

Abstract
It is well established that both an increase in reactive oxygen species (ROS: i.e. O2•−, H2O2 and OH•), as well as protein aggregation, accompany ageing and proteinopathies such as Parkinson's and Alzheimer's disease. However, it is far from clear whether there is a causal relation between the two. This review describes how protein aggregation can be affected both by redox signalling (downstream of H2O2), as well as by ROS-induced damage, and aims to give an overview of the current knowledge of how redox signalling affects protein aggregation and vice versa. Redox signalling has been shown to play roles in almost every step of protein aggregation and amyloid formation, from aggregation initiation to the rapid oligomerization of large amyloids, which tend to be less toxic than oligomeric prefibrillar aggregates. We explore the hypothesis that age-associated elevated ROS production could be part of a redox signalling-dependent-stress response in an attempt to curb protein aggregation and minimize toxicity.
Keywords: amyloid, protein aggregation, reactive oxygen species, redox signalling
Introduction
Both the loss of proteostasis and ROS production as a consequence of mitochondrial dysfunction are among the Hallmarks of Ageing [1]. While there is plenty of evidence that these two hallmarks are tightly intertwined, their cause and effect relationships remain unclear. This might be partly due to the fact that ROS, in the form of H2O2, itself plays a dual role. While at lower levels H2O2 acts as a second messenger in redox signalling, which is absolutely required for physiology and for lifespan extension in model systems [2], at higher levels H2O2 and other ROS could lead to random damage including for instance protein unfolding and aggregation, and the latter has been proposed to accelerate the aging process [3–10]. But there is also evidence of functional redox signalling-dependent protein aggregation, for instance providing a means to (temporarily) inactivate or alter the function of proteins. Redox-dependent protein aggregation is also often reversible. But there are also many examples of protein aggregation-induced enhanced ROS production, which may eventually contribute to cellular dysfunction and cell death.
A hypothesis that could unite both H2O2 as a signalling molecule and ROS as a driver of age-related protein aggregation was posed by Hekimi et al. [2]. It proposes that age-related damage triggers stress response pathways that could depend on redox signalling and hence produce H2O2 (either directly or indirectly through O2•− followed by dismutation) as a second messenger in an attempt to regain homeostasis. Over time, when more damage accumulates, H2O2 produced to further boost this stress response surpasses levels that are merely involved in signalling, leading to a build-up of H2O2 and ROS associated damage. This eventually would lead to a vicious cycle in which ROS-dependent damage triggers a redox signalling-dependent-stress response, leading to a further increase in ROS. This hypothesis would also fit with the observations that several types of aggregates can trigger ROS production.
In this review, we will focus on examples of the interplay between redox signalling and protein aggregation. We review the current knowledge and try to illuminate possible relationships between redox signalling and proteostasis.
Protein folding
Proteins are synthesized as linear peptide chains on ribosomes and must fold into 3D structures to execute their biological functions. Protein folding is driven for a large part by the spontaneous burial of nonpolar amino acids in the folding core, but also guided by hydrogen bonds, van der Waals- and electrostatic interactions. The many weak, noncovalent and often distant (in sequence) interaction possibilities complicate the conformational possibilities. The stability of natively folded proteins depends on local environmental factors such as pH and temperature.
Most (∼70%) protein folding takes place at the ribosome during translation. Several mechanisms are in place to ensure correct folding: the sequential folding of domains emerging from the ribosome, the spatial restrictions of the ribosomal exit channel, the rate of translation as well as the ribosome-associated chaperones RAC and NAC (ribosome-associated complex and nascent-chain associated complex, respectively) [11,12]. More downstream of the ribosome, the HSP70 system of chaperones prevents undesirable domain interactions. Moreover, HSP70 functions as a binding interface for other chaperones like HSP90 and chaperonins, which aid in de novo folding by recognizing exposed hydrophobic residues and promoting ATP-dependent refolding.
Proteins need to overcome considerable energy barriers to reach their final, stable conformation, inherently leading to the accumulation of folding intermediates (Figure 1) [13]. Examples of slow steps in protein folding include disulfide bond formation and prolyl isomerization [14]. Partially folded proteins are at high risk of misfolding and aggregation, due to non-native interactions through for instance the exposure of hydrophobic residues [15,16]. Other reasons for faulty protein accumulation are mutations or polymorphisms, translation errors and the structurally dynamic characteristic of proteins. Misfolded conformations are quasi-stable, making them more prone to aggregation when proteostasis control systems are saturated. The latter happens increasingly during ageing [17]. The high plasticity of partially folded intermediates is in contrast with extremely structured aggregates like amyloid fibrils.
Figure 1. Energy landscape in proteostasis.

Newly synthesized peptides sample different conformations during protein folding, on their way downhill to the most thermodynamically favourable state. Energetically trapped, partially unfolded or sub-optimally folded intermediates may accumulate as they need to cross energy barriers to reach their native, low energy state. Non-native interactions may lead to protein aggregation, thereby interfering with the protein folding process. The proteostasis network can assist in lowering energy barriers and preventing non-native interactions. As indicated by the yellow circles, H2O2-mediated redox signaling or ROS-dependent damage helps to overcome the transition state between intermediates of the proteostasis network.
Another hallmark of ageing, the failure to maintain proteostasis, presents itself as an accumulation of misfolded proteins and aggregates. Like folding, aggregation is predominantly driven by hydrophobic interactions, which is why aggregation prone regions (APRs) in proteins are generally distinguished by their high hydrophobicity, low net charge and high β-sheet propensity [18,19]. During aggregation, the hydrophobic interactions are mostly intermolecular, and therefore aggregation is concentration dependent. Due to the similarity between the composition of APRs and protein regions driving hydrophobic core formation during folding, aggregation and folding pathways constantly compete. The aggregation has long been considered only as a sign of degeneration and dysfunction. However, despite strong selective pressure against protein aggregation, numerous APRs remain in the proteome, which is in line with the notion that protein aggregation can play a functional or regulatory role [20,21]. The number of possible conformations for aggregation intermediates is large, and they need to overcome free energy barriers on their way to mature aggregates. This means that aggregation intermediates are energetically trapped and thus accumulate (Figure 1), allowing more non-native interactions to occur. Typically, these non-specific interactions between polypeptides form a disordered assembly without a specified shape termed amorphous aggregates.
While most aggregates are amorphous, examples of more structured aggregate types are oligomeric aggregates and the extremely structured β-amyloid fibrils, and the latter are characterized by a cross-β-structure (in which β-strands lie perpendicular to the fibril axis). Amyloid fibrillization consists of a slow lag phase directed by intermolecular interactions during which misfolded polypeptides congregate into nuclei and form oligomers containing β-sheets. During the subsequent exponential growth phase, oligomers cluster further with these nuclei and grow rapidly into prefibrils and protofibrils with a cross-β-structure. During the final saturation phase, 2–6 protofibrils assemble into mature multistrand amyloid fibrils which can adopt several polymorphic structures by twisting or lateral association [22]. Amyloid fibrils are one of the most thermodynamically stable and stiff protein arrangements known [23].
Amyloids are a hallmark of (age-related) proteinopathies such as ALS, Alzheimer's, prion disease and cataract. Early studies focused on the mature aggregate deposits as toxic species. The toxicity of amyloids is complex, however, and several intermediates and oligomers but also mature amyloid fibrils have now been linked to pathogenesis [11–13]. Some reports even suggest an inverse correlation between oligomer size and toxicity of aggregates [24–27]. This complexity is also thought to be one of the reasons for clinical trial failures in proteinopathies, with hardly any effective therapy available for treatment [13,28]. Adding to the complexity is the fact that many types of amyloids are shown to be functional with roles reported in chemical storage, structure, signalling and inactivation of the soluble protein [29]. Another type of functional aggregation is controlled by protein domains lacking rigid 3D structures under physiological conditions, present in 15–30% of proteins. These intrinsically disordered regions (IDRs) have a high conformational plasticity and susceptibility to modifications, enabling a multitude of interactions [30]. With these interactions, IDRs drive misfolding and aggregation of (partially) disordered proteins as well as liquid–liquid phase separation (LLPS), thereby forming membrane-less compartments which are important for the concentration and segregation of biochemical reactions [31]. However, LLPS condensates have also been reported to catalyze amyloid fibrillization [32,33].
Clearance of protein aggregates
There are three quality control networks to ensure continuous surveillance of the proteome: (i) chaperones that mediate (re)folding, (ii) the ubiquitin-proteasome system (UPS) and (iii) autophagy to clear misfolded proteins and aggregates.
As mentioned previously, chaperones aid in de novo protein folding by lowering energy barriers between folding intermediates. During aggregation, chaperones instead raise the energy barriers toward aggregation by preventing intermolecular interactions. Besides preventing aggregation, chaperones play an important role in active disaggregation [34]. Recent reports show that almost all types of aggregates are reversible [35,36]. The co-ordinated action of small HSPs (sHSPs), HSP40, HSP70 and HSP110 can even disaggregate amyloid fibrils by fragmentation and depolymerization into both monomeric and oligomeric species. The activity of chaperones is, therefore, twofold: on the one hand to prevent aggregation, on the other to disaggregate (intermediate) aggregates.
One of the major protein degradation systems is the UPS. There are several constitutions of the proteasome, but the 20S and 26S are most prominent. Ubiquitinated proteins and insoluble aggregates are pulled through the proteasomal ring-like structure, to be broken down by proteolysis. Not surprisingly, proteasomal dysfunction is associated with ageing and leads to the accumulation of aggregates [5–7,9,10], catalyzing a chain reaction in which aggregates block the proteasome which causes further dysfunction [37].
A third mechanism by which cells can clear and recycle cellular components is by autophagy. Many types of protein aggregates are cleared by autophagy, including τ, Aβ, α-synuclein, huntingtin, SOD1 and p16INK4A [38–43], with autophagy defects leading to neurodegenerative disease [44,45].
Defects in and decreased activity of any of these proteostasis surveillance systems are associated with ageing and proteinopathies, underpinning their importance in maintaining proteostasis.
