

Abstract
Purpose
Multiple acyl-CoA dehydrogenase deficiency (MADD) is a life-threatening, ultrarare inborn error of metabolism. Case reports described successful D,L-3-hydroxybutyrate (D,L-3-HB) treatment in severely affected MADD patients, but systematic data on efficacy and safety is lacking.
Methods
A systematic literature review and an international, retrospective cohort study on clinical presentation, D,L-3-HB treatment method, and outcome in MADD(-like) patients.
Results
Our study summarizes 23 MADD(-like) patients, including 14 new cases. Median age at clinical onset was two months (interquartile range [IQR]: 8 months). Median age at starting D,L-3-HB was seven months (IQR: 4.5 years). D,L-3-HB doses ranged between 100 and 2600 mg/kg/day. Clinical improvement was reported in 16 patients (70%) for cardiomyopathy, leukodystrophy, liver symptoms, muscle symptoms, and/or respiratory failure. D,L-3-HB appeared not effective for neuropathy. Survival appeared longer upon D,L-3-HB compared with historical controls. Median time until first clinical improvement was one month, and ranged up to six months. Reported side effects included abdominal pain, constipation, dehydration, diarrhea, and vomiting/nausea. Median D,L-3-HB treatment duration was two years (IQR: 6 years). D,L-3-HB treatment was discontinued in 12 patients (52%).
Conclusion
The strength of the current study is the international pooling of data demonstrating that D,L-3-HB treatment can be effective and safe in MADD(-like) patients.
Keywords: D,L-3-hydroxybutyrate treatment; fatty acid oxidation; inborn error of metabolism; ketone bodies; multiple acyl-CoA dehydrogenase deficiency
INTRODUCTION
Multiple acyl-CoA dehydrogenase deficiency (MADD; also known as glutaric aciduria type II, OMIM 231680) is an ultrarare (i.e., <1:50,000) inborn error of metabolism (IEM). MADD can be primary, caused by a genetic defect in the electron transfer flavoproteins (ETF) or in ETF dehydrogenase (ETFDH), or secondary, resulting from genetic defects of riboflavin transport (RFVT) or flavin adenine dinucleotide (FAD) synthesis (i.e., MADD-like disease). The impairment of mitochondrial fatty acid oxidation (FAO) and amino acid metabolism causes energy deficiency and the accumulation of toxic metabolites, such as medium-chain and long-chain length plasma acylcarnitines, urinary organic acids (e.g., isovaleric-, isobutyric-, 2-methylbutyric-, glutaric-, ethylmalonic-, 3-hydroxyisovaleric-, 2-hydroxyglutaric-, 5-hydroxyhexanoic-, and several dicarboxylic acids) and urinary acylglycines (e.g., isovalerylglycine, isobutyrylglycine, and 2-methylbutyrylglycine).1
Historically, MADD patients are classified into three categories: patients with a severe, neonatal onset with or without congenital anomalies (type I or II, respectively), and patients with a relatively mild, later onset (type III).1 Type I and II patients often demonstrate life-threatening symptoms including metabolic derangements, cardiomyopathy, leukodystrophy, and severe hypotonia. The clinical course in type III patients can vary from recurrent hypoglycemia to lipid storage myopathy and exercise intolerance.1 Treatment options include dietary fat and protein restriction, fasting avoidance, and supplementation with carnitine, glycine, and/or riboflavin, when riboflavin responsive. Despite early diagnosis and treatment, morbidity and mortality remain high in neonatal onset patients.1
Upon prolonged fasting, hepatic mitochondrial FAO fuels synthesis of ketone bodies (KB) acetoacetate and 3-hydroxybutyrate, as important alternative energy sources for the brain, skeletal muscle, and heart.2–4 Patients with mitochondrial FAO disorders, such as MADD, demonstrate multiorgan dysfunction especially during catabolism.2 Administration of exogenous KB might bypass the disturbed ketogenesis. Several case reports described successful treatment of severely affected MADD patients with racemic D,L-3-hydroxybutyrate (D,L-3-HB).5–9 The lack of systematic data on efficacy and safety of D,L-3-HB hampers the treatment of seriously ill patients and prevents D,L-3-HB reimbursement. Therefore, we performed a twofold study including a systematic literature review and an international, retrospective cohort study to describe the clinical presentations of MADD(-like) patients treated with D,L-3-HB, the details of D,L-3-HB treatment methods, and outcomes.
MATERIALS AND METHODS
The Medical Ethical Committee of the University Medical Center Groningen confirmed that the Medical Research Involving Human Subjects Act does not apply and that official approval of this study by the Medical Ethical Committee was not required (METc code 2016/470). The study protocol was performed in compliance with the Declaration of Helsinki and approved for waiver of consent by all participating institutes or performed conforming to the laws and regulations of the respective countries and institutes.
Systematic literature review
To identify all reported IEM patients treated with D,L-3-HB and their health-care providers, a comprehensive search strategy for relevant publications before 21 December 2016 was performed in PubMed and EMBASE public databases. Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines were followed as accurately as possible. The detailed search strategy, a protocol of the screening process and data extraction, a flowchart, and the PRISMA-P 2015 checklist are presented in Supplementary Data 1. Articles were included based on the presence of detailed patient data concerning D,L-3-HB- or KB treatment, as well as a confirmed diagnosis by biochemical (acylcarnitine or urinary organic acid profile), DNA, or enzymatic analysis. Exclusion criteria were (1) no detailed patient data described, (2) lack of accessibility of the abstracts or articles, and (3) no availability in English or Dutch language.
