat#A31553 |
| Donkey anti-rat Alexa 488 | ThermoFisher | Cat#A21208 |
| Goat anti-chicken Alexa 488 | ThermoFisher | Cat#A11039 |
| Goat anti-rabbit Alexa 555 | ThermoFisher | Cat#A21428 |
| Goat anti-rat Alexa 568 | ThermoFisher | Cat#A11077 |
| Goat anti-rabbit Alexa 568 | ThermoFisher | Cat#A11036 |
| Goat anti-guinea pig Alexa 594 | ThermoFisher | Cat#A11076 |
| Goat anti-rat Alexa 594 | Jackson ImmunoResearch | Cat#112-585-167 |
| Donkey anti-rabbit Alexa 647 | ThermoFisher | Cat#A31573 |
| Donkey anti-mouse Alexa 647 | Jackson ImmunoResearch | Cat#715-605-150 |
| Goat anti-mouse Alexa 647 | Jackson ImmunoResearch | Cat#115-605-166 |
| Donkey anti-guinea pig Alexa 647 | Jackson ImmunoResearch | Cat#706-605-148 |
| Goat anti-mouse Atto 647N | Sigma | Cat#50185 |
| Goat anti-rat IRDye 800CW | LI-COR | Cat#925-32219 |
| Goat anti-rabbit IRDye 800CW | LI-COR | Cat#926-32211 |
| Goat anti-rabbit IRDye 680RD | LI-COR | Cat#926-68071 |
| Bacterial and Virus Strains | ||
| E. coli NEB Stable | NEB | Cat#C3040 |
| E. coli One Shot Stbl3 | ThermoFisher | Cat#C737303 |
| AAV2/1 encapsulated HiUGE vectors | This paper | N/A |
| AAV2/9 encapsulated HiUGE vectors | This paper | N/A |
| AAV2-retro encapsulated HiUGE vectors | This paper | N/A |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-AP5 | Tocris | Cat#0106 |
| Cytosine arabinoside | Sigma | Cat#C1768 |
| Polyethylenimine (PEI) | Polysciences | Cat#24765 |
| Benzonase | Novagen | Cat#70664 |
| Lipofectamine 3000 | ThermoFisher | Cat#L3000008 |
| Normal goat serum | Sigma | Cat#G9023 |
| Optiprep density gradient | Sigma | Cat#D1556 |
| Critical Commercial Assays | ||
| BCA protein assay | Pierce | Cat#23227 |
| InFusion cloning kit | TaKaRa | Cat#638910 |
| TOPO TA Cloning Kit for Sequencing | ThermoFisher | Cat#K457501 |
| CanGetSignal solution | TOYOBO | Cat#NKB-101T |
| MyTaq Extraction-PCR kit | Bioline | Cat#BIO-21127 |
| PureLink Genomic DNA Mini Kit | ThermoFisher | Cat#K182001 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| MyTaq HS Mix | Bioline | Cat#BIO-25045 |
| MyTaq HS Red Mix | Bioline | Cat#BIO-25047 |
| PowerUp SYBR Green Master Mix | ThermoFisher | Cat#A25741 |
| SensiFAST Real-Time PCR Kit | Bioline | Cat#BIO-86005 |
| Genome Walker kit | Clontech | Cat#636406 |
| Amicon Ultra-15 filtration unit | Millipore | Cat#UFC910008 |
| Spin-X centrifuge tube filter | Costar | Cat#8162 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | CRL-11268 |
| NIH3T3 cells | ATCC | CRL-1658 |
| HeLa cells | ATCC | CCL-2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh | Jackson Laboratory | 024857 |
| Mouse: C57BL/6J | Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| Donor Recognition Sequences (DRS), see Table S1 | This paper | N/A |
| Guide RNA (gRNA) targets, see Table S1 | This paper | N/A |
| Donor sequences, see Table S1 | This paper | N/A |
| Primers, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| AAV:ITR-U6-sgRNA(backbone)-hSyn-Cre-2A-EGFP-KASH-WPRE-shortPA-ITR | Feng Zhang (Platt et al., 2014) | Addgene Plasmid #60231 |