Redox signalling
Increased markers for ROS-induced damage and mitochondrial ROS generation have long been associated with age and neurodegenerative disorders, which has often been regarded as evidence for a causal link between oxidative damage and ageing [46–54]. This is in contrast with the unexpected observation that increased ROS levels may extend lifespan in yeast and Caenorhabditis elegans [55,56], while increased ROS does not accelerate ageing in mice [57]. The key to resolve this apparent paradox probably lies in the notion that ROS in the form of H2O2 has become widely recognized as the second messenger in so-called redox signalling, which has been shown to be involved in a plethora of cellular responses [46]. Central to redox signalling is the reversible oxidation/reduction of the nucleophilic thiol side chain of specific cysteine residues. Oxidation of cysteines in proteins commonly causes structural changes and functional interactions through disulfide bond formation, such as (hetero)dimerization, oligomerization, and even aggregation, and thereby provides an important molecular switch for protein activity or function. For a comprehensive review see [58].
Compared with superoxide anions and hydroxyl radicals, the other main cellular ROS, H2O2, has a relatively low reactivity and allows for specific rather than random oxidation of dedicated cysteines. Redox signalling starts with the production of H2O2: either directly (i.e. by DUOX enzymes or ERO1-dependent protein folding in the ER) or after dismutation of superoxide produced by the leakage of electrons from complex I and III of the electron transport chain during mitochondrial respiration, or by NADPH-dependent oxidases (NOXs). For a comprehensive review on subcellular sources of ROS see [59].
Superoxide and H2O2 are efficiently scavenged by antioxidant systems and the balance between ROS production and antioxidant capacity determines the redox state of cellular compartments [60,61]. When the amount of ROS surpasses the levels required for signalling, non-specific cellular damage (oxidative stress) can occur due to their high reactivity with biomolecules. Furthermore, H2O2 may be converted by more reactive species in the presence of transition metal ions, thereby changing its role from messenger to damaging agent. In line with this, it has been suggested that oxidative damage exists merely as a side product of cellular signalling, and that ROS are in fact part of the stress response to for instance proteotoxic stress [2,62]. A good distinction between ROS as a signalling molecule versus ROS as oxidative stress has been proposed by Sies et al. [59]. Throughout this review, we will try to use the term ‘ROS’ in case the exact species is unclear or in the case of random damage rather than signalling, whereas redox signalling and reversible cysteine oxidation are considered regulated processes downstream of H2O2.
Other amino acid side chains besides cysteine and methionine are also subject to oxidation. For example, Tyr and Trp phenoxyl radicals, carbonylated Lys/His/Cys, SNO-/SSG-modifications as well as lipid peroxidation and its aldehyde byproduct 4-hydroxynonenal (HNE) are all ROS-induced and have been associated with proteinopathies [63,64]. Collectively, the dual roles of H2O2/ROS in signalling and damage make it difficult to understand whether the increase in ROS in ageing represents a causal link with oxidative damage, redox signalling or both. In this review, we will focus mostly on the effects of reversible cysteine oxidation-dependent redox signalling but will also include examples of how random oxidative damage affects protein aggregation.
Redox signalling and protein aggregation
Protein aggregation can be affected or directly regulated by redox signalling or the cellular redox state. In general, cysteine oxidation results in structural changes, for instance through disulfide formation, that affect protein function [58]. These structural changes can also provide a molecular switch to partially unfold and subsequently aggregate. Interestingly, many proteins are predicted to contain conditionally disordered regions that could be redox sensitive and thereby facilitate the transition from disorder-to-order or order-to-disorder dependent on oxidation or reduction[65]. Similar to IDPs, this transition is driven by the multiplicity of possible interactions. Conversely, it is also known that misfolded proteins are more sensitive to oxidation, which has been suggested to tag proteins for proteolysis [66].
Functional redox-dependent aggregation
As mentioned earlier, several amyloid fibrils have functional roles in humans. Amyloid fibril formation can also be reversible [67,68] and reversible oxidation of cysteines can provide the molecular switch that regulates fibrillization. For example, the tumour suppressor p16INK4A is readily oxidized on its only cysteine in an oxidizing environment, causing disulfide-linked homodimerization and subsequent rapid but reversible β-amyloid fibrillization. This redox-dependent change from a soluble monomeric protein into insoluble but reversible β-amyloid fibrils leads to the inactivation of the protein, allowing reactivation of CDK4/6 proteins otherwise inactivated by high p16INK4A expression [69]. These observations fit with the notion that redox signalling can regulate S-phase entry and cellular proliferation [70,71].
Another illustration of redox-dependent functional protein aggregation is provided by tryptophan hydroxylase 2 (TPH2), the rate-limiting enzyme in serotonin neurotransmitter production. TPH2 aggregates upon oxidation of any out of 13 cysteines and subsequent intra- and intermolecular disulfide bond formation, thereby reversibly inhibiting protein activity. TPH2 catalytic activity correlates directly with the number of cysteines that are oxidized [72,73]. Although the direct purpose for redox regulation of serotonin biosynthesis is unknown, it provides a link between neurological function and redox signalling, a concept that is widely accepted in the molecular regulation of circadian rhythm [74].
In a similar way, redox status is linked to intracellular calcium concentrations through oxidation-dependent protein aggregation of visinin-like protein-1 (VSNL1), a neuronal calcium sensor important that can activate guanylyl cyclase (GC). Under low calcium concentrations, GC produces the second messenger cGMP that can activate calcium channels. When bound to calcium, structural rearrangements in VSNL1 make the C-terminal C187 available for reversible oxidation and subsequent homodimerization and aggregation. These disulfide cross-linked aggregates are reversible upon treatment with reducing agents. As a result of aggregation, functional levels of VSNL1 are decreased, possibly providing a second mechanism to keep GC inactive besides calcium levels. Furthermore, VSNL1 aggregates are found in amyotrophic lateral sclerosis (ALS)-associated deposits, linking them to neuronal impairment [75–79].
Conversely, there are also examples where cysteine oxidation prevents functional aggregation rather than trigger it. For example, yeast ataxin2 spontaneously forms liquid-like droplets that can convert into β-amyloid fibrils. Oxidation of ataxin1 regulates this process by melting the droplets, a process that can be reversed by methionine sulfoxide reductases [80]. Phase-separated ataxin2 is an inhibitor of TORC1 during respiratory growth, thereby stimulating autophagy. Reactivation of TORC1 under oxidizing conditions though regulation of ataxin2, in combination with nutrient starvation thereby allows the coupling of mitochondrial function to TORC1-mediated metabolism [81].
This combination of findings suggests a more general mechanism, where reversible redox-regulated protein aggregation directly dictates protein activity. It also has significant implications for our understanding of aggregated proteins, which are not solely a waste product of misfolded proteins but rather a temporary conformation linked to protein activity.
Cysteine oxidation-driven protein aggregation in disease
Cysteine oxidation is also involved in toxic protein aggregation. This is illustrated by the amorphous aggregation of γ-crystallins, known to cause cataract. Reduced antioxidant capacity induces the formation of an intramolecular disulfide bond between C32 and C41, which is both necessary and sufficient to induce irreversible aggregation through the destabilization of the N-terminus [82,83]. This is especially interesting in an age-related context since the reducing capacity of the eye diminishes with age [84].
Another detrimental type of aggregation is caused by mutations resulting in an uneven number of cysteines in the transmembrane receptor NOTCH3. NOTCH3 contains a highly conserved even number of cysteines forming disulfide bridges that maintain structural integrity. Any unpaired cysteine stimulates multimerization and aggregation through the loss of a structural disulfides and exposure of an oxidation-prone cysteine that can form non-native disulfide [85,86]. Aggregated NOTCH3 accumulates in the vascular wall, leading to the rare systemic vasculopathology CASADIL.
Oxidation can also occur on other amino acids like methionine which can drastically alter protein structure. In this way, oxidation of a surface-exposed vital residue results in misfolding and aggregation. Several proteins follow this order of events. Examples include: GAPDH [87], γ-synuclein [88], interferon β1a [89], human growth hormone [90], κ-casein [91], FasL [92], transthyretin [93], apolipoprotein AI [94], AMPK [95], PrP [96] and huntingtin [97].
Interestingly, oxidation also triggers the aggregation of direct redox modulators. Cu,Zn-superoxide dismutase (SOD1) is an enzyme important for the dismutation of superoxide to H2O2 and O2, and its misfolding and fibrillization plays a crucial role in the aetiology of the familial form of ALS. The enzymatic product of SOD1, H2O2, can directly oxidize the surface-exposed C111 in SOD1 which triggers its amyloid fibrillization. In a different manner, higher concentrations of H2O2 overoxidize C111 to SO2/3H causing amorphous aggregation [98–102]. Additionally, mutations causing conformational changes in SOD1 expose its four cysteines, making the structural disulfide bond between C57 and C146 accessible to reduction by the TRX and GSH-GRX systems [101]. Subsequent oxidation leads to the formation of non-native disulfides that cause the formation of insoluble SOD1 multimers and aggregates. Contributing to the cytotoxicity of SOD1 aggregates, SOD1 oxidation has been shown to co-occur with cytoplasmic mislocalization and fibrillization of TAR-DNA-binding protein TDP-43, thereby inducing apoptosis [102].
Besides facilitating aggregation, cysteine oxidation can also prevent it. This is evident for human amylin (hIAPP), which forms a disulfide bridge between C2 and C7 upon oxidation. Oxidized hIAPP stabilizes an α-helical structure at the N-terminus, protecting the peptide from amyloid formation and safeguarding its activity in insulin and glucagon secretion as well as reducing food intake and gastric emptying [103,104]. A similar mechanism has been published for β-microglobulin and endostatin, for which two disulfide bonds guard its native folded conformation [105–107].
Furthermore, oxidation does not only trigger aggregation. Post-aggregation oxidation of proteins seems to be widespread, changing the structural conformation of established aggregates of proteins including huntingtin and β-microglobulin [106,107]. Besides providing an explanation for the abundance of oxidative modifications found in aggregate deposits, this finding suggests that insoluble aggregates are not inert protein disposals but can still alter their structure and interactions due to redox-dependent post-aggregation modifications. This view is supported by the concept that distinct structures of aggregated proteins also cause distinct phenotypes [108,109]. Targeting post-misfolding oxidation might also offer therapeutic opportunities. For example, thiol-reactive compounds can force refolding and reactivation of mutant p53 tumour suppressor (be it direct or indirect) [110,111], a concept that could be meaningful in proteinopathies.