Retrospective cohort study
In February 2017 health-care providers with experience in D,L-3-HB treatment of MADD(-like) patients were invited to collaborate in this study by contacting (1) the first and/or corresponding authors of previous publications, identified in our systematic literature study; (2) clinicians who have previously contacted the authors (J.L.K.V.H. or T.G.J.D.) on this topic; (3) several professional organizations and networks, including a list server for the metabolic community (Metab-l), Society for the Study of Inborn Errors of Metabolism (SSIEM), Society for Inherited Metabolic Disorders (SIMD), and the European Reference Network for Hereditary Metabolic Diseases (MetabERN).
D,L-3-HB treatment has been reported in at least two MADD-like patients who were later found to have RFVT defects.10,11 Patients with genetic defects of RFVT (i.e., SLC52A1, SLC52A2, SLC52A3 [alias C20orf54]) and FAD metabolism (i.e., SLC25A32, FLAD1) were, therefore, included in this study in addition to those with ETF or ETF dehydrogenase defects. MADD(-like) patients were eligible for enrollment in case of a diagnosis confirmed by biochemical, DNA, or enzymatic analysis, performed conforming to local protocols. Outcome parameters included data on clinical presentation, laboratory and molecular parameters, D,L-3-HB treatment method, and (long-term) outcome. Data was collected via an anonymous questionnaire in Microsoft Word to be completed by health-care providers involved. Data inclusion was concluded in December 2018, after which data from all completed questionnaires were summarized. The STROBE checklist for reporting observational studies is presented in Supplementary Data 2.
Statistical analysis
Data analysis was performed using Microsoft Excel and GraphPad Prism, version 5.0 (GraphPad Software, La Jolla, CA). Descriptive statistics were used to summarize the data. Categorical and continuous variables are presented as numbers (percentages) or median (interquartile range [IQR]), respectively. Kaplan–Meier plots were used to estimate the survival and visualize the data on time until first reported clinical improvement and D,L-3-HB treatment duration. The survival of MADD(-like) patients treated with D,L-3-HB was compared with survival data from historical controls who were not reported to have been treated with D,L-3-HB, as collected in a previous meta-analysis.12 Mann–Whitney U test was used to analyze the significance of differences between groups. If data were missing, the analysis was performed on data from the remaining patients. A p value of <0.05 was considered statistically significant.
RESULTS
Systematic literature review
Supplementary Table 1 summarizes data from 14 references on D,L-3-HB treatment in 16 patients with MADD(-like) disease.2,5–8,10,11,13–19 Additionally, D,L-3-HB treatment was reported in 18 patients with other IEMs in which ketogenesis is disturbed, demonstrating potential indications of the compound. These IEMs included carnitine–acylcarnitine translocase deficiency (n = 1),17,18 glycogen storage disease type III (n = 3),20,21 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency (n = 4),17,18,22,23 mitochondrial complex IV deficiency (n = 2),20 persistent hyperinsulinemic hypoglycemia of infancy (n = 6),24,25 propionic acidemia (n = 1),20 and very long-chain acyl-CoA dehydrogenase deficiency (n = 1).20
Retrospective cohort study
Patient characteristics
In total, 23 MADD(-like) patients treated with D,L-3-HB treatment were identified, including 14 novel cases. The individual patient characteristics are presented in Table 1. Median age at clinical ascertainment was two months (IQR: 8 months). Nine patients (39%) had a neonatal disease onset and all presented clinically during the first week of life. Structural congenital anomalies were not reported. Hence, they were classified as type II patients. The 14 remaining patients (61%) could be categorized as type III patients, including two with a clinical onset during adulthood. Abnormal population newborn screening results were observed in 14 patients (61%) of whom eight (57%) developed clinical symptoms and signs during the first week of life.
Table 1.
Diagnostic characteristics of patients included in the retrospective cohort study.
