

Abstract
Purpose
Spinal muscular atrophy (SMA), caused by loss of the SMN1 gene, is a leading cause of early childhood death. Due to the near identical sequences of SMN1 and SMN2, analysis of this region is challenging. Population-wide SMA screening to quantify the SMN1 copy number (CN) is recommended by the American College of Medical Genetics and Genomics.
Methods
We developed a method that accurately identifies the CN of SMN1 and SMN2 using genome sequencing (GS) data by analyzing read depth and eight informative reference genome differences between SMN1/2.
Results
We characterized SMN1/2 in 12,747 genomes, identified 1568 samples with SMN1 gains or losses and 6615 samples with SMN2 gains or losses, and calculated a pan-ethnic carrier frequency of 2%, consistent with previous studies. Additionally, 99.8% of our SMN1 and 99.7% of SMN2 CN calls agreed with orthogonal methods, with a recall of 100% for SMA and 97.8% for carriers, and a precision of 100% for both SMA and carriers.
Conclusion
This SMN copy-number caller can be used to identify both carrier and affected status of SMA, enabling SMA testing to be offered as a comprehensive test in neonatal care and an accurate carrier screening tool in GS sequencing projects.
Keywords: spinal muscular atrophy (SMA), carrier screening, copy-number analysis, genome sequencing (GS), bioinformatics
INTRODUCTION
Spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disorder characterized by loss of alpha motor neurons, causes severe muscle weakness and atrophy presenting at or shortly after birth.1,2 SMA is the leading genetic cause of infant death after cystic fibrosis.3 The incidence of SMA is 1 in 6000–10,000 live births, and the carrier frequency is 1:40–80 among different ethnic groups.4–7 Four clinical types of SMA are classified based on age of onset and severity of the disease:1 very weak infants unable to sit unsupported (type I), weak sitters but unable to stand (type II), ambulant patients with weaker legs than arms (type III), and adult onset SMA (type IV). Early detection of SMA can be critical for long term quality of life due to the availability of two early treatments, nusinersen8 and Zolgensma,9 which have received FDA approval for the amelioration of SMA symptoms.
The disease causing gene, SMN1, and its paralog SMN2, reside in a ~2-Mb region on 5q with a large number of complicated segmental and inverted segmental duplications. SMN2 is 875 kb away from SMN1 and is created by an ancestral gene duplication that is unique to the human lineage.10,11 The genomic region around SMN1/2 is subject to unequal crossing over and gene conversion, resulting in variable copy numbers (CNs) of SMN1 and SMN2.7,12 Importantly, SMN2 has >99.9% sequence identity to SMN1 and one of the base differences, NM_000344.3:c.840C>T in exon 7, has a critical functional consequence. By interrupting a splicing enhancer, c.840T promotes skipping of exon 7, resulting in the vast majority of SMN2-derived transcripts (70–85%, depending on tissue13) being unstable and not fully functional. Approximately 95% of SMA cases result from biallelic absence of the functional c.840C nucleotide14 caused by either a deletion of SMN1 or gene conversion to SMN2 (c.840T). In the remaining 5% of SMA cases, patients also have other pathogenic variants in SMN1.15 SMN2 can produce a small amount of functional protein, and the number of SMN2 copies in an individual modifies the disease severity and is highly correlated with the clinical types described above.16
Due to the high incidence rate and disease severity, population-wide SMA screening is recommended by the American College of Medical Genetics and Genomics.17 The utility of population-wide carrier screening has been demonstrated in pilot studies.18 The key to screening for SMA is (1) determining the copy number of SMN1 for SMA diagnosis and carrier testing and (2) determining the copy number of SMN2 for clinical classification and prognosis. Traditionally, SMA testing and carrier testing are done with polymerase chain reaction (PCR) based assays, such as quantitative PCR (qPCR),19 multiplex ligation-dependent probe amplification (MLPA),20,21 and digital PCR.12,22 These methods primarily determine the copy number of SMN1 based on the c.840C>T site that differs between SMN1 and SMN2.
With recent advances in next-generation sequencing (NGS), it is now possible to profile a large number of genes or even the entire genome at high throughput and in a clinically relevant timeframe. Driven by these advances, many countries are undertaking large scale population sequencing efforts23–25 wherein testing for rare genetic disorders including carrier status will be one of the primary drivers. Demonstrating that GS can meet or exceed the performance of PCR-based SMA tests would indicate that both current and future precision medicine initiatives could leverage genome data for population-level screening. Replicating the current SMA testing regime poses a problem for high throughput GS due to the almost perfect sequence identity between SMN1 and SMN2. Furthermore, it is thought that frequent gene conversion between SMN1 and SMN2 leads to hybrid genes. These challenges demand an informatics method specifically designed to overcome the difficulties of this region.