The previously described cases of protein aggregation and dysfunction induced by cysteine oxidation seem in line with a causal role for ROS in proteinopathies. However, it has been suggested that there is an inverse correlation between the size and toxicity of aggregates, meaning that an increase in size from toxic oligomeric intermediates to insoluble aggregates means a decrease in detrimental effects (Figure 2). In that line of reasoning, stimulating the formation of insoluble aggregates might be a cellular means to confine toxic oligomers. In accordance with this, several types of smaller soluble oligomeric aggregates (including Aβ, α-synuclein and huntingtin) actually impair the 20S/26S proteasome by stabilizing its closed conformation [37]. This could mean that the formation of the more toxic soluble oligomers from natively folded proteins triggers a unidirectional switch, forming insoluble aggregates due to the inability of the proteasome to break down misfolded proteins. Redox signalling-dependent protein oxidation could, therefore, be a facilitator of aggregation and actually partake in the proteotoxic stress response. In support of this, it was shown that promoting the formation of large insoluble aggregates is protective against amyloid-induced ROS production [112].
Figure 2. The proteostasis network.

Cells employ several mechanisms to maintain proteins integrity and minimize non-native or harmful protein conformations. Mechanisms are in place for proteome quality control. Redox signalling participates in proteostasis by modulating the folding, misfolding, (dis)aggregation and the extent of toxicity of protein aggregates. References to examples of the various steps are indicated in numbers. Note that in some cases it is not clear from the literature what exact step in aggregation is affected, or whether multiple steps are affected, and in this case, the reference is denoted at the first transition from native to partially unfolded.
In summary, the cellular redox environment and protein aggregation show a strong association. With examples of protein oxidation both inducing and preventing aggregation (summarized in Table 1), having functional as wells as pathological consequences and occurring both before and after aggregation, there seems no unifying role for protein oxidation in protein aggregation. However, many proteins do seem to follow the same sequence of events upon oxidation, which includes a partial unfolding and subsequent aggregation step.
Table 1. Summary of redox-regulated protein aggregates.
| Protein | Abbreviation | Normal function | Involved residue (Cys) | Modification | Consequence(s) | Type of aggregation | Related pathologies | Reference |
|---|---|---|---|---|---|---|---|---|
| AMP-activated protein kinase | AMPK-alpha | Energy metabolism | C130 and C174 | Disulfide | Inactivation | Soluble aggregates | Cardiopathologies, energy starvation | [95] |
| Apolipoprotein A-I | APOA1 | Cholesterol transport | Methionines | Methionine oxidation | Partial unfolding, fibrillization and inactivation | Amyloid | APOA1 amyloidoses and atherosclerosis | [94] |
| APP/Amyloid-β | Aβ | Unknown | M35 | Methionine oxidation | Required for pro-oxidative activity | Amyloid | Alzheimer's | [119] |
| Ataxin-2 | ATXN2 | TORC1 inhibition | Methionines | Methionine oxidation | Oxidation reverses aggregation, functional regulation of activity | LLPS, amyloid | Spinocerebellar atrophy, ALS | [80] |
| Cellular prion protein | PrPC and PrPSC | Synaptic function | C179 and C214 | Disulfide exchange | Reduction of PrPC induces aggregation of PrPSC polymer | Amyloid | Transmissible spongiform encephalopathies | [96] |
| Cyclin-dependent kinase inhibitor 2A | p16INK4A | Cell cycle regulation, senescence | C72 | Homodimerisation | Inactivation | Amyloid | Cancer | [69] |
| Endostatin | COL18A1 | Angiogenesis inhibition | C33, C135, C165 and C173 | Disulfide | Prevents aggregation | Amyloid | Alzheimer's | [105] |
| Fas ligand | FasL | Apoptosis, inflammation | Methionines | Methionine oxidation | Multimerization and aggregation enhanced biological activity | Unknown | Acute lung injury | [92] |
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | Glycolysis | M46 | Methionine oxidation, disulfides | Local conformational change promotes disulfide cross-linking and aggregation | Amyloid | Alzheimer's, motor neuron disease | [87] |
| Growth hormone (recombinant) | hGH | (Therapeutic protein production) | M14 and M125 | Methionine oxidation | Lower stability | Unknown | GH deficiency | [90] |
| human Islet Amyloid Polypeptide | hIAPP | Insulin/glucagon secretion, gastric emptying | C2 and C7 | Disulfide | Prevents aggregation | Amyloid | Type 2 diabetes | [103] |
| Huntingtin | HTT | Unknown | M8 | Post-aggregation methionine oxidation | Controls interaction between aggregates | Amyloid | Huntington disease | [106] |
| C115 and C119 | Disulfide mediated oligomerization | Oxidation-dependent soluble toxic oligomers, slower clearance | Soluble oligomeric aggregates | [97] | ||||
| Interferon-β1a (recombinant) | IFNβ1a | (Therapeutic protein production) | Many residues (M, H, F, W, Y) | Cross-linking | Lower stability | Unknown | [89] | |
| Mitochondrial GrpE protein homologue | MGE1 | Proteostasis, HSP70 cochaperone | M155 | methionine oxidation | Inactive HSP70 but targeting it to unfolded proteins | Amyloid | Myopathies | [141] |
| Sequestosome-1 | SQSTM1/p62 | Autophagy | C105 and C113 | Disulfide mediated oligomerization | More autophagy, cell survival | Insoluble aggregates, LLPS | ALS | [170] |
| Superoxide dismutase 1 | SOD1 | Dismutation of superoxide | C111 | Disulfide-linked dimerization | Oligomerization and subsequent fibril formation | Oligomeric, amyloid and amorphous | ALS | [102] |
| Transthyretin | TTR | Thyroid hormone binding | C10, M1 and M13 | Cysteine sulfonic acid, methionine sulfoxide | Tetramer dissociation and aggregation | Amyloid | Senile systemic amyloidosis | [93] |
| Tryptophan hydroxylase 2 | TPH2 | Serotonin biosynthesis | Any out of 13 cysteines | Disulfide, cross-linking | Misfolding, intra- and intermolecular disulfide bond formation, protein inactivation | Unknown, disulfide cross-linked oligomers | Parkinson's | [72] |
| Vinisin-like protein 1 | VSNL1 | Calcium sensing | C187 | Disulfide-linked homodimerization | Reduced levels of functional protein | Amyloid, disulfide cross-linked oligomers | ALS, AD | [75] |
| y-synuclein | SNCG | Neurofilament network integrity | M38 and Y39 | Oxidation-dependent oligomerization | Aggregation and seeding for α-synuclein aggregation | Amyloid | Parkinson's | [88] |
| β2-microglobulin | β2M | MHCI light chain | C25 and C80 | Disulfide reduction, disulfide exchange, post-aggregation oxidation | Aggregation, post-aggregation stabilization | Amyloid | Hemodialysis-related amyloidosis | [107] |
| γ-crystallins | CRYG | Lens transparency | C32 and C41 | Intramolecular disulfide | Destabilizes its N-terminal domain, stabilizes an intermediate which is prone to aggregation | Amorphous | Cataract | [82] |
| κ-casein | CSN3 | Milk protein | M95 and M106 | Methionine oxidation | Increased aggregation, increased toxicity | Amyloid | Corpora amylacea | [91] |
Effects of aggregation on the cellular redox state
Protein aggregation can also modulate the cellular redox state, and, as mentioned, this could either be part of the proteotoxic stress response, cellular dysfunction as a result of the accumulation of protein aggregates, or both. For example, based on thiol-disulfide redox couples and redox sensors it has been determined that under basal conditions mitochondria, the cytosol and nucleus are in a relatively reduced state as compared with the ER. This reverses radically when protein aggregation induces proteasomal dysfunction, resulting in a more oxidizing cytosol and reducing ER [60,113]. S-nitrosylation could be a possible contributor to this change due to the overactivation of the N-methyl-d-aspartate receptor (NMDAR, an inducer of neuronal nitric oxide synthase (nNOS)) or Aβ-dependent iNOS activation in neurons and glial cells of AD patients [114–116]. A shift to a more oxidizing cellular redox state upon proteotoxic stress is further supported by many reversible cysteine modifications that change in models for proteinopathies like AD [117,118].
A more direct explanation for the redox changes that occur upon aggregation can be found in the direct redox-dependent aggregation of redox modulators. As discussed above, SOD1 aggregation is directly triggered by oxidation of C111, thereby inactivating its function. This leads to the accumulation of superoxide at the expense of H2O2, where the former is more associated with random damage and the latter with redox signalling.
Another effect of aggregation on the cellular redox state is exemplified by the transmembrane Aβ protein precursor (AβPP), which is the precursor for the archetypical amyloidogenic Aβ peptide and the main component of amyloid plaques in brains of AD patients. Interestingly, monomeric Aβ is suggested to have an antioxidant activity by hydrophilic chelation of transition metals, thereby preventing lipoprotein oxidation [119–122]. However, this chelation of metals also promotes the aggregation of Aβ, and redox-active metal ions like Cu(II) and Fe(III) can catalyze the production of ROS when bound to aggregated Aβ. Aβ binding results in the reduction of the metal's oxidation state, which then converts O2 into H2O2, superoxide and hydroxyl radicals via Fenton chemistry [123–126]. Hence, the binding of Aβ to metals changes its properties from an antioxidant to a pro-oxidant [120]. ROS-induced o,o′-dityrosine covalent cross-linking then catalyzes further aggregation of Aβ [127]. Interestingly, a similar interplay between metals and oligomers has been reported for α-synuclein [128], pointing potentially at a more general mechanism in which misfolded oligomeric proteins in conjunction with metal ions induce the production of ROS. Metal concentrations, aggregation as well as ROS production all increase with age and even without knowing the exact cause, the consequence is faster neurodegeneration [129].
Furthermore, many aggregates can directly cause mitochondrial dysfunction, resulting in metabolic stress, enhanced ROS production and eventually cell death. For example, amyloid oligomers are well-known for their permeabilization of membranes, which is considered a main toxicity event. A rapid influx of intracellular Ca2+, as well as the direct permeabilization of the mitochondrial membrane, causes an increase in mitochondrial ROS production [130–133].
