

Abstract
The combination of lentiviruses with techniques such as CRISPR-Cas9 has resulted in efficient and precise processes for targeted genome modification. An often-limiting aspect, however, is the efficiency of cell transduction. Low efficiencies with particular cell types and/or the high complexity of lentiviral libraries can cause insufficient representation. Here, we present a protocol that yielded substantial increases in transduction efficiency in various cell lines in comparison to several other procedures.
Keywords: lentivirus, cell transduction, CRISPR-Cas, shRNA
Introduction
Genomic screens and targeted modifications have become more and more interesting to the scientific community. They permit the identification of regulatory elements and pathways that are involved in cellular processes, with relevance even for elucidating the causes of particular diseases and the identification of potential remedies. Most of these studies are based on inhibitory RNA or CRISPR-Cas techniques. Particularly the latter have an enormous impact on genome engineering as they facilitate highly accurate genetic as well as epigenetic modifications and allow to obtain engineered cell lines or animal models in a relatively short time frame compared to alternative approaches [1–3], hence rapidly moving towards clinical application [4, 5]. Lentiviruses have become an important tool for such genomic manipulation, allowing the introduction of a specific DNA fragment into cells and the study of the functional consequences in both in vitro and in vivo systems. The methodology is utilized in a wide range of applications, such as performing genetic and synthetic lethality screens [6, 7] or studying the effects of specific genetic variations [8, 9]. Lentiviruses integrate their RNA genome into the infected host cells’ genomes, a property that is frequently used for the delivery and stable expression of transgenes [10], small hairpin RNA (shRNA) constructs [11] or single-guide RNA (sgRNA) expression cassettes that target sequences in a CRISPR-Cas mediated process [12].
A challenge for the lentiviral delivery of CRISPR-Cas systems into target cells is the packaging limit of lentiviruses [13]. Especially, when co-delivering sgRNA expression cassettes with the typically quite large Cas enzymes, such as the >4 kb-long Cas9 from Streptococcus pyogenes, packaging limits are quickly reached. This issue is further amplified by the addition of reporter genes used for the selection of successfully transduced target cells, such as fluorescence or antibiotic resistance marker genes. Virus production with such large constructs typically results in mediocre viral titres, which is a major limitation for the delivery of present genome engineering tools as well as complex sgRNA libraries into target cells. Also in transductions with individual constructs, low efficiencies could increase the risk of selecting a particular cell line sub-population rather than real representatives of the cell population, thus possibly yielding biased results [14, 15]. Since there are major differences between particular cell types with respect to transduction efficiency, modulating some of them could become experimentally limiting. In order to address such shortcomings, we developed and optimized a protocol by which highly efficient lentiviral production and transduction delivery are achieved.
Material and methods
Cell lines
The following cells were used in the analysis: BxPC3 (ATCC CRL-1687), adherent culturing; Capan-1 (ATCC HTP-79), adherent culturing; CCRF-CEM (ATCC CCL-119), suspension culturing; HEK-293T (ATCC CRL-3216), adherent culturing; Jurkat (ATCC TIP-152), suspension culturing; NCI-H1299 (ATC CRL-5803), adherent culturing; Suit-2 (JCRB1094), adherent culturing. Cell line authentication was performed at the DKFZ Genomics and Proteomics Core Facility by profiling single nucleotide polymorphism or analysis of short tandem repeats. All cells were regularly checked for mycoplasma contamination and all experiments were performed with mycoplasma-free cells.
Cell culture
CCRF-CEM, Jurkat and NCI-H1299 cells were cultured in RPMI medium (Life Technologies, Darmstadt, Germany) supplemented with 10% foetal bovine serum (FBS, Life Technologies). Cell lines Suit-2 and Capan-1 were cultured in IMDM medium (Life Technologies), 10% FBS. HEK-293T was cultured in IMDM medium, 10% FBS. All cells lines were kept at 37°C, 95% humidity and 5% CO2. Suspension cells were kept at 106 cells/ml and cultured in suspension flasks (Greiner Bio One, Frickenhausen, Germany). Adherent cells were kept at 70–80% confluency and cultured in standard cell culture flasks (Greiner Bio One) while expanding the cell lines.