| Patient | Sex | Age at clinical onset | Affected gene | Variant allele 1 | Variant allele 2 | Enzyme activity (nmol/min/mg protein) | ||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | cDNA | Protein | |||||
| 1a,b(7) | F | 0 days | ETFA | c.1–40G>A | c.1–40G>A | |||
| 2 | M | 0 days | ETFA | c.797C>T | p.T266M | c.797C>T | p.T266M | ETF:<0.01 (<1% of C) |
| 3c | F | 1 day | ETFA | c.797C>T | p.T266M | c.797C>T | p.T266M | |
| 4ab,c(17,18) | M | 1 day | ETFA | c.370G>A | p.A124T | c.370G>A | p.A124T | |
| 5 | F | 1 day | ETFA | c.200T>C | p.L67P | c.854A>T | p.Q285L | |
| 6a(17,18) | M | 7 days | ETFAe | c.365G>A | p.R122K | c.809–811del | p.V270del | |
| 7 | F | 1 day | ETFDH | c.896T>C | p.L299S | c.1842C>A | p.Y614X | |
| 8b,c | F | 3 days | ETFDH | c.1141G>C | p.G381R | c.1141G>C | p.G381R | ETF-QO: 0.05 (C: 0.22 ± 0.09) |
| 9 | F | 1 month | ETFDH | c.34G>C | p.A12P | c.1234G>T | p.E412X | |
| 10 | M | 2 months | ETFDH | c.1001T>C | p.L334P | c.1074G>C | p.R358S | ETF-QO: 0.71 (C: 0.8–2.4) |
| 11c | M | 4 months | ETFDH | c.820G>T | p.G274X | c.1601C>T | p.P534L | ETF: 1.23 (C: 0.79–2.1) ETF-QO: 0.96 (C: 0.8–2.4) |
| 12a(17,18) | M | 5 months | ETFDH | c.858G>A | p.W286X | c.1099A>G | p.N367D | |
| 13a(15) | F | 6 months | ETFDH | c.51dupT | p.A18Cfs | c.940G>A | p.E314K | ETF-QO: 0.44 (C 0.8–2.4) |
| 14 | M | 10 months | ETFDH | c.463A>G | p.R155G | c.463A>G | p.R155G | |
| 15 | F | 1 year 3 months | ETFDH | c.665A>C | p.Q222P | c.665A>C | p.Q222P | ETF-QO: 0.07 (C: 0.31 ± 0.19)f |
| 16b,d | F | 1 year 8 months | ETFDH | c.1693G>C | p.V565L | c.1693G>C | p.V565L | |
| 17a,b(8) | M | 2 years 7 months | ETFDH | c.1106G>C | p.G369A | c.1106G>C | p.G369A | |
| 18 | M | 25 years 11 months | ETFDH | c.1367C>T | p.P456L | c.1367C>T | p.P456L | |
| 19 | F | 19 years | ETFDH | c.1774T>C | p.C592R | ETF: 1.82 (C: 0.79–2.1) ETF-QO: 0.21 (C: 0.8–2.4) | ||
| 20a,c(17,18) | F | 7 days | NF | |||||
| 21c | F | 1 month | ||||||
| 22a(10) | M | 5 months | SLC52A3 | c.639C>G | p.Y213X | c.678–680del | p.L227del | ETF: 1.27 (C: 1.25 ± 0.32) ETF-QO: 0.06 (C: 0.22 ± 0.09) |
| 23a(11) | F | 6 months | SLC52A3 | c.49T>C | p.W17R | c.639C>G | p.Y213X | ETF: 1.11 (C: 1.25 ± 0.32) ETF-QO: 0.17 (C: 0.22 ± 0.09) |
C control, cDNA complementary DNA, NF no variant found.
aPatient has been published previously in relation to D,L-3-HB treatment; see corresponding reference.
bConsanguinity.
cDeceased.
dDiagnosed prenatally due to family history.
eDNA analysis also demonstrated compound heterozygous variants in ETFB (c.217–4G>T and c.438+20C>T), classified as variant of uncertain significance and likely benign, respectively.
fAnalysis only performed in sister.
Diagnosis was molecularly confirmed in 20 cases (87%) (pathogenic variants in ETFA [n = 4], ETFDH [n = 6]; compound heterozygosity in ETFA [n = 2], ETFDH [n = 6], SLC52A3 [n = 2]). In one patient in whom DNA analysis was inconclusive, the results of an enzyme assay were indicative of MADD. All reported acylcarnitine profiles (n = 18) and urinary organic acid profiles (n = 22) at diagnosis demonstrated at least mild abnormalities consistent with MADD.
D,L-3-hydroxybutyrate treatment method
In our cohort of patients, D,L-3-HB was prescribed as a food supplement (KetoForce) in one patient, and as hospital pharmacy constituted formulation or prepared by a caregiver in others, after being obtained from various suppliers including Huddersfield Pharmacy Specials, Inresa, M2i, Sigma-Aldrich, and Special Products Ltd (Veriton Pharma). The most reported formulation involved a racemic sodium salt. Currently, in The Netherlands, the D,L-3-HB is magistrally prepared as a 593.3 mg/mL (4.7 M) solution in distilled water. The D,L-3-HB is acquired by Sigma-Aldrich and this treatment costs €0.0040/mg (price for the active ingredient only and a simple product formulation) averaging approximately €3.60/kg/day at an assumed starting dose of 900 mg/kg/day.
The median age at start of D,L-3-HB treatment was seven months (IQR: 4.5 years). Prescribed doses ranged between 100 and 2600 mg/kg/day, divided in one to six daily doses. Five patients (22%) received D,L-3-HB continuously during the night and one patient (4%) continuously for 24 hours per day. The D,L-3-HB was administered orally or via nasogastric/gastrostomy tube, usually combined with nutrition or before/after the meal.