To date, two NGS-based tests for SMA carrier detection have been reported.26,27 Larson et al.26 used a Bayesian hierarchical model to calculate the probability that the fraction of SMN1-derived reads is equal to or smaller than 1/3 at three base differences between SMN1 and SMN2. Since this method does not perform copy-number calling, it is not an ideal solution for carrier screening. Conversely, Feng et al.27 developed a copy-number caller for both SMN1 and SMN2 based on targeted sequencing data that closely mimics the current PCR-based method. Their method is designed for targeted sequencing and thus requires specialized normalization that limits their method to one assay at one site. More importantly, the relatively low depths of GS (~30×) cannot provide reliable bp-resolution CN calls (see Figure S1) and since this method relies on a single locus, it is not amenable to accurate CN calls with GS. Precision medicine initiatives will need a way to diagnose and detect carriers of SMA from GS data.
Here, we report a novel method that detects the CN of both SMN1 and SMN2 using GS data. While most conventional assays only test for the absence of c.840C as a proxy for the “exon 7 deletion” (either a true deletion or as a product of gene conversion with SMN2), here we describe a tool that can more fully characterize the variability in the region including (1) DNA deletions, including both whole-gene deletion/duplication and a partial deletion of a region that includes exons 7 and 8; and (2) small variant detection including the NM_000344.3:c.*3+80T>G (also referred to as g.27134T>G in literature) single-nucleotide polymorphism (SNP) that is correlated with “silent” carriers of SMA (two copies of SMN1 on the same haplotype).28 To demonstrate the accuracy of this method, we compared CN calls using digital PCR and MLPA with our GS-based calls and showed a concordance of 99.8% for SMN1 and 99.7% for SMN2. Additionally, we applied this method to 2504 unrelated samples from the 1000 Genomes Project29 (1kGP) and 10,243 unrelated samples from the NIHR BioResource Project (The NIHR BioResource, in revision) to report on the population distributions of SMN1 and SMN2 copy numbers. The carrier frequencies for SMA using this method agreed with those reported by previous PCR-based studies.5,6 In addition to demonstrating the accuracy of our method to quantify variants in the SMN region, we also highlight the importance of using ethnically diverse populations when developing novel informatic methods to resolve difficult clinically relevant regions of the genome.
MATERIALS AND METHODS
Samples and data processing
Samples validated using digital PCR were procured from the Motor Neuron Diseases Research Laboratory (Nemours Alfred I. duPont Hospital for Children) collection and were generated from cell lines as described previously.12,30 Historical patient samples with known SMA or carrier status measured by MLPA were obtained from Cambridge University. GS was performed on 73 samples with digital PCR results, 45 samples with MLPA results, and 12,747 population samples from the 1000 Genomes Project29 (1109 of which have MLPA calls), the National Institute for Health Research (NIHR) BioResource Rare Diseases project and the Next Generation Children (NGC) project.31 The sequencing and processing of this data was done using a variety of sample preparation methods, Illumina sequencers, and read aligners. A full description of this data can be found in the Supplementary Methods.
SMN copy-number analysis by orthogonal methods
SMN1 and SMN2 CNs were measured for 73 samples using the QuantStudio 3D Digital PCR System (Life Technologies) using allele-specific exon 7 probes as described previously.12 SMN1 and SMN2 copy numbers were normalized against those for RPPH1 (RNase P). MLPA CN calls for 1109 of the 1kGP samples were available from Vijzelaar et al.32 Forty-five historical patient samples were previously tested in a clinically accredited laboratory by MLPA for SMN1/2 exons 7 and 8 copy number (SALSA MLPA P060 SMA Carrier probemix, MRC-Holland). Additionally, two samples from the Next Generation Children project were confirmed using MLPA.
Copy-number calling for intact and truncated SMN
Two common copy-number variants (CNVs) involve the SMN1 and SMN2 loci, the whole-gene CNV, and a partial gene deletion of exons 7 and 8 (SMN2∆7–8)32,33 (see “Results”). We first count reads that align to either SMN1 or SMN2. Read counts in a 22.2-kb region encompassing exon 1 to exon 6 are used to calculate the total SMN (SMN1, SMN2, and SMN2∆7–8) CN, and read counts in the 6.3-kb region including exons 7 and 8 are used to calculate the CN of intact SMN (SMN1 and SMN2). Read counts are normalized and converted into copy numbers using a one-dimensional mixture of Gaussian distributions (see Supplementary Information). The copy number of truncated SMN (SMN2∆7–8) is derived by subtracting the intact SMN CN from total SMN CN.