Transduction analysis
For fluorescence-activated cell sorting (FACS) analysis, 106 cells were counted for each sample. Cells were spun down at 500 × g at 4°C for 3 min and washed twice with 500 µl PBS (Life Technologies). Subsequently, the cells were resuspended in FACS buffer (PBS supplemented with 4% FBS) in a total volume of 300 µl and analysed on an LSRFortessa flow cytometry machine (BD Biosciences, San Jose, USA). Un-transduced cells were used as the negative control (unstained) to set the right voltage for the mCherry detecting channel. The cell population was selected plotting forward scatter (FCS) versus side scatter (SSC); doublets were excluded from the analysis by plotting FCS width versus FCS area. The percentage of cells positive for mCherry fluorescence reflected the transduction efficiency.
Infectious titres were calculated with the following formula: titre = [(F × Cn)/V] × DF; F: frequency of mCherry-positive cells; Cn: total number of target cells; V: volume of inoculum; DF: virus dilution factor.
For microscopy, cells were analysed using a Cell Observer microscope (Carl Zeiss, Jena, Germany). Data was acquired by overlaying the brightfield image with that of the mCherry detection filter. Since the wavelength of mCherry is located in the far-red, we used the burner (a xenon lamp) to increase the power of the signal in order to avoid underestimation of the transduction efficiency. Un-transfected cells were used to determine the background.
Detailed protocol for an optimal cell transduction with lentiviruses
Virus production
HEK-293T cells were cultured in IMDM growth medium supplemented with 10% FBS. Cells were at 80% confluency before seeding; passage number was ≤12. Cells were seeded in culture dishes with 150 mm diameter (Corning, Kaiserslautern, Germany) at a cell density of 0.6 × 105 cells/cm2 in 15 ml of medium. After 24 h, cells were transfected as follows: A DNA solution was made up by mixing 17 µg plasmids of lentiviral sgRNA library consisting of >260 000 molecules [16] in vector sgLenti containing mCherry (Addgene, Watertown, USA). About 17 µg psPAX2 (lentiviral packaging plasmid; Addgene), 5.7 µg pMD2.G (VSV-G envelope expressing plasmid; Addgene), water to a total volume of 60 μl, 140 µl P3000 reagent, and 2.1 ml Opti-MEM reduced serum medium (Thermo Fisher Scientific, Waltham, USA). The lipofectamine mix consisted of 160 µl lipofectamine LTX (Thermo Fisher Scientific), 40 µl lipofectamine 3000 (Thermo Fisher Scientific), and 2.1 ml Opti-MEM medium (Thermo Fisher Scientific).
The DNA solution was added to the lipofectamine mix and incubated for 30 min at room temperature. The solution was then added dropwise to the HEK-293T cells with the 15 ml growth medium still in place. The cell culture dish was gently shaken in order to distribute the lipofectamine-DNA complex evenly. After 24 h cell growth, the medium was replaced with fresh IMDM, 10% FBS, and 1% penicillin-streptomycin solution. Supernatant containing viruses was collected 72 h post-transfection, loaded onto a 20 ml syringe and filtered through a 0.45 µm filter (Merck, Darmstadt Germany). Finally, the virus was concentrated 10-fold via centrifugation using Vivaspin 10 000 MWCO columns (Sartorius, Göttingen, Germany). The columns were centrifuged at 1000 × g for 20–30 min, the time necessary to reduce the volume to 1 ml.
Cell transduction
For cell transduction, 25 000 cells per well were seeded in 96-well U-bottom microtitre plates (Greiner-Bio-One). The cells were immediately pelleted by centrifugation at 500 × g and 4°C for 3 min; the supernatant was discarded by careful pipetting. A volume of 12.5 µl of the concentrated viral solution (equivalent to 125 µl of non-concentrated supernatant) was added to the cells and the volume of each well was adjusted to 50 µl with RPMI medium supplemented with 10% FBS without resuspending the cells. Polybrene (Merck) was added to a final concentration of 10 µg/ml. Cells were incubated at 37°C, 95% humidity, 5% CO2 for 7–9 h. Afterwards, cells were transferred to 48-well plates (Greiner-Bio-One); the medium was replaced with 1 ml fresh RPMI, 10% FBS and 1% penicillin-streptomycin (Life Technologies). Transduction efficiency was established via flow cytometry (FACS) 4 days after viral infection.
