

Abstract
Background:
Apolipoprotein M (APOM) mediates the physical interaction between high-density lipoprotein (HDL) particles and sphingosine-1-phosphate (S1P). APOM exerts anti-inflammatory and cardio-protective effects in animal models.
Methods:
In a subset of Penn-HF study (PHFS) participants (n=297), we measured APOM by ELISA. We also measured total S1P by liquid chromatography-mass spectrometry and isolated HDL particles to test the association between APOM and HDL-associated S1P. We confirmed the relationship between APOM and outcomes using modified aptamer-based APOM measurements, among 2,170 adults in the PHFS and 2 independent cohorts: the Washington University HF registry (n=173) and a subset of the TOPCAT trial (n=218). Finally, we examined the relationship between APOM and ~5000 other proteins (SomaScan assay) to identify biological pathways associated with APOM in HF.
Results:
In the PHFS, APOM was inversely associated with the risk of death (Standardized Hazard Ratio=0.56; 95%CI=0.51–0.61; P<0.0001) and the composite of death/ventricular assist device or heart transplant (Standardized HR=0.62; 95%CI=0.58–0.67; P<0.0001). This relationship was independent of HDL-C or APOA-I levels. APOM remained associated with death (HR=0.78; 0.69–0.88; P<0.0001) and the composite of death/ventricular assist device/heart transplant (HR=0.85; 95%CI=0.76–0.94; P=0.001) in models that adjusted for multiple confounders. This association was present in both HF with reduced (HFrEF) and preserved (HFpEF) ejection fraction, and was replicated in the Washington University cohort and a HFpEF-only cohort (TOPCAT). The S1P and APOM content of isolated HDL particles strongly correlated (R=0.81; P<0.0001). The top canonical pathways associated with APOM were inflammation (negative association), the coagulation system (negative association) and LXR/RXR activation (positive association). The relationship with inflammation was validated with multiple inflammatory markers measured with independent assays.
Conclusions:
Reduced circulating APOM is independently associated with adverse outcomes across the spectrum of human HF. Further research is needed to assess whether the APOM/S1P axis is a suitable therapeutic target in HF.
Keywords: heart failure, apolipoprotein M, high-density lipoprotein, survival, sphingosine-1-phosphate
Background
Apolipoprotein M (APOM) is a lipocalin primarily secreted by the liver, and is present in ~5% of high-density lipoprotein (HDL) and <2% of low-density lipoprotein (LDL) particles.1–3 APOM exerts multiple pleiotropic effects, including anti-inflammatory effects, antioxidant and anti-atherogenic effects4, 5; it promotes endothelial protection6, 7 and enhances cell survival8. APOM contains a hydrophobic binding pocket for sphingosine-1-phosphate (S1P), a small signaling sphingolipid. S1P activates multiple G-protein coupled receptors on cell types including endothelial cells and cardiomyocytes. Multiple studies suggest APOM and S1P attenuate ischemic injury, but little is known about the role of APOM or S1P in heart failure (HF) progression.9–11
Mechanistically, the APOM/S1P axis inhibits inflammation and attenuates the effect of tumor necrosis factor (TNF)-α on gene expression, limiting monocyte adhesion to the endothelium and maintaining endothelial barrier integrity.12 APOM also protects animals from non-ischemic insults, such as lipopolysaccharide-induced death and organ injury via anti-inflammatory and anti-apoptotic effects, as well as modulation of the coagulation system.13 APOM enhances endothelial nitric oxide production and vasodilation, and increases cardiomyocyte survival.9, 14 Despite the strong rationale for a cardioprotective role of APOM, the relevance of APOM in human HF has not been previously investigated.
Based on the multiple described protective functions of APOM and S1P in cell and animal models, we hypothesized that reduced levels of APOM are associated with worse outcomes in human HF. Specifically, we test the hypothesis that reduced circulating APOM is associated with the risk of death, a composite of death/ventricular assist device (VAD) implantation/heart transplant, and a composite of death/HF-related hospitalization among adults with HF enrolled in a large multi-center cohort study (Penn Heart Failure Study, PHFS), with stratified analyses in HF with reduced (HFrEF) and preserved (HFpEF) ejection fraction, and subsequent replication in 2 independent cohorts. We also aimed to identify biologic pathways associated with APOM using large scale plasma proteomics.
Methods
Study populations
We primarily analyzed data from participants enrolled in the PHFS and replicated our results in 2 independent cohorts: (1) the Washington University HF Registry, and (2) the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist Trial (TOPCAT). The data, analytic methods, and study materials are not publicly available for purposes of reproducing the results or replicating the procedures. Such data may be made available to other researchers for collaborative research, through the establishment of appropriate data sharing agreements and regulatory approvals. The parent TOPCAT trial data are available to other researchers through the National Institutes of Health Biolincc website.
PHFS:
The PHFS design has been previously published.15–18 Briefly, the PHFS was a prospective cohort study of ambulatory patients with chronic HF recruited between 2003–2011 at the University of Pennsylvania (Philadelphia, PA), Case Western Reserve University (Cleveland, OH), and the University of Wisconsin (Madison, WI). Patients with a clinical diagnosis of HF as determined by a HF specialist were enrolled. Each participant provided written informed consent. At the time of study entry, standardized questionnaires were administered to participants and their physicians to obtain detailed clinical data. Participants with expected mortality of 6 months or less from a non-cardiac condition, , mechanical circulatory support, or inability to provide informed consent were excluded. Venous blood samples were obtained at enrollment and stored at −80 °C for later analysis. An institutional review board from each participating center approved the protocol.
Washington University HF Registry:
This is a prospective registry of patients with a clinical diagnosis of HF evaluated at Washington University School of Medicine (Barnes Jewish Hospital, St. Louis, MO). As previously described,19 detailed patient information was prospectively collected, including heart disease onset, etiology, clinical stage,severity, medications, device therapies, co-morbidities, demographics, and health status. All patients provided informed consent, and the study was approved by the Washington University Institutional Review Board.
TOPCAT:
TOPCAT data and samples were obtained from the National Heart, Lung, and Blood Institute. TOPCAT was a multi-center, international, randomized, double-blinded, placebo-controlled trial of spironolactone that enrolled 3,445 adults with HFpEF across 6 countries from 2006–2012. The primary goal of the trial was to determine if spironolactone was associated with a reduction in the composite outcome of cardiovascular mortality, aborted cardiac arrest, or HF hospitalization. The design, general characteristics of the study population and primary results of the trial have been previously published.20–22 Due to substantial differences regarding subject recruitment and study implementation in Russia and Georgia21, 23, 24, we only performed measurements from subjects enrolled in the Americas who had available plasma samples for de novo measurements of APOM levels (n=218).
APOM level determination
Our a priori hypothesis was initially tested in a subset of study participants from the PHFS (n=297), among whom APOM levels were determined by single-plate ELISA using a human APOM antibody, as previously described.25 Subsequently, we analyzed APOM levels measured with a modified aptamer assay (SomaScan® assay), as previously described26–29 in PHFS cohort (n=2170), the Washington University HF Registry (n=173) and TOPCAT (n=218). The SomaScan APOM measurement has been previously validated by mass spectrometry,26 as detailed in the online supplement.
Plasma Proteomics and pathway analyses
We utilized the SomaScan® assay version 4, which is a multiplexed, modified aptamer-based binding-assay for PHFS and TOPCAT assays. This assay includes 4,979 modified aptamer reagents to 4,776 unique protein targets. The SomaScan assay utilizes Slow-Off-rate Modified Aptamer (SOMAmer) reagents, which are chemically modified nucleotides, to bind and quantify target proteins in relative fluorescent units directly proportional to the amount of target protein in the sample. We performed knowledge-based pathway analysis to assess the correlates of APOM. First, we assessed the correlation between levels of APOM and all proteins measured in the SomaScan assay, after Box-Cox transformation. We adjusted for multiple comparisons based on the principal components underlying the variability of all measured proteins, as previously described30–32. Associations between APOM and individual proteins that were significant, with an adjusted P value <0.01 were then utilized to perform pathway analyses, using Ingenuity® Pathway Analysis (IPA) software (Qiagen; Hilden, Germany; www.qiagen.com/ingenuity). The full list of proteins and their associations with APOM is provided in the supplemental excel file. Proteins were identified according to their UniProt ID annotation. The totality of proteins included in the SomaScan assay was used as the reference set and both direct and indirect experimentally confirmed relationships from all species were included. The analysis calculates a P value (Fisher exact test and right tailed), quantifying the overlap, and a Z score, quantifying the likelihood and direction (up or downregulated), between the plasma proteomics pattern and known canonical pathways. For Washington University HF registry samples, serum samples were analyzed using the SomaScan protein array platform version Plasma_4.2_20161012_1.5k, which included 1,306 total analytes, including APOM.
S1P determination
We measured S1P in a subset of PHFS subjects (n=206). PHFS participants with LVEF <50% and ischemic cardiomyopathy etiology were matched to participants with non-ischemic etiology based on a propensity score that included age, sex, hypercholesterolemia, statin use, and an interaction term between sex and hypercholesterolemia. Matching was performed by means of nearest neighbor matching, with a caliper width of 0.05 selected for a target sample size of approximately 200, using the MatchIt extension package in the R programming environment. Serum samples (10 μL) were diluted with 55 μL tris-buffered saline (50 mM Tris-HCL pH 7.5, 0.15 mM NaCl). Precipitation solution (200 μL methanol containing 20 nM internal standard) was added to diluted human serum samples, followed by 30 seconds of vortexing and subsequent centrifugation at 17,000g for 2 minutes. 5 μL of supernatant were injected for liquid chromatography tandem mass spectrometric (LC-MS/MS) analysis33.
Additional assays (inflammatory biomarkers, APOA-I measurements, and HDL particle isolation)
To confirm the association between APOM and the top canonical pathway identified by pathway analysis (inflammation), we analyzed the relationship between APOM and high-sensitivity C-reactive protein, a well-established marker of inflammation, in the PHFS. We measured hsCRP using standard ARCHITECT immunoassays (Abbott Laboratories, Abbott Park, IL). We also measured various inflammatory biomarkers (TNF, TNFRI, TNFRII, IL1-beta, IL-6, IL-8, pentraxin 3 and myeloperoxidase) using a Luminex® Bead-Based multiplexed assay (Bristol-Myers-Squibb; Ewing Township, NJ).
In order to discern whether the relationship between APOM and outcomes is independent of its association with HDL-cholesterol and/or APOA-I, we measured APOA-I by immunonephelometry among 201 participants, as previously described.34 HDL-cholesterol was measured using the enzymatic colorimetric method.35
In order to assess the correlation between HDL-APOM and HDL-S1P, HDL was isolated from 151 PHFS participants via gradient density ultracentrifugation. Briefly, lipoproteins are separated according to density in sequential ultracentrifugation spins. HDL was isolated by adjusting the density to 1.21 g/ml. , as previously described36.
