

Abstract
Understanding the population structure and mechanisms of taxa diversification is important for organisms responsible for the transmission of human diseases. Two vectors of West Nile virus, Culex pipiens pipiens and Cx. p. molestus, exhibit epidemiologically important behavioral and physiological differences, but the whole-genome divergence between them was unexplored. The goal of this study is to better understand the level of genomic differentiation and population structures of Cx. p. pipiens and Cx. p. molestus from different continents. We sequenced and compared the whole genomes of 40 individual mosquitoes from two locations in Eurasia and two in North America. Principal Component, ADMIXTURE, and neighbor joining analyses of the nuclear genomes identified two major intercontinental, monophyletic clusters of Cx. p. pipiens and Cx. p. molestus. The level of genomic differentiation between the subspecies was uniform along chromosomes. The ADMIXTURE analysis determined signatures of admixture in Cx. p. pipens populations but not in Cx. p. molestus populations. Comparison of mitochondrial genomes among the specimens showed a paraphyletic origin of the major haplogroups between the subspecies but a monophyletic structure between the continents. Thus, our study identified that Cx. p. molestus and Cx. p. pipiens represent different evolutionary units with monophyletic origin that have undergone incipient ecological speciation.
Subject terms: Ecology, Evolution, Genetics
Introduction
The advent of genomics has provided new insights into population divergence between incipient taxa, changing our vision about the mechanisms of adaptation and speciation1. Experimental data demonstrate highly heterogenous patterns of population differentiation in various groups of organisms2. In addition to the classical model of allopatric speciation, when incipient taxa are isolated geographically3, it becomes obvious that ecological speciation or the development of reproductive isolation between populations as a result of adaptation to different environments is feasible and common in nature4–6. Understanding the population structure and the mechanisms of taxa diversification in the changing environment is extremely important if the studied organisms are related to the transmission of human diseases7. Members of the Culex pipiens complex are globally distributed throughout Europe, Asia, America, Africa, and Australia and represent competent vectors of the lymphatic filariasis parasite and encephalitis viruses, including the widely spread West Nile virus8–11. However, despite the fact that Cx. pipiens was the first mosquito species described by C. Linnaeus in his “Systema Naturae”12, mosquitoes from the Cx. pipiens complex still represent “one of the major outstanding problems in mosquito taxonomy” because the members of the complex can mate and produce viable progeny in nature13,14. Thus, the mechanisms of genetic differentiation in the members of this complex remain poorly understood.
The “Systematic Catalog of Culicidae” maintained by the Walter Reed Biosystematics Unit at the Smithsonian Institution15, recognizes the following species as members of the Cx. pipiens complex: Cx. pipiens, Cx. quinquefasciatus, Cx. australicus, and Cx. globocoxitus16. Among these species, Cx. pipiens and Cx. quinquefasciatus spread globally in temperate and tropical/subtropical regions, respectively. Distribution of Cx. australicus and Cx. globocoxitus is restricted to Australia. In addition to these species, the Cx. pipiens complex includes a subspecies, Cx. p. pallens, found in Japan and the Far East of Eurasia, and two additional members, Cx. p. pipiens and Cx. p. molestus, that, according to this catalog, represent two physiological forms. Indeed, Cx. p. pipiens and Cx. p. molestus exhibit important physiological and ecological differences8,16. Cx. p. pipiens mates in open spaces, feeds on birds, and requires a blood meal for oviposition. During the winter, females of Cx. p. pipiens undergo diapause. In contrast, Cx. p. molestus mates in confined spaces, feeds on mammals, can lay eggs without a blood meal, and females of this subspecies cannot enter diapause. Cx. p. molestus is also known as an underground mosquito because it invades basements, sewers, and underground railways. This fact explains why Cx. p. molestus can survive in a cold climate without the ability to enter diapause8,17. Since Cx. p. pipiens has strong bias toward feeding on birds, while Cx. p. molestus readily bites humans and other mammals, hybrids between the two forms can act as a bridge vector between the bird reservoirs and susceptible mammalian hosts18–20. Thus, understanding the abilities of Cx. p. pipiens and Cx. p. molestus to produce viable progeny in natural populations is of medical importance.
Historically, P. Forskal described Cx. molestus as a separate species from Egyptian specimens in 1775, but, later, this species was synonymized with Cx. pipiens13. Following the concept of E. Mayr3, P. Mattingly proposed considering the Cx. pipiens complex as a polytypic species; thus, the status of Cx. molestus was reduced to the subspecies Cx. p. molestus21,22. Later, E. Vinogradova considered Cx. p. pipiens and Cx. p. molestus as ecophysiological forms because of the differences in their behavior17. R. Harbach argued that Cx. p. molestus is a “phenotypic, ecological, and physiological variant” of Cx. p. pipiens and should not be considered a subspecies because of the absence of clear morphological differences and the presence of hybrids13. However, a molecular analysis based on microsatellite markers determined that Cx. p. pipiens and Cx. p. molestus from different continents cluster together in a subspecies specific manner and more likely represent two separate taxonomic units or species with a unique evolutionary history19. Similar results regarding the monophyletic origin of Cx. p. pipiens and Cx. p. molestus were obtained by the analysis of single nucleotide polymorphisms (SNPs) in the mitochondrial Cytochrome Oxidase I (COI) gene of European populations23. These conclusions were strongly supported by another work using amplified fragment length polymorphism markers that identified discrete genomic differences between the subspecies and, thus, considered Cx. p. pipiens and Cx. p. molestus to be distinct evolutionary entities that are likely in the process of incipient speciation24.
Sequencing the Cx. quinquefasciatus genome offered exciting opportunities for comparative genomic studies of the Cx. pipiens complex25. Here, for the first time, we used whole-genome analysis of individual mosquitoes to investigate the level of genomic differentiation and population structures of Cx. p. pipiens and Cx. p. molestus from different continents. We compared sequences of 40 samples from field collected mosquitoes from Eurasia (Minsk, Republic of Belarus and Mailuu-suu, Kyrgyz Republic) and North America (Washington, D.C., USA) and two laboratory colonies of Cx. p. pipiens and Cx. p. molestus derived from Chicago, IL, USA. In our study, we follow the classical nomenclature that was proposed by Mattingly in 1951 and consider Cx. p. pipiens and Cx. p. molestus as subspecies21,22.
