

Abstract
Development of efficient and selective C–N bond-forming reactions from abundant feedstock chemicals remains a central theme in organic chemistry, due to the key roles of amines in synthesis, drug discovery and materials science. Here, we present a dual catalytic system for N-alkylation of diverse aromatic carbocyclic and heterocyclic amines directly with carboxylic acids, by-passing preactivation as redox-active esters. The reaction that is enabled by visible light-driven, acridine-catalyzed decarboxylation provides access to N-alkylated secondary and tertiary anilines, and N-heterocycles. Additional examples, including double alkylation, installation of metabolically robust deuterated methyl groups and tandem ring formation further demonstrate the potential of the direct decarboxylative alkylation (DDA) reaction.
Keywords: Amination, Carboxylic acids, Copper catalysis, Photocatalysis, Visible light
Graphical Abstract
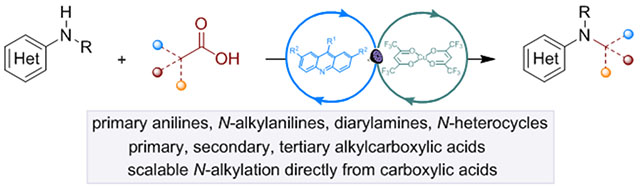
Carbocyclic and heterocyclic aromatic amines undergo decarboxylative N-alkylation directly with carboxylic acids. The directional character of the acridine photocatalysis enables the challenging decarboxylation of unactivated carboxylic acids in the presence of more readily oxidizable anilines, resulting in a synthetic platform that encompasses a wide range of structurally diverse amines and acids.
Introduction
Aromatic carbocyclic and heterocyclic amines are among the most privileged motifs in medicinal chemistry, materials science and catalysis.[1] Given the preeminence of aromatic amines, several key methods for construction of C(sp2)–N bonds have been developed, primarily from aryl halides and boronic acids, e.g., the Cu-catalyzed Ullmann-Goldberg[2] and Chan-Evans-Lam[3] couplings, and the Pd-catalyzed Buchwald-Hartwig amination.[4] In contrast, construction of C(sp3)–N bonds as a strategy for accessing aromatic amines is comparatively less explored, and few alternatives exist to the well-established reductive amination[5] and nucleophilic substitution methods.[6] In this context, the groups of Hu and MacMillan recently demonstrated that carboxylic acids can serve as coupling partners for the construction of C(sp3)–N bonds via alkyl radicals produced in photocatalytic decarboxylation of pre-formed activated carboxylic acid derivatives, e.g., iodine(III) carboxylates or redox-active esters.[7,8]
Decarboxylative generation of alkyl radicals from redox-active esters has emerged as a practical platform for the development of new carbon–carbon and carbon–heteroatom bond-forming transformations.[9] Redox-active esters are widely used because they can be prepared from carboxylic acids that are abundant industrial and bioderived feedstocks. Additionally, decarboxylation of redox-active esters can be initiated in the presence of a suitable reductant, e.g., a photoredox catalyst or a reducing metal, providing a workaround for the challenging direct decarboxylation of carboxylic acids. The direct decarboxylation is attractive from a synthetic perspective, as it obviates formation and isolation of redox-active derivatives, but it remains problematic because of the relatively high oxidation potentials of carboxylic acids and carboxylates (e.g., Eox = 1.06 V vs SCE for tetrabutylammonium cyclohexanecarboxylate), necessitating strong photooxidants that are incompatible with more easily oxidizable substrates, for example anilines (e.g., Eox = 0.52 V vs SCE for N-methyl-p-anisidine).
We recently described new acridine photocatalysts A1 and A2 for the direct visible light-driven decarboxylation of a broad range of carboxylic acids that produces alkyl radicals by a photoinduced proton-coupled electron transfer (PCET) process in the acridine–carboxylic acid hydrogen bond complex (Figure 1),[10] i.e., by a different mechanism than the structurally related N-alkyl-or N-arylacridinium salts.[11]
Figure 1.
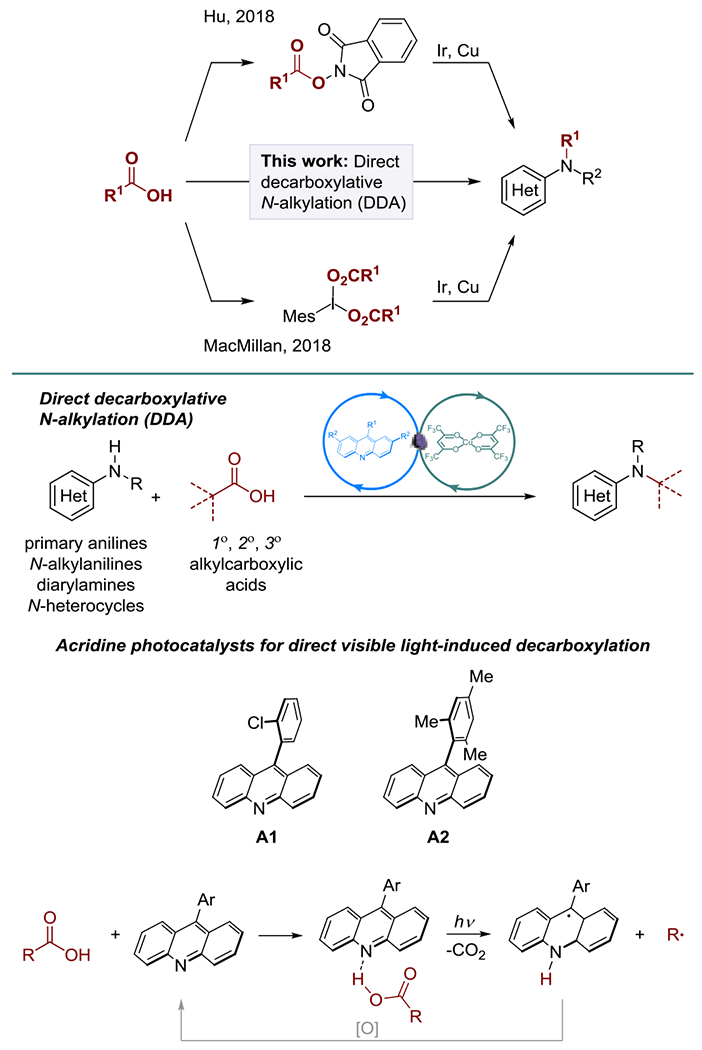
Decarboxylative N-alkylation of anilines.