Statistical analysis
Participant characteristics were summarized using mean (SD) for continuous variables with a symmetric distribution and median (interquartile range) for continuous variables with a skewed distribution. Categorical variables are expressed as counts (percentages).
We stratified the study populations according to tertiles of APOM and compared various clinical characteristics between the strata. We used analysis of variance (ANOVA) for symmetric variables, the Kruskal-Wallis test for skewed variables, and the chi-square or Fisher’s exact test, as appropriate, for categorical data. We computed Kaplan-Meier survival curves for tertiles of APOM and compared them with the log-rank test. We assessed the relationship between APOM and: (A) All-cause death; (B) a composite outcome of death, VAD implantation, or heart transplantation. Because VAD implantation and heart transplantation are not part of the standard therapeutic approach to HFpEF, for analyses restricted to participants with HFpEF, we assessed all-cause mortality, as well as the composite outcome of death or HF-related hospitalization, which is increasingly utilized in HFpEF studies.37
We further assessed the relationship between APOM and the risk of outcomes using Cox regression models. To perform unit-independent analyses that can be easily compared between measurement assays and between biomarkers (i.e., APOM vs. S1P), we express hazard ratios (HRs) per-standard-deviation increase (i.e., 1-point increase in the z-score, after Box-Cox transformation to improve the normality of data distribution).
We built unadjusted survival models and models that adjusted for confounders, including: (1) potential clinical confounders and/or any characteristics that significantly differed between APOM tertiles; (2) All covariates in adjusted model 2 plus BNP or NT-proBNP levels. For analyses in smaller cohorts (ELISA-based APOM measurements in PHFS, stratified analyses in HFpEF vs. HFrEF, and analyses in our 2 validation cohorts), we performed unadjusted survival analyses and models adjusted for: (1) The MAGGIC risk score, a single variable that incorporates multiple demographic, clinical and laboratory parameters38, in order to prevent model overfitting; (2) Analyses that adjust for the MAGGIC risk score plus BNP or NT-proBNP levels. The time of HF diagnosis, one of the components of the MAGGIC risk score, was not available, and no points were given to any subject for this component during the score computation.
To confirm the association between APOM and the top canonical pathway identified by pathway analyses (inflammation), we compared inflammatory biomarkers across tertiles of APOM using analysis of variance. Given the non-normal distribution of most biomarkers, we compared geometric means (means obtained after log-transformation). All values are expressed in the native scale. To assess whether the relationship between APOM and outcomes is dependent of its relationship with inflammation, we built Cox models in which the association with outcomes was assessed with and without adjustment for the inflammatory biomarkers mentioned above, to assess the extent to which this adjustment attenuated the relationship between APOM and incident events.
Statistical significance was defined as a 2-tailed P value<0.05. All probability values presented are 2-tailed. Analyses were performed using the MATLAB statistics and machine learning toolbox (Matlab 2016b, the Mathworks; Natwick, MA) and R Statistical Software v3.5.2 (Foundation for Statistical Computing, Vienna, Austria).
Results
Relationship between APOM measured by ELISA and the risk of adverse outcomes in the PHFS
The general characteristics of PHFS subjects are shown in Online Table 1. To test our a priori hypothesis, we measured APOM by ELISA in the PHFS. In this population, mean APOM was 0.92±0.28 μM. During follow-up, 94 deaths occurred and129 participants reached the composite endpoint of VAD implantation, heart transplant or death. Figure 1 shows Kaplan-Meier survival curves for participants stratified by tertiles of APOM, for all-cause death (Figure 1A) and for VAD implantation, heart transplant or death (Figure 1B). There was a highly significant difference between the tertiles, with the lowest tertile of APOM (≤0.79 μM) exhibiting the highest risk.
Figure 1. Risk of adverse outcomes among Penn Heart Failure Study participants and textiles APOM, measured by ELISA.
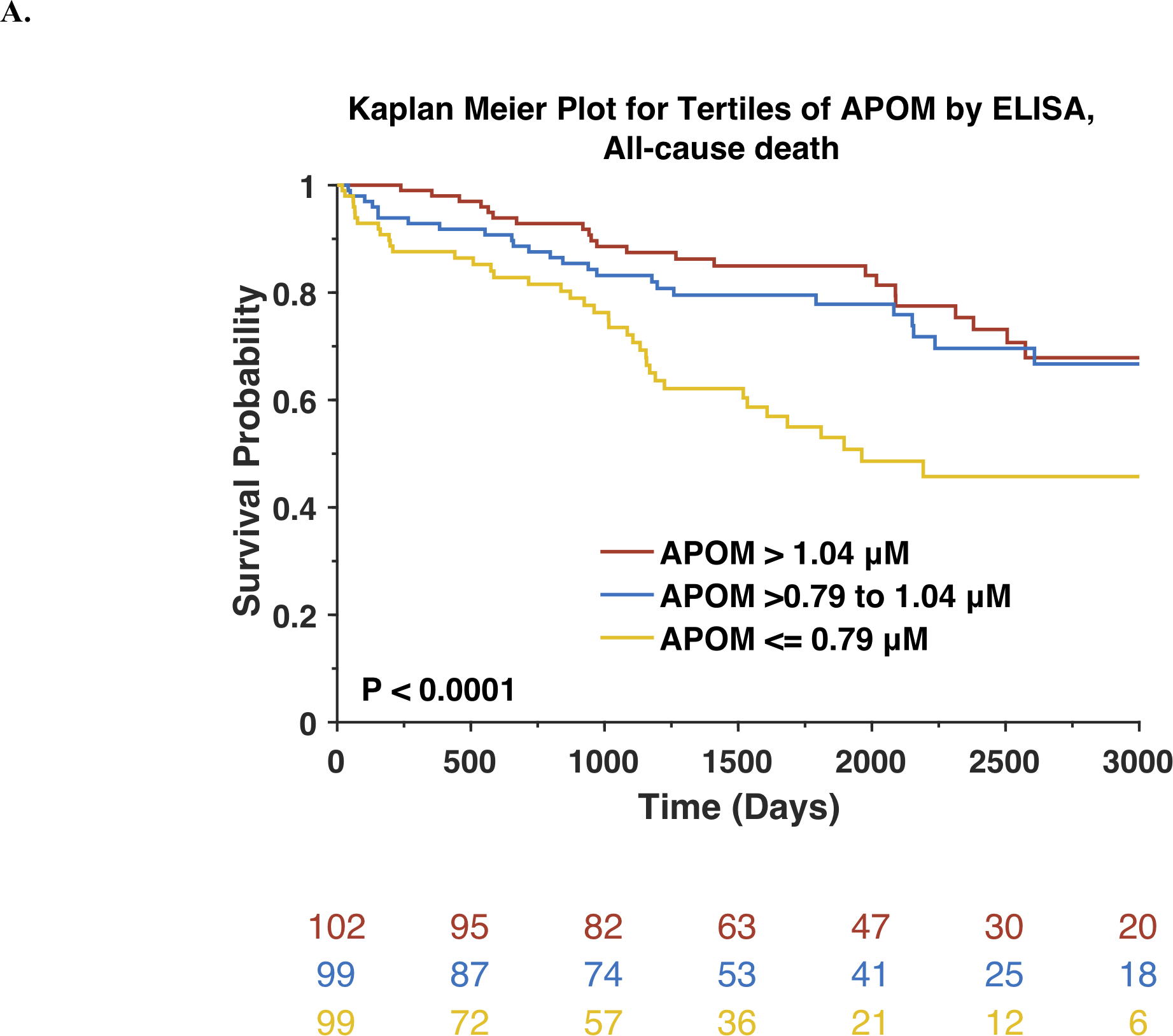
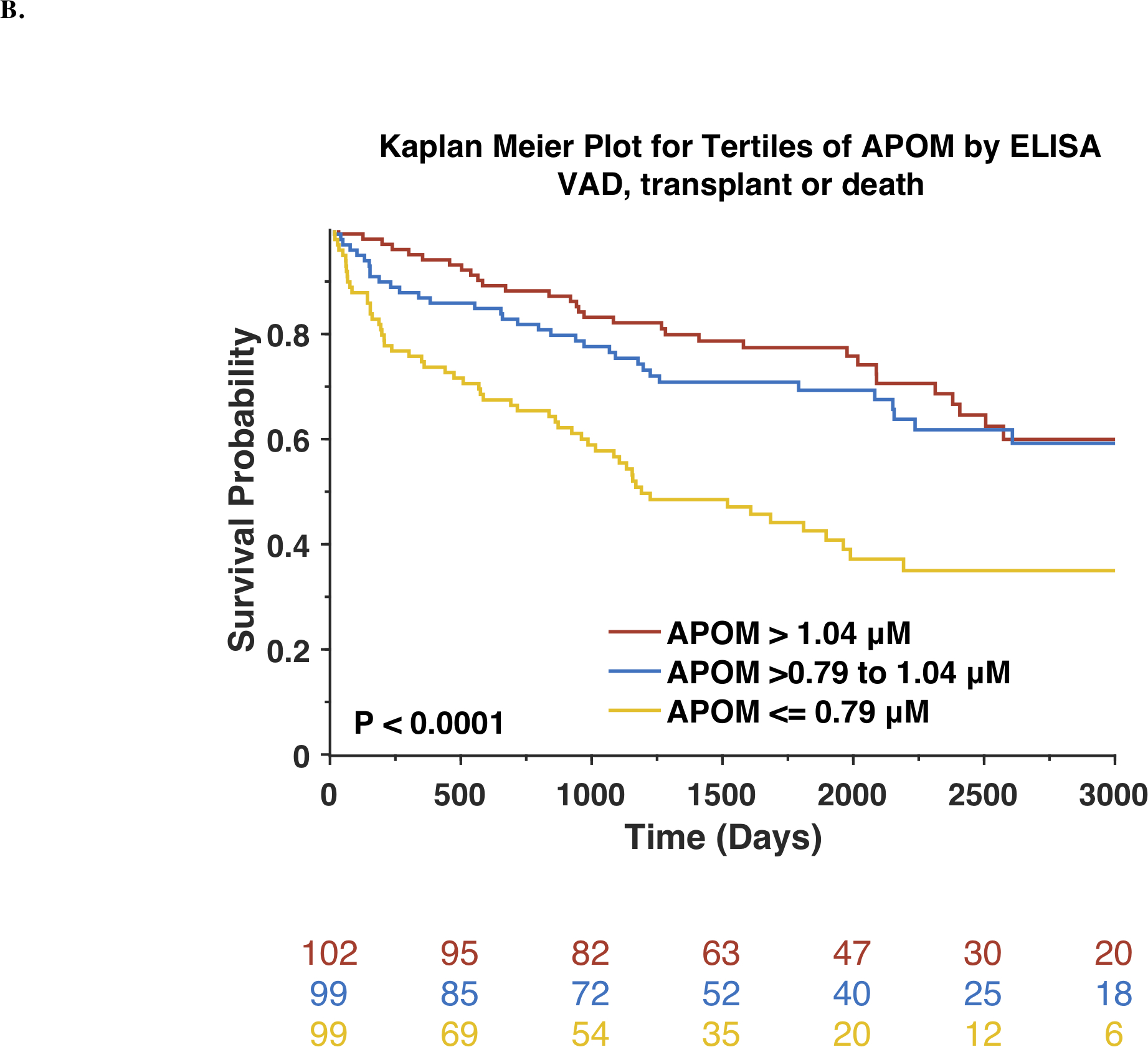
Kaplan-Meier survival curves for all-cause mortality (A) or the composite outcome of death, ventricular assist device or heart transplant (B) are shown. The number of patients at risk at each timepoint is presented below the graph.