When unfolded proteins accumulate in the ER, a condition called ER stress triggers the unfolded protein response (UPR) in an attempt to restore homeostasis. Protein aggregates like oligomeric Aβ have been shown to trigger ER stress and the UPR [134,135]. Prolonged ER stress is known to evoke intracellular ROS production at the ER through several mechanisms. These include overactivation of Ero1 oxidoreductases through a futile cycle of forming and repairing mismatched disulfides, thereby producing H2O2 and oxidizing GSH, respectively [132,136,137]. In addition, ER stress causes superoxide production though activation of NOXs and the release of Ca2+ which increases electron leakage from mitochondria (for a review see [138]). Hence, since ROS are both a trigger and a consequence of ER stress, this further aggravates the imbalance accompanying ER stress [139].
Redox regulation of aggregate clearance
To cope with the challenges caused by protein aggregation, cells are equipped with several mechanisms aimed at the clearance of misfolded proteins and aggregates, which themselves are also shown to be redox-regulated, adding another level of the complex interdependency of proteostasis and ROS.
Besides their role in folding of newly synthesized proteins, molecular chaperones are also part of this cellular disaggregation machinery. There are many types of chaperones, but a general initial step seems to be the initial coverage of aggregates with HSP70 [140]. An example of the redox regulation of HSPs is through the reversible oxidation of MGE1, a mitochondrial nucleotide exchange factor of HSP70, on M155. The consequential structural change from an active homodimer to monomer leads to MGE1 amyloid formation, which prevents the activation of HSP70. Interestingly, oxidized MGE1 is suggested to increases the binding affinity of the inactive HSP70 for unfolded substrates. As HSP70 is essential in protein folding and proteostasis, MGE1 might act as an initial sensor of protein aggregation, priming the chaperone system for resolution of protein aggregates [141,142].
Peroxiredoxins are more unconventional chaperones. With their abundance and exceptional reactivity to H2O2, peroxiredoxins are important H2O2 scavengers. Their highly conserved active site consists of two catalytic cysteine residues. Besides their antioxidant activity, peroxiredoxins are known for their ability to oxidize protein thiols by a redox relay as well as for their chaperone activity [143]. Assembled as a high molecular mass complex, peroxiredoxins can form ring-like chaperone structures with holdase activity that bind and prevent aggregation of unfolded proteins [144,145]. Among others, hyperoxidation of the peroxiredoxin catalytic cysteine particularly stimulates an oligomeric chaperone structure, whereas glutathionylation inhibits it [146–149]. Thereby peroxiredoxins can sense high and low ROS levels and switch their function accordingly from antioxidant and redox signalling mediator to chaperone.
Redox control of the UPS is complex, with studies claiming both inhibitory and activating effects. In general, the 20S, but not 26S proteasome is thought to specifically degrade oxidized proteins [150–152]. Elevated ROS and mitochondrial dysfunction shift the proteasome population from 26S to 20S, thereby adapting to the proteolytic need for clearance of oxidized substrates [153]. In support of this, proteasomal degradation has been shown to be stimulated >10-fold upon exposure to H2O2 or superoxide, while simultaneously abolishing 26S-mediated degradation [154–156]. The proteasome is also subject to direct redox modifications. For example, the 20S proteasome can be glutathionylated, resulting in its opening and activation [156,157]. Thus, under oxidizing conditions, the 20S proteasome is stimulated to clear oxidized substrates, but this shift away from 26S permits the accumulation of otherwise misfolded proteins. This, however, has been debated by the notion that a high NAD+/NADH ratio, correlating with an oxidizing cellular state, opens and activates the 26S proteasome [158,159]. Also, the lipid peroxidation byproduct HNE was found to inhibit proteasomal activity for the breakdown of oxidized proteins [160,161]. In summary, oxidizing conditions result in a shift to the 20S proteasome, accompanied by a possible decrease in 26S activity. This might favour the accumulation of non-oxidized misfolded proteins. Moreover, the interaction between the cellular redox state and the proteasome is bidirectional. The blockage of the proteasome, for instance, leads to an increase in ROS, which in turn might cause a vicious cycle of protein oxidation and aggregation [162,163].
It has been suggested that both aggregation and cellular redox state are coupled through their regulation of autophagy [164]. Autophagy is thought to be activated in more oxidizing conditions [165–168], although this might not always be straightforward as glutathione reductase loss, resulting in oxidizing conditions, was recently shown to suppress autophagy and enhance aggregation [169]. The recruitment of ubiquitinylated substrates to autophagosomes is mediated by receptors like SQSTM1/p62. Interestingly, components of the autophagy system have also been found to aggregate in a redox-dependent manner themselves. For example, oxidation of p62 at C105/113 in the N-terminal disordered region causes oligomerization and subsequent aggregation of p62. This stimulates autophagy in response to ROS, possibly in an attempt to maintain cellular homeostasis [170]. In addition, aggregated p62 occupies the NRF2-binding site in KEAP1, allowing the stabilization of NRF2. NRF2 nuclear translocation causes the expression of antioxidant- and stress response genes, among which p62 itself [171]. Redox-dependent p62 aggregation, therefore, ensures a robust stress response involving both autophagy and antioxidant response. Accordingly, mutations perturbing the redox-sensitivity of the NRF2 pathway are linked to ALS. Of note, mutant KRAS induced lung tumours in mice have been shown to depend on the NRF2 pathway for their survival and outgrowth [172], likely because it facilitates antioxidant- and autophagy-dependent clearance of cancer-associated proteotoxic and metabolic stress.
Taken together, protein aggregation has been shown to modulate ROS levels in several ways. Some of the increases in ROS production upon the gradual accumulation of aggregates might, therefore, be an attempt to regain homeostasis through modulation of the stress response. This eventually reaches a turning point when ROS reaches toxic levels and triggers a stress response on its own [2]. The notion that oxidation by ROS can facilitate protein aggregation and that amyloids themselves can trigger ROS production could in principle also constitute a feed-forward loop in which a small change in either ROS or protein aggregation could rapidly lead to a toxic cellular catastrophe, making redox-regulated protein aggregation an irreversible process [173]. But extensive random damage induced by ROS production either from an overactive stress response or from gross cellular dysfunction resulting in for instance lipid peroxidation might also have an evolutionary benefit and serve to actively eliminate damaged cells through the induction of ferroptosis [174].
Conclusions
In this review, we have discussed the reciprocal regulation of redox signalling and protein aggregation. In short, oxidation of specific residues causes conformational changes and causes partial unfolding of a protein. This exposes residues that can participate in non-native interactions. Further structural rearrangements drive the oligomerization and subsequent formation of insoluble aggregates. We can distinguish two types of cysteine residues here: structural residues that form disulfide bonds for correct protein folding, and cysteines that can be reversibly oxidized which serve as important signalling switches. When reversible disulfide bond formation changes the protein structure such that it directly corresponds to protein function, it can be both a structural and a regulatory residue. Although the underlying processes seem somewhat similar, the functional consequences of redox-dependent protein aggregation are not. Besides simply resulting in toxic protein aggregates, oxidative aggregation can result in reversible (in)activation of a protein which allows regulation. This can alter protein function and may serve as a protective or pro-survival mechanism. Although little is known about functional aggregation, it is thought that most aggregates are actually reversible, benefiting the regulatory possibilities. This is also reflected in the suggestion that insoluble aggregates are not inert protein disposals but can still alter its structure and morphology according to redox-dependent post-aggregation modifications and that distinct structures of aggregated proteins also cause distinct phenotypes.
On the other hand, there is the hypothesis that ROS-induced aggregation might be a cellular strategy to clear the more toxic soluble aggregates. Many of the examples we discussed in this review support this; they are part of a positive feedback system in which high ROS levels promote aggregation and the aggregates themselves promote ROS production. In this way, redox signalling creates a bistable switch between functional proteins and insoluble aggregates without the accumulation of toxic intermediates. Therefore, modulating ROS levels to promote rather than inhibit aggregation could be a more sensible therapeutic approach.
Almost all the processes involved in protein aggregation are redox regulated (Figure 3). However, it is hard to determine whether altered redox signalling is a cause or consequence of protein aggregation. Often times it seems like it is both: altered redox signalling favours aggregation, but the mutual amplification of the systems also provides a feed-forward loop, consequently altering the cellular redox state. One clue might lie in the hypothesis stating that H2O2 is produced as a signalling molecule in response to damage, such as an accumulation of aggregates. A gradual increase in aggregation is accompanied by an increased H2O2 production, which intensifies over time. Eventually, the H2O2 might reach toxic levels, which itself may lead to damage. Understanding whether H2O2 is produced as a stress response in order to regain homeostasis might change our view of proteinopathies.
Figure 3. Redox regulation of proteostasis.
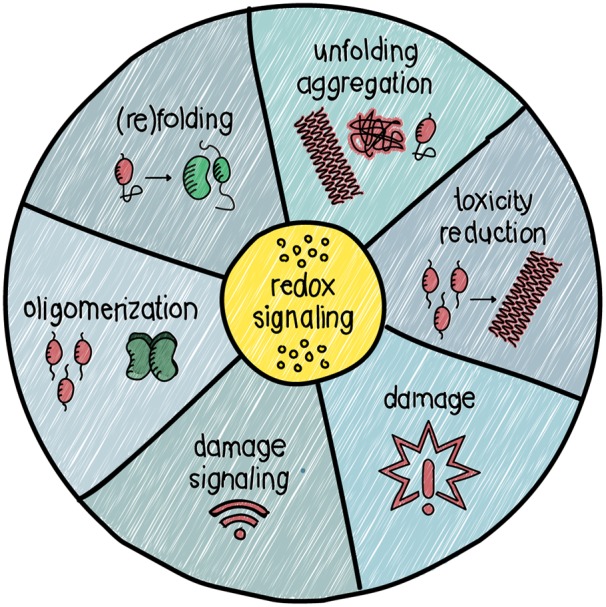
Redox signalling modulates proteostasis in many ways. Among its roles are the regulation of protein folding, unfolding/aggregation, toxicity reduction and damage response.
Why aggregation is so much more prominent in aged individuals is not entirely clear. The age-dependent increase of aggregation, oxidation and mitochondrial dysfunction, together with a decline in multiple aggregation clearance systems could cumulatively cause a destabilizing environment from which protein aggregates can no longer recover [175,176]. The diverse effects of oxidative aggregation on proteins suggest that in order to regulate proteins by oxidative aggregation, the reversibility of the process is essential in some cases. Little is known about the reversibility of oxidized aggregates. For example, when the reducing capacity of a cell is restored, can the aggregates fall apart into functional monomers again by reduction of the oxidative modifications? It is also not clear if and how cellular systems to clear protein aggregates such as the proteasome and autophagy can distinguish between ‘functional’ aggregates that are a temporary, regulated protein state, and toxic protein aggregates that need clearance.