Genotyping SMN1/2 copy number using differentiating bases
We call the number of chromosomes carrying the SMN1 and SMN2 bases by combining the total SMN CN with the read counts supporting each of the gene-specific bases. At each SMN1/2 differentiating base (Table S1), based on the called copy number of intact SMN, the caller iterates through all possible combinations of SMN1 and SMN2 copy numbers and derives the combination that produces the highest posterior probability for the observed number of SMN1 and SMN2 supporting reads. The SMN1 CNs called at single bases are then combined to make the aggregate SMN1 CN call based on a consensus rule (see Supplementary Methods). In addition to calling the CN of bases that are specific to either SMN1 or SMN2, this method can be applied to variant positions to identify the copy number of bases known to be specific to one of the two genes, e.g., c.*3+80T>G as described in “Results”.
Ethics approval and consent
The 13,343 individuals from the NIHR BioResource Rare Diseases project were recruited through NHS Cambridge University Hospitals Foundation Trust under Cambridge South Research Ethics Committee approval 13/EE/0325.
The 65 patient-derived DNA samples from the Motor Neuron Diseases Research Laboratory were isolated from established fibroblast or lymphoblastoid cell lines. For those cell lines obtained from noncommercial sources, biospecimens were obtained after written consent or assent and parental permission. This study was approved by the Nemours/Alfred I. duPont Hospital for Children Institutional Review Board. These samples were de-identified so that no protected health information is known for these lines.
RESULTS
Common CNVs involving the SMN1/2 loci
While existing PCR- or NGS-based methods focus primarily on the c.840C>T site, we adopted a copy-number approach based on the sequencing data from the full genes. We examined the read depth across the ~30-kb homologous region harboring SMN1 and SMN2 genes in 1kGP samples (Fig. 1a). The depth profile shows that this entire region can be deleted or duplicated in the population. The exact breakpoints of this CNV are expected to vary from sample to sample due to the extensive sequence homology within and beyond this region and can only be resolved in high resolution with long read sequencing. We restricted our CN analysis to the (~30 kb) regions that include the SMN genes (SMN1 or SMN2).
Fig. 1. Common copy-number variants (CNVs) affecting the SMN1/2 loci.

a Depth profiles across the SMN1/SMN2 regions. Samples with a total SMN1 + SMN2 copy number of 2, 3, 4, and 5 (derived from average read depth in the two genes) are shown as green, blue, black, and orange dots, respectively. Depth from 50 samples are summed up for each CN category. Each dot represents normalized depth values in a 100-bp window. Read counts are calculated in each 100-bp window, summing up reads from both SMN1 and SMN2, and normalized to the depth of wild-type samples (CN = 4). The SMN exons are represented as purple boxes. The two x-axes show coordinates (hg19) in SMN1 (bottom) and SMN2 (upper). b Depth profiles aggregated from 50 samples carrying a deletion of exons 7 and 8 are shown as red dots. Read depths are calculated in the same way as in (a).
In addition to whole-gene CNVs, we also found a 6.3-kb partial gene deletion encompassing both exons 7 and 8 (Fig. 1b, Figure S2) that was recently described in another study.33 The sequences at the breakpoint are identical between SMN1 and SMN2, so this deletion occurs at either chr5: 70244114–70250420 in SMN1 or chr5: 69368689–69375000 in SMN2 (Figure S2, hg19). However, about 500 bp downstream from the breakpoint that defines the end of this deletion there are three base differences between the SMN1 and SMN2 loci (70250881A>69375425C, 70250981A>69375525G, 70250991A>69375535G). Among the 1kGP samples that contain this deletion, we identified 245 read pairs from 237 samples where one spanned the breakpoint and the mate spanned at least two of the three SMN-differentiating bases. Analysis of these read pairs revealed that 100% were consistent with the deletion occurring on the SMN2 sequence background. We named this truncated form of SMN2 “SMN2∆7–8”. Since both exons 7 and 8 are deleted, SMN2∆7–8 most likely has limited or no biological function. Therefore, SMN2∆7–8 is an important variant that any SMN CN caller should take into account.
After searching for anomalous read pairs in the 1kGP samples, we did not identify any other common CNVs in the SMN region. Combining this information together, we called CNs of the SMN genes to specifically identify the number of intact and truncated forms by dividing the genes into two regions: the 6.3-kb region that includes exons 7–8 and the 22.2-kb region that includes exons 1–6. The CN calculated from the exon 7–8 region provided the number of intact SMN genes. Samples with SMN2∆7–8 have a higher CN call from the exon 1–6 region compared with the CN call from the exon 7–8 region, and this difference represents the CN of SMN2∆7–8. Figure 2 shows the results of this calculation for 12,747 samples where we identified 2144 instances of SMN2∆7–8 including 140 samples with two copies of SMN2∆7–8 and one sample with three copies of SMN2∆7–8.
Fig. 2.