Cox proportional hazard analyses demonstrated that APOM was significantly associated with the risk of death (Standardized HR=0.63; 95%CI=0.51–0.76; P<0.0001). This association remained significant after adjustment for the MAGGIC risk score (Standardized HR=0.71; 95%CI=0.56–0.90; P=0.0044). Compared to a base model containing the MAGGIC risk score, the Harrel’s c index increased from 0.686 to 0.697 for the prediction of death.
As shown in Online Table 2, APOM was significantly associated with the risk of VAD implantation, heart transplant or death (Standardized HR=0.67; 95%CI=0.57–0.79; P<0.0001). This association remained significant after adjustment for the MAGGIC risk score (Standardized HR=0.77; 95%CI=0.63–0.94; P=0.0110). Compared to a base model containing the MAGGIC risk score, the Harrel’s c index increased from 0.683 to 0.692 for the prediction of VAD, transplant and death. APOM was also associated with the risk of death and the risk of VAD implantation, heart transplant or death after further adjustment for BNP).
Prognostic value of APOM vs HDL-cholesterol or APOA-I
To assess whether the relationship between APOM and outcomes is independent of its association with HDL-cholesterol and/or APOA-I, we built models in which the relationship between APOM and outcomes was assessed with or without adjustment for HDL-C or APOA-I (Online Figures 1 and 2). In contrast to APOM, HDL-C was not significantly associated with the risk of death (Online Figure 1A) and the risk of VAD, heart transplant or death (Online Figure 1B). In models that included both APOM and HDL-C, APOM (but not HDL-C) was associated with the risk of death and of VAD, heart transplant or death, and its relationship with these outcomes was not attenuated.
In unadjusted analyses, APOA-I was significantly associated with the risk of death (Online Figure 2A) or the risk of VAD, heart transplant or death (Online Figure 2B). In models that included both APOM and APOA-I, APOM was associated with risk of death and the risk of VAD, heart transplant or death, whereas APOA-I was not independently associated with these outcomes.
Relationship between APOM by a modified aptamer assay and outcomes in the PHFS measured
Among subjects with available APOM levels measured by the SomaScan (n=2,170), follow-up data were available for 2,135 subjects The SomaScan aptamer for APOM was previously validated by mass spectrometry from human serum samples29. In the subset of PHFS participants with available ELISA APOM measurements, we found a linear relationship between APOM measured by ELISA vs. APOM measured by SomaScan, with a Pearson correlation coefficient value of 0.73 (P<0.0001), as shown in Online Figure 3. The majority (95%) of APOM is anchored to HDL by its retained signal peptide39, 40. The correlation between APOM, measured by SomaScan, and HDL-C (r=0.36, P <0.00001) was highly consistent with the previously described association between APOM, measured by ELISA, and HDL-C (r=0.37)25.
The general characteristics of PHFS subjects stratified by APOM tertiles are shown in Table 1. Lower APOM levels were associated with older age, male sex, a higher body mass index (BMI), higher serum creatinine, lower blood pressures, ischemic etiology, history of coronary revascularization, diabetes mellitus, history of pacemaker implantation, a lower left ventricular EF, more advanced New York Heart Association (NYHA) functional class, greater B-type natriuretic peptide (BNP) levels, greater use of aspirin, hydralazine and organic nitrates, digoxin, loop diuretics, mineralocorticoid receptor antagonists, and statins, as well as lower use of lower angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers.
Table 1.
General Characteristics of PHFS Participants stratified by tertiles of APOM (n=2170)
|
Lowest tertile (n=723) |
Middle tertile (n=724) |
Highest tertile (n=723) |
P value | |
|---|---|---|---|---|
| APOM (AU) | 614 (547,665) | 804 (761,850) | 1027 (962,1134) | ---- |
| Age (years) | 61.3 (53.2,68.8) | 58.3 (48.1,66.1) | 53.3 (43.6,62.1) | <0.0001 |
| Male sex | 525 (72.61%) | 459 (63.40%) | 451 (62.38%) | <0.0001 |
| Race/Ethnicity | ||||
| Caucasian | 517 (75.58%) | 519 (74.78%) | 544 (77.05%) | 0.6026 |
| African American | 154 (22.51%) | 157 (22.62%) | 149 (21.10%) | 0.7467 |
| Other | 13 (1.90%) | 18 (2.59%) | 13 (1.84%) | 0.5551 |
| BMI, (kg/m2) | 30.6 (26.2,36.2) | 29.3 (25.2,34.4) | 27.5 (24.1,31.1) | <0.0001 |
| Systolic BP (mmHg) | 110 (98,125) | 114 (100,130) | 114 (100,128) | 0.0034 |
| Diastolic BP (mmHg) | 68 (60,76) | 70 (62,78) | 70 (62,78) | <0.0001 |
| Ischemic etiology | 317 (44.27%) | 204 (28.33%) | 141 (19.61%) | <0.0001 |
| History of PCI | 223 (30.84%) | 139 (19.20%) | 106 (14.66%) | <0.0001 |
| History of CABG | 194 (26.83%) | 121 (16.71%) | 77 (10.65%) | <0.0001 |
| Current smoking | 59 (8.16%) | 64 (8.84%) | 73 (10.10%) | 0.4277 |
| Diabetes | 317 (43.85%) | 194 (26.80%) | 111 (15.35%) | <0.0001 |
| Atrial fibrillation or flutter | 313 (43.29%) | 261 (36.05%) | 207 (28.63%) | <0.0001 |
| History of pacemaker | 61 (8.44%) | 42 (5.80%) | 34 (4.70%) | 0.0111 |
| History of ICD | 162 (22.41%) | 140 (19.34%) | 154 (21.30%) | 0.3487 |
| History of Biventricular pacer | 216 (29.88%) | 173 (23.90%) | 118 (16.32%) | <0.0001 |
| Serum creatinine | 1.3 (1,1.8) | 1.1 (0.97,1.41) | 1.01 (0.9,1.3) | <0.0001 |
| LV EF (%) | 25 (20,40) | 30 (20,45) | 35 (20,45.8) | 0.0003 |
| EF Category | ||||
| Reduced EF | 507 (82.84%) | 534 (81.65%) | 525 (79.31%) | 0.2555 |
| Recovered EF | 52 (8.50%) | 71 (10.86%) | 80 (12.08%) | 0.1076 |
| Preserved EF | 53 (8.66%) | 49 (7.49%) | 57 (8.61%) | 0.6888 |
| NYHA Class | <0.0001 | |||
| NYHA 1 | 69 (9.69%) | 129 (17.92%) | 176 (24.38%) | |
| NYHA 2 | 343 (48.17%) | 465 (64.58%) | 534 (73.96%) | |
| NYHA 3 | 365 (51.26%) | 349 (48.47%) | 345 (47.78%) | |
| NYHA 4 | 142 (19.94%) | 164 (22.78%) | 195 (27.01%) | |
| BNP (pg/mL) | 345 (100,1040) | 158 (49,530) | 91 (26,283) | <0.0001 |
| Medication Use | ||||
| Beta Blocker | 649 (89.76%) | 649 (89.64%) | 625 (86.45%) | 0.0790 |
| Aspirin | 482 (66.67%) | 412 (56.91%) | 340 (47.03%) | <0.0001 |
| ACEI/ARBs | 597 (82.57%) | 626 (86.46%) | 633 (87.55%) | 0.0182 |
| Hydralazine | 91 (12.59%) | 67 (9.25%) | 25 (3.46%) | <0.0001 |
| Organic Nitrates | 157 (21.72%) | 130 (17.96%) | 56 (7.75%) | <0.0001 |
| Digoxin | 294 (40.66%) | 263 (36.33%) | 217 (30.01%) | 0.0001 |
| Loop diuretic | 598 (82.71%) | 515 (71.13%) | 411 (56.85%) | <0.0001 |
| MRA | 264 (36.51%) | 251 (34.67%) | 224 (30.98%) | 0.0777 |
| Statin | 454 (62.79%) | 379 (52.35%) | 294 (40.66%) | <0.0001 |
| CCBs | 58 (8.02%) | 80 (11.05%) | 62 (8.58%) | 0.1056 |
APOM=apolipoprotein M; ARB=angiotensin receptor blocker; ACE: angiotensin converting enzyme; CCB: calcium channel blocker; BNP = b-type natriuretic peptide; ICD=Implanted Cardioverter Defibrillator; LV EF= left ventricular ejection fraction; BMI=body mass index; NYHA=New York Heart Association; MRA=mineralocorticoid receptor antagonist.
During a median follow-up of 5.02 years, 523 participants died and 716 experienced the composite endpoint of VAD, heart transplantation or death. Lower APOM levels were significantly associated with an increased risk of death. Kaplan-Meier survival plots for the study population stratified by tertiles of APOM are shown in Figure 2A. Results of unadjusted and adjusted Cox models are shown in Table 2. Each standard-deviation decrease in APOM was associated with nearly a doubling in the mortality risk (Standardized HR=0.56; 95%CI=0.51–0.61; P<0.0001). In a model that adjusted for age, sex, race, enrollment site, history of PCI, CABG, atrial fibrillation or flutter, pacemaker, bi-ventricular pacer implantation, LV ejection fraction, NYHA class, ischemic vs. non-ischemic etiology, systolic and diastolic blood pressure, body mass index, history of diabetes mellitus, serum creatinine, ACE inhibitor/ARB use, digoxin, hydralazine, loop diuretic, organic nitrate and statin use, APOM remained significantly associated with the risk of death (Standardized HR=0.73; 95%CI=0.65–0.81; P<0.0001). APOM was also associated with the risk of death after further adjustment for BNP levels (Standardized HR=0.78; 95%CI=0.69–0.88; P<0.0001).
Figure 2. Risk of adverse outcomes among Penn Heart Failure Study participants stratified by tertiles of APOM.