Perspectives
Importance of the field: Both redox signalling and correct protein folding are essential for maintaining healthy cellular homeostasis. Accordingly, loss of proteostasis and ROS production as a consequence of mitochondrial dysfunction have been described as hallmarks of ageing. This becomes especially clear in proteinopathies, where both aberrant redox signalling and protein aggregation are associated with severe neurodegenerative problems.
Summary of the current thinking: Collectively, these studies outline the complex relationship between ROS, redox signalling and proteostasis, where cause and consequence are often hard to differentiate. Whereas protein oxidation can trigger aggregation, increases in H2O2 production upon the gradual accumulation of aggregates might be a stress response by itself. A small change in redox state or aggregation can in this way rapidly lead to a feed-forward loop, making redox-regulated protein aggregation an irreversible process.
Future directions: Going forward, it is important to better understand whether or not H2O2 is produced as a signal in response to proteotoxic stress in order to restore homeostasis, and whether increased H2O2 levels promote aggregation as a cellular strategy to clear the more toxic oligomeric aggregates. Understanding the multifaceted mechanisms regulating protein aggregation will pave the way for novel therapeutic windows to combat proteinopathies.
Acknowledgements
T.B.D. is supported by grants (UU2014-6902 and Alpe/Unique High Risk #11077) from the Dutch Cancer Society (KWF Kankerbestrijding).
Abbreviations
- ALS
amyotrophic lateral sclerosis
- APRs
aggregation prone regions
- AβPP
amyloid-β protein precursor
- C
cysteine
- DUOX
dual oxidase
- H2O2
hydrogen peroxide
- IDRs
intrinsically disordered regions
- iNOS
inducible nitric oxide synthase
- LLPS
liquid–liquid phase separation
- M
methionine
- NMDAR
N-methyl-d-aspartate receptor
- nNOS
neuronal nitric oxide synthase
- NOX
NADPH oxidase
- O2•−
superoxide anion
- OH•
hydroxyl radical
- ROS
reactive oxygen species
- SOD1
Cu,Zn-superoxide dismutase
- TPH2
tryptophan hydroxylase 2
- UPR
unfolded protein response
- UPS
ubiquitin-proteasome system
- VSNL1
visinin-like protein-1
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Author Contributions
All authors contributed to, read and approved the final version of the manuscript.
References
- 1.López-Otín C., Blasco M.A., Partridge L., Serrano M. and Kroemer G. (2013) The hallmarks of aging. Cell 153, 1194–1217 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hekimi S., Lapointe J. and Wen Y. (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576 10.1016/j.tcb.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hipp M.S., Kasturi P. and Hartl F.U. (2019) The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 10.1038/s41580-019-0101-y [DOI] [PubMed] [Google Scholar]
- 4.Walther D.M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P. et al. (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932 10.1016/j.cell.2015.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsui H., Ito H., Taniguchi Y., Inoue H., Takeda S. and Takahashi R. (2010) Proteasome inhibition in medaka brain induces the features of Parkinson's disease. J. Neurochem. 115, 178–187 10.1111/j.1471-4159.2010.06918.x [DOI] [PubMed] [Google Scholar]
- 6.Kitajima Y., Tashiro Y., Suzuki N., Warita H., Kato M., Tateyama M. et al. (2014) Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 127, 5204–5217 10.1242/jcs.150961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R., Zhao J., Zhang J., Liu W., Zhao M., Li J. et al. (2015) Effect of lysosomal and ubiquitin-proteasome system dysfunction on the abnormal aggregation of α-synuclein in PC12 cells. Exp. Ther. Med. 9, 2088–2094 10.3892/etm.2015.2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koga H., Kaushik S. and Cuervo A.M. (2011) Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215 10.1016/j.arr.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visscher M., Henau S.D., Wildschut M.H.E., Es van R.M., Dhondt I., Michels H. et al. (2016) Proteome-wide changes in protein turnover rates in C. elegans models of longevity and age-related disease. Cell Rep. 16, 3051 10.1016/j.celrep.2016.08.025 [DOI] [PubMed] [Google Scholar]
- 10.Kumar V., Singh D., Singh B.K., Singh S., Mittra N., Jha R.R. et al. (2018) Alpha-synuclein aggregation, ubiquitin proteasome system impairment, and l-Dopa response in zinc-induced Parkinsonism: resemblance to sporadic Parkinson's disease. Mol. Cell. Biochem. 444, 149–160 10.1007/s11010-017-3239-y [DOI] [PubMed] [Google Scholar]
- 11.Wilson D.N. and Beckmann R. (2011) The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr. Opin. Struct. Biol. 21, 274–282 10.1016/j.sbi.2011.01.007 [DOI] [PubMed] [Google Scholar]
- 12.Balchin D., Hayer-Hartl M. and Hartl F.U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 13.Brockwell D.J. and Radford S.E. (2007) Intermediates: ubiquitous species on folding energy landscapes? Curr. Opin. Struct. Biol. 17, 30–37 10.1016/j.sbi.2007.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braakman I. and Hebert D.N. (2013) Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5, a013201, 10.1101/cshperspect.a013201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weids A.J., Ibstedt S., Tamás M.J. and Grant C.M. (2016) Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 6, srep24554 10.1038/srep24554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uversky V.N. and Fink A.L. (2004) Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta 1698, 131–153 10.1016/j.bbapap.2003.12.008 [DOI] [PubMed] [Google Scholar]
- 17.Gupta R., Kasturi P., Bracher A., Loew C., Zheng M., Villella A. et al. (2011) Firefly luciferase mutants as sensors of proteome stress. Nat. Methods 8, 879–884 10.1038/nmeth.1697 [DOI] [PubMed] [Google Scholar]
- 18.Bouziane H. and Chouarfia A. (2020) Sequence- and structure-based prediction of amyloidogenic regions in proteins. Soft Comput. 24, 3285–3308 10.1007/s00500-019-04087-z [DOI] [Google Scholar]
- 19.Chiti F., Stefani M., Taddei N., Ramponi G. and Dobson C.M. (2003) Rationalization of the effects of mutations on peptide andprotein aggregation rates. Nature 424, 805 10.1038/nature01891 [DOI] [PubMed] [Google Scholar]
- 20.Monsellier E. and Chiti F. (2007) Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 8, 737–742 10.1038/sj.embor.7401034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy E.D., De S. and Teichmann S.A. (2012) Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. Proc. Natl. Acad. Sci. U.S.A. 109, 20461–20466 10.1073/pnas.1209312109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Annamalai K., Gührs K., Koehler R., Schmidt M., Michel H., Loos C. et al. (2016) Polymorphism of amyloid fibrils in vivo. Angew. Chem. Int. Ed. 55, 4822–4825 10.1002/anie.201511524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knowles T.P., Fitzpatrick A.W., Meehan S., Mott H.R., Vendruscolo M., Dobson C.M. et al. (2007) Role of intermolecular forces in defining material properties of protein nanofibrils. Science 318, 1900–1903 10.1126/science.1150057 [DOI] [PubMed] [Google Scholar]
- 24.Sengupta U., Nilson A.N. and Kayed R. (2016) The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. Ebiomedicine 6, 42–49 10.1016/j.ebiom.2016.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M. et al. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glabe C.G. (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 10.1016/j.neurobiolaging.2005.04.017 [DOI] [PubMed] [Google Scholar]
- 27.Soeda Y., Saito M., Maeda S., Ishida K., Nakamura A., Kojima S. et al. (2019) Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer's disease. J. Alzheimer's Dis. 68, 1677–1686 10.3233/JAD-181001 [DOI] [PubMed] [Google Scholar]
- 28.Mijnders M., Kleizen B. and Braakman I. (2017) Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr. Opin. Pharmacol. 34, 83–90 10.1016/j.coph.2017.09.014 [DOI] [PubMed] [Google Scholar]
- 29.Otzen D. and Riek R. (2019) Functional amyloids. Cold Spring Harb. Perspect. Biol. 11, a033860 10.1101/cshperspect.a033860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uversky V.N., Oldfield C.J. and Dunker A.K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 10.1146/annurev.biophys.37.032807.125924 [DOI] [PubMed] [Google Scholar]
- 31.Banani S.F., Lee H.O., Hyman A.A. and Rosen M.K. (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peskett T.R., Rau F., O'Driscoll J., Patani R., Lowe A.R. and Saibil H.R. (2018) A liquid to solid phase transition underlying pathological huntingtin Exon1 aggregation. Mol. Cell 70, 588–601.e6 10.1016/j.molcel.2018.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ambadipudi S., Biernat J., Riedel D., Mandelkow E. and Zweckstetter M. (2017) Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein tau. Nat. Commun. 8, 275 10.1038/s41467-017-00480-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wentink A., Nussbaum-Krammer C. and Bukau B. (2019) Modulation of amyloid states by molecular chaperones. Cold Spring Harb. Perspect. Biol. 11, a033969 10.1101/cshperspect.a033969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X., Carroni M., Nussbaum-Krammer C., Mogk A., Nillegoda N.B., Szlachcic A. et al. (2015) Human Hsp70 disaggregase reverses Parkinson's-linked α-synuclein amyloid fibrils. Mol. Cell 59, 781–793 10.1016/j.molcel.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cereghetti G., Saad S., Dechant R. and Peter M. (2018) Reversible, functional amyloids: towards an understanding of their regulation in yeast and humans. Cell Cycle 17, 1545–1558 10.1080/15384101.2018.1480220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thibaudeau T.A., Anderson R.T. and Smith D.M. (2018) A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 9, 1097 10.1038/s41467-018-03509-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caccamo A., Ferreira E., Branca C. and Oddo S. (2016) P62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 22, 865 10.1038/mp.2016.139 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Cho M.-H., Cho K., Kang H.-J., Jeon E.-Y., Kim H.-S., Kwon H.-J. et al. (2014) Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10, 1761–1775 10.4161/auto.29647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T. and Sulzer D. (2004) Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- 41.Ravikumar B., Duden R. and Rubinsztein D.C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- 42.Yung C., Sha D., Li L. and Chin L.-S. (2016) Parkin protects against misfolded SOD1 toxicity by promoting its aggresome formation and autophagic clearance. Mol. Neurobiol. 53, 6270–6287 10.1007/s12035-015-9537-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coryell P.R., Goraya S.K., Griffin K.A., Redick M.A., Sisk S.R. and Purvis J.E. (2019) Autophagy regulates the localization and degradation of p16INK4a. Biorxiv, 521682 10.1101/521682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y., Liu H., Guan Y., Wang Q., Zhou F., Jie L. et al. (2015) The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am. J. Trans. Res. 7, 1574–1587 PMID: [PMC free article] [PubMed] [Google Scholar]