The directional character of the acridine–carboxylic acid interaction in combination with the PCET enabled a triple catalytic biointerfaced conversion of esters to alkenes that was not possible with photoredox catalysts operating by a photoinduced oxidation of carboxylate anions.[10] It was therefore hypothesized that the acridine-catalyzed, visible light-driven direct decarboxylation of carboxylic acids may be compatible with easily oxidizable anilines, and may be coupled with a carbon–nitrogen bond forming catalytic cycle, resulting in a direct decarboxylative alkylation (DDA) of anilines. The distinct mechanism of acridine-catalyzed photodecarboxylation was further expected to enable a decarboxyaltive N-alkylation of a broad range of anilines, including N-alkylanilines, diarylamines and N-heterocycles that currently cannot be accessed via decarboxylation of activated carboxylic acid derivatives.
Advantageously, acridines A1 and A2 are readily accessible on gram scales in one and two steps from inexpensive precursors.[10]
Results and Discussion
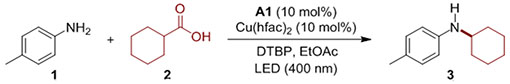
After initial optimization studies with aniline 1 and acid 2, secondary alkyl aniline 3 was obtained in high yield with the dual catalytic system comprising acridine A1 and Cu(hfac)2 (hfac = hexafluoroacetylacetonate) with di-tert-butyl peroxide (DTBP) as a readily available oxidant (Table 1). Both catalysts as well as the oxidant and the irradiation with light were necessary to effect the direct decarboxylative alkylation (entries 2–5). Acridine A2 was also an effective photocatalyst. Other photocatalysts did not effect the direct decarboxylative amination, pointing to the enabling role of the acridine photocatalysis (see SI). Cu(hfac)2 was substantially more efficient than other copper(II) salts (e.g., entry 7). Other solvents (toluene and trifluorotoluene) were also potentially suitable, and LED light with λmax = 420 nm could also be used. Additionally, the reaction was not sensitive to air, underscoring the practicality of the acridine-photocatalyzed decarboxylation, and in agreement with prior mechanistic conclusions that acridines effect the photocatalytic decarboxylation via the singlet excited state.[10]
Table 1.
Reaction conditions for the direct decarboxylative alkylation (DDA) of anilines.[a]
 | ||
|---|---|---|
| Entry | Change from optimal conditions | Yield, %[b] |
| 1 | No change | 99 (97[c]) |
| 2 | No light | 0 |
| 3 | No A1 | 0 |
| 4 | No Cu(hfac)2 | 0 |
| 5 | No DTBP | 7 |
| 6 | A2 instead of A1 | 86 |
| 7 | Cu(acac)2 instead of Cu(hfac)2 | 38 |
| 8 | PhCF3 instead of EtOAc | 78 |
| 9 | PhCH3 instead of EtOAc | 76 |
| 10 | 420 nm instead of 400 nm LED | 86 |
| 11 | Under air | 94 |
Reaction conditions: aniline (0.3 mmol), carboxylic acid (0.75 mmol), A1 (10 mol%), Cu(hfac)2 (10 mol%), DTBP (0.6 mmol), EtOAc (4.5 mL), LED (400 nm), 12 h.
Determined by 1H NMR with 1,4-dimethoxybenzene as an internal standard.
Isolated yield.
The scope of the DDA reaction was explored next (Scheme 1, 3-54). The reaction performed equally well with electron-deficient and electron-rich primary anilines (4-32). Substituents in o-, m-, and p- positions were tolerated. Medicinally relevant fluorine-containing anilines afforded N-alkylated products in good yields (9, 10, 16, 18, 19, 22). Ester, sulfonyl, boryl, cyano, and halogen-substituted anilines were also readily converted to the corresponding products 7, 8, 11-14, 22-24, 27.
Scheme 1.