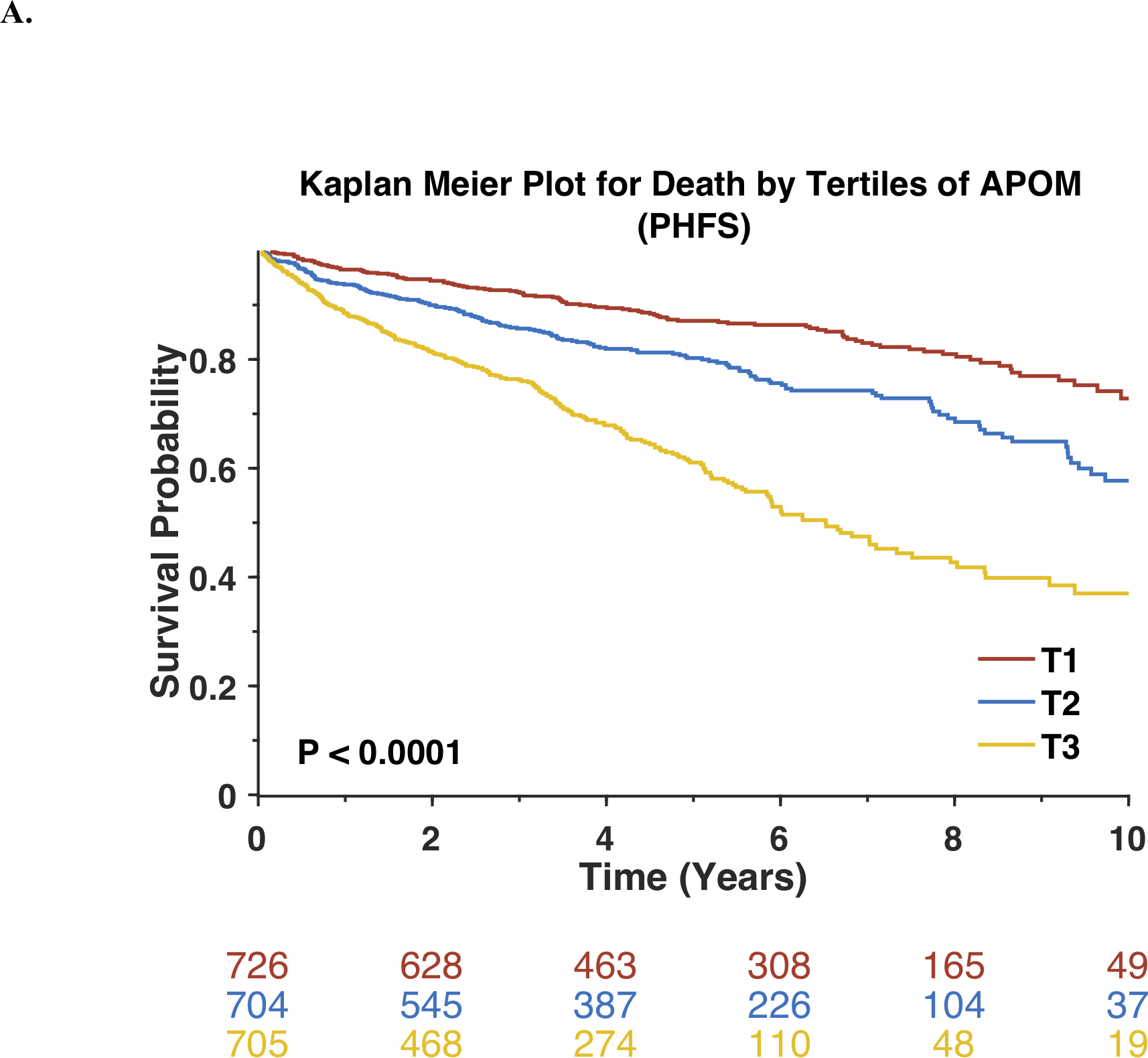
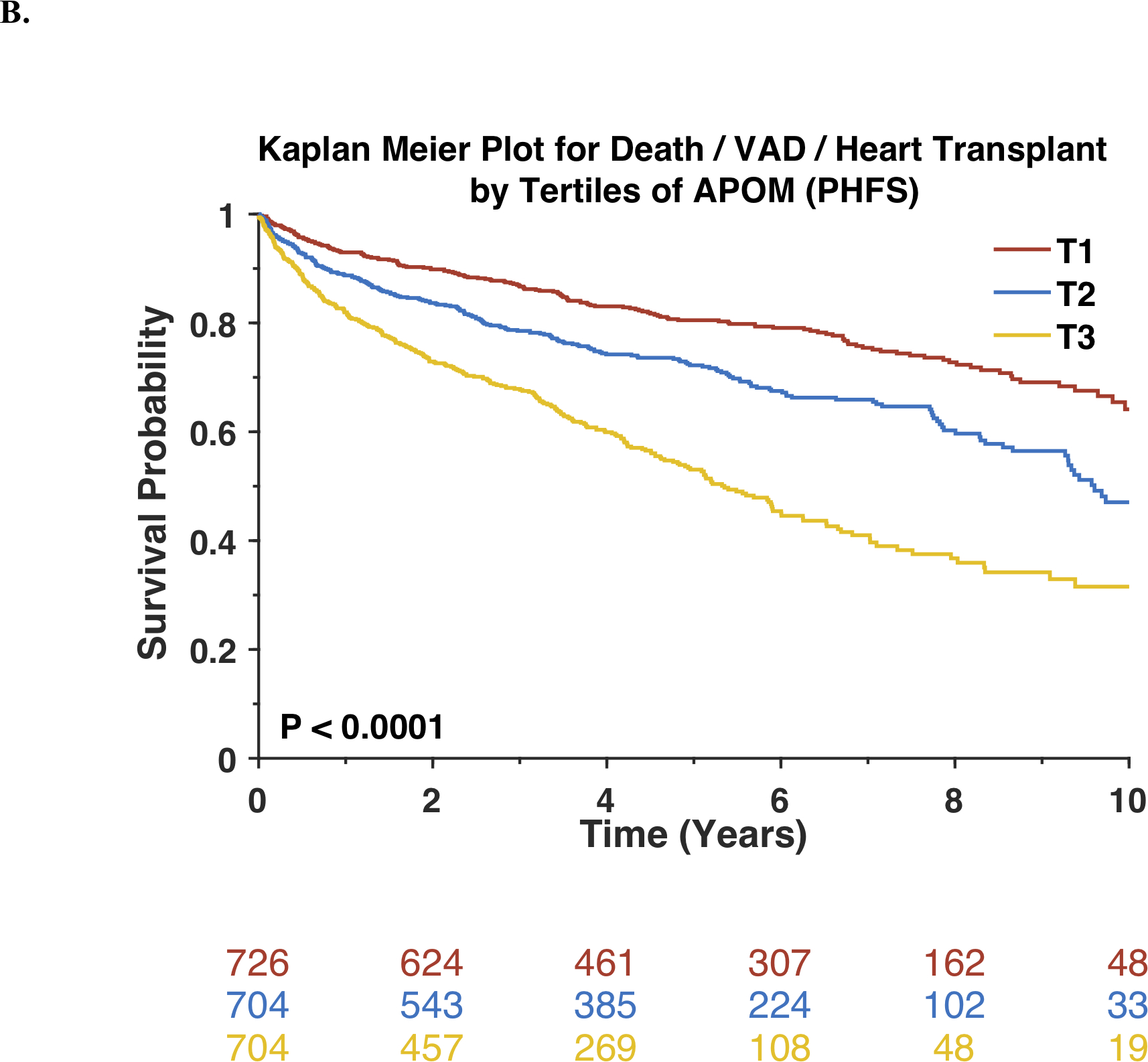
Kaplan-Meier survival curves for all-cause mortality (A) or the composite outcome of death, ventricular assist device or heart transplant (B) are shown. The number of patients at risk at each timepoint is presented below the graph.
Table 2.
Hazard Ratios for death per standard deviation increase in APOM in the PHFS
| APOM measured by modified aptamer assay (n=2,135) | ||
|---|---|---|
| All-cause death (NE=523) | ||
| Non-adjusted | 0.56 (0.51–0.61) | <0.0001 |
| Adjusted for clinical factors1 | 0.73 (0.65–0.81) | <0.0001 |
| Adjusted for clinical factors plus BNP | 0.78 (0.69–0.88) | <0.0001 |
| Death / VAD / Heart Transplant (NE=716) | ||
| Non-adjusted | 0.62 (0.58–0.67) | <0.0001 |
| Adjusted for clinical factors | 0.79 (0.72–0.87) | <0.0001 |
| Adjusted for clinical factors plus BNP | 0.85 (0.76–0.94) | 0.0014 |
NE= number of events; VAD=ventricular assist device; BNP=b-type natriuretic peptide.
Clinical factors included in the model were age, sex, race, enrollment site, history of PCI, CABG, atrial fibrillation or flutter, pacemaker, bi-ventricular pacer implantation, LV ejection fraction, NYHA class, ischemic vs. non-ischemic etiology, systolic and diastolic blood pressure, body mass index, history of diabetes mellitus, serum creatinine, ACE inhibitor/ARB use, digoxin, hydralazine, loop diuretic, organic nitrate and statin use.
Similarly, lower APOM levels were significantly associated with an increased risk of the composite endpoint of death, VAD implantation or heart transplantation (Standardized HR=0.62; 95%CI=0.58–0.67; P<0.0001; Table 2). Kaplan-Meier survival plots for APOM tertiles are shown in Figure 2B. The association between APOM and the composite endpoint remained significant after adjustment for multiple potential confounders, clinical risk factors, and BNP (Table 2). Similarly, APOM was also associated with risk of death (Standardized HR=0.80; 95% CI=0.71–0.91; P=0.0007) or with the risk of death, VAD implantation, or heart transplantation (Standardized HR=0.88; 95% CI=0.79–0.99; P=0.0165) after adjustment for APOA-I and APOB.
Interactions with ischemic etiology and key clinical and demographic factors
There was no significant interaction between APOM and ischemic vs. non-ischemic etiology for either death, death or heart-failure related hospitalization or the composite of death, ventricular assist device implantation or heart transplant (Online Table 3). Accordingly, APOM was associated with these outcomes in both ischemic and non-ischemic HF (Online Table 4).
Although we observed associations between APOM protein levels and age, sex, diabetes, and renal function, we did not observe any significant interaction between these variables, APOM, and HF outcomes. There was a significant interaction between APOM and African-American ethnicity for the outcome of death (P for interaction=0.0153) and VAD, heart transplant or death (P for interaction=0.004). Among African-Americans, the HR for death was 0.67 (95%CI=0.56–0.80; P<0.0001), whereas among non-African Americans, the HR was 0.53 (95%CI-0.48–0.59; P<0.0001). Similarly, among African-Americans, the HR for VAD, heart transplant or death was 0.73 (95CI=0.62–0.86; P=0.0001), whereas among non-African-Americans, the HR was 0.59 (95%CI=0.55–0.65).
Analyses stratified by HFrEF vs HFpEF
In analyses restricted to subjects with HFrEF (n=1761, Table 3), APOM was associated with death (Standardized HR=0.57; 95%CI=0.52–0.62; P<0.0001) and the composite endpoint of death, VAD implantation or heart transplantation (Standardized HR=0.62; 95%CI=0.51–0.76; P<0.0001). These associations remained after adjustment for the MAGGIC risk score and adjustment for both the MAGGIC risk score and BNP (Table 3).