For representative analyses with the highly complex CRISPR-library of some 260 000 sgRNAs [16], 10 million rather than 25 000 cells were used per microtitre well and 200 µl of the concentrated virus solution and polybrene were added. After a 7–9 h incubation, cells were transferred to a 1 l flask for cell growth. In total, 600 million cells were used, equivalent to 60 microtitre plate wells.
Alternative protocols for virus production
The other protocols used for producing lentiviruses in HEK-293T cells in comparison were as follows:
HEPES-buffered saline protocol
HEK293T cells were seeded at a density of 0.6 × 105 cells/cm2 in 6 cm2 culture dishes in 4 ml IMDM growth medium supplemented with 10% FBS and grown for 24 h. A DNA solution was made up by mixing 6.42 µg plasmids of lentiviral sgRNA library [16], 6.42 µg psPAX2, 2.14 µg pMD2.G, water to a total volume of 438 μl, and 62 μl 2 M CaCl2. Dropwise, 500 μl of 2× HBS (HEPES-buffered saline) buffer (50 mM HEPES (Sigma-Aldrich, Taufkirchen, Germany), 280 mM NaCl, 1.5 mM Na2HPO4, pH 7.0; sterilized by filtration) were added to the DNA solution while mixing it by blowing air through the solution with a Pasteur pipette at the bottom of the tube. The resulting 1 ml transfection solution was added dropwise to the HEK-293T cells while shaking the cell culture dish gently to distribute the solution evenly. After 10 h, the growth medium was replaced with 5 ml of fresh IMDM, 10% FBS, 1% penicillin-streptomycin solution. The supernatant containing viruses was collected 72 h post-transfection, loaded onto a 20 ml syringe and filtered through a 0.45 µm filter.
Polyethyenimine protocol
HEK-293T cells were seeded at a density of 0.6 × 105 cells/cm2 in 10 cm2 culture dishes in 10 ml IMDM growth medium supplemented with 10% FBS and grown for 24 h. A DNA solution was made up by mixing 8 µg plasmids of lentiviral sgRNA library [16], 4 µg psPAX2, 4 µg pMD2.G, and Opti-MEM medium to a total volume of 250 μl. For a polyethyenimine (PEI): DNA ratio of 3:1, 48 µl of PEI solution (1 µg/µl; Polysciences, Hirschberg, Germany) were added to 202 µl Opti-MEM. The DNA solution was added to the PEI solution, mixed well, spun briefly to collect all the liquid and incubated at room temperature for 20 min. Then, the transfection solution was added dropwise to the HEK-293T cells while shaking the cell culture dish gently to distribute the solution evenly. After 24 h, the growth medium was replaced with 10 ml of fresh IMDM, 10% FBS, 1% penicillin-streptomycin solution. The supernatant containing viruses was collected 72 h post-transfection, loaded onto a 20 ml syringe and filtered through a 0.45 µm filter.
Other protocols
All other transfections were performed as suggested by the respective reagent provider using the same cells and plasmids as above.
Lipofectamine 3000 protocol https://www.thermofisher.com/de/de/home/brands/product-brand/lipofectamine/lipofectamine-3000.html
Lipofectamine LTX protocol https://www.thermofisher.com/de/de/home/brands/product-brand/lipofectamine/lipofectamine-ltx-reagent.html
jetPRIME protocol https://www.polyplus-transfection.com/products/jetprime/#files-btn
TransIT-LT1 protocol https://www.mirusbio.com/products/transfection/transit-lt1-transfection-reagent
Statistics
All measurements were done as entirely independent replicates between 3 and 9 times. For analysis, two-tailed Student’s t-test was applied. P-values of <0.05 were defined as being significant: *P < 0.05; **P < 0.01; ***P < 0.001.