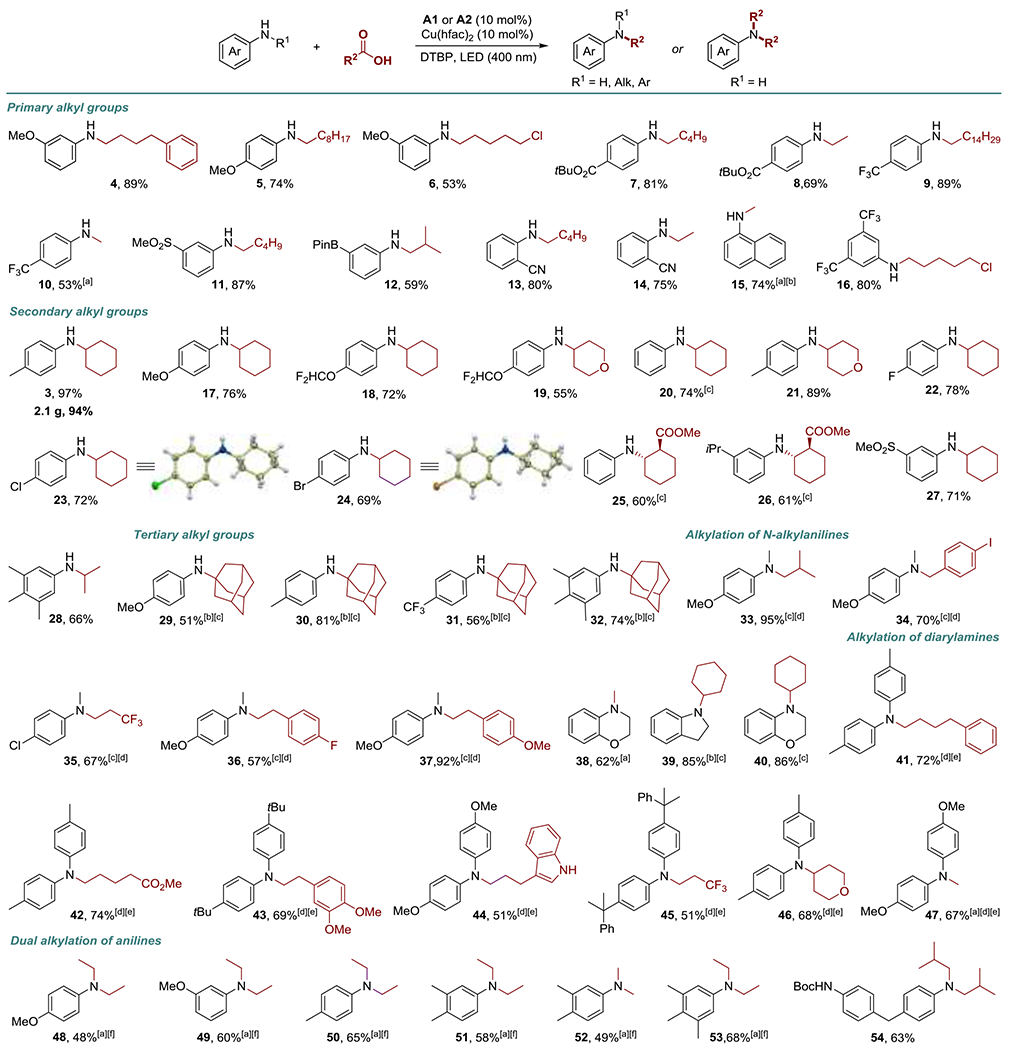
Scope of the direct decarboxylative N-alkylation. Method A: aniline (0.3 mmol), carboxylic acid (0.75 mmol), A1 (10 mol%), Cu(hfac)2 (10 mol%), DTBP (0.6 mmol), EtOAc (4.5 mL), LED (400 nm). Method A was used unless otherwise specified. [a] Carboxylic acid (1.5 mmol). [b] DCP (0.45 mmol). [c] Method B: aniline (0.75 mmol), carboxylic acid (0.3 mmol), A1 (10 mol%), Cu(hfac)2 (10 mol%), DTBP (0.6 mmol), EtOAc (4.5 mL), LED (400 nm). [d] PhCF3 (4.5 mL). [e] A2 (10 mol%). [f] A1 (15 mol%).
The acridine-catalyzed DDA reaction also provided the first example of decarboxylative alkylation of secondary anilines. Both cyclic and acyclic N-alkylanilines were efficiently converted to the corresponding tertiary anilines 33-40. Furthermore, diarylamines that typically require deprotonation with strong bases to effect N-alkylation also proved to be suitable substrates for the synthesis of the corresponding tertiary anilines (41-47) under the conditions that, remarkably, do not require protection of an indole nitrogen (44). This result indicates that a substantial chemoselectivity can be achieved even for closely related nitrogen nucleophiles.
Double alkylation of primary anilines is a synthetically valuable reaction that provides a one-step access to tertiary anilines. Gratifyingly, minor adjustment of the DDA reaction conditions allowed for double alkylation of primary anilines (48-54).
The scope of carboxylic acids was also examined. Primary carboxylic acids demonstrated excellent reactivity with a variety of primary and secondary anilines (4-16, 33-38, 41-45, 47). Secondary alkylcarboxylic acids were equally reactive, and a range of amines bearing secondary alkyl groups were synthesized, unaffected by steric or electronic effects of substituents in o-, m-, or p-positions in the aromatic ring (3, 17-28, 39, 40, 46). Notably, trans-substituted β-amino esters 25 and 26 were accessed as single diastereomers from the corresponding trans-substituted acid, pointing to the potential of the reaction to effect stereoselective alkylations. Several N-tert-alkyl-substituted anilines were also synthesized (29-32). The DDA reaction can be carried out with either acid or aniline as a limiting reagent (cf. methods A and B in Scheme 1), providing additional synthetic flexibility.