Table 3.
Standardized Hazard Ratios for adverse outcomes per standard deviation increase in APOM in the PHFS in analyses restricted to HFrEF (n=1761) and HFpEF (n=249)
| HFrEF (n=1761) | ||||
|---|---|---|---|---|
| Model | Standardized Hazard Ratio | 95%CI, LB | 95%CI, UB | P value |
| All-cause death (NE=474) | ||||
| Non-adjusted | 0.57 | 0.52 | 0.62 | <0.0001 |
| Adjusted for the MAGGIC Risk Score | 0.75 | 0.67 | 0.83 | <0.0001 |
| Adjusted for the MAGGIC Risk Score plus BNP | 0.74 | 0.66 | 0.82 | <0.0001 |
| Death, VAD or Heart /Transplant (NE= 672) | ||||
| Non-adjusted | 0.62 | 0.51 | 0.76 | <0.0001 |
| Adjusted for the MAGGIC Risk Score | 0.81 | 0.74 | 0.89 | <0.0001 |
| Adjusted for the MAGGIC Risk Score plus BNP | 0.81 | 0.74 | 0.90 | <0.0001 |
| HFpEF (n=249) | ||||
| Model | Standardized Hazard Ratio | 95% CI, LB | 95%CI, UB | P value |
| All-cause death (NE=58) | ||||
| Non-adjusted | 0.44 | 0.34 | 0.57 | <0.0001 |
| Adjusted for the MAGGIC Risk Score | 0.61 | 0.43 | 0.85 | 0.0035 |
| Adjusted for the MAGGIC Risk Score plus BNP | 0.52 | 0.35 | 0.77 | 0.001 |
| Death or HFA (NE=110) | ||||
| Non-adjusted | 0.62 | 0.50 | 0.75 | <0.0001 |
| Adjusted for the MAGGIC Risk Score | 0.68 | 0.53 | 0.88 | 0.0034 |
| Adjusted for the MAGGIC Risk Score plus BNP | 0.72 | 0.55 | 0.94 | 0.0162 |
NE= number of events.
In analyses restricted to subjects with HFpEF (n=249), APOM was inversely associated with death. Kaplan-Meier survival plots for this subset of participants stratified by tertiles of APOM are shown in Figure 3A. Each standard-deviation decrease in APOM was associated with a >2-fold increase in mortality risk (Standardized HR=0.44; 95%CI=0.34–0.57; P<0.0001). APOM was also associated with the composite endpoint of death or HF-related hospitalization (Standardized HR=0.62; 95%CI=0.50–0.75; P<0.0001). Figure 3B shows Kaplan-Meier survival plots for the composite endpoint of death or HF-related hospitalization among this subset of participants, stratified by tertiles of APOM. Among participants with HFpEF, APOM was also associated with these endpoints independently of the MAGGIC risk score and BNP (Table 3).
Figure 3. Risk of adverse outcomes among Penn Heart Failure Study participants with HFpEF stratified by tertiles of APOM.
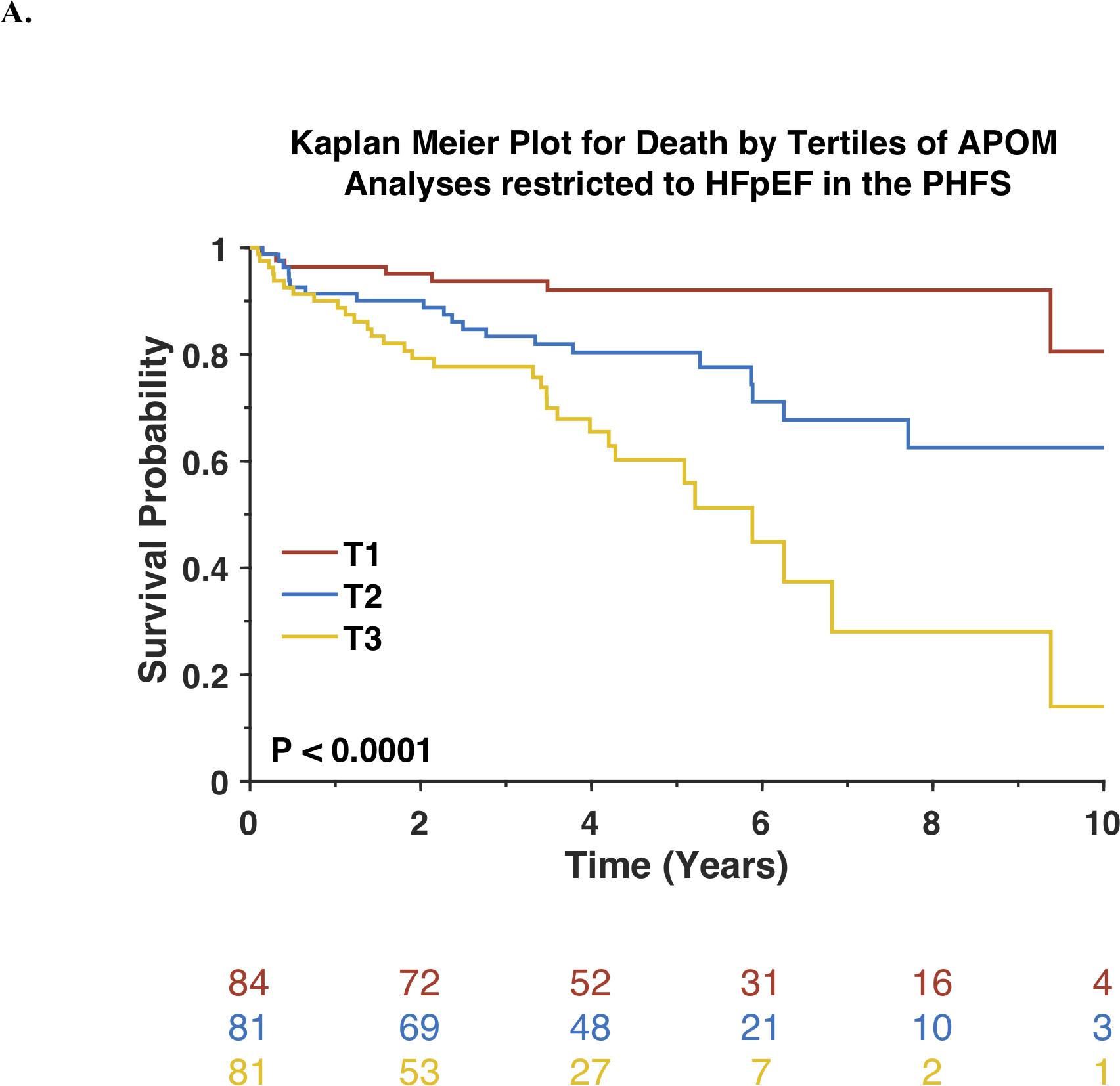
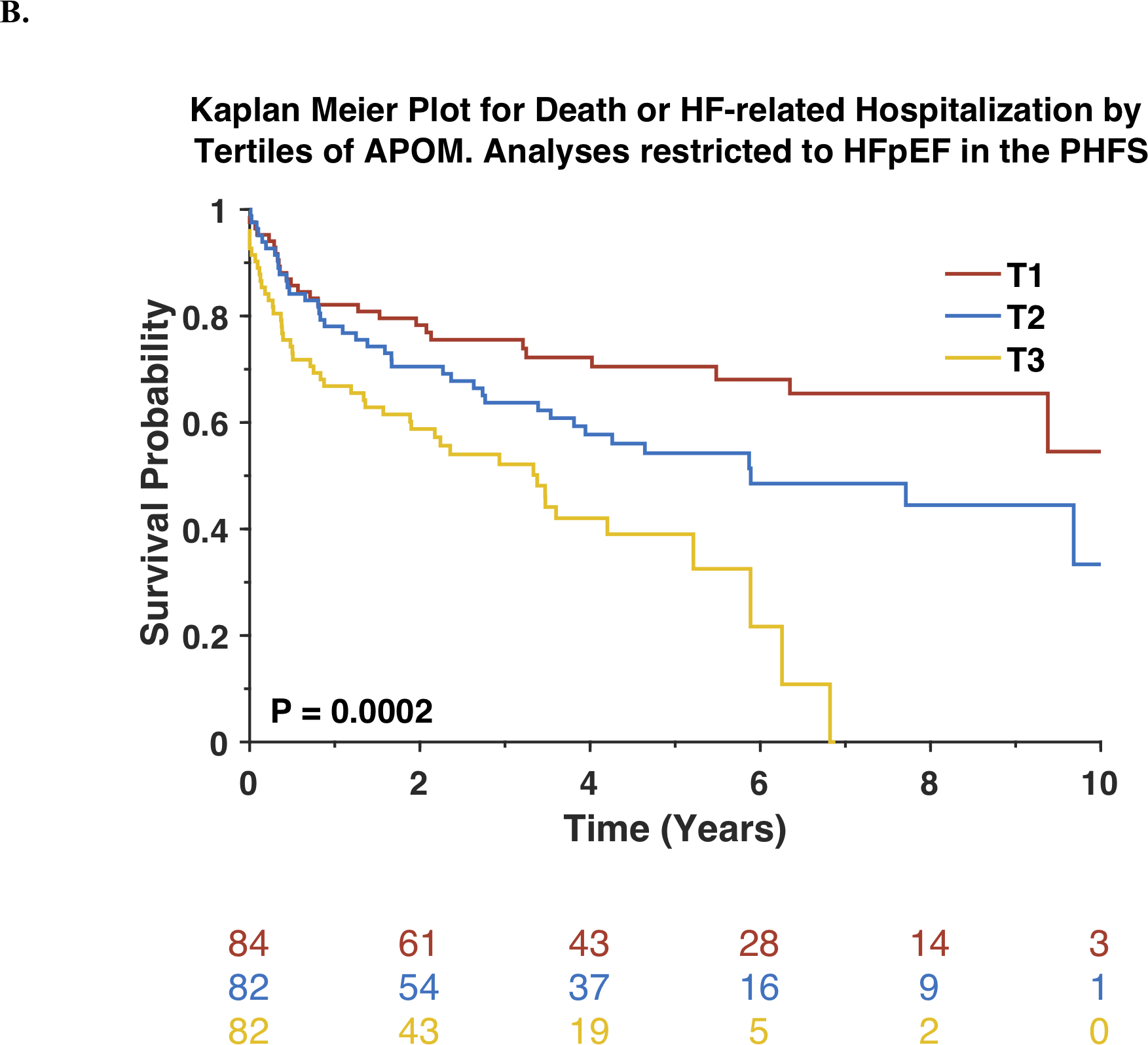
Kaplan-Meier survival curves for all-cause mortality (A) or the composite outcome of death or heart failure-related hospitalization (B) are shown. The number of patients at risk at each timepoint is presented below the graph.