Results
Mosquito collections
In this study, we sequenced whole genomes of 40 individual mosquitoes from 4 different locations—two from Eurasia and two from North America (Table 1, Fig. 1). All mosquitoes were collected from the urban environment. Cx. p. molestus mosquitoes were collected at the larval stage from an underground water source in two locations (the Kyrgyz Republic and the Republic of Belarus). Cx. p. pipiens were also collected at the larval stage in an open water reservoir in the Kyrgyz Republic, but at the adult stage in the basement of a multi-floor building in the Republic of Belarus. All field collected specimens from Eurasia were identified as Cx. pipiens by larval morphology and then tested to subspecies level using the COI assay, which is based on SNPs in a specific position26. Mosquitoes from the Washington, D.C., USA samples were collected as egg rafts from two different urban environments—an underground parking lot and an open water reservoir. Both egg collections were used to establish mosquito colonies that were kept in the laboratory for several generations. At the larval stage, mosquitoes from both collections were identified morphologically as Cx. pipiens. However, molecular assays based on COI SNP26 and polymorphism in the flanking region of a microsatellite locus27 failed to identify them at the subspecies level. The latter method was considered unreliable for the diagnosis of Cx. p. molestus and Cx. p. pipiens in California28. In addition, we performed a physiological assay and identified that mosquitoes from the parking lot colony were able to lay eggs without a blood meal. Mosquitoes from the open water collection needed a blood meal for egg development. Thus, we called our samples from Washington, D.C, USA Cx. pipiens autogenetic (WPA) and Cx. pipiens anautogenic (WPN). In addition, we used two colonies of Cx. p. molestus and Cx. p. pipiens that were derived from the Chicago area in the USA24.
Table 1.
Collection of the Cx. pipiens mosquitoes identified and used for sequencing.
| Sample ID | Country | City, state | GPS coordinates | Habitat | Species/subspecies, stage, physiology | Identification method | Sample size |
|---|---|---|---|---|---|---|---|
| BP | Republic of Belarus | Minsk | 54.00, 27.00 | Basement, multi floor building | Cx. p. pipiens, adults | COI25, | 5 |
| BM | Republic of Belarus | Minsk | 54.00, 27.00 | Basement, multi floor building | Cx. p. molestus, larvae | COI25, | 5 |
| KP | Kyrgyz Republic | Mailuu-suu II | 41.00, 72.00 | Aboveground open water pool | Cx. p. pipiens, larvae | COI25, | 5 |
| KM | Kyrgyz Republic | Mailuu-suu | 41.00, 72.00 | Underground barrel | Cx. p. molestus, larvae | COI25, | 5 |
| CP | USA | Chicago, IL | N/A | colony | Cx. p. pipiens, larvae | 23 | 5 |
| CM | USA | Chicago, IL | N/A | colony | Cx. p. molestus, larvae | 23 | 5 |
| WPN | USA | Washington, DC | 38.25, −76.76 | Aboveground puddle | Cx. pipiens, larvae, anautogenic | morphology | 5 |
| WPA | USA | Washington, DC | 38.25, −76.76 | Underground parking lot | Cx. pipiens, larvae, autogenic | morphology | 5 |
Figure 1.

Mosquito collection sites in North America (a) and Eurasia (b). Worldwide distribution of Cx. pipiens and Cx. quinquefasciatus is indicated by blue color and green lines. The overlap between the sheds represents a species hybrid zone indicating that collections in the USA (Chicago, IL and Washington, D.C.) were made within the hybrid zone. The map was created using program ESRI ArcGis Pro v.2.4 (https://www.esri.com/). The species distributions are shown according to previously published data16.
Nuclear genome analysis
After the sequencing the 40 individual genomes, we performed neighbor joining analysis. The Cx. quinquefasciatus genome assembly25,29 was used as a reference genome and an outgroup. The Neighbor-Joining (NJ) and Maximum-Likelihood (ML) analyses identified two major monophyletic clusters that segregated in a subspecies-specific manner with high bootstrap support (Fig. 2, Supplementary Figs. 1–3). Cx. p. molestus and Cx. p. pipiens clusters included field collected mosquitoes from the Republic of Belarus and the Kyrgyz Republic as well as colonies from Chicago, IL, USA. In addition, we determined the presence of a third cluster that had a polyphyletic nature and included mosquitoes from Cx. pipiens autogenic and Cx. pipiens anautogenic colonies from Washington, D.C., USA and Cx. quinquefasciatus, based on the genome assembly of this species25,29. In fact, the Washington, DC area is located within the Cx. pipiens - Cx. quinqufasciatus hybrid zone (Farajiollahi, et al. 2011) and, thus, these mosquitoes may represent hybrids between the latter species not included in the analysis.
Figure 2.

Neighbor joining tree based on K2P distances and autosomal genome-wide SNVs. Samples of Cx. p. pipiens and Cx. p. molestus from the Republic of Belarus (Belarus), the Kyrgyz Republic (Kyrgyzstan), and the USA (Chicago, IL) demonstrate their monophyletic origin except for the autogenic (WPA) and anautogenic (WPN) samples from Washington, D.C., USA. Cx. quinquefasciatus is used for the outgroup species. All the nodes with bootstrap support less than 90% were collapsed.
Principle Component Analysis (PCA) demonstrated groupings along the first principal component (13.57% of variance explained) with a closer relationship among Cx. p. molestus and Cx. p. pipiens subspecies rather than between geographical groups (Fig. 3a). The first and third principal components (11.65% and 7.53%, respectively) grouped mainly geographical populations (Fig. 3b). In this analysis, both autogenic and anautogenic mosquitoes from Washington, D.C. grouped together with Cx. p. pipiens from other locations. The ADMIXTURE analysis demonstrated that Cx. p. molestus and Cx. p. pipiens from the Republic of Belarus, the Kyrgyz Republic, and Chicago, IL, USA formed subspecies-specific clusters at K = 2 (the optimal number of genetic clusters according to the ΔLogLikelihood statistics, Supplementary Fig. 4b), demonstrating the major pattern of population differentiation (Fig. 4, Supplementary Fig. 4). The specimens from Washington, DC clustered together with Cx. p. pipiens at K = 2–3. We observed some putative signatures of admixture at K = 2–3 in all Cx. p. pipens samples from the Republic of Belarus, the Kyrgyz Republic, in 2 out of 5 individuals in the Cx. p. pipens Chicago colony, and in all autogenic and 4 out of 5 anautogenic mosquitoes from Washington, D.C. Overall, the introgression signature between the subspecies was lower in northern populations of Cx. p. pipiens in the Republic of Belarus and Chicago, IL than in southern locations in the Kyrgyz Republic and Washington, D.C. In the Washington, D.C. samples, the admixture levels were higher in autogenic strains overall than in anautogenic strains. Interestingly, the signature of admixture in all Cx. p. molestus samples was very low or even absent, suggesting restricted gene flow from Cx. p. pipiens to Cx. p. molestus. At a higher level of clustering (K = 6–9), the local populations of the subspecies formed distant well-defined clusters without high overlap, especially for Cx. p. molestus. Thus, we concluded that both autogenic and anautogenic samples from Washington, D.C. represent Cx. p. pipiens or Cx. p. pipiens - Cx. quinqufasciatus hybrids with some admixture of Cx. p. molestus, which is more pronounced in autogenic samples.