D,L-3-hydroxybutyrate treatment outcome
Patient and treatment characteristics according to outcome are summarized in Table 2. Individual D,L-3-HB treatment characteristics and outcome are presented in Supplementary Table 2. In total, clinical improvement upon D,L-3-HB was reported in 16 patients (70%) for cardiomyopathy, leukodystrophy, liver symptoms (i.e., hyperammonemia, hypoglycemia, liver dysfunction or failure, and metabolic acidosis), muscle symptoms (i.e., exercise intolerance, hypotonia, myopathy, and rhabdomyolysis), and/or respiratory failure. D,L-3-HB treatment was effective in 6/9 type II patients (67%). The efficacy was questionable in two type II patients (22%) in whom D,L-3-HB was used as a preventive measure, which complicated the interpretation of treatment outcomes. Clinical improvement was also reported in 10/14 type III patients (71%), including a patient with RFVT3 deficiency. The efficacy was questionable in one type III patient (7%) because the duration of treatment was only three months at the time of data analysis. Additional follow-up demonstrated that the patient remained clinically stable without further deterioration of the leukodystrophy. D,L-3-HB was ineffective in 1 type II (11%) and 3 type III patients (21%), of whom one patient was diagnosed with RFVT3 deficiency. The treatment indications in those patients included muscle and liver symptoms, respiratory failure, and neuropathy with the maximum prescribed doses ranging between 750 and 1800 mg/kg/day. Figure 1a presents the summarized organ-based D,L-3-HB treatment indications and efficacy. Symptom-specific indications and efficacy are demonstrated in Supplementary Fig. 1. Compared with data from historical controls (i.e., 26 type II patients), the survival appeared longer in type II MADD(-like) patients treated with D,L-3-HB, as shown in Fig. 1b. The median interval from start of D,L-3-HB to first reported clinical improvement was one month (IQR: 3 months) and ranged up to six months, as demonstrated in Fig. 1c. Three of four patients (75%) in the group of nonresponders and 1/3 patients (33%) in the group with questionable efficacy had a treatment duration of more than six months.
Table 2.
Summarized patient and D,L-3-hydroxybutyrate treatment characteristics according to outcome.
| Clinical improvement upon D,L-3-HB treatment | |||
|---|---|---|---|
| Yes (n = 16; 70%) | Questionable (n = 3; 13%) | No (n = 4; 17%) | |
| Gender | M:F = 9:7 | M:F = 1:2 | M:F = 0:4 |
| Alive | 12 (75%) | 2 (67%) | 3 (75%) |
| Current age | 13 years (6.5 years) | 3 years (1.5 years) | 13.5 years (10.5 years) |
| Age at death | 1.5 years (8 years) | 8 months | 10 days |
| Age at onset | 3 months (8 months) | 3 days (5 months) | 3 months (5 years) |
| Congenital anomalies | - | - | - |
| Positive NBS results | 8 (50%) | 3 (100%) | 2 (50%) |
| Genetic analysis | 14 (88%) | 3 (100%) | 4 (100%) |
| ETFA | 5a | - | 1 |
| ETFB | - | - | - |
| ETFDH | 8 | 3 | 2b |
| SLC52A3 | 1 | - | 1 |
| Enzyme assay | 6 (38%) | 1 (33%) | 2 (50%) |
| ETF deficiency | 1 | - | - |
| ETF-QO deficiency | 4c | 1 | 1 |
| D,L-3-HB treatment | |||
| Age at start | 1.5 years (6 years) | 6 months (2 years) | 5 months (6.5 years) |
| Minimum D,L-3-HB dose (mg/kg/day) | 330 (215) | 200 (105) | 490 (215) |
| Maximum D,L-3-HB dose (mg/kg/day) | 650 (400) | 395 (925) | 905 (330) |
| Maximum number of doses/day | 4 (0.3)d | 4 (1.5) | 4 (0.5)e |
| D,L-3-HB discontinuation | 7 (44%) | 2 (33%) | 3 (75%) |
| Age at discontinuation | 6 years (17 years) | 1 year (5 months) | 3.5 years (13 years) |
| D,L-3-HB treatment duration | 3 years (7.5 years) | 6 months (5 months) | 2 years (3.5 years) |
Values are presented as number of patients or median (interquartile range [IQR]).
NBS newborn screening.
aIn one patient, DNA analysis also demonstrated compound heterozygous variants in ETFB (c.217–4G>T and c.438+20C>T), which were classified as variant of uncertain significance and likely benign, respectively.
bIn one patient only one pathogenic variant identified.
cIn one patient only performed in sister.
dContinuous nocturnal administration (n = 4).
eContinuous nocturnal administration (n = 1) and continuous 24-hour administration (n = 1).
Fig. 1. Efficacy of D,L-3-hydroxybutyrate treatment.

a Proportion of multiple acyl-CoA dehydrogenase deficiency (MADD)(-like) patients with organ based indication and efficacy of D,L-3-hydroxybutyrate treatment, with the numbers presented in the columns. Clinical improvement regarding liver symptoms included hyperammonemia, hypoglycemia, and/or metabolic acidosis; clinical improvement regarding muscle symptoms included exercise intolerance, hypotonia, myopathy, and/or rhabdomyolysis. b Kaplan–Meier curve of the survival in type II (n = 9) and type III (n = 14) MADD(-like) patients treated with D,L-3-HB compared with the survival in historical controls from literature (type I [n = 16], type II [n = 26] and type III [n = 156] MADD) who were not reported to have been treated with D,L-3-HB. c Kaplan–Meier plot that demonstrates the cumulative proportion of MADD(-like) patients with reported clinical improvement upon initiation of D,L-3-HB treatment over time. Calculated from a total of 21 patients with sufficient data. HC historical control. x = censored patient.
The following side effects of D,L-3-HB were reported in 8 patients (35%): abdominal pain, constipation, dehydration, diarrhea, and vomiting or nausea. Detailed data on D,L-3-HB safety are presented in Fig. 2. D,L-3-HB treatment related (treatment duration >1 day) side effects appeared to be dose dependent with a median maximum dose of 600 mg/kg/day (IQR: 410 mg/kg/day) in patients without side effects compared with 950 mg/kg/day (IQR: 555 mg/kg/day) in patients with side effects (p = 0.0544). In four patients (17%), the D,L-3-HB dose was titrated based upon biochemical parameters, including (peak) concentrations of D-3-HB in plasma, blood, and urine, and ammonia concentrations.