Replication in the Washington University HF registry
The general characteristics of Washington University HF registry participants are shown in Online Table 5. During a follow-up of 2 years, 21 deaths occurred, and 29 participants reached the composite outcome of death, LVAD implantation or heart transplantation. In this cohort, lower APOM levels were significantly associated with an increased risk of death (Online Table 6). Similar to findings in our primary cohort, each standard-deviation decrease in APOM was associated with nearly a doubling in the mortality risk (Standardized HR=0.54; 95%CI=0.34–0.84; P=0.0064). APOM was also associated with the composite endpoint of death, VAD implantation or heart transplantation (Standardized HR=0.60; 95%CI=0.41–0.87; P=0.0077). APOM remained associated with these outcomes after adjustment for the MAGGIC risk score and after adjustment for the MAGGIC risk score and BNP (Online Table 6).
Replication in the TOPCAT trial
The general characteristics of TOPCAT trial participants included in this analysis are shown in Online Table 7. During a follow-up of 3.42 years, 48 deaths occurred, and 77 participants reached the composite outcome of death or HF-related hospitalization (Online Table 6). In this cohort, lower APOM levels were significantly associated with an increased risk of death (Standardized HR=0.76; 95%CI=0.58–0.99; P=0.0419) and an increased risk of the composite endpoint of death/HF-related hospital admission (Standardized HR=0.65; 95%CI=0.51–0.82; P=0.0002). APOM remained associated with these outcomes after adjustment for the MAGGIC risk score and after adjustment for the MAGGIC risk score and BNP (Online Table 6).
APOM, S1P, and HF outcomes
Online Figure 6 shows the correlation between HDL-associated APOM and S1P among PHFS participants (R=0.81; P<0.0001). Serum S1P levels were associated with death (Standardized HR=0.65; 95%CI=0.49–0.85; P=0.0021). This association remained after adjustment for the MAGGIC risk score (Standardized HR=0.70; 95%CI=0.53–0.93; P=0.0129).
There was a weak but significant correlation between S1P and APOM (R=0.25; P=0.00038). In a model that included both APOM and S1P, both were associated with death (Standardized HR for APOM =0.70; 95%CI=0.54–0.90; P=0.0051; Standardized HR for S1P =0.75; 95%CI=0.57–0.98; P=0.0361). There was a significant interaction between S1P and APOM (P=0.0355), indicating that their association with mortality risk was more pronounced at lower levels of APOM and/or S1P.
Accordingly, among participants with S1P levels below 50th percentile, APOM was associated with death (Standardized HR=0.64; 95%CI=0.47–0.88; P=0.006), whereas among those ≥50th percentile for S1P, APOM was not significantly associated (Standardized HR=0.71; 95%CI=0.48–1.07; P=0.10). Similarly, among participants with APOM levels <50th percentile, S1P was significantly associated with death (Standardized HR=0.61; 95%CI=0.47–0.80; P=0.0004), whereas among those ≥50th percentile for APOM, S1P was not significantly associated with mortality (Standardized HR=0.53; 95%CI=0.53–1.08; P=0.12).
Pathway analysis
The top canonical pathways associated with APOM are shown in Figure 4 and listed in Online Table 8. The results of regression analyses between APOM and all other proteins, utilized for pathway analysis, can be accessed as an online file at https://ahajournals.org/journal/circ. APOM protein levels were found to be negatively associated with inflammatory pathways, coagulation pathways, and a number of other biologically plausible pathways including renin-angiotensin signaling. The top pathways positively associated with APOM plasma protein were the liver-X-receptor (LXR) and complement pathways.
Figure 4. Top 20 canonical pathways associated with APOM.
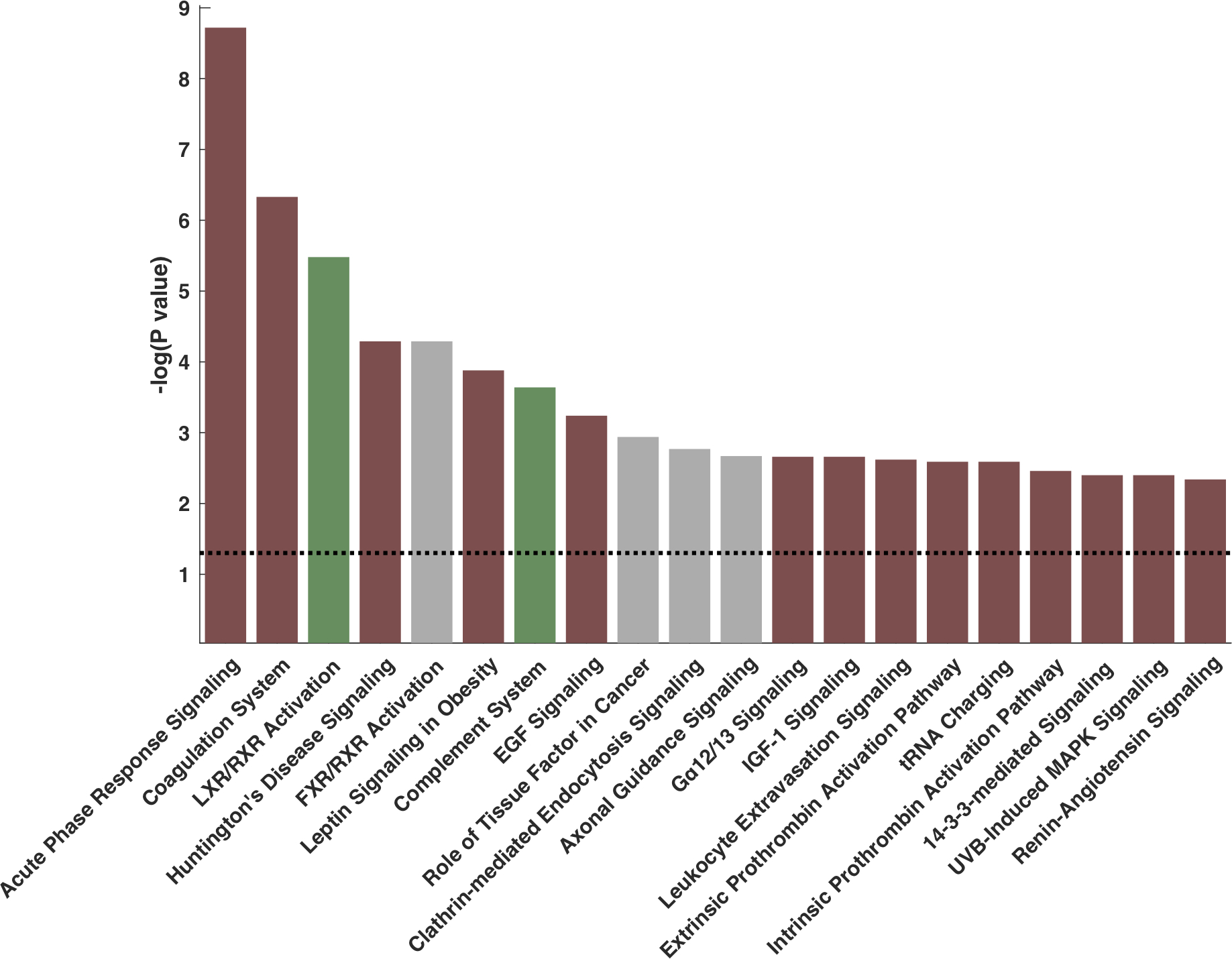
Red bars indicate a negative z-score (negative correlation). Green bars indicate positive z-scores (i.e., positive correlations). Gray bars denote pathways in which there is significant overlap with ApoM, but the directionality of the relationship is unclear.
We confirmed the association between APOM and the top canonical pathway (inflammation) using inflammatory biomarker levels measured by independent assays. Online Figure 5 shows a comparison of various inflammatory biomarkers between tertiles of APOM in the PHFS. Lower levels of APOM were associated with higher levels of multiple inflammatory biomarkers, including TNF-α, TNF-RI, TNF-RII, IL-6, IL-8, pentraxin-3, myeloperoxidase, and hsCRP.
To assess the degree to which the relationship between APOM and outcomes may depend on inflammation, we constructed survival models in which the relationship between APOM and outcomes was compared with vs. without adjustment for all the inflammatory biomarkers shown in Online Figure 6. In general, adjustment for these inflammatory biomarkers only partially attenuated the relationship between APOM and outcomes, suggesting that both inflammation and inflammation-independent factors underlie this relationship.
Discussion
We demonstrate that reduced APOM plasma protein levels are associated with the risk of adverse outcomes across the spectrum of human HF, by ELISA and SomaScan in the PHFS, a large cohort study performed at three academic centers with many years of follow-up and a large number of adjudicated events. In this cohort, we demonstrate that APOM is associated with adverse outcomes in the overall population, and in analyses restricted to HFpEF and HFrEF. We validated our findings in 2 independent cohorts, including a mixed HF cohort (Washington University HF registry), and in a HFpEF-only cohort (TOPCAT). The use of multiple cohorts across multiple centers and different methods to measure APOM provides convincing evidence that reduced levels of circulating APOM protein are independently associated with adverse outcomes (including increased mortality) in both HFpEF and HFrEF. Our findings are novel and provide information that, interpreted in the context of accumulating mechanistic animal data, supports a role for APOM in human HF.
What are the mechanisms by which APOM may promote improved outcomes in human HF? APOM is a chaperone for S1P, a sphingolipid that activates G-protein coupled receptors and the PI3-kinase signaling pathway14, 39. Animal studies suggest that APOM mediates S1P signaling to promote anti-inflammatory effects, survival of cardiomyocytes and improved endothelial function10, 12, 13 (Figure 5). We measured S1P by LC/MS-MS and observed an inverse association between S1P concentration and survival. We also observed a significant interaction between S1P and APOM, such that their relationship with outcomes was more pronounced when levels of both were reduced. APOM may target S1P to endothelial receptors to reduce endothelial-leukocyte adhesion12. Alternatively, APOM/S1P may restrain lymphopoiesis via direct effects on inflammatory cells40. Further study of the mechanisms by which APOM mediates cardioprotection will be a crucial step forward both in APOM biology and therapeutics targeting this pathway, particularly because S1P receptors are expressed on many cell types and have a multitude of effects. Increasing APOM may bias S1P signaling, allowing preferential activation of endothelial, anti-inflammatory and cardioprotective pathways while avoiding the potential toxicities of indiscriminately increasing S1P. Furthermore, the recent development of APOM peptides highlights the therapeutic potential of directly targeting APOM11. Interestingly, the relationship between APOM and death was not fully dependent on the levels of S1P, as APOM remained significantly associated with death after adjustment for S1P levels. We found that APOM protein levels are associated with multiple markers of baseline disease status, while our pathway analyses suggest a link between APOM and inflammation. Whether the anti-inflammatory effects of APOM are direct, or related to APOM’s role in modulating endothelial protection, will be the subject of future studies.
Figure 5. Proposed protective effects of apolipoprotein M in heart failure.