| pAAV-nEFCas9 | Juan Belmonte (Suzuki et al., 2016) | Addgene Plasmid #87115 |
| rAAV2-retro helper | Alla Karpova & David Schaffer (Tervo et al., 2016) | Addgene Plasmid #81070 |
| pCAG-smFP HA | Loren Looger (Viswanathan et al., 2015) | Addgene Plasmid #59759 |
| pAAV-HiUGE-GS-gRNA vectors | This paper | N/A |
| pAAV-HiUGE-Donor vectors | This paper | N/A |
| pAAV-HiUGE-GS-gRNA vectors with intein-split-Cas9 | This paper | N/A |
| pAAV-HiUGE-Donor vectors with intein-split-Cas9 | This paper | N/A |
| Software and Algorithms | ||
| FIJI | (Schindelin et al., 2012; Schneider et al., 2012) | https://fiji.sc |
| CRISPOR | (Haeussler et al., 2016) | http://crispor.tefor.net |
| Cas-OFFinder | (Bae et al., 2014) | http://www.rgenome.net/cas-offinder |
| Trim Galore Toolkit (v 0.5.0) | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| Trimmomatic | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| Cutadapt (v 1.18) | (Martin, 2011) | https://pypi.org/project/cutadapt/ |
| FastQC (v 0.11.5 or v 0.11.8) | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| bwa-mem (v0.7.17) | (Li and Durbin, 2009) | https://sourceforge.net/projects/bio-bwa/files/ |
| SAMtools (v1.9) | (Li et al., 2009) | https://github.com/samtools/samtools |
| Picard | Broad Institute | http://broadinstitute.github.io/picard |
| GATK HaplotypeCaller (v3.8-1-0-gf15c1c3ef) | (McKenna et al., 2010) | https://software.broadinstitute.org/gatk/ |
| FLASH | (Magoc and Salzberg, 2011) | https://ccb.jhu.edu/software/FLASH/ |
| EMBOSS Needle | (Chojnacki et al., 2017) | https://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html |
| CRISPResso | (Pinello et al., 2016) | http://crispresso.rocks/ |
| R | The R Foundation | https://www.r-project.org/ |
| JMP Pro | SAS | https://www.jmp.com |
| Other | ||
| Zeiss LSM 880 | Zeiss | N/A |
| Zeiss LSM 710 | Zeiss | N/A |
| Zeiss Imager.M2 with Apotome.2 | Zeiss | N/A |
| Leica STED microscope | Leica | N/A |
| Odyssey Fc Imager | LI-COR | N/A |
Highlights.
HiUGE, an AAV-based CRISPR-mediated method for universal insertional genome editing
Arrayed gRNAs and premade donors enable high-throughput endogenous protein labeling
“Plug and play” selection of donors at each target locus enables diverse applications
HiUGE provides a powerful method to analyze proteomic candidates in vitro and in vivo
Acknowledgments
We thank Drs. Nicolas Devos, David Corcoran, and Jennifer Modliszewski for performing deep sequencing and subsequent bioinformatic analyses. We thank Shataakshi Dube and Riley Mangan for performing STED imaging. We thank Drs. Cagla Eroglu and Anne West for their critical reading and comments. This work was supported by the following grants: R01MH103374 (S.H.S.), R01NS102456 (S.H.S.), Kahn Neurotechnology Award, and the Duke Bryan Scholar Award (S.H.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.H.S. and Y.G. have filed a patent application related to this work.
S.H.S. is a founder of CasTag Biosciences and a member of its scientific advisory board.