Fig. 2. Safety of D,L-3-hydroxybutyrate treatment.
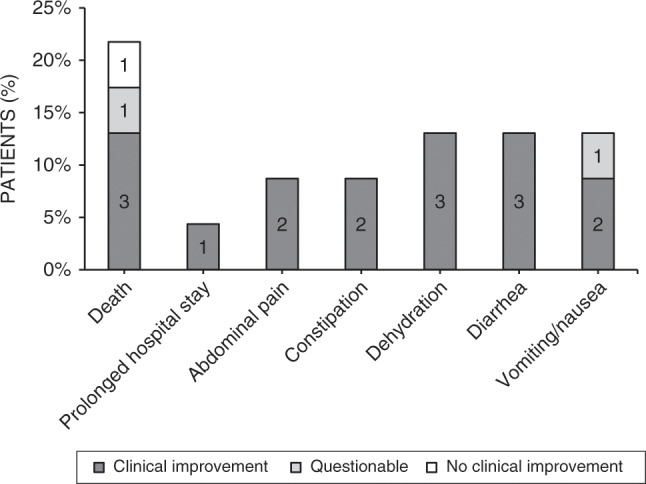
The (serious) adverse effects that occurred in a total of 13 multiple acyl-CoA dehydrogenase deficiency (MADD)(-like) patients during the course of D,L-3-HB treatment.
The median duration of D,L-3-HB treatment in the whole cohort was two years (IQR: 6 years) and is demonstrated in Fig. 3, categorized according to D,L-3-HB treatment efficacy. Treatment was discontinued in 12 patients (52%) due to (combinations of) clinical improvement after which further D,L-3-HB treatment was deemed unnecessary (n = 2), lack of clinical improvement (n = 2), death irrespective of the cause (n = 5), side effects (n = 1), noncompliance (n = 1), and costs (n = 2). The median age at D,L-3-HB discontinuation was three years (IQR: 13.5 years). The median treatment duration before D,L-3-HB discontinuation was 10 months (IQR: 1.5 years), while in the patients who continued D,L-3-HB treatment, the median treatment duration was 6.5 years (IQR: 7 years). In patients who died during the course of D,L-3-HB treatment, the median D,L-3-HB treatment duration was 8 months (IQR: 4 months).
Fig. 3. D,L-3-Hydroxybutyrate treatment duration.

Kaplan–Meier plot that demonstrates the cumulative proportion of multiple acyl-CoA dehydrogenase deficiency (MADD)(-like) patients receiving D,L-3-HB treatment over time, categorized according to D,L-3-HB treatment efficacy. Calculated from a total of 23 patients with sufficient data; x = censored patient.
DISCUSSION
D,L-3-HB treatment is unlicensed but has been reported in at least eight IEMs in which exogenous KB treatment may be indicated. In 70% of the presented cohort of 23 MADD(-like) patients, we observed clinical improvement of cardiomyopathy, leukodystrophy, liver symptoms, muscle symptoms, and respiratory failure upon start of D,L-3-HB treatment. D,L-3-HB treatment appeared to be ineffective for neuropathy. Side effects occurred in 35% of the patients but were never a reason to discontinue supplementation in patients who experienced clinical improvement.
To date, there are no clinical or laboratory parameters predicting clinical efficacy of D,L-3-HB treatment, such as phenotype, genotype, or age at start of D,L-3-HB treatment. In our study, symptom improvement is observed up to six months after commencing D,L-3-HB. It was not possible to relate the (timespan of) D,L-3-HB treatment efficacy to age at clinical onset, D,L-3-HB dosing, or to a specific organ because in a number of patients there were several concurrent treatment indications. The authors emphasize the importance of a relatively long evaluation period for assessment of efficacy, because clinical improvement occurs in weeks or months rather than days after starting D,L-3-HB treatment. Furthermore, studies are warranted to identify MADD (bio)markers that correlate with clinical severity and can be used as outcome parameters during prospective trials.12
Biochemical monitoring of D,L-3-HB treatment was performed in only four patients (17%), all of whom experienced clinical improvement. D,L-3-HB dose titration toward at least detectable concentrations in blood, plasma, or urine can indicate that a sufficient amount of exogenous KB is supplied. Stable isotope infusion studies demonstrated an increased endogenous KB production in fasting healthy newborns compared with healthy adults.26,27 Thus, when endogenous KB production rates are insufficient, it may be hypothesized that exogenous requirements are higher in infants. Prospective in vitro and (stable isotope) in vivo metabolic flux studies may help guide the (individualized) dose response curves and relations to symptoms and signs.