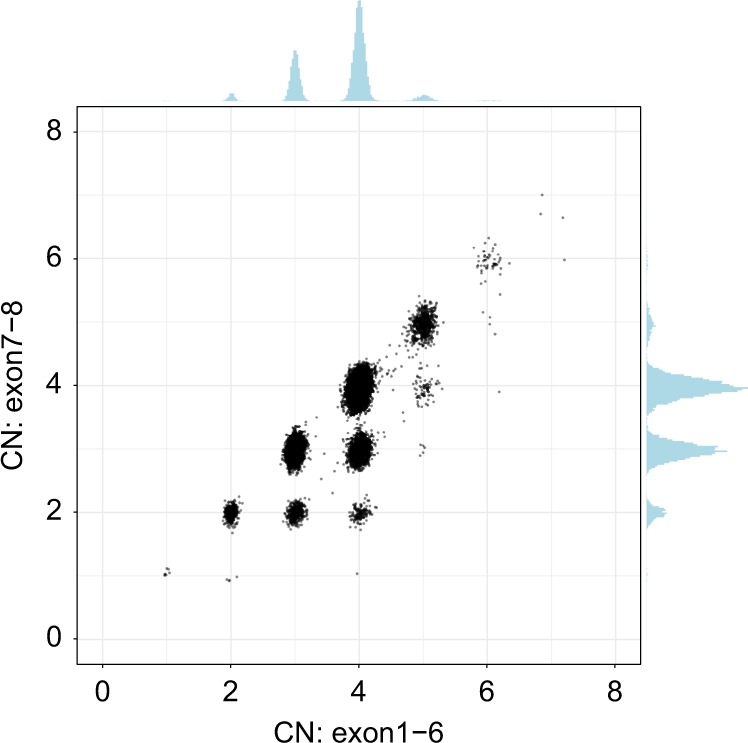
Scatterplot of total SMN (SMN1, SMN2, and SMN2∆7–8) copy number (CN) (x-axis, called by read depth in exons 1–6) and intact SMN copy number (y-axis, called by read depth in exons 7–8).
SMN1/2 CN calls using differentiating bases
We called the CNs of SMN1 and SMN2 at the 16 base difference sites between SMN1 and SMN2 (see “Materials and Methods”, Table S1) in 1kGP samples, and compared the CN calls for each position with the CN calls at the splice variant site (Fig. 3a, Figure S3). There was a notable difference between the concordance of calls in the African and non-African populations (Fig. 3a). Excluding the African samples, there were 13 sites that had high (>85%) CN concordance with the splice variant site. Conversely, for the African samples there were only seven sites that had high CN concordance with the splice variant site, and the concordance values were lower than in non-African populations. This is consistent with within-gene variation at many of these positions in the African population. We selected the splice variant site and the seven positions that were highly concordant with the splice variant site in both African and non-African populations to make SMN1 and SMN2 CN calls based on the consensus of CN calls at the selected sites (see Supplementary Information for the detailed rules and in-depth analysis on the variability of the 16 sites).
Fig. 3. Distribution of SMN1/SMN2/SMN2∆7–8 copy numbers in the population.

a Percentage of samples showing copy number (CN) call agreement with c.840C>T across 16 SMN1–SMN2 base difference sites in African and non-African populations. Coordinates of these 16 sites are given in Table S1 and site 13* is the c.840C>T splice variant site. The black horizontal line denotes 85% concordance. b Histogram of the distribution of SMN1, SMN2, and SMN2∆7–8 copy numbers across five populations in 1kGP and the National Institute for Health Research (NIHR) BioResource cohort. c SMN1 CN vs. total SMN2 CN (intact SMN2 + SMN2∆7–8). d Two trios with an SMA proband detected by the caller and orthogonally confirmed in the Next Generation Children project cohort. CNs of SMN1, SMN2, and SMN2∆7–8 are phased and labeled for each member of the trios.
Validation of the SMN copy-number caller
After developing this method, we tested its accuracy against results from orthogonal methods. We sequenced 73 samples with known SMN1 and SMN2 CNs measured by digital PCR; 45 samples with known results measured by MLPA; and also compared our CN calls with MLPA calls published by Vijzelaar et al.33 on 1109 samples from the 1kGP. These validation data include 64 SMA probands, 45 SMA carriers, and 1118 samples with an SMN1 CN larger than 1. Our SMN1 CN calls agreed with digital PCR results in 99.8% of the samples, the SMN2 CN calls agreed in 99.7% of the samples, and SMN2∆7–8 calls agreed in 100% of the samples (Table 1, Table S2). Combined, our recall is 100% for SMA (SMN1 CN = 0, 64/64), 97.8% for carriers (SMN1 CN = 1, 44/45, the one missed carrier does not show read evidence supporting the carrier status, see Supplementary Information), and our precision is 100% for both SMA (1160/1160) and carriers (1179/1179). We also analyzed the consistency of SMN1/SMN2/SMN2∆7–8 CN calls in 258 trios from the Next Generation Children project cohort. There is no Mendelian error in any of the calls (Table S3).