Given the growing role of heterocyclic nitrogen-containing motifs in drug discovery,[12] we were interested to explore the scope of the DDA reaction with various heterocyclic aromatic amines (Scheme 2). We were pleased to observe that heterocyclic amines were suitable substrates (55-68). Indoles and carbazoles typically require strong bases to effect deprotonation and subsequent alkylation,[13] and development of mild methods of N-alkylation of azoles remains an area of unmet need. Gratifyingly, the DDA reaction proceeded smoothly, providing access to N-substituted indoles and carbazoles (55-59) in good yields. Other heterocycles also gave the alkylation products in good yields, including quinolines (60-63), indazoles (64, 65) and thiophenes (66-68).
Scheme 2.
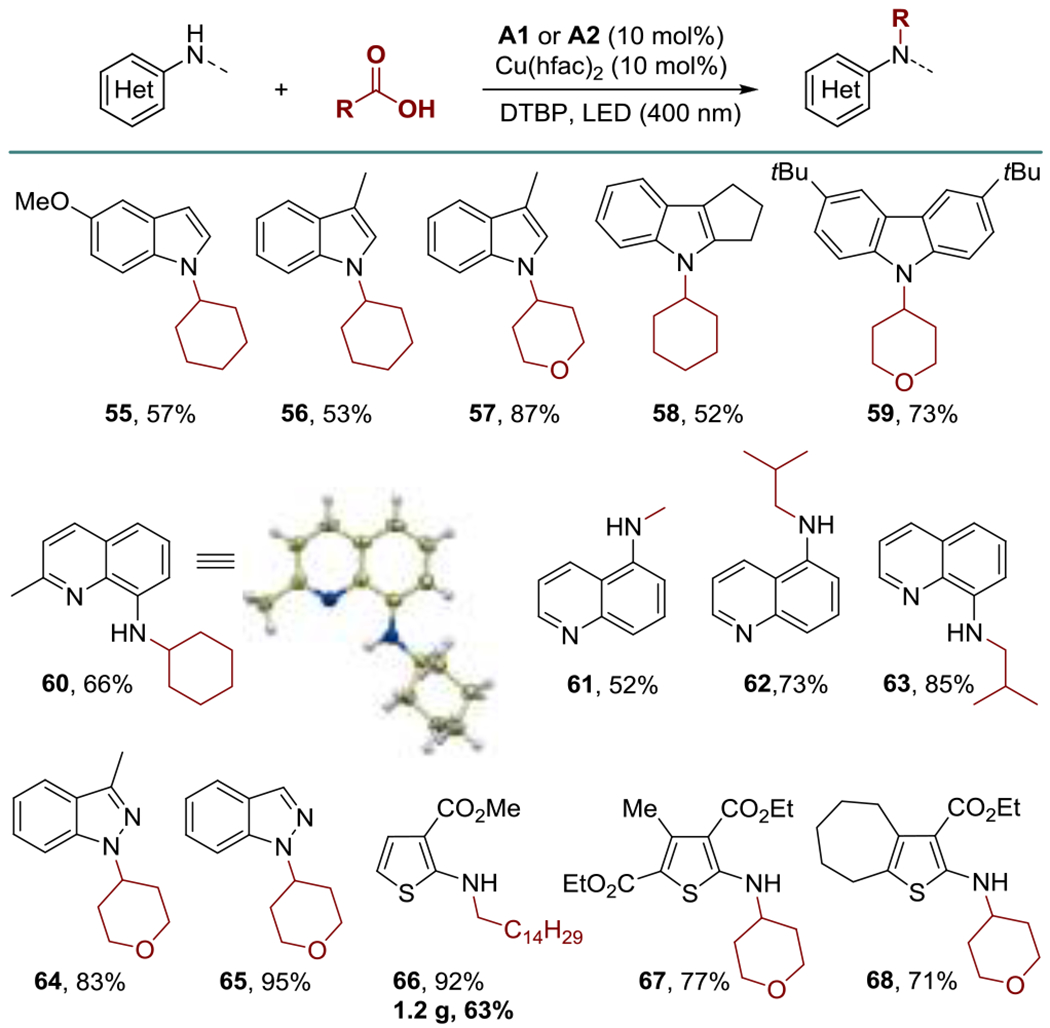
N-Alkylation of heteroaromatic amines.
The DDA reaction was further evaluated with several types of natural products and drugs (Scheme 3). Aromatase inhibitor aminoglutethimide was readily alkylated in good yields (69-72). Naturally occurring aleuritic and oleic acids were also used with success (73-75). Furthermore, N-alkylanilines derived from cholic, deoxycholic, and chenodeoxycholic acids were synthesized (76-82). Significantly, the DDA reaction tolerated unprotected imide and alcohol functionalities (69-72, 75-79, 81, 82). In another demonstration of functional group tolerance, unprotected 5-hydroxyindole was directly N-alkylated without concomitant oxidation or alkylation of the phenolic hydroxy group. Indole 83 is a precursor to a potent anti-cancer histone demethylase inhibitor 84.[14]
Scheme 3.