Results
We started our analysis with the T-cell acute lymphoblastic leukaemia (T-ALL) suspension cell line CCRF-CEM, which exhibits particularly poor transducibility. We specifically focussed on the optimization of three aspects: (i) identification of the best performing transfection reagent for lentivirus production; (ii) increasing lentivirus concentration prior to transduction without compromising the viral particles and thus their functionality; and (iii) achieving a higher recipient cell concentration without compromising cell viability (Fig. 1). The enhanced performance of the established protocol for efficient lentiviral transduction of CCRF-CEM was then confirmed in additional cell lines. In the analysis, we used a lentiviral sgRNA library consisting of >260 000 molecules [16]. By virtue of the mCherry fluorescence marker gene in its sgLenti vector, cells could be analysed by flow cytometry in order to determine quantitatively the percentage of transduced cells.
Figure 1:

Schematic overview of the various steps of the optimized transduction process. The steps in blue-labelled frames are different from standard protocols and jointly increase transduction efficiency substantially.
Virus production
First, we compared the performance of transfection reagents commonly used for virus production in HEK-293T cells: polyethyenimine (PEI), HEPES-buffered saline (HBS), lipofectamine 3000 and lipofectamine LTX, alone and in combination, as well as jetPRIME and TransIT-LT1. For all transfection reagents, the protocols recommended by their manufacturers were used, unless otherwise stated in the Materials and Methods section. Transduction yields obtained with virus supernatants from the respective HEK-293T cultures varied widely (Fig. 2). In cell suspension, best results were obtained with jetPRIME and PEI with transduction efficiencies of slightly above or below 4%, respectively. However, mixing lipofectamine 3000 and lipofectamine LTX for a combined use actually led to a substantial rise in transduction efficiency to ∼14% (Fig. 2A).
Figure 2:

Comparison of transduction efficiencies with CCRF-CEM cells. Viruses were produced by transfecting HEK-293T cells with virus constructs using the reagents indicated in panels A–C. The virus-containing cell supernatant was then used for transduction of CCRF-CEM cells. This was done by directly adding the virus to the cell suspension (A) or by gently pelleting the cells before adding the virus (B). In panel C, results are shown that were obtained by using a mixture of lipofectamines 3000 and LTX for virus production. Prior to transduction of CCRF-CEM cells, the virus was either used directly as supernatant as in panels A and B (labelled as no action; red = cell suspension; blue = pelleted cells) or concentrated via ultracentrifugation or spin-column. Panels D and E: Microscopic images that were acquired 4 days after transduction; red signals indicate successfully transduced cells. Results are shown of a transduction of CCRF-CEM cells with the optimized protocol (D) and with viruses that had been concentrated by ultracentrifugation rather than spin-column (E). *P < 0.05; ***P < 0.001.
In order to improve yields further, we expected that a higher concentration of CCRF-CEM cells during transduction might boost the process. Cells were spun down in a 96-well, U-bottom microtitre plate at 500 × g and 4°C for 3 min. The supernatant was removed and the lentivirus then added to the cell pellet. This led to transduction efficiencies in an overall range as without cell pelleting (Fig. 2B). However, the order of performance changed in comparison to the results with cell suspension. In particular, viruses produced with jetPRIME exhibited much lower yields. As before, however, viruses produced with a mixture of lipofectamines 3000 and LTX yielded the best results by a wide margin.
Target cell transduction
Since the lipofectamine 3000 and LTX mixture had consistently exhibited much superior performance in comparison to all other transfection reagents, we focussed our further efforts on the optimization of yields with viruses produced with this compound mix. We contemplated that concentrating the virus could improve transduction further. In one approach, the virus supernatant produced with HEK-293T cells—as always filtered through pores of 0.45 µm for the removal of cell debris and other contaminants—was centrifuged at 200 000 × g at 4°C for 2 h and the pellet resuspended in 200 µl PBS. Alternatively to ultracentrifugation, the virus solution was loaded onto an ultrafiltration column (Vivaspin, 10.000 MWCO), followed by centrifugation at 1000 × g at 4°C for typically 25 min until the remaining volume was ∼1 ml. In all measurements, an equivalent number of viruses was used for transduction. To our surprise, ultracentrifugation decreased transduction efficiency, while virus concentration by spin-column improved transduction markedly (Fig. 2C). Differences were such pronounced that they were directly visible looking at relatively few cells microscopically (e.g. Fig. 2D and E). Interestingly, adding column-concentrated virus to pelleted cells further enhanced transduction efficiency significantly to almost 30% (Fig. 2C).