Table 1.
Validation against samples with known SMN1/SMN2 copy numbers (CNs).
| CN by orthogonal method | Total | Concordant | Discordant | Agreement | |
|---|---|---|---|---|---|
| SMN1 | 0 | 64 | 64 | 0 | 100.0% |
| 1 | 45 | 44 | 1 | 97.8% | |
| 2 | 897 | 897 | 0 | 100.0% | |
| 3 | 174 | 174 | 0 | 100.0% | |
| 4 | 43 | 43 | 0 | 100.0% | |
| 6 | 1 | 0 | 1 | 0.0% | |
| Total | 1224 | 1222 | 2 | 99.8% | |
| SMN2 | 0 | 117 | 117 | 0 | 100.0% |
| 1 | 466 | 465 | 1 | 99.8% | |
| 2 | 541 | 539 | 2 | 99.6% | |
| 3 | 60 | 60 | 0 | 100.0% | |
| 4 | 9 | 8 | 1 | 88.9% | |
| Total | 1193 | 1189 | 4 | 99.7% | |
| SMN2∆7–8 | 0 | 1089 | 1089 | 0 | 100.0% |
| 1 | 80 | 80 | 0 | 100.0% | |
| 2 | 4 | 4 | 0 | 100.0% | |
| Total | 1173 | 1173 | 0 | 100.0% |
Further inspection of the five discrepant samples does not show evidence of errors in our calls except in one sample (see Supplementary Information). The validation for the 73 and 45 samples was done using two sets of probes targeting exons 7 and 8 of SMN1 and SMN2, while the validation for the 1109 samples used a newer version of the MLPA probemix that includes an additional 17 probes used to determine the SMN1 + SMN2 CN in exons 1–6 and exons 7–8, which helped refine the individual SMN1 and SMN2 CN calls. Correspondingly our concordance is significantly higher (p value = 0.009223 for SMN1, 2.651e-05 for SMN2, Fisher’s exact test) measured against the newer version of MLPA (100% concordance for SMN1 and SMN2) than other methods (98.3% for SMN1 and 95.4% for SMN2).
Copy number of SMN1, SMN2, and SMN2∆7–8 by population
Given the high accuracy demonstrated by our validation against orthogonal methods, we next applied this method to high depth (all samples sequenced to at least 30×) GS data from 12,747 unrelated samples from the 1kGP and the NIHR BioResource Project (Table S4). We analyzed the CN distributions by population (Europeans, Africans, East Asians, South Asians, and admixed Americans consisting of Colombians, Mexican Americans, Peruvians, and Puerto Ricans). Figure 3b shows the histogram of the number of individuals with different CNs of intact SMN1, intact SMN2, and SMN2∆7–8. The distributions are similar between the 1kGP samples and the NIHR BioResource samples (Figure S4, Table S5). In general, individuals have more copies of SMN1 than SMN2. The most common combinations of SMN1/SMN2 copy number are 2/2 (44.9%) and 2/1 (33.4%). The variability of SMN1 copy number is much lower than that of SMN2 copy number in most populations, while the Africans show higher variability in both SMN1 and SMN2 CN. Conversely, 54.7% of Africans have three or more copies of SMN1, which is more than double what is observed in any of the other four populations (Fig. 3b, Table 2). There is an inverse relationship between the copy number of SMN1 and SMN2 (Fig. 3c, correlation coefficient -0.344, p value <2.2e-16). This observation is consistent with a mechanism where gene conversion occurs between SMN1 and SMN2.34,35 The observed higher SMN1 CN relative to SMN2 CN could be a result of a bias toward SMN2-to-SMN1 conversion or selection against a low SMN1 CN. Africans have significantly lower SMN2 CN than the other populations (Wilcoxon test, p value < 2.2e-16).
Table 2.
SMN1 copy number (CN) and c.*3+80T>G frequency by population.