- 45.Bordi M., Berg M.J., Mohan P.S., Peterhoff C.M., Alldred M.J., Che S. et al. (2016) Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483 10.1080/15548627.2016.1239003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stadtman E.R. (1988) Protein modification in aging. J. Gerontol. 43, B112–B120 10.1093/geronj/43.5.B112 [DOI] [PubMed] [Google Scholar]
- 47.Evans A.R., Gu L., Guerrero R. and Robinson R.A.S. (2015) Global cPILOT analysis of the APP/PS-1 mouse liver proteome. Proteomics Clin. Appl. 9, 872–884 10.1002/prca.201400149 [DOI] [PubMed] [Google Scholar]
- 48.Abdul H.M., Sultana R., Clair D.K.S., Markesbery W.R. and Butterfield D.A. (2008) Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic. Biol. Med. 45, 1420–1425 10.1016/j.freeradbiomed.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinclair A.J., Bayer A.J., Johnston J., Warner C. and Maxwell S.R.J. (1998) Altered plasma antioxidant status in subjects with Alzheimer's disease and vascular dementia. Int. J. Geriatr. Psychiatry 13, 840–845 [DOI] [PubMed] [Google Scholar]
- 50.Smith M.A., Rottkamp C.A., Nunomura A., Raina A.K. and Perry G. (2000) Oxidative stress in Alzheimer's disease. Biochim. Biophys. Acta 1502, 139–144 10.1016/S0925-4439(00)00040-5 [DOI] [PubMed] [Google Scholar]
- 51.Gu L. and Robinson R.A.S. (2016) High-throughput endogenous measurement of S-nitrosylation in Alzheimer's disease using oxidized cysteine-selective cPILOT. Analyst 141, 3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson R.A.S., Cao Z. and Williams C. (2013) Oxidative stress in CD90+ T-cells of AβPP/PS-1 transgenic mice. J. Alzheimer's Dis. 37, 661–666 10.3233/JAD-130665 [DOI] [PubMed] [Google Scholar]
- 53.Sohal R.S. and Sohal B.H. (1991) Hydrogen peroxide release by mitochondria increases during aging. Mech. Ageing Dev. 57, 187–202 10.1016/0047-6374(91)90034-W [DOI] [PubMed] [Google Scholar]
- 54.Asensi M., Sastre J., Pallardo F.V., Lloret A., Lehner M., Asuncion J.G. et al. (1999) [23] Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 299, 267–276 10.1016/S0076-6879(99)99026-2 [DOI] [PubMed] [Google Scholar]
- 55.Raamsdonk J.M.V. and Hekimi S. (2009) Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361 10.1371/journal.pgen.1000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mesquita A., Weinberger M., Silva A., Sampaio-Marques B., Almeida B., Leão C. et al. (2010) Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. U.S.A. 107, 15123–15128 10.1073/pnas.1004432107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pérez V.I., Remmen H.V., Bokov A., Epstein C.J., Vijg J. and Richardson A. (2009) The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75 10.1111/j.1474-9726.2008.00449.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holmström K.M. and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- 59.Sies H., Berndt C. and Jones D.P. (2017) Oxidative stress. Annu. Rev. Biochem. 86, 715–748 10.1146/annurev-biochem-061516-045037 [DOI] [PubMed] [Google Scholar]
- 60.Go Y.-M. and Jones D.P. (2008) Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 1780, 1273–1290 10.1016/j.bbagen.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaludercic N., Deshwal S. and Lisa F.D. (2014) Reactive oxygen species and redox compartmentalization. Front. Physiol. 5, 285 10.3389/fphys.2014.00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y. and Hekimi S. (2015) Mitochondrial dysfunction and longevity in animals: untangling the knot. Science 350, 1204–1207 10.1126/science.aac4357 [DOI] [PubMed] [Google Scholar]
- 63.Cheignon C., Tomas M., Bonnefont-Rousselot D., Faller P., Hureau C. and Collin F. (2018) Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 14, 450–464 10.1016/j.redox.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sultana R., Perluigi M. and Butterfield D.A. (2013) Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169 10.1016/j.freeradbiomed.2012.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erdős G., Mészáros B., Reichmann D. and Dosztányi Z. (2019) Large-scale analysis of redox-sensitive conditionally disordered protein regions reveals their widespread nature and key roles in high-level eukaryotic processes. Proteomics 19, 1800070 10.1002/pmic.201800070 [DOI] [PubMed] [Google Scholar]
- 66.Dukan S., Farewell A., Ballesteros M., Taddei F., Radman M. and Nyström T. (2000) Protein oxidation in response to increased transcriptional or translational errors. Proc. Natl. Acad. Sci. U.S.A. 97, 5746–5749 10.1073/pnas.100422497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Audas T.E., Audas D.E., Jacob M.D., Ho J.J.D., Khacho M., Wang M. et al. (2016) Adaptation to stressors by systemic protein amyloidogenesis. Dev. Cell 39, 155–168 10.1016/j.devcel.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saad S., Cereghetti G., Feng Y., Picotti P., Peter M. and Dechant R. (2017) Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 19, 1202–1213 10.1038/ncb3600 [DOI] [PubMed] [Google Scholar]
- 69.Göbl C., Morris V.K., van Dam L., Visscher M., Polderman P.E., Hartlmüller C. et al. (2019) Cysteine oxidation triggers amyloid fibril formation of the tumor suppressor p16INK4A. Redox Biol. 28, 101316 10.1016/j.redox.2019.101316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pervaiz S. and Clement M.-V. (2007) Superoxide anion: oncogenic reactive oxygen species? Int. J. Biochem. Cell Biol. 39, 1297–1304 10.1016/j.biocel.2007.04.007 [DOI] [PubMed] [Google Scholar]
- 71.Hoffman A., Spetner L.M. and Burke M. (2008) Ramifications of a redox switch within a normal cell: its absence in a cancer cell. Free Radic. Biol. Med. 45, 265–268 10.1016/j.freeradbiomed.2008.03.025 [DOI] [PubMed] [Google Scholar]
- 72.Kuhn D.M., Sykes C.E., Geddes T.J., Jaunarajs K.L.E. and Bishop C. (2011) Tryptophan hydroxylase 2 aggregates through disulfide cross-linking upon oxidation: possible link to serotonin deficits and non-motor symptoms in Parkinson's disease. J. Neurochem. 116, 426–437 10.1111/j.1471-4159.2010.07123.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuhn D.M. and Geddes T.J. (1999) Peroxynitrite inactivates tryptophan hydroxylase via sulfhydryl oxidation. J. Biol. Chem. 274, 29726–29732 10.1074/jbc.274.42.29726 [DOI] [PubMed] [Google Scholar]
- 74.Bothwell M.Y. and Gillette M.U. (2018) Circadian redox rhythms in the regulation of neuronal excitability. Free Radic. Biol. Med. 119, 45–55 10.1016/j.freeradbiomed.2018.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liebl M.P., Kaya A.M., Tenzer S., Mittenzwei R., Koziollek-Drechsler I., Schild H. et al. (2014) Dimerization of visinin-like protein 1 is regulated by oxidative stress and calcium and is a pathological hallmark of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 72, 54 10.1016/j.freeradbiomed.2014.04.008 [DOI] [PubMed] [Google Scholar]
- 76.Groblewska M., Muszyński P., Wojtulewska-Supron A., Kulczyńska-Przybik A. and Mroczko B. (2015) The role of visinin-like protein-1 in the pathophysiology of Alzheimer's disease. J. Alzheimer's Dis. 47, 17–32 10.3233/JAD-150060 [DOI] [PubMed] [Google Scholar]
- 77.Youn H., Jeoung M.K., Koo Y., Ji H., Markesbery W.R., Ji I. et al. (2007) Kalirin is under-expressed in Alzheimer's disease hippocampus. J. Alzheimer's Dis. 11, 385–397 10.3233/JAD-2007-11314 [DOI] [PubMed] [Google Scholar]
- 78.Lederer C.W., Torrisi A., Pantelidou M., Santama N. and Cavallaro S. (2007) Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics 8, 26 10.1186/1471-2164-8-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Braunewell K., Riederer P., Spilker C., Gundelfinger E.D., Bogerts B. and Bernstein H.G. (2001) Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement Geriatr. Cogn. Disord. 12, 116 10.1159/000051244 [DOI] [PubMed] [Google Scholar]
- 80.Kato M., Yang Y.-S., Sutter B.M., Wang Y., McKnight S.L. and Tu B.P. (2019) Redox state controls phase separation of the yeast ataxin-2 protein via reversible oxidation of its methionine-rich low-complexity domain. Cell 177, 711–721.e8 10.1016/j.cell.2019.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Y.-S., Kato M., Wu X., Litsios A., Sutter B.M., Wang Y. et al. (2019) Yeast ataxin-2 forms an intracellular condensate required for the inhibition of TORC1 signaling during respiratory growth. Cell 177, 32 10.1016/j.cell.2019.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Serebryany E., Woodard J.C., Adkar B.V., Shabab M., King J.A. and Shakhnovich E.I. (2016) An internal disulfide locks a misfolded aggregation-prone intermediate in cataract-linked mutants of human γD-crystallin. J. Biol. Chem. 291, 19172–19183 10.1074/jbc.M116.735977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thorn D.C., Grosas A.B., Mabbitt P.D., Ray N.J., Jackson C.J. and Carver J.A. (2018) The structure and stability of the disulfide-linked γS-crystallin dimer provide insight into oxidation products associated with lens cataract formation. J. Mol. Biol. 431, 483–497 10.1016/j.jmb.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 84.Lou M.F. (2003) Redox regulation in the lens. Prog. Retin. Eye Res. 22, 657–682 10.1016/S1350-9462(03)00050-8 [DOI] [PubMed] [Google Scholar]