Figure 3.
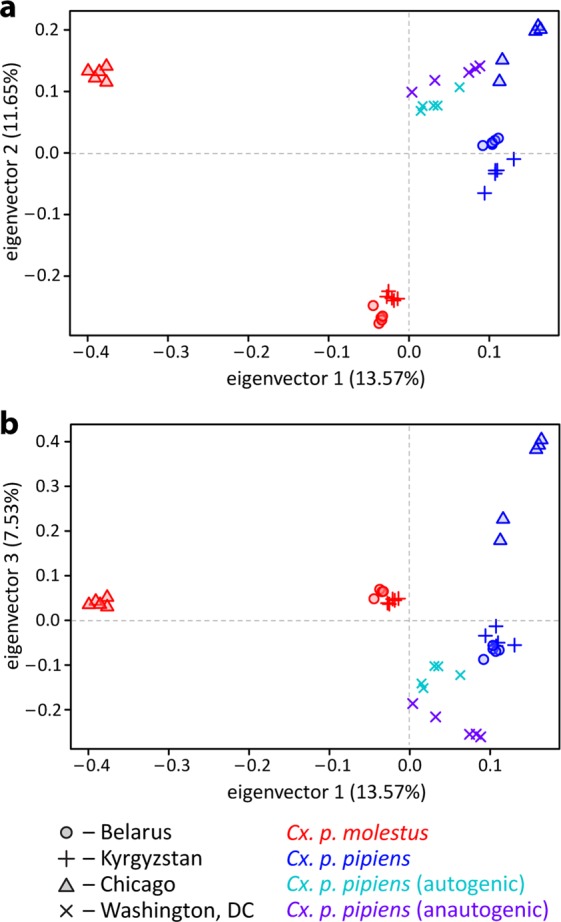
Principal component analysis of the individual samples for PC1-2 (a) and PC1-3 (b). PC1 separates the subspecies Cx. p. pipiens and Cx. p. molestus and PC2 with PC3 into separate geographic regions. Samples are from the Washington, D.C., USA group with other samples of Cx. p. pipiens.
Figure 4.

ADMIXTURE plot of the samples grouped by subspecies and regions. The major pattern of clustering is formed by subspecies level at K = 2–3 and local populations at K = 6–9. Samples are from the Washington, D.C., USA group with other samples of Cx. p. pipiens.
To get more insight into the nature of genomic differentiation between subspecies/populations, we calculated genome-wide pairwise Fst values and found that they varied between 0.08 and 0.25 and were highly significant for all comparisons (Fig. 5). The most diverged group was Cx. p. molestus from the Chicago strain, which can be explained by the very low genetic diversity of this strain caused by its longtime cultivation and possible bottleneck, which accelerated the genetic drift (Fig. 7). Fst analysis along the chromosomes demonstrated relatively uniform patterns of genetic differentiation across the genome without clear signs of “islands of divergence” (Fig. 6), which can indicate infrequent hybridization in nature and/or prezygotic instead of postzygotic reproductive isolation as major mechanisms of the subspecies differentiation. The Fst levels dropped around centromeres probably because a small number of markers are present in these regions due to a highly repetitive sequence composition. Overall, Fst values were higher between the subspecies than between autogenic and anautogenic Cx. p. pipiens from Washington, D.C. The level of genomic diversity did not differ significantly between the subspecies demonstrating an overall trend of slightly lower genetic diversity in Cx. p. molestus; however, this was not significant in pairwise comparisons except for the cultivated colonies from Chicago, IL (Fig. 7), which demonstrated a strong depletion of genetic diversity in Cx. p. molestus.
Figure 5.

Fst matrix between the studied species/locations. Cx. p. molestus is the most divergent group due to its high level of genetic drift and low diversity. All values are highly significant.
Figure 7.
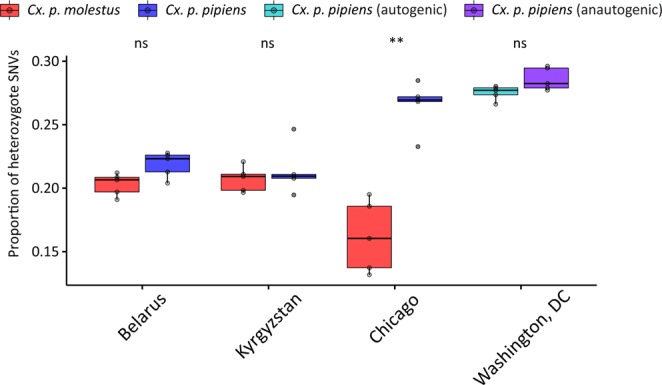
Level of individual genetic diversity estimated as a proportion of heterozygous SNVs per genome. Samples of Cx. p. molestus from Chicago, IL, USA demonstrate the lowest level of diversity and are significantly different from Cx. p. pipiens from the same location. **Indicate significant differences (P-value < 0.01).
Figure 6.

Fst values between the subspecies in different locations plotted along chromosomes. The patterns of genetic differentiation across the genome are more or less uniform. Overall, Fst values are higher between the subspecies than between autogenic and anautogenic Cx. p. pipiens from Washington, D.C., USA. P and q stand for short and long chromosome arms, respectively.
Mitochondrial genome analysis
In addition, to nuclear genome comparisons, we performed NJ and ML tree analyses using almost complete mitochondrial genomes for all 40 specimens recovered from the whole genome sequencing data. The patterns of phylogenetic structure were very different from the nuclear counterpart (Fig. 8, Supplementary Fig. 5) and showed the paraphyletic origin of the major haplogroups among the subspecies but a monophyletic structure between the continents. The mitochondrial genome of Cx. quinquefasciatus grouped inside the American haplogroup along with the samples from Washington, D.C. and did not demonstrate any significant divergence. Samples of Cx. p. molestus from the Republic of Belarus and the Kyrgyz Republic formed a diverged monophyletic haplogroup. Interestingly, two samples of Cx. p. pipiens from the Republic of Belarus represented a very diverged haplogroup, which may be reminiscent of the past species diversity or has been introgressed from other diverged populations/species. We specifically checked the quality of alignment and calling for this haplogroup but did not identify any abnormalities. Overall, we concluded that mitochondrial genome comparison does not reflect the true evolutionary history of the subspecies/species, probably due to multiple introgression events.
Figure 8.