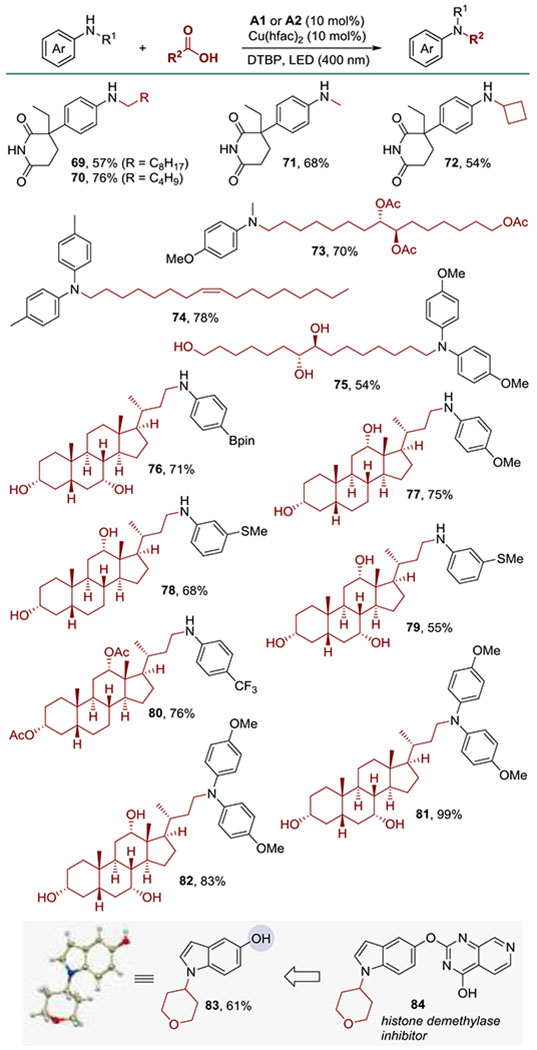
N-Alkylation of natural products and drugs.
N-Methyl groups have the propensity to undergo metabolic oxidation, and replacement of α-hydrogens with deuterium has recently gained traction as a means of reducing metabolic liabilities of redox active groups.[15] Installation of trideuteriomethyl group is not straightforward and typically requires several steps.[15]
Given the widespread availability of acetic acid-d4 as a solvent for NMR spectroscopy, we explored the reaction of acetic acid-d4 with aromatic amines as a simple, yet hitherto unavailable, one-step method of N-trideuteriomethylation (Scheme 4). Gratifyingly, acetic acid-d4 mediated the desired N-trideuteriomethylation, and a variety of N-CD3 anilines 85-98 were accessed from the corresponding aniline precursors, establishing a straightforward N-trideuteriomethylation route directly from a readily available deuterated solvent.
Scheme 4.
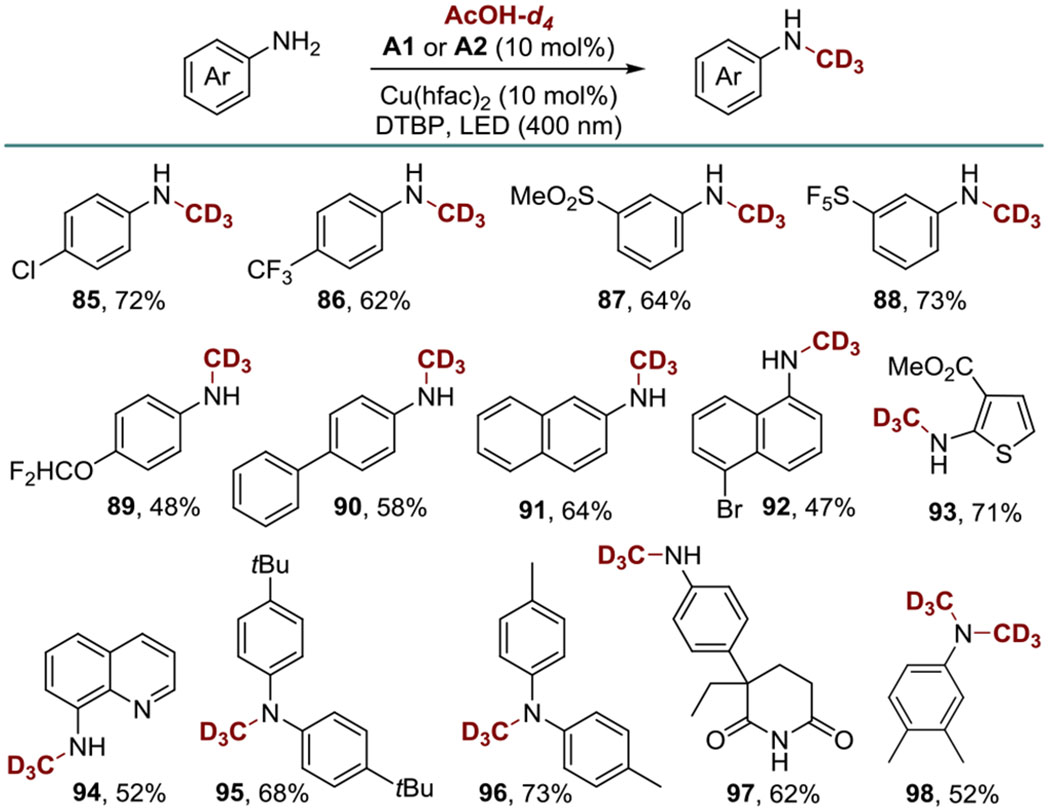
N-Trideuteromethylation by DDA with AcOH-d4.