In this study, the risk of side effects due to D,L-3-HB appeared to increase with dose. However, frequently reported side effects such as abdominal pain, constipation, dehydration, diarrhea, and vomiting or nausea are difficult to discriminate from the natural course of the underlying disorder and from intercurrent illness. In addition, it is important to realize that the salt-free dose between different compounds can differ. It is unclear whether the adverse effects would be caused by the high amount of D,L-3-HB or by the associated cation load. A relatively low dose of 600 mg/kg/day of sodium-D,L-3-HB provides 4.8 mmol/kg/day of sodium, and a high dose of 2600 mg/kg/day provides 20.6 mmol/kg/day of sodium, compared with the normal sodium intake of 1 and 3–4 mmol/kg/day for adults and infants and young children, respectively. Nevertheless, it should be emphasized that in our study the benefits of D,L-3-HB appeared to outweigh the side effects. Recently, D,L-3-HB treatment in the form of a sodium or calcium salt was described in an MADD patient. Severe alkalosis and hypernatremia were reported after D,L-3-HB doses above 1400 mg/kg/day.9 Hypothetically, the alkalosis might be caused by the high cation load or the conjugate base excess of dissolved D,L-3-HB.9,28 The high sodium load is also associated with increased calcium loss and alkalization of urine, which can lead to nephrocalcinosis and renal stones.9,28 Sufficient hydration is recommended for these associated electrolyte challenges. Future studies are warranted to investigate the influence of D,L-3-HB on fluid, electrolyte, and acid–base homeostasis.
The mode of action of D,L-3-HB treatment is incompletely understood and several mechanisms likely act simultaneously. In mitochondrial FAO disorders, next to intracellular energy deficiency and accumulation of toxic metabolites, shortage of KB impairs cholesterol synthesis, which is required for myelination.29 Endogenous 3-hydroxybutyrate also has several direct and indirect signaling functions including gene expression and activation of hydroxycarboxylic acid receptor 2, which is associated with reduced lipolysis as well as anti-inflammatory and neuroprotective effects.30,31 Presumably, this all targets the complex pathophysiology and clinical manifestations in MADD patients, such as cardiomyopathy, leukodystrophy, and myopathy. In MADD-like disorders, a different working mechanism can be proposed. D,L-3-HB treatment was also effective in one patient suffering from RFVT3 deficiency in whom the treatment indication included respiratory failure due to diaphragm paralysis and muscle symptoms. It can be hypothesized that D,L-3-HB treatment acts on the glutamate excitotoxicity and generation of reactive oxygen species, which are potentially induced by riboflavin deficiency and mitochondrial dysfunction.30,32–35 Additionally, exogenous D,L-3-HB can provide a therapeutic option in selected cases of other IEMs in which ketogenesis is impaired, such as mitochondrial FAO disorders, defects of FAD metabolism, glycogen storage disease, mitochondrial respiratory chain disorders, organic acidurias, or hyperinsulinism, and as an additive for patients using a ketogenic diet.
3-Hydroxybutyrate is a chiral molecule with two enantiomers: D-3-hydroxybutyrate and L-3-hydroxybutyrate. Compared with D-3-hydroxybutyrate, utilization of L-3-hydroxybutyrate appears slower and through different routes.28,36 Metabolism of D-3-hydroxybutyrate yields two molecules of acetyl-CoA, which enter the Krebs cycle.31 After mitochondrial import likely via monocarboxylate transporter 1,37 L-3-hydroxybutyrate is activated to L-3-hydroxybutyryl-CoA by a specific coenzyme A ligase and becomes a substrate for short-chain acyl-CoA dehydrogenase.38–41 Although effects that depend on 3-hydroxybutyrate catabolism might primarily be induced by D-3-hydroxybutyrate, L-3-hydroxybutyrate may have its own specific utility. The primary use of L-3-hydroxybutyrate seems to be in the central nervous system, where the key enzymes are most expressed.36 In rats, L-3-hydroxybutyrate seems to be the preferred substrate for synthesis of fatty acids and sterols in spinal cord, brain, and kidney, while D-3-hydroxybutyrate is favored for oxidation.36,39 Future studies should evaluate if L-3-hydroxybutyrate may have a specific therapeutic role toward neurological symptoms. In addition to optimal dosing, the most advantageous ratio between D-3-hydroxybutyrate and L-3-hydroxybutyrate may need to be determined based on (organ-specific) treatment indications.
Several methodological limitations concerning this study should be considered. As this study concerned a retrospective study collecting data over a period of >20 years, it was not possible to consistently capture all detailed data, for instance regarding specific time points. Second, the lack of standardized clinical and biochemical outcome parameters made a detailed study of efficacy difficult. Third, the assignment of outcome to D,L-3-HB treatment was complicated by concurrent other treatment options, and by a fluctuating natural disease course. Finally, despite their best efforts, the authors have been unable to include all centers with experience in this field.
Since the first publication in 2003,6 the story of D,L-3-HB for MADD has become an excellent example of the long and complex journey toward possible orphan drug designation and registration of an unlicensed compound for the treatment of an ultrarare, life-threatening disease. The strength of the current study is the international pooling of data on the efficacy and safety profile of D,L-3-HB in MADD(-like) patients. FAIR (i.e., findable, accessible, interoperable, reusable),42 evidence-based, and transparent approaches are essential to establish sustainable orphan drugs.43,44 Therefore, the authors included information on preparation and pricing of D,L-3-HB. Our findings may be useful in the pursuit of orphan drug designation and registration. To this aim, organizations such as Fair Medicine,45 which introduces a coalition model that involves all stakeholders in the pharmaceutical development process, can perhaps bring a fresh impetus.
Supplementary information
Acknowledgements
The authors thank Katinka A.M. Mulder, legal advisor—research contracts, for her contribution to the realization of the consortium agreement. Pharmaceutical industries did not play any role in this study. The MD/PhD scholarships of W.J.v.R. and E.A.J. are funded by the Junior Scientific Masterclass from the University of Groningen, University Medical Center Groningen (MD/PhD 15–30, MD/PhD 18–55, respectively). J.V. is supported in part by National Institutes of Health (NIH) grant R01-DK78755. The sources of funding had no involvement in the study design; data collection, analysis, and interpretation; reporting of the results; or in the decision to submit the paper for publication.