Neighbor joining tree based on the almost complete mtDNA genomes derived from WGS data (K2P distance). Patterns showed paraphyletic origins of the major haplogroups among the subspecies but monophyletic structures between the continents. All the nodes with bootstraps lower than 70% were collapsed.
Discussion
Independent monophyletic origin of Culex pipiens pipiens and Culex pipiens molestus
Two alternative hypotheses have been proposed to explain the differences in ecological and physiological strategies of Cx. p. pipiens and Cx. p. molestus19. One hypothesis explains such differentiation as a rapid shift in physiological and behavior traits as an adaptation to an underground environment that is associated with human activities22. In this scenario, the local populations of Cx. p. molestus must be closely related to the local populations of Cx. p. pipiens30. This scenario considers Cx. p. molestus as an eco-physiological variant of Cx. p. pipiens. An alternative hypothesis suggests that the difference between Cx. p. pipiens and Cx. p. molestus is a result of the distinction in their evolutionary history. Ecological and physiological strategies of Cx. p. molestus could have first arisen under the warm climate19. Later, such a strategy appeared to be relevant because of the human activities that created an underground environment and, thus, spread Cx. p. molestus all over the world. This scenario considers Cx. p. molestus and Cx. p. pipiens as separate evolutionary entities.
In our study, we tested these hypotheses using whole genome analysis of 40 Cx. p. molestus and Cx. p. pipiens samples collected from four locations in Eurasia and North America. Our findings rejected the hypothesis of reemergence of Cx. p. molestus from the local populations of Cx. p. pipiens and strongly supported the idea of an independent monophyletic origin of both Cx. p. pipiens and Cx. p. molestus from different continents. NJ and ML analyses separately cluster Cx. p. pipiens and Cx. p. molestus from the Republic of Belarus, the Kyrgyz Republic, and Chicago, IL, USA (Fig. 2, Supplementary Figs. 1–3). The presence of an additional hybrid cluster formed by autogenic and anautogenic field collected specimens from Washington, D.C., USA more likely reflects the existence of a Cx. pipiens – Cx. quinquefasciatus hybrid zone in this area16. However, based on genetic distances, this cluster is much closer related to the Eurasian Cx. p. pipiens rather than to the Cx. p. molestus from Eurasia and the USA (Chicago, IL). Additionally, PCA (Fig. 3) and ADMIXTURE (Fig. 4) analyses identified two distinct genetic clusters that correspond to Cx. p. pipiens and Cx. p. molestus. In both cases, all the specimens from Washington, D.C. clustered together with Cx. p. pipiens from other locations. Genome-wide pairwise Fst values were highly significant for all the comparisons (Fig. 5). We did not identify any specific region of high divergence in the genome (Fig. 6). Overall, Fst values were much higher between Cx. p. molestus and Cx. p. pipiens than between autogenic and anautogenic samples of Cx. p. pipiens from Washington, D.C. The most diverged group, based on all conducted analyses, was the strain of Cx. p. molestus from Chicago, IL, which can be explained by the very low genetic diversity of this strain caused by its longtime colonization and possible bottleneck, which accelerated genetic drift (Fig. 7).
Similar observations about the independent monophyletic origin of Cx. p. molestus and Cx. p. pipiens were obtained using microsatellite analysis in ~600 samples of Cx. pipiens mosquitoes derived from different world-wide locations19. The unrooted distance tree procedure clustered aboveground populations of Cx. p. pipiens and underground populations of Cx. p. molestus from northern and southern Europe separately. Similar to our study, the aboveground populations of Cx. p. pipiens from the USA formed an additional cluster suggesting more intense hybridization between the members of the Cx. pipiens complex in North America. The admixture analysis indicated the presence of three major clusters that corresponded to Cx. p. molestus, Cx. p. pipiens, and Cx. quinquefasciatus. The latter cluster was found in this study only in the USA samples. Another work based on amplified fragment length polymorphism (AFLP) analyses compared samples from southern and northern Europe and strains from Chicago, IL, USA24. The North American and European populations used in this study showed a similar ADMIXTURE pattern in the AFLP genome scan. The analysis of COI genes also indicated a monophyletic origin of Cx. p. pipiens and Cx. p. molestus in Europe, Asia, and Africa26.
Thus, the independent monophyletic origin and high level of genetic divergence between Cx. p. molestus and Cx. p. pipiens suggest that these two members of the Cx. pipiens complex represent distinct phylogenetic entities with independent evolutionary histories prior to human-mediated translocation.
Concepts of speciation and evolution of the Culex pipiens complex
Theoretical models of speciation in animals could be subdivided into two major groups: allopatric or geographical speciation and sympatric or ecological speciation. The first concept of speciation, which was intensively promoted by E. Mayr3, suggests that incipient taxa first become isolated geographically. This situation reduces the gene flow between the populations and may lead to accumulation of mutations that cause genetic incompatibility among the hybrids. An alternative concept of speciation emphasizes ecological barriers between the emerging taxa as major drivers of evolution31. This scenario considers the development of reproductive isolation between the populations as a result of adaptation to different environments without geographical isolation, which usually takes place in the face of gene flow. In this situation, the hybrids between incipient taxa become less fit to the environment that promotes natural selection of any traits that reduce mating between them. We think that overall diversification of the Cx. p. pipiens and Cx. p. molestus subspecies represents a striking example of speciation through isolation-by-ecology mechanisms. In our study, the Fst analysis determined significant levels of genomic divergence between Cx. p. pipiens and Cx. p. molestus (Figs. 5 and 6) across the entire genome without clear islands of speciation. Surprisingly, the levels of differentiation were lower around the centromeres, probably due to the low number of reliable SNVs in these highly repetitive regions. The differentiation was extremely high between the Chicago, IL, USA strains of Cx. p. pipiens and Cx. p. molestus, but was lower between the subspecies in the Eurasian mosquito collections. Overall, these observations suggest significant restriction of gene flow between the subspecies.
Several mechanisms of reproductive isolation between Cx. p. pipiens and Cx. p. molestus have been described17. Two of them are prezygotic acting, before fertilization, and reduce the opportunities for mosquito mating. The first mechanism is related to habitat specialization of the larvae: Cx. p. molestus occupies basements or other underground environments, but Cx. p. pipiens prefers open aboveground water bodies for breeding sites. This reduces chances for the two subspecies to meet and mate at the adult stages. The second isolating mechanism relies on the differences in mating behavior between Cx. p. molestus and Cx. p. pipiens. Cx. p. molestus males usually form homogeneous swarms near the ground and require limited space for mating17. In contrast, Cx. p. pipiens males swarm near the foliage about 2–3 m above the ground. Experimental studies of mating behavior in small cages indicated that in crosses of females and males of Cx. p. molestus copulation success was 90% but in Cx. p. pipiens it was only 3.3%32. In inter-subspecies crosses between Cx. p. molestus and Cx. p. pipiens, the copulation success was also low and varied between 6.6% to 10% depending on the sexes of the subspecies. This study demonstrated that females of both subspecies actively avoid copulation with males from an alternative subspecies. Moreover, Cx. p. pipiens females were unsuccessful in receiving sperm from Cx. p. molestus and, as a result, produced no eggs.