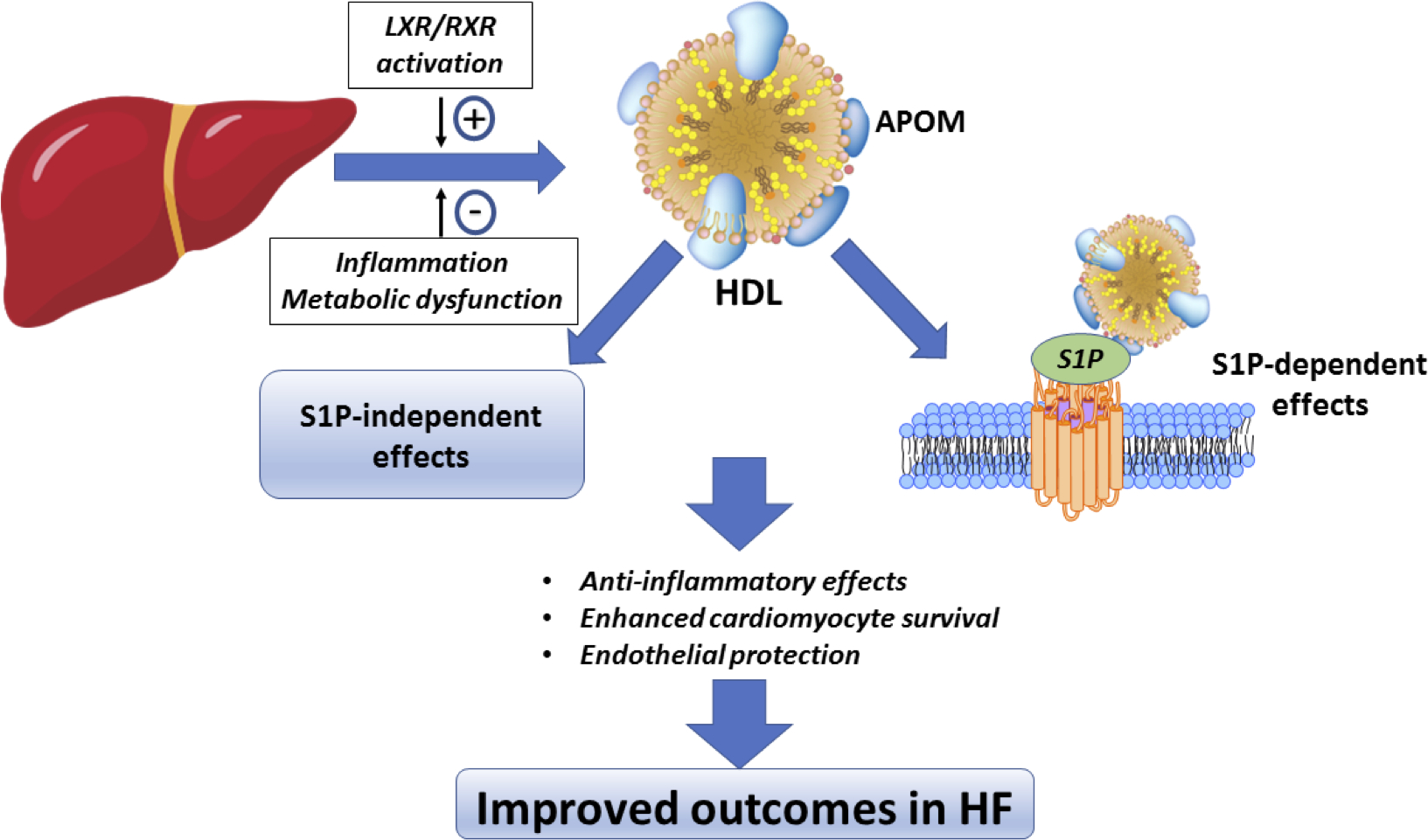
APOM is associated with HDL and binds S1P. S1P signaling enhances cardiomyocyte survival, activates endothelial protective pathways, and is anti-inflammatory. The culmination of these effects may result in improved survival in heart failure. However, whether the relationship between APOM and outcomes is causal remains to be determined.
APOM may also link hepatic and lipoprotein metabolism with HF outcomes. Our pathway analyses are consistent with a positive relationship between APOM and the LXR ligand-activated transcription factors, that have been implicated in metabolic homeostasis, inflammation, hepatic disease, but also HF.41 Although LXR-agonism has been suggested to decrease transcription of APOM in vitro, more recent studies suggest a more nuanced interpretation of these initial findings42. In theory, LXR agonism could contribute to APOM stability via an effect on APOB-containing lipoproteins, which may decrease APOM clearance43,44. In patients with advanced HF, factors including inflammation, metabolic disease, hepatic dysfunction, and reduced lipoprotein levels could contribute to further reductions in APOM secretion or increases in clearance, creating a feed-forward mechanism leading to progressive mortality.
Our findings are relevant to observations linking HDL subsets to adverse outcomes in HF45 and prior studies demonstrating that reduced circulating lipoproteins are associated with increased mortality risk in HF patients46.. There are multiple potential mechanisms by which HDL may play a future role in the treatment of HF. Rodent models support the use of reconstituted HDL47–49 in HF models, but also chimeric APOM peptides that are not HDL-associated11 to prevent cardiac ischemic injury. To account for possible confounding due to reductions in lipoprotein levels or APOA-I, we adjusted our analyses for HDL-C and APOA-I, the major protein constituent of HDL. While our findings support prior studies that reduced levels of APOA-I are associated with HF mortality50, we observed an independent association between APOM protein levels and all-cause mortality. Mechanistically, APOA-I interacts with scavenger receptor BI, loss of which has been implicated in HF pathogenesis in rodent models51. Further basic research must explore the interactions of APOM with APOA-I and SR-BI, whether APOM requires APOA-I or SR-BI, or whether there might be a synergistic effect of increasing both APOM and APOA-I. These questions will need to be tested in pre-clinical, rodent models. Although the possibility remains that APOM is a marker of improved HDL function, or improves HDL-functionality by an indirect mechanism, increasing APOM may be one mechanism of HDL-mediated cardio-protection.
Our study should be interpreted in the context of its strengths and limitations. Our study was focused on the role of APOM in the prognosis of patients with existing HF; whether APOM is associated with incident HF incidence is a separate question. Second, we have not intended to compare APOM across cohorts, and this is a technical limitation as well because the aptamer-specific fluorescence signal depends on the characteristics and the number of other aptamers in the assay, which in turn varies with specific versions of the SomaScan. We cannot rule out that both ELISA and SomaScan measurements of apolipoproteins may be affected by unmeasured biological confounders; nonetheless, the consistent directionality of both assays provides a high degree of confidence in our results. Despite comprehensive adjustments to the extent possible, we cannot rule out residual confounding. Our analyses adjusted for the presence of diabetes mellitus, but not for hemoglobin A1c levels (which were not available in these cohorts). Our analyses did not discern the relationship between APOM and cardiovascular vs. non-cardiovascular death. Because our study is observational in nature, we cannot rule out that APOM and S1P may be inversely associated with increased mortality, but not causally related to it, a hypothesis that should be strictly tested with interventions designed to increase APOM.
Additionally, our studies were performed in a large number of patients within a well-characterized primary cohort, with long-term follow-up and a large number of well-adjudicated events, and 2 independent secondary cohorts for validation of our findings regarding APOM and outcomes. We also convincingly demonstrate that the relationship between APOM and outcomes is present in both HFpEF and HFrEF. After utilizing ELISA to determine that reductions in APOM were associated with worse outcomes in PHFS, we subsequently utilized SomaScan analyses from 3 separate cohorts at different timepoints and different institutions.. Furthermore, we directly measured S1P to better discern the relationships between APOM, S1P and mortality, and demonstrated that in HF patients, HDL-APOM is associated with HDL-S1P. Finally, we performed pathway analysis using a broad proteomic scan, revealing relationships between APOM and specific biologic pathways. The mechanistic underpinnings and high biologic plausibility, based on animal and pre-clinical data, increase the relevance and generalizability of our findings. While our work is an important first step, further insights from human genetics, animal studies, and randomized clinical trials will be required to gain further insights into the mechanisms by which APOM is associated with mortality in human HF.
In conclusion, we have identified that reduced circulating APOM protein levels are associated with an increased risk of death across the spectrum of human HF. The APOM/S1P axis merits further exploration as a therapeutic target in patients with HF.
Supplementary Material
Clinical Perspective.
What Is New?
Reduced APOM plasma protein levels are associated with adverse outcomes in HF (including HFpEF and HFrEF).
The relationship between reduced APOM and outcomes in HF is particularly pronounced when concentrations of its binding partner, sphingosine-1-phosphate, are also reduced.
APOM protein levels are associated with inflammation in human HF.
What Are the Clinical Implications?
APOM represents a risk marker in human HF.
APOM is associated with inflammation in HF, but its relationship with risk appears to be only partially dependent with this association.
Further studies are needed to assess whether targeting APOM/S1P can improve outcomes in HF.
Acknowledgements:
We would like to thank Eric and Jill Becker for their kind support.
Funding: JAC is supported by NIH grants R01 HL 121510-01A1, R61-HL-146390, R01-AG058969, 1R01HL104106, P01HL094307 and R56 HL136730. BD is supported by Swedish Research Council. DLM is supported by NIH grants R01HL107594-06, NIH HHSN268201100026C, NIH RC2-HL102222, LA was supported by T32 HL007081. JSP is supported by R01 HL119962. KBM and TPC are supported by U10 HL110338. AJ is supported by K08HL138262,P30DK056341.
Footnotes
Disclosures: J.A.C. has received consulting honoraria from Sanifit, Microsoft, Fukuda-Denshi, Bristol-Myers Squibb, OPKO Healthcare, Ironwood Pharmaceuticals, Pfizer, Akros Pharma, Merck, Edwards Lifesciences and Bayer. J.A.C. is named as inventor in University of Pennsylvania patent applications for the use of inorganic nitrates/nitrites for the treatment of HF and Preserved Ejection Fraction, for the use of novel neoepitope biomarkers of tissue fibrosis, and for novel methods of arterial pulse wave analysis. KMB has received research funding from Merck, Sharp and Dohme, Sanofi-Aventis and Glaxo-Smith-Kline. Other authors report no disclosures.