| Ethnicity | Total | SMN1 CN = 1 | SMN1 CN = 2 | SMN1 CN = 3 | SMN1 CN = 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Count | c.*3+80T>G+ | Count | c.*3+80T>G+ | Count | c.*3+80T>G+ | Count | c.*3+80T>G+ | ||
| African | 902 | 4 | 0 (0.0%) | 404 | 134 (33.2%) | 373 | 315 (84.5%) | 121 | 112 (92.6%) |
| European | 9648 | 212 | 0 (0.0%) | 8899 | 4 (0.04%) | 524 | 22 (4.2%) | 13 | 2 (15.38%) |
| South Asian | 1199 | 20 | 0 (0.0%) | 965 | 1 (0.1%) | 195 | 5 (2.56%) | 19 | 1 (5.26%) |
| East Asian | 593 | 8 | 0 (0.0%) | 552 | 1 (0.18%) | 33 | 1 (3.03%) | 0 | 0 (NA) |
| Admixed American | 341 | 7 | 0 (0.0%) | 296 | 7 (2.36%) | 36 | 9 (25.0%) | 2 | 1 (50.0%) |
The number of SMA carriers identified across populations is summarized in Table 2 and Table S6. In 12,683 individuals with confident SMN1/SMN2 CN calls, Europeans have the highest carrier frequency at 2.2%, followed by admixed Americans (2.05%), South Asians (1.67%), and East Asians (1.35%). Africans have the lowest carrier frequency (0.44%) by SMN1 CN, and this carrier frequency does not include the 2 + 0 SMA silent carriers that are more common in Africans (see “Detection of ‘silent’ carriers”). The CN frequency distributions observed in this study are consistent with previous studies of SMN1/SMN2 CN distribution in the general population.5,6 In addition, we also report the frequency of the exon 7–8 deletion (SMN2∆7–8) across populations: 21.2% of Europeans and 11.5% of admixed Americans have at least one copy of SMN2∆7–8, while the frequency is lower in South Asians (3.35%), Africans (1.1%), and East Asians (0.34%).
In the Next Generation Children project cohort, we identified SMA in two neonatal probands from trio analysis, which were confirmed independently. SMN1, SMN2, and SMN2∆7–8 CNs are phased for each trio member (Fig. 3d).
Detection of “silent” carriers
The c.*3+80T>G SNP has been reported to be associated with the 2 + 0 SMA silent carrier status where one chromosome carries two copies of SMN1 (either by SMN1 duplication or gene conversion of SMN2 to SMN1) and the other chromosome has no copies of SMN1.28 Our method can also detect the presence of this SNP and thus can be used to screen for potential silent carriers. In the population samples, this SNP is differentially associated with two-copy and one-copy SMN1 haplotypes and can be used to identify silent carriers (Table 2). We estimate the sensitivity improvement (with the biggest improvement from 70.5% to 91.8% in Africans) and residual risks for calling SMA carriers using the combination of SMN1 CN and c.*3+80T>G SNP calling (see Supplementary Information, Table S7, Table S8). One of our validation samples is a silent carrier and we correctly called SMN1 CN = 2 and the presence of c.*3+80T>G SNP (Table S2).
DISCUSSION
Due to the high sequence homology between SMN1 and SMN2, the SMN region is difficult to resolve with both short and long read sequencing and thus far this important region has been excluded from standard GS analysis. Here, we demonstrate an algorithm that can resolve the CNs of SMN1 and SMN2 independently using short-read GS data, filling in an important gap in SMA diagnosis and carrier screening for precision medicine initiatives. Accurate measurement of SMN1 and SMN2 CNs is essential not only for the diagnosis of SMA but is also a prognostic indicator and the basis of therapeutic options.36 SMN2 CN has been used as a criterion for many clinical trials for SMA, including nusinersen8 and Zolgensma.9
As a demonstration of this algorithm, we made CN calls for SMN1 and SMN2 using sequencing data from 12,747 samples covering five distinct subpopulations. We identified a total of 251 samples with SMN1 losses (less than two copies) and 1317 with SMN1 gains (more than two copies), 6241 samples with SMN2 losses and 1274 with SMN2 gains, and 2144 samples carrying one or more copies of the truncated form SMN2∆7–8. We cannot quantify the role that deletions, duplications, or gene conversion play to drive the CN changes in this region but we see evidence supporting all three mechanisms including (1) 3853 samples with total (SMN1 + SMN2) CN <4 (deletions), (2) 670 samples with total CN >4 (duplications), and (3) a strong inverse correlation between the SMN1 and SMN2 CN (gene conversion, Fig. 3c). Additionally, we identified a carrier frequency between 1:45 and 1:225 depending on ancestral population (Table 2). Comparing the CN frequencies by population shows that they are highly different and our per-population results agree with previous population studies.5,6 While this provides qualitative support for the accuracy of our method, we also directly assessed its accuracy by comparing our CN calls against the results from digital PCR or MLPA. In this direct comparison, 99.8% of our SMN1 and 99.7% of our SMN2 CN calls agreed with the digital PCR-based or MLPA-based results. Importantly, our recall is 100% for SMA and 97.8% for carriers, and our precision is 100% for both SMA and carriers.
In this study, we optimized our CN calling to work for individuals of any ancestry and thus limited SMN1/2 differentiation to the functionally important splice variant plus seven sites in high concordance with the splice variant across all populations (Fig. 3a). By quantifying the concordance between all of the reference differences and the splice variant, we were able to identify variations at these sites that, if not accounted for properly (i.e., removed from our analysis), could lead to errors in our CN calls. This would be especially problematic in analyzing Africans because they harbor more diverse haplotypes. Future population genetic studies, possibly including using long read sequencing, will help profile the haplotypic diversity across populations more directly and identify new variant sites that could further improve the accuracy of SMN1/SMN2 differentiation.