Two other reproductive isolating mechanisms described for Cx. p. pipiens and Cx. p. molestus are postzygotic, they act after the mating and result in decreased fitness of the hybrids. One of the mechanisms is related to the inheritance of diapause in hybrids of Cx. p. molestus and Cx. p. pipiens as a recessive trait8. F1 hybrids and a significant portion of F2 hybrids are unable to develop diapause and cannot survive under winter conditions. This mechanism, perhaps, could explain the higher introgression levels in Cx. p. pipiens in southern locations19,23,24. Finally, the members of the Cx. pipiens complex are exposed to cytoplasmic incompatability of hybrids infected with different strains the rickettsial parasite Wolbachia pipientis. Despite cytoplasmic introgression of this parasite through hybridization between the members of the Cx. pipiens complex33, cytoplasmic incompatibility could significantly limit survival rates of the hybrids. For example, W. pipientis infection significantly reduced hybridization between Cx. pipiens and Cx. quinquefasciatus in South Africa34. Another study demonstrated that in Eurasian populations Cx. p. molestus was only infected by one strain of W. pipientis but Cx. p. pipiens by two different strains35. Moreover, the specimens of Cx. p. molestus and Cx. p. pipiens, which we used in our study, were infected with the same strains of W. pipientis in the south of the Kyrgyz Republic but by different strains in the north of the Republic of Belarus. Thus, it may explain the differences in introgression from Cx. p. molestus to Cx. p. pipiens that was more pronounced in the Republic of Belarus than in the Kyrgyz Republic. In fact, an interesting example of infectious speciation was described in the South American Drosophila paulistorum complex. In this complex, six semi-species with overlapping geographic distribution became reproductively isolated as a result of premating and postmating isolation triggered by the Wolbachiae infection36.
Recent genomic studies conducted on different organisms including Drosophila simulans37, Rhagoletis fruit flies38,39, and Heliconius butterflies40,41 provide additional evidence that ecological speciation occurs in nature. The genomic patterns of speciation can be very different2 ranging from small genomic islands of speciation40 to significant levels of divergence across the entire genome. Widespread genomic divergence was identified between the incipient species Anopheles gambiae and An. coluzzii in the An. gambiae complex42. These species were originally identified by differences in the structure of their ribosomal DNA as S and M forms43, but later their taxonomic status was elevated to the species level44. An. gambiae and An. coluzzii are believed to develop reproductive barriers in sympatry as a result of the differences in their ecological preferences45. The An. gambiae larval stage is associated with small rain pools. In contrast, An. coluzzi exploits persistent water reservoirs associated with rice cultivation. Although, premating barriers between the species are incomplete46, they developed differences in their swarming behavior47,48 and divergent song types49.
Thus, a high level of genome-wide divergence, a striking difference in adaptation to distinct ecological environments and evidence of prezygotic and postzygotic barriers for mating suggest that Cx. p. pipiens and Cx. p. molestus represent distinct ecological units that undergo incipient ecological speciation.
Hybridization in the Culex pipiens complex
The most intriguing observation of the members of the Cx. pipiens complex is that, despite differences in ecology, physiology, behavior, and geographic distribution, they still can hybridize and produce viable progeny in nature, indicating that reproductive isolation between them is not complete. Our study also demonstrated significant hybridization events between the subspecies Cx. p. molestus and Cx. p. pipiens. Whole genome comparison indicated that most of the Cx. p. pipiens samples represent individuals with some level of introgression from Cx. p. molestus (Fig. 4). We observed high discrepancy between the nuclear and mitochondrial phylogenies (Figs. 2, 8), which indicates that, historically, the transmission of mitochondrial genomes can happen between the subspecies. At the same time, we did not observe haplogroups shared between the local populations of the subspecies. In the continents, all the mitochondrial phylogenies were strongly monophyletic, which points to male-mediated dispersal and hybridization. The admixture with Cx. p. molestus was higher in southern populations of Cx. p. pipiens in the Kyrgyz Republic and in Washington, D.C., USA, but lower in northern populations in the Republic of Belarus and Chicago, IL, USA. This could be related to an incapability of the hybrids to develop diapause in cold climates. For comparison, microsatellite analysis revealed a modest level of hybridization between Cx. p. pipiens and Cx. p. molestus of ~8% in the northern cities of the USA (Chicago, IL and New York, NY)30,50. In southern Europe, where Cx. p. pipiens and Cx. p. molestus could both be found aboveground, the hybridization levels between them were similar and was estimated at 8–10%51. Much higher levels of hybridization were found in southern populations in eastern USA, where ~40% of all samples were identified as hybrids between Cx. p. molestus and Cx. p. pipiens19. Thus, overall hybridization rates between the members of Cx. pipiens were higher in North America than in the Old World. For comparison, hybridization between cryptic species in the An. gambiae complex (An. gambiae and An. coluzzi) significantly varied between 1% in Mali46 to >20% in Guinea Bissau52, which is comparable to overall hybridization levels between Cx. p. molestus and Cx. p. pipiens.
Intriguingly, in our study, we did not determine a Cx. p. pipiens admixture signature in any Cx. p. molestus samples from the Republic of Belarus, the Kyrgyz Republic, and Chicago, IL, USA (Fig. 4). These findings demonstrate very limited or no gene flow from Cx. p. pipiens to Cx. p. molestus. Similar findings of asymmetric introgression from Cx. p. molestus to Cx. p. pipiens was shown by previous studies20,51. The mechanism of asymmetric introgression is currently unknown. One hypothesis suggests that males of Cx. p. molestus, which can mate in confined spaces, can hybridize with both Cx. p. molestus and Cx. p. pipiens females. In contrast, Cx. p. pipiens males, which require space for swarming, have a higher disposition to mate with Cx. p. pipiens females51. However, more population genetic and experimental studies are needed to explain this phenomenon.
Finally, our study demonstrated that Cx. p. pipiens can develop autogeny as a result of adaptive introgression of the genetic material from Cx. p. molestus. In the field collected samples from aboveground and underground environments in Washington, D.C., USA, we selected mosquitoes for autogeny. The underground mosquitoes were autogenic but the aboveground mosquitoes were anautogenic. However, mosquitoes from both autogenic and anautogenic colonies formed a single cluster when neighbor joining analysis was applied (Fig. 2). Moreover, PCA (Fig. 3) and ADMIXTURE (Fig. 4) approaches cluster these samples together with Cx. p. pipiens from other locations. Populations with mixed characteristics were found in Europe51 and in the USA53. In Portugal, an unusual pattern of blood feeding behavior on birds was also found in Cx. p. molestus54.