Validation with other cell lines
After getting such strikingly improved results with our overall protocol on CCRF-CEM cells (T-ALL, suspension culturing), we studied its effect on five other cancer cell lines: Jurkat (another T-ALL cell line often used in leukaemia studies and grown in suspension culture), BxPC3 (pancreatic ductal adenocarcinoma, primary tumour, adherent culturing), Capan-1 (pancreatic ductal adenocarcinoma, liver metastasis, and adherent culturing), Suit-2 (pancreatic ductal adenocarcinoma, liver metastasis, and adherent culturing) and NCI-H1299 (lung cancer, lymph node metastasis, adherent culturing). The lipofectamine 3000 and LTX transfection for virus production was combined with concentrating the resulting lentivirus supernatant by means of an ultrafiltration spin-column as well as gentle pelleting of the recipient cells prior to transduction; yields were compared to results obtained with a commonly applied protocol using PEI (e.g. [17]) (see ‘Materials and methods’ section). The new protocol achieved almost 100% transduction efficiency with NCI-H1299 and around 90% with BxPC3 and Suit-2, up from ∼50% and 30% with PEI, respectively (Fig. 3). Transduction of Capan-1, known to be a cell line that is difficult to transduce, most likely due to its high level of mucin expression [18, 19], yielded 28% transduction efficiency with our method while 17% were obtained in the control experiment. For Jurkat cells, the difference was about 5-fold with 50% versus 9%.
Figure 3:
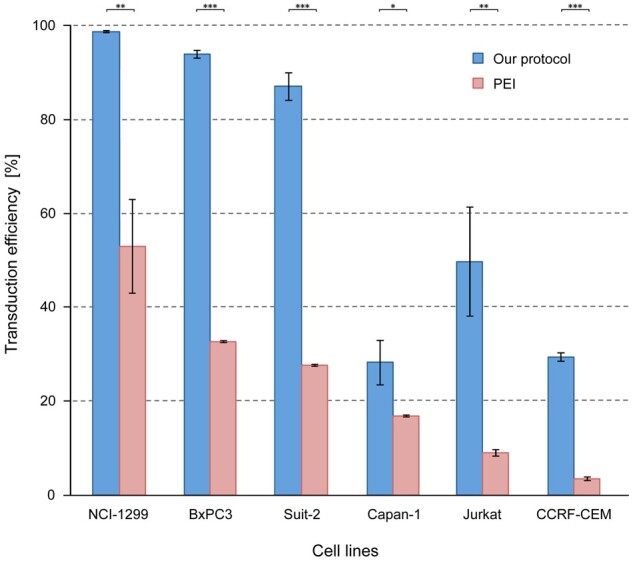
Comparison of transduction efficiencies in different cell lines. Blue columns represent the results obtained with the method of mixing lipofectamine 3000 and lipofectamine LTX to transfect HEK-293T cells, concentrating the resulting virus supernatant through Vivaspin 10 000 MW columns, and gently pelleting the recipient cells prior to cell transduction. In comparison, red columns stand for results obtained with a commonly used PEI protocol without concentrating viruses and cells. *P < 0.05; **P < 0.01; ***P < 0.001.
Functional information
For identification of possible reasons for the observed differences, we studied transfection and transduction separately. Transfection into HEK-293T cells was performed with either PEI, lipofectamine LTX, lipofectamine 3000, or the mixture of lipofectamines LTX and 3000. By FACS analysis, the percentage of transfected cells was determined. The combination of lipofectamines yielded results that were better than with the two reagents alone by a factor of two or three, respectively (Fig. 4). The PEI protocol, however, produced a percentage of transfected HEK-293T cells that was identical to those obtained with the lipofectamine combination (Fig. 4).