An important area for improvement is the detection of “silent” carriers. One type of silent carrier occurs when an individual has two copies of the SMN1 gene but they are both on the same haplotype. A SNP (c.*3+80T>G) has been used to identify individuals who are at an increased risk of being carriers when SMN1 CN is two but the risk associated with this SNP can vary greatly between studies and populations (Table S7, Table S8). When an individual has just one copy of SMN1 they can be definitively identified as a carrier, but this variant only indicates a 2–8% chance of being a carrier when SMN1 CN is two (Table S8). With GS, it would be possible to catalog the different variants that occur with different CN combinations of SMN1 and SMN2 and possibly identify additional markers that could be used to improve our ability to identify these silent carriers. In addition, the loss of the c.840C splice variant currently explains around 95% of SMA cases and the remaining cases include other pathogenic variants. These other pathogenic variants represent another type of silent carrier and as more of them are identified, we will extend this software to directly genotype these as part of the testing process, further improving the ability to detect SMA carriers and cases.
While there exist difficult regions in the genome where normal GS pipelines do not deliver variant calls, here we demonstrate the ability to apply GS paired with a targeted informatics approach to solve one such difficult region. So far, this targeted strategy (GS+ specialized informatics) has been applied successfully to a number of difficult variants, such as repeat expansions37 and CYP2D6.38 Traditionally, it is not cost effective to perform all of the known genetic tests and carrier screenings on every individual, so candidates for specific genetic testing are identified using information such as the carrier rate and family history. However, this process means that many people without a family history who would benefit from knowing their SMA status do not routinely have access to this data. Once GS analysis can detect all single-nucleotide variants (SNVs) and CNVs in all clinically relevant genes accurately then a more general and population-wide genetic testing strategy will be feasible with a single test. Improving GS to become economical as a substitute for one current genetic test will help facilitate the integration of more genetic tests and carrier screens into GS, allowing more general access to genetic testing population-wide. GS provides a valuable opportunity to assess the entire genome for genetic variation and the continued development of more targeted informatics solutions for difficult regions with GS data will help bring the promise of personalized medicine one step closer to reality.
Supplementary information
Acknowledgements
This work was supported by the Cambridge Biomedical Research Centre and the NIHR for the NIHR BioResource (grant number RG65966), the National Institute of General Medical Sciences of the National Institutes of Health (P30GM114736 and P20GM103446; to M.E.R.B.) and the Nemours Foundation (to M.E.R.B.). We thank the New York Genome Center (supported by National Human Genome Research Institute grant 3UM1HG008901–03S1), and the Coriell Institute for Medical Research for generating and releasing the 1kGP GS data. We thank NIHR BioResource volunteers for their participation, and gratefully acknowledge NIHR BioResource centers, NHS Trusts, and staff for their contribution. We thank the NIHR and NHS Blood and Transplant. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Software and data availability
The SMN copy-number caller described here can be downloaded from https://github.com/Illumina/SMNCopyNumberCaller.
The 1kGP data can be downloaded from https://www.ncbi.nlm.nih.gov/bioproject/PRJEB31736/. Data from the NIHR BioResource participants have been deposited in European Genome-phenome Archive (EGA) at the EMBL European Bioinformatics Institute (accession codes available at https://www.biorxiv.org/content/10.1101/507244v1). Those participants from the NIHR BioResource who enrolled for the 100,000 Genomes Project–Rare Diseases Pilot can be accessed by seeking access via Genomics England Limited following the procedure outlined at https://www.genomicsengland.co.uk/about-gecip/joining-research-community. The Bam files from the NGC individuals have been deposited in EGA under accession number EGAD00001004357.
Disclosure
X.C., Z.K., A.C., A.L.H., R.J.T., D.R.B. and M.A.E. are employees of Illumina Inc. The other authors declare no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41436-020-0754-0) contains supplementary material, which is available to authorized users.