Thus, the presence of ongoing hybridization between members of the Cx. pipiens complex suggests that the speciation process between them is not complete and postzygotic barriers of reproductive isolation are not fully formed. Overall, we believe that members of the Cx. pipiens complex represent a remarkable model for studying different aspects of geographical and ecological speciation in the face of ongoing gene flow between them and local adaptations to diverse environments.
Materials and Methods
Mosquito collections
In this study, we used field collected mosquitoes from Minsk, Republic of Belarus, Mailuu-suu, Kyrgyz Republic, and Washington, D.C., USA (Table 1, Fig. 1). The map on Fig. 1. was created using program ESRI ArcGis Pro v.2.4 (https://www.esri.com/). The species distributions are shown according to previously published data16. Mosquitoes from the Republic of Belarus were collected from an urban environment in Minsk, the capital city of the Republic. Cx. p. pipiens mosquitoes were collected at the adult stage in the basement of a multi-floor building. Cx. p. molestus were collected at the larval stage by dipping on the ground floor of a clinical hospital. In the Kyrgyz Republic, all samples were collected in an urban environment at the larval stage by dipping. Cx. p. pipiens were found in an open water pool, while Cx. p. molestus were found in an underground water barrel. Mosquitoes were identified as Cx. p. molestus and Cx. p. pipiens by the COI assay26. In Washington, D.C., USA mosquitoes were collected in an urban environment as egg rafts from an open water puddle and an underground parking lot. Mosquitoes from both locations were successfully colonized and fed on fish pellet food at the larval stage. Morphologically, mosquito larvae from both collection sites were identified as Cx. pipiens. Adult mosquitoes were kept at 26 °C and offered 10% sucrose solution. Mosquitoes from both sites were tested for autogeny. Individuals from the parking lot demonstrated the ability to lay eggs without blood feeding, while individuals from the open water reservoir did not. Anautogenic mosquitoes were fed on artificial membrane blood feeders 4–5 days after emerging. Five mosquitoes from the second generation of both colonies were selected for sequencing. Finally, we used laboratory colonies of Cx. p. molestus and Cx. p. pipiens from Chicago, IL, USA. These colonies were established from mosquitoes collected in the Chicago, IL area. Cx. p. molestus was collected by sampling a drainage sump using aspirators and larval dipping in January 2009 (Mutebi and Savage, 2009) and Cx. p. pipiens was collected as egg rafts in above ground breeding sites in Evanston and Northfield, IL in August of 2016.
DNA extraction and sequencing
DNA was extracted from individual mosquitoes for all Eurasian samples using the DNA Invisorb Spin Tissue Mini Kit (Invitek, Berlin, Germany) and for all American samples using the Qiagen Blood and Tissue Kit (Qiagen, Germantown, MD, USA). DNA concentration was determined by nanodrop; ~50 ng of DNA from each individual mosquito was sent for sequencing to the Fasteris company (Fasteris Inc., Switzerland). Sequencing was done using Illumina HiSeq. 2 ×150 single index configuration with a 12–17X coverage for individual mosquito samples.
Genomic analysis: variant calling
Individual genomes for each population were aligned against the Cx. quinquefasciatus reference genome29 using BWA-MEM software v0.7.17 (r1188)55 (https://github.com/lh3/bwa). The resulting BAM files were sorted and PCR duplicates were removed using Samtools v1.956 (https://github.com/samtools/samtools/releases/). To call Single Nucleotide Variations (SNVs), we used the bcftools mpileup and call multisample (-m) functions of the bcftools package v1.957 (https://github.com/samtools/bcftools/releases/) and alignments with the quality of mapping equal to at least 40 and a base quality not less than 20. All of the BAM files were called simultaneously. The raw VCF file was filtered with vcftools v0.1.1658 (https://github.com/vcftools/vcftools/releases) to remove all variants with a quality less than 500 (–minQ 500), more than two alleles per position, and were monomorphic with missing genotypes in more than four individuals.
To call mitochondrial DNA (mtDNA) variants, the whole genome sequence (WGS) reads were aligned to the nearly complete mitochondrial genome of Cx. quinquefasciatus with BWA-MEM and only unique, properly paired alignments with the highest quality (60) were left for the sorting and deduplication steps. The mitochondrial variants were called using the bcftools consensus algorithm (bcftools call -c)57 considering only bases with Phred scale quality >25 (-Q 25). The resulting VCF file with only polymorphic positions was converted into FASTA format and used then for the phylogenic analysis (NJ and ML, below).
Population genetic analysis
To reduce the influence of the linkage between neighboring loci on population genetic analysis, we removed all loci located closer than 10 Kbp from each other using vcftools (–thin 10000). The resulting pruned dataset with all individuals from both subspecies simultaneously was used to compute PCA using SNPrelate software v1.20.159 (https://bioconductor.org/packages/release/bioc/html/SNPRelate.html) and unsupervised clustering using ADMIXTURE v1.360 (http://software.genetics.ucla.edu/admixture/download.html) for K = 2–9. To find the most optimal number of ADMIXTURE clusters we employed Cross-Validation Error estimation and ΔLogLikelihood which uses a relative gain of LogLikelihood for each successive step (estimated as LogLikelihood K = x − LogLikelihood K = x − 1). Pairwise Fst values61 between locations/subspecies were calculated with the Stacks package v2.462 (http://catchenlab.life.illinois.edu/stacks/). To profile the Fst values between subspecies along the chromosomes, we used the sliding window Fst approach implemented in vcftools with a window size equal to 50 Kbp and steps equal to 5 Kbp.
To construct neighbor-joining trees for the autosomal and mtDNA datasets, we used RapidNJ software v2.3.263 (https://birc.au.dk/software/rapidnj/) with K2P distance64 and 1000 bootstrap replications. Additionally, we used ML approach realized in RaxML software v8.2.1265 (https://github.com/stamatak/standard-RAxML/releases) with GTRGAMMA model, rooting to Cx. quinquefasciatus and 500 bootstrap replications to corroborate the NJ analysis for nuclear WGS dataset. A ML approach for mitochondrial dataset was used in MEGA X software v10.1.766 (https://www.megasoftware.net) with K2P distances and 1000 bootstrap replicates. To find an optimal model of substitutions we used Bayesian information criteria in jModelTest 2 software67. Finally, all the nodes with bootstrap values lower 90% and 70% for nuclear and mitochondrial phylogenies respectively were collapsed. A part of figures was produced using R software v3.5.3 (https://cran.r-project.org/mirrors.html)68.