- 85.Duering M., Karpinska A., Rosner S., Hopfner F., Zechmeister M., Peters N. et al. (2011) Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum. Mol. Genet. 20, 3256–3265 10.1093/hmg/ddr237 [DOI] [PubMed] [Google Scholar]
- 86.Wollenweber F.A., Hanecker P., Bayer-Karpinska A., Malik R., Bäzner H., Moreton F. et al. (2015) Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke 46, 786–792 10.1161/STROKEAHA.114.007472 [DOI] [PubMed] [Google Scholar]
- 87.Samson A.L., Knaupp A.S., Kass I., Kleifeld O., Marijanovic E.M., Hughes V.A. et al. (2014) Oxidation of an exposed methionine instigates the aggregation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 289, 26922–26936 10.1074/jbc.M114.570275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Surgucheva I., Sharov V.S. and Surguchov A. (2012) γ-Synuclein: seeding of α-synuclein aggregation and transmission between cells. Biochemistry 51, 4743–4754 10.1021/bi300478w [DOI] [PubMed] [Google Scholar]
- 89.Torosantucci R., Sharov V.S., van Beers M., Brinks V., Schöneich C. and Jiskoot W. (2013) Identification of oxidation sites and covalent cross-links in metal catalyzed oxidized interferon beta-1a: potential implications for protein aggregation and immunogenicity. Mol. Pharm. 10, 2311–2322 10.1021/mp300665u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mulinacci F., Poirier E., Capelle M.A.H., Gurny R. and Arvinte T. (2013) Influence of methionine oxidation on the aggregation of recombinant human growth hormone. Eur. J. Pharm. Biopharm. 85, 42–52 10.1016/j.ejpb.2013.03.015 [DOI] [PubMed] [Google Scholar]
- 91.Koudelka T., Dehle F.C., Musgrave I.F., Hoffmann P. and Carver J.A. (2012) Methionine oxidation enhances κ-casein amyloid fibril formation. J. Agric. Food Chem. 60, 4144–4155 10.1021/jf205168t [DOI] [PubMed] [Google Scholar]
- 92.Herrero R., Kajikawa O., Matute-Bello G., Wang Y., Hagimoto N., Mongovin S. et al. (2011) The biological activity of FasL in human and mouse lungs is determined by the structure of its stalk region. J. Clin. Invest. 121, 1174–1190 10.1172/JCI43004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao L., Buxbaum J.N. and Reixach N. (2013) Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 52, 1913–1926 10.1021/bi301313b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong Y.Q., Binger K.J., Howlett G.J. and Griffin M.D.W. (2010) Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 107, 1977–1982 10.1073/pnas.0910136107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S. et al. (2014) A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metab. 19, 232–245 10.1016/j.cmet.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sideri T.C., Stojanovski K., Tuite M.F. and Grant C.M. (2010) Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc. Natl. Acad. Sci. U.S.A. 107, 6394–6399 10.1073/pnas.1000347107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fox J.H., Connor T., Stiles M., Kama J., Lu Z., Dorsey K. et al. (2011) Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J. Biol. Chem. 286, 18320–18330 10.1074/jbc.M110.199448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cozzolino M., Amori I., Pesaresi M.G., Ferri A., Nencini M. and Carrì M.T. (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 866–874 10.1074/jbc.M705657200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karch C.M., Prudencio M., Winkler D.D., Hart P.J. and Borchelt D.R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7779 10.1073/pnas.0902505106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen X., Shang H., Qiu X., Fujiwara N., Cui L., Li X.M. et al. (2012) Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem. Res. 37, 835–845 10.1007/s11064-011-0679-8 [DOI] [PubMed] [Google Scholar]
- 101.Álvarez-Zaldiernas C., Lu J., Zheng Y., Yang H., Blasi J., Solsona C. et al. (2016) Cellular redox systems impact the aggregation of Cu,Zn superoxide dismutase linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 291, 17197–17208 10.1074/jbc.M115.708230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu W.-C., Liang J.-Z., Li C., He Z.-X., Yuan H.-Y., Huang B.-Y. et al. (2018) Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 9, 67 10.1038/s41419-017-0106-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Camargo D.C.R., Tripsianes K., Buday K., Franko A., Göbl C., Hartlmüller C. et al. (2017) The redox environment triggers conformational changes and aggregation of hIAPP in type II diabetes. Sci. Rep. 7, 11 10.1038/s41598-017-00052-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paulsson J.F., Andersson A., Westermark P. and Westermark G.T. (2006) Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 49, 1237–1246 10.1007/s00125-006-0206-7 [DOI] [PubMed] [Google Scholar]
- 105.He Y., Zhou H., Tang H. and Luo Y. (2006) Deficiency of disulfide bonds facilitating fibrillogenesis of endostatin. J. Biol. Chem. 281, 1048–1057 10.1074/jbc.M507745200 [DOI] [PubMed] [Google Scholar]
- 106.Mitomi Y., Nomura T., Kurosawa M., Nukina N. and Furukawa Y. (2012) Post-aggregation oxidation of mutant huntingtin controls the interactions between aggregates. J. Biol. Chem. 287, 34764–34775 10.1074/jbc.M112.387035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu C., Sawaya M.R. and Eisenberg D. (2011) β2-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat. Struct. Mol. Biol. 18, 49 10.1038/nsmb.1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tanaka M., Chien P., Naber N., Cooke R. and Weissman J.S. (2004) Conformational variations in an infectious protein determine prion strain differences. Nature 428, 323 10.1038/nature02392 [DOI] [PubMed] [Google Scholar]
- 109.Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T. and Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 10.1073/pnas.0812083106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Q., Bykov V.J.N., Wiman K.G. and Zawacka-Pankau J. (2018) APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 9, 439 10.1038/s41419-018-0463-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Q., Bergman J., Wiman K.G. and Bykov V.J.N. (2018) Role of thiol reactivity for targeting mutant p53. Cell Chem. Biol. 25, 1219–1230.e3 10.1016/j.chembiol.2018.06.013 [DOI] [PubMed] [Google Scholar]
- 112.Carija A., Navarro S., de Groot N.S. and Ventura S. (2017) Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 12, 699–711 10.1016/j.redox.2017.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kirstein J., Morito D., Kakihana T., Sugihara M., Minnen A., Hipp M.S. et al. (2015) Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 34, 2334–2349 10.15252/embj.201591711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Combs C.K., Karlo J.C., Kao S.-C. and Landreth G.E. (2001) β-amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 21, 1179–1188 10.1523/JNEUROSCI.21-04-01179.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao Q.-F., Yu J.-T. and Tan L. (2015) S-nitrosylation in Alzheimer's disease. Mol. Neurobiol. 51, 268–280 10.1007/s12035-014-8672-2 [DOI] [PubMed] [Google Scholar]
- 116.Floden A.M., Li S. and Combs C.K. (2005) β-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor α and NMDA receptors. J. Neurosci. 25, 2566–2575 10.1523/JNEUROSCI.4998-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Correani V., Francesco L.D., Cera I., Mignogna G., Giorgi A., Mazzanti M. et al. (2015) Reversible redox modifications in the microglial proteome challenged by beta amyloid. Mol. Biosyst. 11, 1584–1593 10.1039/C4MB00703D [DOI] [PubMed] [Google Scholar]
- 118.Gu L. and Robinson R.A.S. (2016) A simple isotopic labeling method to study cysteine oxidation in Alzheimer's disease: oxidized cysteine-selective dimethylation (OxcysDML). Anal. Bioanal. Chem. 408, 2993–3004 10.1007/s00216-016-9307-4 [DOI] [PubMed] [Google Scholar]
- 119.Kontush A., Berndt C., Weber W., Akopyan V., Arlt S., Schippling S. et al. (2001) Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med. 30, 119–128 10.1016/S0891-5849(00)00458-5 [DOI] [PubMed] [Google Scholar]
- 120.Kontush A. (2001) Amyloid-β: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer's disease. Free Radic. Biol. Med. 31, 1120–1131 10.1016/S0891-5849(01)00688-8 [DOI] [PubMed] [Google Scholar]
- 121.Atwood C.S., Obrenovich M.E., Liu T., Chan H., Perry G., Smith M.A. et al. (2003) Amyloid-β: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-β. Brain Res. Rev. 43, 1–16 10.1016/S0165-0173(03)00174-7 [DOI] [PubMed] [Google Scholar]
- 122.Baruch-Suchodolsky R. and Fischer B. (2009) Aβ 40, either soluble or aggregated. Is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 48, 4354–4370 10.1021/bi802361k [DOI] [PubMed] [Google Scholar]
- 123.Huang X., Atwood C.S., Hartshorn M.A., Multhaup G., Goldstein L.E., Scarpa R.C. et al. (1999) The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal Ion reduction. Biochemistry 38, 7609–7616 10.1021/bi990438f [DOI] [PubMed] [Google Scholar]
- 124.Hensley K., Carney J.M., Mattson M.P., Aksenova M., Harris M., Wu J.F. et al. (1994) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 3270–3274 10.1073/pnas.91.8.3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith D.G., Cappai R. and Barnham K.J. (2007) The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim. Biophys. Acta 1768, 1976–1990 10.1016/j.bbamem.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 126.Barnham K.J., Ciccotosto G.D., Tickler A.K., Ali F.E., Smith D.G., Williamson N.A. et al. (2003) Neurotoxic, redox-competent Alzheimer's β-amyloid Is released from lipid membrane by methionine oxidation. J. Biol. Chem. 278, 42959–42965 10.1074/jbc.M305494200 [DOI] [PubMed] [Google Scholar]
- 127.Atwood C.S., Perry G., Zeng H., Kato Y., Jones W.D., Ling K.-Q. et al. (2004) Copper mediates dityrosine cross-linking of Alzheimer's amyloid-β. Biochemistry 43, 560–568 10.1021/bi0358824 [DOI] [PubMed] [Google Scholar]
- 128.Deas E., Cremades N., Angelova P.R., Ludtmann M.H.R., Yao Z., Chen S. et al. (2016) Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxid. Redox Signal. 24, 376–391 10.1089/ars.2015.6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lovell M.A., Robertson J.D., Teesdale W.J., Campbell J.L. and Markesbery W.R. (1998) Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 158, 47–52 10.1016/S0022-510X(98)00092-6 [DOI] [PubMed] [Google Scholar]