References:
- Aurnhammer C, Haase M, Muether N, Hausl M, Rauschhuber C, Huber I, Nitschko H, Busch U, Sing A, Ehrhardt A, and Baiker A (2012). Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 23, 18–28. [DOI] [PubMed] [Google Scholar]
- Bae S, Park J, and Kim JS (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KE, and Parker R (2004). Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol 16, 293–299. [DOI] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, and Gertler FB (2000). Negative regulation of fibroblast motility by Ena/VASP proteins. Cell 101, 717–728. [DOI] [PubMed] [Google Scholar]
- Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, Persson A, Ottosson J, Wernerus H, Nilsson P, et al. (2008). A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics 7, 2019–2027. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury A, and Pluckthun A (2015). Reproducibility: Standardize antibodies used in research. Nature 518, 27–29. [DOI] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, and Jaenisch R (2001a). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27, 327–331. [DOI] [PubMed] [Google Scholar]
- Chen ZY, He CY, Ehrhardt A, and Kay MA (2003). Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther 8, 495–500. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Yant SR, He CY, Meuse L, Shen S, and Kay MA (2001b). Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol Ther 3, 403–410. [DOI] [PubMed] [Google Scholar]
- Cheriyan M, Pedamallu CS, Tori K, and Perler F (2013). Faster protein splicing with the Nostoc punctiforme DnaE intein using non-native extein residues. J Biol Chem 288, 6202–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki S, Cowley A, Lee J, Foix A, and Lopez R (2017). Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res 45, W550–W553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, et al. (2011). An assessment of histone-modification antibody quality. Nat Struct Mol Biol 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet M-C, Friocourt G, McDonnell N, Reiner O, Kahn A, et al. (1999). Doublecortin Is a Developmentally Regulated, Microtubule-Associated Protein Expressed in Migrating and Differentiating Neurons. Neuron 23, 247–256. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, and Reiner O (2004). DCX, a new mediator of the JNK pathway. EMBO J 23, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, and Liu J (2010). Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogins J, Crawford DC, Zorumski CF, and Mennerick S (2011). Excitotoxicity triggered by Neurobasal culture medium. PLoS One 6, e25633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, and Liu DR (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, and Caceres JF (2016). Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res 44, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsaro JJ, Huang L, Gutierrez L, and MacDonald RI (2008). Molecular epitopes of the ankyrin-spectrin interaction. Biochemistry 47, 7452–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, and Svitkina TM (2016). Axon Initial Segment Cytoskeleton: Architecture, Development, and Role in Neuron Polarity. Neural Plast 2016, 6808293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D, Roberts BL, Richardson WD, and Smith AE (1984). A short amino acid sequence able to specify nuclear location. Cell 39, 499–509. [DOI] [PubMed] [Google Scholar]
- Kennedy SP, Warren SL, Forget BG, and Morrow JS (1991). Ankyrin binds to the 15th repetitive unit of erythroid and nonerythroid beta-spectrin. J Cell Biol 115, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, and Soderling SH (2013). Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33, 6081–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, and Joung JK (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, and Yanagawa H (2009). Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem 284, 478–485. [DOI] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, and Corbett AH (2007). Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem 282, 5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, and Salzberg SL (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Wieland T, and Tsujimoto G (2009). How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch Pharmacol 379, 385–388. [DOI] [PubMed] [Google Scholar]
- Mikuni T, Nishiyama J, Sun Y, Kamasawa N, and Yasuda R (2016). High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell 165, 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Niopek D, Benzinger D, Roensch J, Draebing T, Wehler P, Eils R, and Di Ventura B (2014). Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat Commun 5, 4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H, et al. (2018). Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Nishiyama J, Mikuni T, and Yasuda R (2017). Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 96, 755–768 e755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N, and Helleday T (2004). Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res 32, 3683–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Honing K, Ebert TS, and Hornung V (2016). CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat Commun 7, 12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, and Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, and Zhang F (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence EF, and Soderling SH (2015). Actin Out: Regulation of the Synaptic Cytoskeleton. J Biol Chem 290, 28613–28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DJ, Kuhner K, Kuhn R, Werfel S, Engelhardt S, Wurst W, and Ortiz O (2015). Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res 43, 6450–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, Weinberg RJ, and Soderling SH (2016). Identification of an elaborate complex mediating postsynaptic inhibition. Science 353, 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S, Williams ME, Bloss EB, Stasevich TJ, Speer CM, Nern A, Pfeiffer BD, Hooks BM, Li WP, English BP, et al. (2015). High-performance probes for light and electron microscopy. Nat Methods 12, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, and Rasband MN (2007). betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, and Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, and Tsien RY (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 DNA Sequences of gRNA Targets, Donor Payloads, and Primers, Related to the STAR Methods.
Supplemental Video S1 Live Imaging of Neuron with HiUGE-mediated mCherry Labeling of Pyruvate Dehydrogenase E1 Component Subunit Alpha (Pdha1), Related to Figure 6.