Supplementary information
Acknowledgements
We thank Igor V. Sharakhov for the productive discussion of the manuscript, Dmitri A. Karagodin for the help with formatting figures, and Janet Webster for proofreading the text. This project was supported by the Russian Science Foundation grant № 19-14-00130 to MVS. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author contributions
M.V.S. and A.A.Y. designed the experiments. R.A.M., M.L.F., A.K.S. and N.V.K. performed mosquito collections and species identification. A.A.Y. conducted statistical and bioinformatics analysis. M.L.F. provided mosquito colonies. M.V.S., A.A.Y. and R.A.M. wrote the manuscript. All authors read and approved the manuscript.
Data availability
Genomic sequencing data will be available via NCBI upon the publication of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Andrey A. Yurchenko and Reem A. Masri.
Supplementary information
is available for this paper at 10.1038/s41598-020-63305-z.
References
- 1.Arias CF, Van Belleghem S, McMillan WO. Genomics at the evolving species boundary. Curr Opin Insect Sci. 2016;13:7–15. doi: 10.1016/j.cois.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Seehausen O, et al. Genomics and the origin of species. Nat Rev Genet. 2014;15:176–192. doi: 10.1038/nrg3644. [DOI] [PubMed] [Google Scholar]
- 3.Mayr, E. Animal species and evolution. (Harvard University Press, 1963).
- 4.Ortiz-Barrientos D, Engelstadter J, Rieseberg LH. Recombination rate evolution and the origin of species. Trends Ecol Evol. 2016;31:226–236. doi: 10.1016/j.tree.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 5.Shafer AB, Wolf JB. Widespread evidence for incipient ecological speciation: a meta-analysis of isolation-by-ecology. Ecol Lett. 2013;16:940–950. doi: 10.1111/ele.12120. [DOI] [PubMed] [Google Scholar]
- 6.Butlin RK, Galindo J, Grahame JW. Review. Sympatric, parapatric or allopatric: the most important way to classify speciation? Philos Trans R Soc Lond B Biol Sci. 2008;363:2997–3007. doi: 10.1098/rstb.2008.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell, J. R. Genetic variation in insect vectors: death of typology? Insects9, 10.3390/insects9040139 (2018). [DOI] [PMC free article] [PubMed]
- 8.Vinogradova, E. B. Culex pipiens pipiens mosquitoes: taxonomy, distribution, physiology, genetics, applied importance and control. (Pensoft, 2000).
- 9.Turell MJ. Members of the Culex pipiens complex as vectors of viruses. J Am Mosq Control Assoc. 2012;28:123–126. doi: 10.2987/8756-971X-28.4.123. [DOI] [PubMed] [Google Scholar]
- 10.Turell MJ, Dohm DJ, Fonseca DM. Comparison of the potential for different genetic forms in the Culex pipiens complex in North America to transmit Rift Valley fever virus. J Am Mosq Control Assoc. 2014;30:253–259. doi: 10.2987/14-6441R.1. [DOI] [PubMed] [Google Scholar]
- 11.Calistri P, et al. Epidemiology of west nile in europe and in the mediterranean basin. Open Virol J. 2010;4:29–37. doi: 10.2174/1874357901004020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linnaeus, C. Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis., Vol. 1. Holmiae (1758).
- 13.Harbach RE. Culex pipiens: species versus species complex taxonomic history and perspective. J Am Mosq Control Assoc. 2012;28:10–23. doi: 10.2987/8756-971X-28.4.10. [DOI] [PubMed] [Google Scholar]
- 14.Harbach RE, Dahl C, White GB. Culex (Culex) pipiens Linnaeus (Diptera, Culicidae): concepts, type designations, and description. Proc Entomol Soc Wash. 1985;87:1–24. [Google Scholar]
- 15.Catalog of the mosquitoes of the world, http://www.wrbu.si.edu.
- 16.Farajollahi A, Fonseca DM, Kramer LD, Marm Kilpatrick A. “Bird biting” mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology. Infect Genet Evol. 2011;11:1577–1585. doi: 10.1016/j.meegid.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vinogradova EB. Ecophysiological and morphological variations in mosquitoes of the Culex pipiens complex (Diptera:Culicidae) Acta Soc. Zool. Bohem. 2003;67:41–50. [Google Scholar]
- 18.Ciota AT, Chin PA, Kramer LD. The effect of hybridization of Culex pipiens complex mosquitoes on transmission of West Nile virus. Parasit Vectors. 2013;6:305. doi: 10.1186/1756-3305-6-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fonseca DM, et al. Emerging vectors in the Culex pipiens complex. Science. 2004;303:1535–1538. doi: 10.1126/science.1094247. [DOI] [PubMed] [Google Scholar]
- 20.Gomes B, et al. Distribution and hybridization of Culex pipiens forms in Greece during the West Nile virus outbreak of 2010. Infect Genet Evol. 2013;16:218–225. doi: 10.1016/j.meegid.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Mattingly PF, et al. The Culex pipiens complex T. Royal Entomol. Soc. London. 1951;102:331_382. [Google Scholar]
- 22.Mattingly PF. The systematics of the Culex pipiens complex. Bull. World Health Organ. 1965;37:257–382. [PMC free article] [PubMed] [Google Scholar]
- 23.Shaikevich EV, Vinogradova EB, Bouattour A, Gouveia de Almeida AP. Genetic diversity of Culex pipiens mosquitoes in distinct populations from Europe: contribution of Cx. quinquefasciatus in Mediterranean populations. Parasit Vectors. 2016;9:47. doi: 10.1186/s13071-016-1333-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes B, et al. Limited genomic divergence between intraspecific forms of Culex pipiens under different ecological pressures. BMC Evol Biol. 2015;15:197. doi: 10.1186/s12862-015-0477-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arensburger P, et al. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;330:86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaikevich EV. PCR-RFLP of the COI gene reliably differentiate Culex pipiens, Cx. pipiens f. molestus, and Cx. torrentium of the Pipiens complex. Europian Mosquito Bulletin. 2007;23:25–30. [Google Scholar]
- 27.Bahnck CM, Fonseca DM. Rapid assay to identify the two genetic forms of Culex (Culex) pipiens L. (Diptera: Culicidae) and hybrid populations. Am J Trop Med Hyg. 2006;75:251–255. doi: 10.4269/ajtmh.2006.75.2.0750251. [DOI] [PubMed] [Google Scholar]
- 28.Cornel A, et al. Culex pipiens sensu lato in California: a complex within a complex? J Am Mosq Control Assoc. 2012;28:113–121. doi: 10.2987/8756-971X-28.4s.113. [DOI] [PubMed] [Google Scholar]
- 29.Dudchenko O, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kothera L, Godsey M, Mutebi JP, Savage HM. A comparison of above-ground and below-ground populations of Culex pipiens pipiens in Chicago, Illinois, and New York City, New York, using 2 microsatellite assays. J Am Mosq Control Assoc. 2012;28:106–112. doi: 10.2987/8756-971X-28.4.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schluter, D. In Endless forms (eds D.J. Howard & S.H. Berlocher) 114–129 (Oxford University Press, 1998).