References
- 1.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 2.Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11:443–452. doi: 10.1016/S1474-4422(12)70061-3. [DOI] [PubMed] [Google Scholar]
- 3.Prior TW. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med. 2010;12:145–152. doi: 10.1097/GIM.0b013e3181c5e713. [DOI] [PubMed] [Google Scholar]
- 4.Ogino S, Leonard DGB, Rennert H, Ewens WJ, Wilson RB. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002;110:301–307. doi: 10.1002/ajmg.10425. [DOI] [PubMed] [Google Scholar]
- 5.Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46:641–644. doi: 10.1136/jmg.2009.066969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Genet. 2012;20:27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDonald WK, Hamilton D, Kuhle S. SMA carrier testing: a meta-analysis of differences in test performance by ethnic group. Prenat Diagn. 2014;34:1219–1226. doi: 10.1002/pd.4459. [DOI] [PubMed] [Google Scholar]
- 8.Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 9.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 11.Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet. 2001;108:255–266. doi: 10.1007/s004390100473. [DOI] [PubMed] [Google Scholar]
- 12.Stabley DL, Harris AW, Holbrook J, et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol Genet Genomic Med. 2015;3:248–257. doi: 10.1002/mgg3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Burghes AHM, Beattie CE. Spinal muscular atrophy: why do low levels of SMN make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butchbach MER. Copy number variations in the survival motor neuron genes: implications for spinal Muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. 2016;3:7. doi: 10.3389/fmolb.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prior TW. Carrier screening for spinal muscular atrophy. Genet Med. 2008;10:840–842. doi: 10.1097/GIM.0b013e318188d069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraszewski JN, Kay DM, Stevens CF, et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med. 2018;20:608–613. doi: 10.1038/gim.2017.152. [DOI] [PubMed] [Google Scholar]
- 19.Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arkblad EL, Darin N, Berg K, et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord. 2006;16:830–838. doi: 10.1016/j.nmd.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Scarciolla O, Stuppia L, De Angelis MV, et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics. 2006;7:269–276. doi: 10.1007/s10048-006-0051-3. [DOI] [PubMed] [Google Scholar]
- 22.Zhong Q, Bhattacharya S, Kotsopoulos S, et al. Multiplex digital PCR: breaking the one target per color barrier of quantitative PCR. Lab Chip. 2011;11:2167–2174. doi: 10.1039/c1lc20126c. [DOI] [PubMed] [Google Scholar]
- 23.Ashley EA. The Precision Medicine Initiative: a new national effort. JAMA. 2015;313:2119–2120. doi: 10.1001/jama.2015.3595. [DOI] [PubMed] [Google Scholar]
- 24.The Genome of the Netherlands Consortium. Francioli LC, Menelaou A, et al. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–825. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 25.Turnbull C, Scott RH, Thomas E, et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ. 2018;361:k1687. doi: 10.1136/bmj.k1687. [DOI] [PubMed] [Google Scholar]
- 26.Larson JL, Silver AJ, Chan D, Borroto C, Spurrier B, Silver LM. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet. 2015;16:100.. doi: 10.1186/s12881-015-0246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Y, Ge X, Meng L, et al. The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet Med. 2017;19:936–944. doi: 10.1038/gim.2016.215. [DOI] [PubMed] [Google Scholar]
- 28.Luo M, Liu L, Peter I, et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med. 2014;16:149–156. doi: 10.1038/gim.2013.84. [DOI] [PubMed] [Google Scholar]
- 29.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stabley DL, Holbrook J, Harris AW, et al. Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromuscul Disord. 2017;27:439–446. doi: 10.1016/j.nmd.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French CE, Delon I, Dolling H, et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019;45:627–636. doi: 10.1007/s00134-019-05552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vijzelaar R, Snetselaar R, Clausen M, et al. The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS One. 2019;14:e0220211. doi: 10.1371/journal.pone.0220211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruhno C, McGovern VL, Avenarius MR, et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet. 2019;138:241–256. doi: 10.1007/s00439-019-01983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogino S, Gao S, Leonard DGB, Paessler M, Wilson RB. Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Genet. 2003;11:275. doi: 10.1038/sj.ejhg.5200957. [DOI] [PubMed] [Google Scholar]
- 35.Chen T-H, Tzeng C-C, Wang C-C, et al. Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population. J Neurol Sci. 2011;308:83–87. doi: 10.1016/j.jns.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28:103–115. doi: 10.1016/j.nmd.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Dolzhenko E, van Vugt JJFA, Shaw RJ, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27:1895–1903. doi: 10.1101/gr.225672.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S, Wheeler MM, Patterson K, et al. Stargazer: a software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet Med. 2019;21:361. doi: 10.1038/s41436-018-0054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SMN copy-number caller described here can be downloaded from https://github.com/Illumina/SMNCopyNumberCaller.
The 1kGP data can be downloaded from https://www.ncbi.nlm.nih.gov/bioproject/PRJEB31736/. Data from the NIHR BioResource participants have been deposited in European Genome-phenome Archive (EGA) at the EMBL European Bioinformatics Institute (accession codes available at https://www.biorxiv.org/content/10.1101/507244v1). Those participants from the NIHR BioResource who enrolled for the 100,000 Genomes Project–Rare Diseases Pilot can be accessed by seeking access via Genomics England Limited following the procedure outlined at https://www.genomicsengland.co.uk/about-gecip/joining-research-community. The Bam files from the NGC individuals have been deposited in EGA under accession number EGAD00001004357.