Supplemental Materials
Online Figures 1–6
Online Tables 1–8
Online Excel File
References
- 1.Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH and Dahlback B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res. 2006;47:1833–1843. [DOI] [PubMed] [Google Scholar]
- 2.Xu N and Dahlback B. A novel human apolipoprotein (apoM). J Biol Chem. 1999;274:31286–1290. [DOI] [PubMed] [Google Scholar]
- 3.Liu M, Seo J, Allegood J, Bi X, Zhu X, Boudyguina E, Gebre AK, Avni D, Shah D, Sorci-Thomas MG et al. Hepatic apolipoprotein M (apoM) overexpression stimulates formation of larger apoM/sphingosine 1-phosphate-enriched plasma high density lipoprotein. J biol chem. 2014;289:2801–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsoe S, Ahnstrom J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW, Binder CJ, Bjorkbacka H, Dahlback B and Nielsen LB. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221:91–97. [DOI] [PubMed] [Google Scholar]
- 5.Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlback B and Nielsen LB. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem. 2008;283:1839–1847. [DOI] [PubMed] [Google Scholar]
- 6.Christensen PM, Liu CH, Swendeman SL, Obinata H, Qvortrup K, Nielsen LB, Hla T, Di Lorenzo A and Christoffersen C. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J. 2016;30:2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108:9613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruiz M, Okada H and Dahlback B. HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis. 2017;16:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. [DOI] [PubMed] [Google Scholar]
- 10.Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, Mach F, James RW, Naus CC, Chanson M et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc Res. 2016;109:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swendeman SL, Xiong Y, Cantalupo A, Yuan H, Burg N, Hisano Y, Cartier A, Liu CH, Engelbrecht E, Blaho V, et al. An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal. 2017;10:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz M, Frej C, Holmer A, Guo LJ, Tran S and Dahlback B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler Thromb Vasc Biol. 2017;37:118–129. [DOI] [PubMed] [Google Scholar]
- 13.Kurano M, Tsuneyama K, Morimoto Y, Shimizu T, Jona M, Kassai H, Nakao K, Aiba A and Yatomi Y. Apolipoprotein M Protects Lipopolysaccharide-Treated Mice from Death and Organ Injury. Thromb Haemost. 2018;118:1021–1035. [DOI] [PubMed] [Google Scholar]
- 14.Sattler K, Graler M, Keul P, Weske S, Reimann CM, Jindrova H, Kleinbongard P, Sabbadini R, Brocker-Preuss M, Erbel R et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading. J Am Coll Cardiol. 2015;66:1470–1485. [DOI] [PubMed] [Google Scholar]
- 15.Basuray A, French B, Ky B, Vorovich E, Olt C, Sweitzer NK, Cappola TP and Fang JC. Heart failure with recovered ejection fraction: clinical description, biomarkers, and outcomes. Circulation. 2014;129:2380–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ky B, French B, Ruparel K, Sweitzer NK, Fang JC, Levy WC, Sawyer DB and Cappola TP. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J Am Coll Cardiol. 2011;58:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ky B, Kimmel SE, Safa RN, Putt ME, Sweitzer NK, Fang JC, Sawyer DB and Cappola TP. Neuregulin-1 beta is associated with disease severity and adverse outcomes in chronic heart failure. Circulation. 2009;120:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakshmi Kannan ARC. Thyroid Dysfunction in Heart Failure and Cardiovascular Outcomes. Circ Heart Fail. 2018;11:e005266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph SM, Novak E, Arnold SV, Jones PG, Khattak H, Platts AE, Davila-Roman VG, Mann DL and Spertus JA. Comparable performance of the Kansas City Cardiomyopathy Questionnaire in patients with heart failure with preserved and reduced ejection fraction. Circ Heart Fail. 2013;6:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desai AS, Lewis EF, Li R, Solomon SD, Assmann SF, Boineau R, Clausell N, Diaz R, Fleg JL, Gordeev I et al. Rationale and design of the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial: a randomized, controlled study of spironolactone in patients with symptomatic heart failure and preserved ejection fraction. Am Heart J. 2011;162:966–972 e10. [DOI] [PubMed] [Google Scholar]
- 21.Pfeffer MA, Claggett B, Assmann SF, Boineau R, Anand IS, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation. 2015;131:34–42. [DOI] [PubMed] [Google Scholar]
- 22.Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–1392. [DOI] [PubMed] [Google Scholar]
- 23.de Denus S, O’Meara E, Desai AS, Claggett B, Lewis EF, Leclair G, Jutras M, Lavoie J, Solomon SD, Pitt B et al. Spironolactone Metabolites in TOPCAT - New Insights into Regional Variation. N Engl J Med. 2017;376:1690–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girerd N, Ferreira JP, Rossignol P and Zannad F. A tentative interpretation of the TOPCAT trial based on randomized evidence from the brain natriuretic peptide stratum analysis. Eur J Heart Fail. 2016;18:1411–1414. [DOI] [PubMed] [Google Scholar]
- 25.Axler O, Ahnstrom J and Dahlback B. An ELISA for apolipoprotein M reveals a strong correlation to total cholesterol in human plasma. J Lipid Res. 2007;48:1772–1780. [DOI] [PubMed] [Google Scholar]
- 26.Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, Hoover H, Gudmundsdottir V, Horman SR, Aspelund T et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brody E, Gold L, Mehan M, Ostroff R, Rohloff J, Walker J and Zichi D. Life’s simple measures: unlocking the proteome. J Mol Biol. 2012;422:595–606. [DOI] [PubMed] [Google Scholar]
- 28.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG and Williams SA. Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients With Stable Coronary Heart Disease. JAMA. 2016;315:2532–2541. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Starmer J and Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol. 2008;32:361–69. [DOI] [PubMed] [Google Scholar]
- 31.Auro K, Joensuu A, Fischer K, Kettunen J, Salo P, Mattsson H, Niironen M, Kaprio J, Eriksson JG, Lehtimaki T et al. A metabolic view on menopause and ageing. Nat Commun. 2014;5:4708. [DOI] [PubMed] [Google Scholar]
- 32.Tromp J, Khan MA, Klip IT, Meyer S, de Boer RA, Jaarsma T, Hillege H, van Veldhuisen DJ, van der Meer P and Voors AA. Biomarker Profiles in Heart Failure Patients With Preserved and Reduced Ejection Fraction. J Am Heart Assoc. 2017;6:e003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frej C, Andersson A, Larsson B, Guo LJ, Norstrom E, Happonen KE and Dahlback B. Quantification of sphingosine 1-phosphate by validated LC-MS/MS method revealing strong correlation with apolipoprotein M in plasma but not in serum due to platelet activation during blood coagulation. Anal Bioanal Chem. 2015;407:8533–8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajaj A, Xie D, Cedillo-Couvert E, Charleston J, Chen J, Deo R, Feldman HI, Go AS, He J, Horwitz E et al. Lipids, Apolipoproteins, and Risk of Atherosclerotic Cardiovascular Disease in Persons With CKD. Am J Kidney Dis. 2019;73:827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapman MJ, Goldstein S, Lagrange D and Laplaud PM. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J Lipid Res. 1981;22:339–358. [PubMed] [Google Scholar]
- 37.Lam CSP, Gamble GD, Ling LH, Sim D, Leong KTG, Yeo PSD, Ong HY, Jaufeerally F, Ng TP, Cameron VA et al. Mortality associated with heart failure with preserved vs. reduced ejection fraction in a prospective international multi-ethnic cohort study. Eur Heart J. 2018;39:1770–1780. [DOI] [PubMed] [Google Scholar]
- 38.Pocock SJ, Ariti CA, McMurray JJ, Maggioni A, Kober L, Squire IB, Swedberg K, Dobson J, Poppe KK, Whalley GA et al. Predicting survival in heart failure: a risk score based on 39 372 patients from 30 studies. Eur Heart J. 2013;34:1404–1413. [DOI] [PubMed] [Google Scholar]
- 39.Sattler K and Levkau B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res. 2009;82:201–211. [DOI] [PubMed] [Google Scholar]
- 40.Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M, Proia RL, Steinman L, Han MH and Hla T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature. 2015;523:342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cannon MV, van Gilst WH and de Boer RA. Emerging role of liver X receptors in cardiac pathophysiology and heart failure. Basic Res Cardiol. 2016;111:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di D, Wang Z, Liu Y, Luo G, Shi Y, Berggren-Soderlund M, Nilsson-Ehle P, Zhang X and Xu N. ABCA1 upregulating apolipoproein M expression mediates via the RXR/LXR pathway in HepG2 cells. Biochem Biophys Res Commun. 2012;421:152–156. [DOI] [PubMed] [Google Scholar]
- 43.Christoffersen C, Benn M, Christensen PM, Gordts PL, Roebroek AJ, Frikke-Schmidt R, Tybjaerg-Hansen A, Dahlback B and Nielsen LB. The plasma concentration of HDL-associated apoM is influenced by LDL receptor-mediated clearance of apoB-containing particles. J Lipid Res. 2012;53:2198–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Croyal M, Billon-Crossouard S, Goulitquer S, Aguesse A, Leon L, Fall F, Chetiveaux M, Moyon T, Blanchard V, Ouguerram K et al. Stable Isotope Kinetic Study of ApoM (Apolipoprotein M). Arterioscler Thromb Vasc Biol. 2018;38:255–261. [DOI] [PubMed] [Google Scholar]
- 45.Hunter WG, McGarrah RW 3rd, Kelly JP, Khouri MG, Craig DM, Haynes C, Felker GM, Hernandez AF, Velazquez EJ, Kraus WE et al. High-Density Lipoprotein Particle Subfractions in Heart Failure With Preserved or Reduced Ejection Fraction. J Am Coll Cardiol. 2019;73:177–186. [DOI] [PubMed] [Google Scholar]
- 46.Rauchhaus M, Clark AL, Doehner W, Davos C, Bolger A, Sharma R, Coats AJ and Anker SD. The relationship between cholesterol and survival in patients with chronic heart failure. J Am Coll Cardiol. 2003;42:1933–1940. [DOI] [PubMed] [Google Scholar]
- 47.Aboumsallem JP, Mishra M, Amin R, Muthuramu I, Kempen H and De Geest B. Successful treatment of established heart failure in mice with recombinant HDL (Milano). Br J Pharmacol. 2018;175:4167–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aboumsallem JP, Muthuramu I, Mishra M, Kempen H and De Geest B. Effective Treatment of Diabetic Cardiomyopathy and Heart Failure with Reconstituted HDL (Milano) in Mice. Int J Mol Sci. 2019;20:1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mishra M, Muthuramu I, Aboumsallem JP, Kempen H and De Geest B. Reconstituted HDL (Milano) Treatment Efficaciously Reverses Heart Failure with Preserved Ejection Fraction in Mice. Int J Mol Sci. 2018;19:3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gombos T, Forhecz Z, Pozsonyi Z, Janoskuti L, Prohaszka Z and Karadi I. Long-Term Survival and Apolipoprotein A1 Level in Chronic Heart Failure: Interaction With Tumor Necrosis Factor alpha −308 G/A Polymorphism. J Card Fail. 2017;23:113–120. [DOI] [PubMed] [Google Scholar]
- 51.Muthuramu I, Amin R, Aboumsallem JP, Mishra M, Robinson EL and De Geest B. Hepatocyte-Specific SR-BI Gene Transfer Corrects Cardiac Dysfunction in Scarb1-Deficient Mice and Improves Pressure Overload-Induced Cardiomyopathy. Arterioscler Thromb Vasc Biol. 2018;38:2028–2040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.