Cyclic amines are of growing importance in medicinal chemistry, and methods that can be used to construct cyclic amines in one step have attracted significant attention, as exemplified by Bode’s SnAP methodology.[16] We explored the direct decarboxylative amination reaction of 5-chlorovaleric acid (99), anticipating that a cyclization would take place, possibly facilitated by the copper catalyst, after the completion of the DDA step (Scheme 5). Indeed, the reaction with acid 99 directly afforded N-arylpyrrolidines 100-106 in good yields, demonstrating that the DDA method can be used to install cyclic amines in one step under mild conditions. Notably, alkylations of anilines with alkyl chlorides typically require strong bases and heating.[17]
Scheme 5.
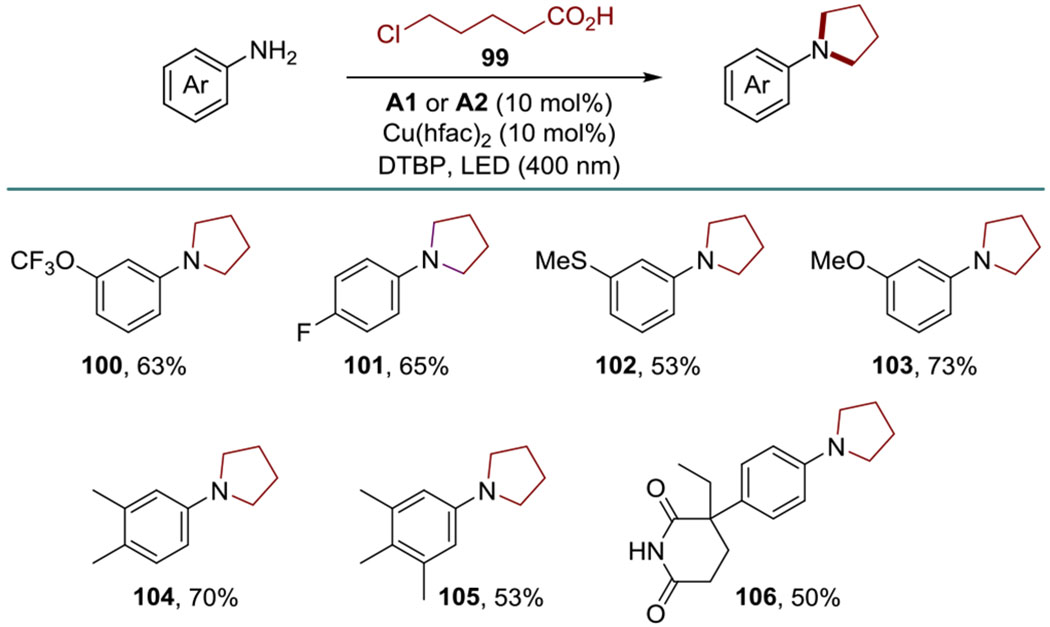
One-step access to N-arylpyrrolidine by DDA with acid 99.
The scalability of the reaction was confirmed in gram-scale batch syntheses of amines 3 and 66.
Prior experimental and computational studies demonstrated that acridines catalyze visible light-induced decarboxylative generation of alkyl radicals in transition metal co-catalyzed dual and triple catalytic reactions.[10] In line with these mechanistic studies, the involvement of alkyl radicals in the DDA reaction was also confirmed in radical trapping experiments, while the absence of carbocation-induced rearrangement by-products indicated that the reaction proceeds by the radical pathway (see SI). The alkyl radical can then be intercepted by a persistent metalloradical (e.g., CoII or CuII) and transferred into a transition metal catalytic cycle.
With the details of acridine photocatalysts previously established, the mechanistic details of the catalysis by Cu(hfac)2 were examined experimentally and computationally.
EPR studies indicated that a complex is formed between Cu(hfac)2 and aniline (Figure 2.a, see also SI) in the absence and in the presence of the carboxylic acid and DTBP. Further UV/vis spectroscopic studies aided by the Job plot analysis revealed the 1:1 stoichiometry of the complex (Figure 2.b, B), while the nonlinear regression analysis produced the respective association constant, Kassoc = 54 M−1, indicating that Cu(hfac)2 primarily exists as monoamine complex B under the reaction conditions. Hammett plot studies showed that the DDA reaction is sensitive to the substituent effects (ρ = −1.1, Figure 2.c), while the moderately negative ρ is in the range reported for other transition metal-catalyzed reactions proceeding via reductive elimination.[18] DFT computational studies further indicated that the formation of complex B is thermodynamically favored by 2.6 kcal/mol, in excellent agreement with the experiment (ΔG = −2.4 kcal/mol). Complex B exergonically cross-terminates with an alkyl radical that is generated in the acridine-catalyzed photodecarboxylation, producing Cu(III) intermediate C. The reductive elimination from intermediate C en route to Cu(I) complex D and anilinium E is also exergonic (–13.6 kcal/mol) with a readily accessible overall barrier of 15.7 kcal/mol from Cu(hfac)2, pointing to the facility of the C–N bond-forming step (see SI for additonal discussion). Subsequent regeneration of Cu(hfac)2 can occur via a PCET process with DTBP assisted by anilinium E, resulting in a kinetically and thermodynamically favorable redox process accompanied by a proton transfer. In contrast, the outer-sphere electron transfer (ET) from D to DTBP was strongly endergonic, underscoring the important role of PCET in the catalyst turnover. The alkoxide radical F can regenerate the acridine catalyst by a highly exergonic hydrogen atom transfer process (HAT) from acridyl intermediate G that is formed in the acridine-catalyzed photodecarboxylation (Figure 1). Thus, the trifecta of the chemoselective acridine-catalyzed photodecarboxylation, the energetically facile Cu(III)/Cu(I) process and the PCET- and HAT-enabled regeneration of the catalysts account for the efficiency of the DDA reaction.