Figure 4:

Comparison of transfection efficiencies. HEK-293T cells were transfected using the protocols indicated by the respective reagents. By FACS analysis, the percentage of transfected cells was measured 72 h after transfection. ns = not significant; ***P < 0.001.
Regarding transduction, we collected virus produced by HEK-293T cells transfected with the four different protocols. Rather than using equivalent virus titres, identical volumes of supernatant were used. To avoid experimental bias, concentration by spin-column was performed for all of them. Both HEK-293T and CCRF-CEM cells were transduced (Fig. 5). Also with the PEI protocol, there was a strongly positive effect on transduction efficiency by spin-column concentration. With PEI-derived virus on CCRF-CEM cells, the transduction yield increased from ∼4% without spin-column concentration (Fig. 3) to ∼14%. Still, the percentage of transduced cells obtained with the lipofectamine LTX and 3000 mixture was twice as high. This superior performance of the lipofectamine mixture is also highlighted by the infectious titres that could be calculated (Fig. 5C). However, titre is a parameter that is only sensible for comparisons between data obtained for one cell line. The transduction saturation seen with larger viral quantities on CCRF-CEM cells, for example, shows that titre is not everything; using more virus and thus a higher titre did not result in more transduction.
Figure 5:

Transduction efficiencies. HEK-293T (A) and CCRF-CEM cells (B) were transduced with equal volumes of virus supernatant obtained from HEK-293T cells that had been infected with the named compounds. In all experiments, supernatants were concentrated by a factor of 10 prior to cell transduction and applied to the cells in an identical volume. The volumes given in the figure indicate the original amount of supernatant from which virus was isolated. Apart from showing the differences between protocols, the analysis also reveals that saturation with virus is achieved much earlier for CCFR-CEM cells than HEK-203T. For comparison, also infectious titres were calculated (C). ns = not significant; ***P< 0.001.
Discussion
We present an effective and scalable protocol for lentivirus production and transduction of cell lines that is applicable to small numbers (e.g. 25 000 cells) as well as high numbers of cells (e.g. 10 million cells) as in the case of library screenings. It yielded higher transduction efficiencies with all analysed cells. Yields were close to 100% for several cell lines and sharply increased for others, which are known to be difficult to transduce. The process consists of mixing lipofectamines 3000 and LTX for transfecting plasmids into HEK-293T cells for virus production as well as concentrating virus and cells prior to transduction by ultrafiltration and carefully pelleting, respectively. The procedure facilitates the delivery of lentiviral cargos into target cells, reducing both cost of reagents and space required for cell culture experiments while maintaining good coverage.
Lipofection as a process and also the lipofectamine class of reagents have been in use for many years already [20, 21]. Also, initial detailed structural and mechanistic information is available [22]. Nevertheless, an explanation of why mixing lipofectamine 3000 and LTX led to a substantial increase in yield as compared to their individual use is made difficult for the lack of specific information on the compounds. However, the mixture consistently produced the best results, while the performance of the other techniques varied markedly with changing experimental conditions. One reason for the superior results is the better transfection of HEK-293T cells as compared to using the two compounds individually. However, there was no difference in the use of the PEI protocol at this level. Therefore, the difference between PEI and the lipofectamine combination in transduction efficiency is likely to be due to more viruses or more active viruses released by the HEK-293T cells.
The positive effect of concentrating lentiviruses by spin-column is well documented, since yielding better transduction with viruses produced by different protocols. The higher concentration should improve mass transport and thus foster interaction of viruses and cells. The use of ultrafiltration or ion-exchange chromatography for improved transduction efficiency has been reported before [23, 24]; yields were similar to that of concentration by ultracentrifugation, however. A direct comparison to our results is difficult since different systems and protocols were used. With our set-up, concentration by spin-column clearly outperformed ultracentrifugation, probably avoiding damage to the viral particles or their clotting. Gently pelleting the cells prior to transduction has a similar effect as long as the cell’s viability or the accessibility of their surfaces are not affected for the worse.