- 130.Hashimoto M., Rockenstein E., Crews L. and Masliah E. (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. Neuromol. Med. 4, 21–35 10.1385/NMM:4:1-2:21 [DOI] [PubMed] [Google Scholar]
- 131.Schubert D., Behl C., Lesley R., Brack A., Dargusch R., Sagara Y. et al. (1995) Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. U.S.A. 92, 1989–1993 10.1073/pnas.92.6.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brookes P.S., Yoon Y., Robotham J.L., Anders M.W. and Sheu S.-S. (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833 10.1152/ajpcell.00139.2004 [DOI] [PubMed] [Google Scholar]
- 133.Curtain C.C., Ali F., Volitakis I., Cherny R.A., Norton R.S., Beyreuther K. et al. (2001) Alzheimer's disease amyloid-β binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–20473 10.1074/jbc.M100175200 [DOI] [PubMed] [Google Scholar]
- 134.Casas-Tinto S., Zhang Y., Sanchez-Garcia J., Gomez-Velazquez M., Rincon-Limas D.E. and Fernandez-Funez P. (2011) The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 20, 2144–2160 10.1093/hmg/ddr100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gerakis Y. and Hetz C. (2017) Emerging roles of ER stress in the aetiology and pathogenesis of Alzheimer's disease. FEBS J. 285, 995–1011 10.1111/febs.14332 [DOI] [PubMed] [Google Scholar]
- 136.Haynes C.M., Titus E.A. and Cooper A.A. (2004) Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15, 767–776 10.1016/j.molcel.2004.08.025 [DOI] [PubMed] [Google Scholar]
- 137.Peng T. and Jou M. (2010) Oxidative stress caused by mitochondrial calcium overload. Ann. NY Acad. Sci. 1201, 183–188 10.1111/j.1749-6632.2010.05634.x [DOI] [PubMed] [Google Scholar]
- 138.Santos C.X.C., Tanaka L.Y., Wosniak J. and Laurindo F.R.M. (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 11, 2409–2427 10.1089/ars.2009.2625 [DOI] [PubMed] [Google Scholar]
- 139.Malhotra J.D. and Kaufman R.J. (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 9, 2277–2294 10.1089/ars.2007.1782 [DOI] [PubMed] [Google Scholar]
- 140.Mogk A., Bukau B. and Kampinga H.H. (2018) Cellular handling of protein aggregates by disaggregation machines. Mol. Cell 69, 214–226 10.1016/j.molcel.2018.01.004 [DOI] [PubMed] [Google Scholar]
- 141.Karri S., Singh S., Paripati A.K., Marada A., Krishnamoorthy T., Guruprasad L. et al. (2018) Adaptation of Mge1 to oxidative stress by local unfolding and altered interaction with mitochondrial Hsp70 and Mxr2. Mitochondrion 46, 140–148 10.1016/j.mito.2018.04.003 [DOI] [PubMed] [Google Scholar]
- 142.Miyata Y., Rauch J.N., Jinwal U.K., Thompson A.D., Srinivasan S., Dickey C.A. et al. (2012) Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem. Biol. 19, 1391–1399 10.1016/j.chembiol.2012.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stöcker S., Maurer M., Ruppert T. and Dick T.P. (2017) A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14, 148–155 10.1038/nchembio.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Weids A.J. and Grant C.M. (2014) The yeast peroxiredoxin Tsa1 protects against protein-aggregate-induced oxidative stress. J. Cell Sci. 127, 1327–1335 10.1242/jcs.144022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Teixeira F., Tse E., Castro H., Makepeace K.A.T., Meinen B.A., Borchers C.H. et al. (2019) Chaperone activation and client binding of a 2-cysteine peroxiredoxin. Nat. Commun. 10, 659 10.1038/s41467-019-08565-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jang H.H., Kim S.Y., Park S.K., Jeon H.S., Lee Y.M., Jung J.H. et al. (2006) Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 580, 351–355 10.1016/j.febslet.2005.12.030 [DOI] [PubMed] [Google Scholar]
- 147.Saccoccia F., Di Micco P., Boumis G., Brunori M., Koutris I., Miele A.E. et al. (2012) Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure 20, 429–439 10.1016/j.str.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Park J.W., Piszczek G., Rhee S.G. and Chock P.B. (2011) Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50, 3204–3210 10.1021/bi101373h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Barranco-Medina S., Lázaro J.-J. and Dietz K.-J. (2009) The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 583, 1816 10.1016/j.febslet.2009.05.029 [DOI] [PubMed] [Google Scholar]
- 150.Shringarpure R., Grune T. and Davies K.J.A. (2001) Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell. Mol. Life Sci. 58, 1442–1450 10.1007/PL00000787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shringarpure R., Grune T., Mehlhase J. and Davies K.J.A. (2003) Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 278, 311–318 10.1074/jbc.M206279200 [DOI] [PubMed] [Google Scholar]
- 152.Grune T., Merker K., Sandig G. and Davies K.J.A. (2003) Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 305, 709–718 10.1016/S0006-291X(03)00809-X [DOI] [PubMed] [Google Scholar]
- 153.Livnat-Levanon N., Kevei É., Kleifeld O., Krutauz D., Segref A., Rinaldi T. et al. (2014) Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 7, 1371–1380 10.1016/j.celrep.2014.04.030 [DOI] [PubMed] [Google Scholar]
- 154.Davies K.J. and Goldberg A.L. (1987) Oxygen radicals stimulate intracellular proteolysis and lipid peroxidation by independent mechanisms in erythrocytes. J. Biol. Chem. 262, 8220–8226 PMID: [PubMed] [Google Scholar]
- 155.Reinheckel T., Ullrich O., Sitte N. and Grune T. (2000) Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch. Biochem. Biophys. 377, 65–68 10.1006/abbi.2000.1717 [DOI] [PubMed] [Google Scholar]
- 156.Silva G.M., Netto L.E.S., Simões V., Santos L.F.A., Gozzo F.C., Demasi M.A.A. et al. (2012) Redox control of 20S proteasome gating. Antioxid. Redox Signal. 16, 1183–1194 10.1089/ars.2011.4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jung T., Höhn A. and Grune T. (2014) The proteasome and the degradation of oxidized proteins: Part III—redox regulation of the proteasomal system. Redox Biol. 2, 388–394 10.1016/j.redox.2013.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tsvetkov P., Myers N., Eliav R., Adamovich Y., Hagai T., Adler J. et al. (2014) NADH binds and stabilizes the 26S proteasomes independent of ATP. J. Biol. Chem. 289, 11272–11281 10.1074/jbc.M113.537175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cagnetta A., Cea M., Calimeri T., Acharya C., Fulciniti M., Tai Y.-T. et al. (2013) Intracellular NAD+ depletion enhances bortezomib-induced anti-myeloma activity. Blood 122, 1243–1255 10.1182/blood-2013-02-483511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jenner P. (2003) Oxidative stress in Parkinson's disease. Ann. Neurol. 53, S26–S38 10.1002/ana.10483 [DOI] [PubMed] [Google Scholar]
- 161.Shringarpure R., Grune T., Sitte N. and Davies K.J.A. (2000) 4-Hydroxynonenal-modified amyloid-β peptide inhibits the proteasome: possible importance in Alzheimer's disease. Cell. Mol. Life Sci. 57, 1802–1809 10.1007/PL00000660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee M., Hyun D., Jenner P. and Halliwell B. (2001) Effect of proteasome inhibition on cellular oxidative damage, antioxidant defences and nitric oxide production. J. Neurochem. 78, 32–41 10.1046/j.1471-4159.2001.00416.x [DOI] [PubMed] [Google Scholar]
- 163.Ling Y.-H., Liebes L., Zou Y. and Perez-Soler R. (2003) Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 278, 33714–33723 10.1074/jbc.M302559200 [DOI] [PubMed] [Google Scholar]
- 164.Mehta N.J., Marwah P.K. and Njus D. (2019) Are proteinopathy and oxidative stress two sides of the same coin? Cells 8, 59 10.3390/cells8010059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L. and Elazar Z. (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760 10.1038/sj.emboj.7601623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bensaad K., Cheung E.C. and Vousden K.H. (2009) Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 28, 3015–3026 10.1038/emboj.2009.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen Y., Azad M.B. and Gibson S.B. (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16, 1040–1052 10.1038/cdd.2009.49 [DOI] [PubMed] [Google Scholar]
- 168.Dodson M., Darley-Usmar V. and Zhang J. (2013) Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic. Biol. Med. 63, 207–221 10.1016/j.freeradbiomed.2013.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Guerrero-Gómez D., Mora-Lorca J.A., Sáenz-Narciso B., Naranjo-Galindo F.J., Muñoz-Lobato F., Parrado-Fernández C. et al. (2019) Loss of glutathione redox homeostasis impairs proteostasis by inhibiting autophagy-dependent protein degradation. Cell Death Differ. 26, 1545–1565 10.1038/s41418-018-0270-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carroll B., Otten E.G.. Manni D., Stefanatos R., Menzies F.M., Smith G.R. et al. (2018) Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 9, 256 10.1038/s41467-017-02746-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y. et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 10.1038/ncb2021 [DOI] [PubMed] [Google Scholar]
- 172.Kerr E.M., Gaude E., Turrell F.K., Frezza C. and Martins C.P. (2016) Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531, 110–113 10.1038/nature16967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Paola D., Domenicotti C., Nitti M., Vitali A., Borghi R., Cottalasso D. et al. (2000) Oxidative stress induces increase in intracellular amyloid β-protein production and selective activation of βI and βII PKCs in NT2 cells. Biochem. Biophys. Res. Commun. 268, 642–646 10.1006/bbrc.2000.2164 [DOI] [PubMed] [Google Scholar]
- 174.Stockwell B.R., Angeli J.P.F., Bayir H., Bush A.I., Conrad M., Dixon S.J. et al. (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Palikaras K. and Tavernarakis N. (2012) Mitophagy in neurodegeneration and aging. Front. Genet. 3, 297 10.3389/fgene.2012.00297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Violi F., Loffredo L., Carnevale R., Pignatelli P. and Pastori D. (2017) Atherothrombosis and oxidative stress: mechanisms and management in elderly. Antioxid. Redox Signal. 27, 1083–1124 10.1089/ars.2016.6963 [DOI] [PubMed] [Google Scholar]