Figure 2.
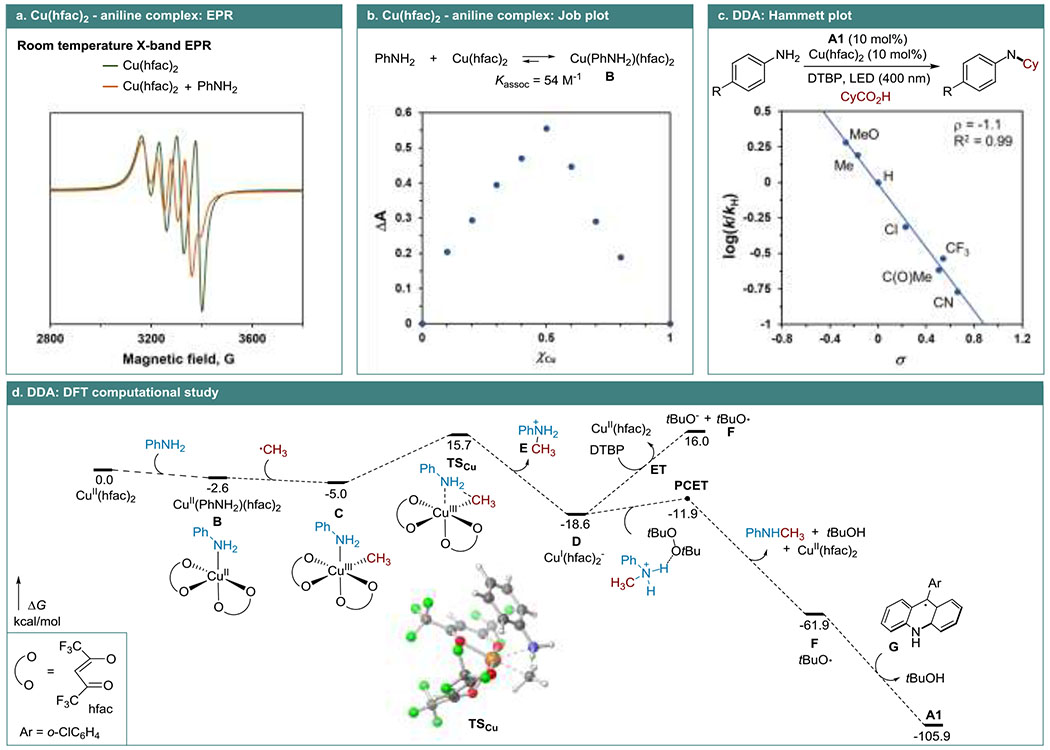
Mechanistic studies of the DDA reaction.
Conclusion
In conclusion, we developed a decarboxylative N-alkylation reaction that allows for construction of C(sp3)–N bonds directly from carboxylic acids. The scope of the reaction includes a variety of anilines, N-heterocycles, and carboxylic acids. Cyclic and N-trideuteriomethyl amines can also be readily obtained. The reaction was enabled by the fine-tuned synchrony of the acridine photocatalysis and the copper-catalyzed C(sp3)–N bond-forming process, whose details were studied experimentally and computationally.
Supplementary Material
Acknowledgements
Financial support by the Welch Foundation (AX-1788), the NSF (CHE-1455061), and NIGMS (GM134371) is gratefully acknowledged. The NMR and X-ray crystallography facilities were supported by the NSF (CHE-1625963 and CHE-1920057). The authors acknowledge the Texas Advanced Computing Center (TACC) at UT Austin for providing computational resources.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Lawrence SA, Amines: Synthesis Properties and Applications, Cambridge University Press, Cambridge, 2004; [Google Scholar]; b) Travis AS in The Chemistry of Anilines, Parts 1 & 2, (Ed.: Rappoport Z), Wiley, Chichester, 2007, pp 715–782. [Google Scholar]; c) Ricci A, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- [2].a) Sambiagio C, Marsden SP, Blacker AJ, McGowan PC, Chem. Soc. Rev. 2014, 43, 3525–3550; [DOI] [PubMed] [Google Scholar]; b) Monnier F, Taillefer M, Angew. Chem. Int. Ed 2009, 48, 6954–6971. [DOI] [PubMed] [Google Scholar]
- [3].a) Qiao JX, Lam PYS in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. (Ed.: Hall DG), Wiley-VCH, Weinheim, 2011, pp. 315–361; [Google Scholar]; b) Lam PYS, in Synthetic Methods in Drug Discovery, Vol 1, (Eds.: Blakemore D, Doyle P, Fobian Y), The Royal Society of Chemistry, 2016, pp. 242–273. [Google Scholar]
- [4].a) Ruiz-Castillo P, Buchwald SL, Chem. Rev 2016, 116, 12564–12649; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hartwig JF, Angew. Chem 1998, 110, 2154–2177; [Google Scholar]; Angew. Chem. Int. Ed. Engl 1998, 37, 2047–2067. [Google Scholar]
- [5].a) Gomez S, Peters JA, Maschmeyer T, Adv. Synth. Catal 2002, 344, 1037–1057; [Google Scholar]; b) Shi F, Cui X, Catalytic Amination for N-Alkyl Amine Synthesis, Academic Press, London, 2018. [Google Scholar]
- [6].Swamy KCK, Khumar NNB, Balaraman E, Kumar KVPP, Chem. Rev 2009, 109, 2551–2651. [DOI] [PubMed] [Google Scholar]
- [7].a) Mao R, Frey A, Balon J, Hu X, Nat. Catal 2018, 1, 120–126; [Google Scholar]; b) Liang Y, Zhang X, MacMillan DWC, Nature 2018, 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Zhao W, Wurz RP, Peters JC, Fu GC, J. Am. Chem. Soc 2017, 139, 12153–12156; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mao R, Balon J, Hu X, Angew. Chem 2018, 130, 9645–9649; [Google Scholar]; Angew. Chem. Int. Ed 2018, 57, 9501–9504; [DOI] [PubMed] [Google Scholar]; c) Sakakibara Y, Ito E, Fukushima T, Murakami K, Itami K, Chem. Eur. J 2018, 24, 9254–9258; [DOI] [PubMed] [Google Scholar]; d) Zhao B, Wang M, Shi Z, J. Org. Chem 2019, 84, 10145–10159; [DOI] [PubMed] [Google Scholar]; e) Tian L, Gao S, Wang R, ; Li