Disclosure
The authors declare no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41436-019-0739-z) contains supplementary material, which is available to authorized users.
References
- 1.Frerman FE, Goodman SI. Chapter 103: defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric acidemia type II. In: Valle D, Beaudet AL, Vogelstein B, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2014. [Google Scholar]
- 2.Olpin SE. Implications of impaired ketogenesis in fatty acid oxidation disorders. Prostaglandins Leukot Essent Fatty Acids. 2004;70:293–308. doi: 10.1016/j.plefa.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Bouteldja N, Andersen LT, Moller N, Gormsen LC. Using positron emission tomography to study human ketone body metabolism: a review. Metabolism. 2014;63:1375–1384. doi: 10.1016/j.metabol.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502. doi: 10.1146/annurev.physiol.64.082201.154705. [DOI] [PubMed] [Google Scholar]
- 5.Bonham JR, Tanner MS, Pollitt RJ, et al. Oral sodium 3-hydroxybutyrate, a novel adjunct to treatment for multiple acyl CoA dehydrogenase deficiency. J Inherit Metab Dis. 1999;22(Suppl 1):101. [Google Scholar]
- 6.Van Hove JL, Grunewald S, Jaeken J, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD) Lancet. 2003;361:1433–1435. doi: 10.1016/S0140-6736(03)13105-4. [DOI] [PubMed] [Google Scholar]
- 7.Van Rijt WJ, Heiner-Fokkema MR, du Marchie Sarvaas GJ, et al. Favorable outcome after physiologic dose of sodium-D,L-3-hydroxybutyrate in severe MADD. Pediatrics. 2014;134:e1224–8. doi: 10.1542/peds.2013-4254. [DOI] [PubMed] [Google Scholar]
- 8.Gautschi M, Weisstanner C, Slotboom J, Nava E, Zurcher T, Nuoffer JM. Highly efficient ketone body treatment in multiple acyl-CoA dehydrogenase deficiency-related leukodystrophy. Pediatr Res. 2015;77:91–98. doi: 10.1038/pr.2014.154. [DOI] [PubMed] [Google Scholar]
- 9.Fischer T, Och U, Marquardt T. Long-term ketone body therapy of severe multiple acyl-CoA dehydrogenase deficiency: a case report. Nutrition. 2018;60:122–128. doi: 10.1016/j.nut.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 10.van Spronsen FJ, de Weerd W, Goorhuis J, et al. Respiratory insufficiency as first presentation of multiple acyl-CoA dehydrogenase deficiency (MADD) J Inherit Metab Dis. 2005;28(Suppl 1):115. [Google Scholar]
- 11.Bosch AM, Abeling NG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–164. doi: 10.1007/s10545-010-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rijt WJ, Ferdinandusse S, Giannopoulos P, et al. Prediction of disease severity in multiple acyl-CoA dehydrogenase deficiency: a retrospective and laboratory cohort study. J Inherit Metab Dis. 2019;42:878–889. doi: 10.1002/jimd.12147. [DOI] [PubMed] [Google Scholar]
- 13.Van Hove J, Jaeken J, Lagae L, Demaerel P, Bourdoux P, Niezen-Koning K. Multiple acyl-CoA dehydrogenase deficiency: acquired leukodystrophy treated with D,L-3-hydroxybutyrate. J Inherit Metab Dis. 2001;24(Suppl 1):72. [Google Scholar]
- 14.Grunewald S, Marek J, Deanfield J, Olpin S, Leonard JV. Five year follow up of D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD) J Inherit Metab Dis. 2008;31(Suppl 1):36. [Google Scholar]
- 15.Al-Hertani W, Mineyko A, Humphreys P, Chakraborty P, Geraghty MT. Glutaric aciduria type II presenting with lipid myopathy, progressive leukodystrophy; intrafamilial variation in two siblings. Presentation at: 58th Annual Meeting of The American Society of Human Genetics; 2008; Philadelphia, PA.