In conclusion, by applying and combining relatively simple procedural modifications—mixing lipofectamines 3000 and LTX for transfecting HEK-293T cells, virus concentration in spin-columns and gently pelleting cells prior to transfection—substantial improvements in transduction yields could be achieved.
Funding
This work was supported by a grant awarded jointly by the German Cancer Research Centre (DKFZ) and the Israeli Ministry of Science and Technology [Ca173 to J.D.H.]. R.O. is supported by a PhD fellowship of the Deutscher Akademischer Austauschdienst (DAAD).
Conflict of interest statement. None declared.
REFERENCES
- 1. Nerys-Junior A, Braga-Dias LP, Pezzuto P. et al. Comparison of the editing patterns and editing efficiencies of TALEN and CRISPR-Cas9 when targeting the human CCR5 gene. Genet Mol Biol 2018;41:167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eid A, Mahfouz MM.. Genome editing: the road of CRISPR/Cas9 from bench to clinic. Exp Mol Med 2016;48:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vojta A, Dobrinic P, Tadic V. et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res 2016;44:5615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lino CA, Harper JC, Carney JP. et al. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv 2018;25:1234–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Z, Zhang Y, Gao F. et al. CRISPR/Cas9 genome-editing system in human stem cells: current status and future prospects. Mol Ther Nucl Acids 2017;9:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joung J, Konermann S, Gootenberg JS. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 2017;12:828–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang T, Yu H, Hughes NW. et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017;168:890–903.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kotler E, Shani O, Goldfeld G. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 2018;71:178–90.e8. [DOI] [PubMed] [Google Scholar]
- 9. Ni X, Ou C, Guo J. et al. Lentiviral vector-mediated co-overexpression of VEGF and Bcl-2 improves mesenchymal stem cell survival and enhances paracrine effects in vitro. Int J Mol Med 2017;40:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kafri T, Van Praag H, Gage FH. et al. Lentiviral vectors: regulated gene expression. Mol Ther 2000;1:516–21. [DOI] [PubMed] [Google Scholar]
- 11. Song H, Yang PC.. Construction of shRNA lentiviral vector. N Am J Med Sci 2010;2:598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chira S, Gulei D, Hajitou A. et al. CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids 2017;7:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar M, Keller B, Makalou N. et al. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther 2001;12:1893–905. [DOI] [PubMed] [Google Scholar]
- 14. Croce MV, Colussi AG, Price MR. et al. Identification and characterization of different subpopulations in a human lung adenocarcinoma cell line (A549). Pathol Oncol Res 1999;5:197–204. [DOI] [PubMed] [Google Scholar]
- 15. Vitor MT, Sart S, Barizien A. et al. Tracking the evolution of transiently transfected individual cells in a microfluidic platform. Sci Rep 2018;8:1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boettcher M, Tian R, Blau JA. et al. Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat Biotechnol 2018;36:170–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang S, Zhou X, Li R. et al. Optimized PEI-based transfection method for transient transfection and lentiviral production. Curr Protoc Chem Biol 2017;9:147–57. [DOI] [PubMed] [Google Scholar]
- 18. Seppen J, Barry SC, Klinkspoor JH. et al. Apical gene transfer into quiescent human and canine polarized intestinal epithelial cells by lentivirus vectors. J Virol 2000;74:7642–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrianifahanana M, Moniaux N, Schmied BM. et al. Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis. Clin Cancer Res 2001;7:4033–40. [PubMed] [Google Scholar]
- 20. Felgner PL, Gadek TR, Holm M. et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci USA 1987;84:7413–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalby B, Cates S, Harris A. et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 2004;33:95–103. [DOI] [PubMed] [Google Scholar]
- 22. Felgner JH, Kumar R, Sridhar CN. et al. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J Biol Chem 1994;269:2550–61. [PubMed] [Google Scholar]
- 23. Sena-Esteves M, Tebbets JC, Steffens S. et al. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Meth 2004;122:131–39. [DOI] [PubMed] [Google Scholar]
- 24. Kutner RH, Zhang XY, Reiser J.. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 2009;4:495–505. [DOI] [PubMed] [Google Scholar]