Y, Tang CL, Shi LL, Fu JK, Chem. Commun 2019, 55, 5347–5350; [DOI] [PubMed] [Google Scholar]; f) Kong D, Moon PJ, Bsharat O, Lundgren RJ, Angew. Chem. Int. Ed 2019, DOI: 10.1002/anie.201912518. [DOI] [PubMed] [Google Scholar]; g) Arshadi S, Ebrahimiasl S, Hosseinian A, Monfared A, Vessally E, RSC Adv 2019, 9, 8964–8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Allen LJ, Cabrera PJ, Lee M, Sanford MS, J. Am. Chem. Soc 2014, 136, 5607–5610; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan CM, Gianatassio R, Schmidt M, Eastgate MD, Baran PS, J. Am. Chem. Soc 2016, 138, 2174–2177; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS, Science 2016, 352, 801–805; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Huihui KM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LK, Weix DJ, J. Am. Chem. Soc 2016, 138, 5016–5019; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jamison CR, Overman LE, Acc. Chem. Res 2016, 49, 1578–1586; [DOI] [PubMed] [Google Scholar]; f) Sandfort F, O’Neill MJ, Cornella J, Wimmer L, Baran PS, Angew. Chem. Int. Ed 2017, 56, 3319–3323; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 3367–3371; [Google Scholar]; g) Jin S, Haug GC, Nguyen VT, Flores-Hansen C, Arman HD, Larionov OV, ACS Catal. 2019, 9, 9764–9774; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Gao L, Wang G, Cao J, Chen H, Gu Y, Liu X, Cheng X, Ma J, Li S, ACS Catal. 2019, 9, 10142–10151; [Google Scholar]; i) Shibutani S, Kodo T, Takeda M, Nagao K, Tokunaga N, Sasaki Y, Ohmiya H, J. Am. Chem. Soc 2020, 142, 1211–1216. [DOI] [PubMed] [Google Scholar]; For a recent review on redox ester-free decarboxylative functionalizations, see:; j) Schwarz J, König B, Green Chem. 2018, 20, 323–361. [Google Scholar]
- [10].Nguyen VT, Nguyen VD, Haug GC, Dang HT, Jin S, Li Z, Flores-Hansen C, Benavides B, Arman HD, Larionov OV, ACS Catal. 2019, 9, 9485–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Tsudaka T, Kotani H, Ohkubo K, Nakagawa T, Tkachenko NV, Lemmetyinen H, Fukuzumi S, Chem. Eur. J 2017, 23, 1306–1317; [DOI] [PubMed] [Google Scholar]; b) Joshi-Pangu A, Levesque F, Roth HG, Oliver SF, Campeau LC, Nicewicz D, DiRocco DA, J. Org. Chem 2016, 81, 7244–7249. [DOI] [PubMed] [Google Scholar]
- [12].Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- [13].Trofimov BA, Nedolya NA in Comprehensive Heterocyclic Chemistry III, Vol. 3 (Eds.: Katritzky AR, Ramsden CA, Scriven EFV, Taylor RJK), Elsevier, Oxford, 2008, pp. 45–268. [Google Scholar]
- [14].Boloor A, Kanouni T, Stafford JA, Veal JM, Wallace MB (Celgene Quanticel Research, Inc.), US 2016/0194323, 2016. [Google Scholar]
- [15].a) Gant TG, J. Med. Chem 2014, 57, 3595–3611; [DOI] [PubMed] [Google Scholar]; b) Pirali T, Serafini M, Cargnin S, Genazzani AA, J. Med. Chem 2019, 62, 5276–5297. [DOI] [PubMed] [Google Scholar]
- [16].Luescher MU, Geoghegan K, Nichols PL, Bode JW, Aldrichim. Acta 2015, 48, 43–48. [Google Scholar]
- [17].a) Schmidt DM, Bonvicino GE, J. Org. Chem 1984, 49, 1664–1666; [Google Scholar]; b) Brown DP, Saklani P, Luo W, J. Heterocycl. Chem 2018, 55, 1815–1821. [Google Scholar]
- [18].Arrechea PL, Buchwald SL, J. Am. Chem. Soc 2016, 138, 12486–12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.