- 16.Marquardt T, Harms E. Ketone body therapy of severe multiple acyl-CoA dehydrogenase deficiency (MADD) Mol Genet Metab. 2009;98:57. [Google Scholar]
- 17.Dalkeith T, Dennison B, Wilcken B, et al. Difficulties in the dietetic management of patients with early childhood onset: multiple acyl co-A dehydrogenase deficiency (MADD) J Inherit Metab Dis. 2010;33(Suppl 1):173. [Google Scholar]
- 18.Dalkeith T, Ellaway C, Thompson S, et al. The use of 3-hydroxybutyrate in patients with fat oxidation disorders. J Inherit Metab Dis. 2013;36(Suppl 2):94. [Google Scholar]
- 19.Hale S, Hahn S, Merritt JLI. Novel therapies in treatment of presumptive multiple acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2011;102:288. [Google Scholar]
- 20.Van I, Landy C, Corriol O, De Lonlay P, Touaty G, Bourget P. Use of sodium D,L-3-hydroxybutyrate in metabolic diseases. Pharm World Sci. 2010;32:219. [Google Scholar]
- 21.Valayannopoulos V, Bajolle F, Arnoux JB, et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III with D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr Res. 2011;70:638–641. doi: 10.1203/PDR.0b013e318232154f. [DOI] [PubMed] [Google Scholar]
- 22.Francois B, Bachmann C, Schutgens RBH. Glucose metabolism in a child with 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency. J Inherit Metab Dis. 1981;4:163–164. [Google Scholar]
- 23.Bhattacharya K, Ho G, Dalkeith T, Dennison B, Thompson S, Christodoulou J. Improvement in severe HMG co-lyase deficiency with fat restriction and 3-hydroxybutyrate therapy. J Inherit Metab Dis. 2010;33(Suppl 1):62. [Google Scholar]
- 24.Bougneres PF, Ferre P, Chaussain JL, Job JC. Glucose metabolism in hyperinsulinemic infants: the effects of fasting and sodium DL-beta-hydroxybutyrate on glucose production and utilization rates. J Clin Endocrinol Metab. 1983;57:1054–1060. doi: 10.1210/jcem-57-5-1054. [DOI] [PubMed] [Google Scholar]
- 25.Plecko B, Stoeckler-Ipsiroglu S, Schober E, et al. Oral beta-hydroxybutyrate supplementation in two patients with hyperinsulinemic hypoglycemia: monitoring of beta-hydroxybutyrate levels in blood and cerebrospinal fluid, and in the brain by in vivo magnetic resonance spectroscopy. Pediatr Res. 2002;52:301–306. doi: 10.1203/00006450-200208000-00025. [DOI] [PubMed] [Google Scholar]
- 26.Bougneres PF, Lemmel C, Ferre P, Bier DM. Ketone body transport in the human neonate and infant. J Clin Invest. 1986;77:42–48. doi: 10.1172/JCI112299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–426. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 28.Stubbs BJ, Cox PJ, Evans RD, et al. On the metabolism of exogenous ketones in humans. Front Physiol. 2017;8:848. doi: 10.3389/fphys.2017.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koper JW, Lopes-Cardozo M, Van Golde LM. Preferential utilization of ketone bodies for the synthesis of myelin cholesterol in vivo. Biochim Biophys Acta. 1981;666:411–417. doi: 10.1016/0005-2760(81)90300-3. [DOI] [PubMed] [Google Scholar]
- 30.Newman JC, Verdin E. Beta-hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51–76. doi: 10.1146/annurev-nutr-071816-064916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262–284. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang SJ, Wu WM, Yang FL, Hsu GS, Huang CY. Vitamin B2 inhibits glutamate release from rat cerebrocortical nerve terminals. Neuroreport. 2008;19:1335–1338. doi: 10.1097/WNR.0b013e32830b8afa. [DOI] [PubMed] [Google Scholar]
- 33.Marashly ET, Bohlega SA. Riboflavin has neuroprotective potential: focus on Parkinson's disease and migraine. Front Neurol. 2017;8:333. doi: 10.3389/fneur.2017.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. 2019;42:598–607. doi: 10.1002/jimd.12053. [DOI] [PubMed] [Google Scholar]
- 35.Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145:256–264. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webber RJ, Edmond J. Utilization of L(+)-3-hydroxybutyrate, D(-)-3-hydroxybutyrate, acetoacetate, and glucose for respiration and lipid synthesis in the 18-day-old rat. J Biol Chem. 1977;252:5222–5226. [PubMed] [Google Scholar]
- 37.Broer S, Schneider HP, Broer A, Rahman B, Hamprecht B, Deitmer JW. Characterization of the monocarboxylate transporter 1 expressed in xenopus laevis oocytes by changes in cytosolic pH. Biochem J. 1998;333(Pt 1):167–174. doi: 10.1042/bj3330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reed WD, Ozand PT. Enzymes of L-(+)-3-hydroxybutyrate metabolism in the rat. Arch Biochem Biophys. 1980;205:94–103. doi: 10.1016/0003-9861(80)90087-9. [DOI] [PubMed] [Google Scholar]
- 39.Swiatek KR, Dombrowski GJ, Jr, Chao KL. The metabolism of D- and L-3-hydroxybutyrate in developing rat brain. Biochem Med. 1984;31:332–346. doi: 10.1016/0006-2944(84)90089-9. [DOI] [PubMed] [Google Scholar]
- 40.Lincoln BC, Des Rosiers C, Brunengraber H. Metabolism of S-3-hydroxybutyrate in the perfused rat liver. Arch Biochem Biophys. 1987;259:149–156. doi: 10.1016/0003-9861(87)90480-2. [DOI] [PubMed] [Google Scholar]
- 41.Desrochers S, David F, Garneau M, Jette M, Brunengraber H. Metabolism of R- and S-1,3-butanediol in perfused livers from meal-fed and starved rats. Biochem J. 1992;285(Pt 2):647–653. doi: 10.1042/bj2850647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2016;3:160018. doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simoens S. Pricing and reimbursement of orphan drugs: the need for more transparency. Orphanet J Rare Dis. 2011;6:42–1172-6-42. doi: 10.1186/1750-1172-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luzzatto L, Hyry HI, Schieppati A, et al. Outrageous prices of orphan drugs: a call for collaboration. Lancet. 2018;392:791–794. doi: 10.1016/S0140-6736(18)31069-9. [DOI] [PubMed] [Google Scholar]
- 45.Fair Medicine. https://www.fairmedicine.eu/en/. Accessed 27 March 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.