- 32.Kim S, Trocke S, Sim C. Comparative studies of stenogamous behaviour in the mosquito Culex pipiens complex. Med Vet Entomol. 2018;32:427–435. doi: 10.1111/mve.12309. [DOI] [PubMed] [Google Scholar]
- 33.Dumas E, et al. Population structure of Wolbachia and cytoplasmic introgression in a complex of mosquito species. BMC Evol Biol. 2013;13:181. doi: 10.1186/1471-2148-13-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornel AJ, et al. Differences in extent of genetic introgression between sympatric Culex pipiens and Culex quinquefasciatus (Diptera: Culicidae) in California and South Africa. J Med Entomol. 2003;40:36–51. doi: 10.1603/0022-2585-40.1.36. [DOI] [PubMed] [Google Scholar]
- 35.Khrabrova NV, Bukhanskaya ED, Sibataev AK, Volkova TV. The distribution of strains of endosymbiotic bacteria Wolbachia pipientis in natural populations of Culex pipiens mosquitoes (Diptera: Culiciadae) Europian Mosquito Bulletin. 2009;27:18–22. [Google Scholar]
- 36.Miller WJ, Ehrman L, Schneider D. Infectious speciation revisited: impact of symbiont-depletion on female fitness and mating behavior of Drosophila paulistorum. PLoS Pathog. 2010;6:e1001214. doi: 10.1371/journal.ppat.1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrigan D, et al. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012;22:1499–1511. doi: 10.1101/gr.130922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doellman, M. M. et al. Genomic differentiation during speciation-with-gene-flow: comparing geographic and host-related variation in divergent life history adaptation in Rhagoletis pomonella. Genes(Basel)9, 10.3390/genes9050262 (2018). [DOI] [PMC free article] [PubMed]
- 39.Doellman, M. M. et al. Geographic and ecological dimensions of host plant-associated genetic differentiation and speciation in the Rhagoletis cingulata (Diptera: Tephritidae) sibling species group. Insects10, 10.3390/insects10090275 (2019). [DOI] [PMC free article] [PubMed]
- 40.Nadeau NJ, et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large-scale targeted sequencing. Philos Trans R Soc Lond B Biol Sci. 2012;367:343–353. doi: 10.1098/rstb.2011.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nadeau NJ, et al. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Mol Ecol. 2013;22:814–826. doi: 10.1111/j.1365-294X.2012.05730.x. [DOI] [PubMed] [Google Scholar]
- 42.Lawniczak MK, et al. Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science. 2010;330:512–514. doi: 10.1126/science.1195755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Favia G, Lanfrancotti A, Spanos L, Siden-Kiamos I, Louis C. Molecular characterization of ribosomal DNA polymorphisms discriminating among chromosomal forms of Anopheles gambiae s.s. Insect Mol Biol. 2001;10:19–23. doi: 10.1046/j.1365-2583.2001.00236.x. [DOI] [PubMed] [Google Scholar]
- 44.Coetzee M, et al. Anopheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex. Zootaxa. 2013;3619:246–274. doi: 10.11646/zootaxa.3619.3.2. [DOI] [PubMed] [Google Scholar]
- 45.Coluzzi M, Sabatini A, della Torre A, Di Deco MA, Petrarca V. A polytene chromosome analysis of the Anopheles gambiae species complex. Science. 2002;298:1415–1418. doi: 10.1126/science.1077769. [DOI] [PubMed] [Google Scholar]
- 46.Tripet F, et al. DNA analysis of transferred sperm reveals significant levels of gene flow between molecular forms of Anopheles gambiae. Mol Ecol. 2001;10:1725–1732. doi: 10.1046/j.0962-1083.2001.01301.x. [DOI] [PubMed] [Google Scholar]
- 47.Diabate A, et al. Spatial swarm segregation and reproductive isolation between the molecular forms of Anopheles gambiae. Proc Biol Sci. 2009;276:4215–4222. doi: 10.1098/rspb.2009.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawadogo PS, et al. Swarming behaviour in natural populations of Anopheles gambiae and An. coluzzii: review of 4 years survey in rural areas of sympatry, Burkina Faso (West Africa) Acta Trop. 2014;132(Suppl):S42–52. doi: 10.1016/j.actatropica.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Pennetier C, Warren B, Dabire KR, Russell IJ, Gibson G. “Singing on the wing” as a mechanism for species recognition in the malarial mosquito Anopheles gambiae. Curr Biol. 2010;20:131–136. doi: 10.1016/j.cub.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 50.Kothera, L. et al. Bloodmeal, host selection, and genetic admixture analyses of Culex pipiens complex (Diptera: Culicidae) mosquitoes in Chicago, IL. J Med Entomol, 10.1093/jme/tjz158 (2019). [DOI] [PMC free article] [PubMed]
- 51.Gomes B, et al. Asymmetric introgression between sympatric molestus and pipiens forms of Culex pipiens (Diptera: Culicidae) in the Comporta region, Portugal. BMC Evol Biol. 2009;9:262. doi: 10.1186/1471-2148-9-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsden CD, et al. Asymmetric introgression between the M and S forms of the malaria vector, Anopheles gambiae, maintains divergence despite extensive hybridization. Mol Ecol. 2011;20:4983–4994. doi: 10.1111/j.1365-294X.2011.05339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelms BM, et al. Phenotypic variation among Culex pipiens complex (Diptera: Culicidae) populations from the Sacramento Valley, California: horizontal and vertical transmission of West Nile virus, diapause potential, autogeny, and host selection. Am J Trop Med Hyg. 2013;89:1168–1178. doi: 10.4269/ajtmh.13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes B, et al. Feeding patterns of molestus and pipiens forms of Culex pipiens (Diptera: Culicidae) in a region of high hybridization. Parasit Vectors. 2013;6:93. doi: 10.1186/1756-3305-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng X, et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 62.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simonsen, M., Mailund, T. & Christian, N. S. In Proceedings of the 8th Workshop in Algorithms in Bioinformatics (WABI), LNBI 5251 113–122 (Springer Verlag, 2008).
- 64.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/bf01731581. [DOI] [PubMed] [Google Scholar]
- 65.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar S, et al. Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/ (2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic sequencing data will be available via NCBI upon the publication of the manuscript.