

Abstract
Genome-wide association studies bring into focus specific genetic variants of particular interest for which validation is often sought in large numbers of study subjects. Practical alternative methods are limiting for the application of genotyping few variants in many samples. A common scenario is the need to genotype a study population at a specific high-value single nucleotide polymorphism (SNP) or insertion-deletion (indel). Not all such variants, however, will be amenable to assay by a given approach. We have adapted a single-nucleotide primer extension (SNuPE) method that may be tailored to genotype a required variant, and implemented it as a useful general laboratory protocol. We demonstrate reliable application for production-scale genotyping, successfully converting 87% of SNPs and indels for assay with an estimated error rate of 0.003. Our implementation of the SNuPE genotyping assay is a viable addition to existing alternative methods; it is readily customizable, scalable, and uses standard reagents and a laboratory plate reader.
Keywords: SNP, polymorphism, primer extension, fluorescence polarization, genotype, genotyping
Introduction
Array and next-generation sequencing technologies gather data on germline genetic variation at a remarkable scale, often motivating follow-up work to further investigate lead genetic variants of particular interest. For example, a candidate genetic variant associated with disease may have been identified by a survey approach within one study population, for which replication is sought in an independent study population. Moreover, array-based imputation to identify such a candidate variant can warrant experimental confirmation. Generation of genotypes for a specific required variant in a large number of samples is not cost-effective by either arrays or sequencing. While a large number of genotyping assay methods have been developed that would be appropriate to serve this application space, few have been adapted for routine use and further demonstrated to be robust in large and diverse projects. Among the more commonly used assays is the 5′ nuclease (commercial TaqMan) assay with detection by plate reader [1], but a significant proportion of genetic variants do not convert successfully for the assay. Basic alternative methods that are also commonly used include restriction fragment length polymorphism, single-strand conformation polymorphism, and allele-specific polymerase chain reaction (PCR) with detection by electrophoresis [2–4].
An additional alternative method is the single-nucleotide primer extension (SNuPE) assay. A primer is hybridized adjacent to the interrogated variant site and is extended by deoxyribonucleic acid (DNA) polymerase to incorporate a complementary base that can be labeled for detection [5]. It was originally described in 1991 [6] and has been adapted for detection by time-of-flight mass spectrometry [7], by capillary gel electrophoresis [8], and by fluorescence polarization [9]. The SNuPE assay can be accomplished without specialized reagents or instruments, but to our knowledge, very few published investigations have successfully adapted it for robust use [10,11]. Here, we update our implementation and apply it in scale to demonstrate practical utility as a general laboratory protocol. The variant-specific reagents are commodity oligos, while a generic set of fluorescently labeled dideoxynucleotides serves as the reporter for all assays. Allelic detection by a general laboratory plate reader uses fluorescence polarization, obviating the need to separate unincorporated reagent from reaction product (Fig. 1). We describe assay development and production-scale assessment, including an approach for rare genetic variants. The SNuPE assay is a viable addition to other methods, and importantly can be tailored to genotype a specific genetic variant of interest that may not be amenable to other methods.
Figure 1:
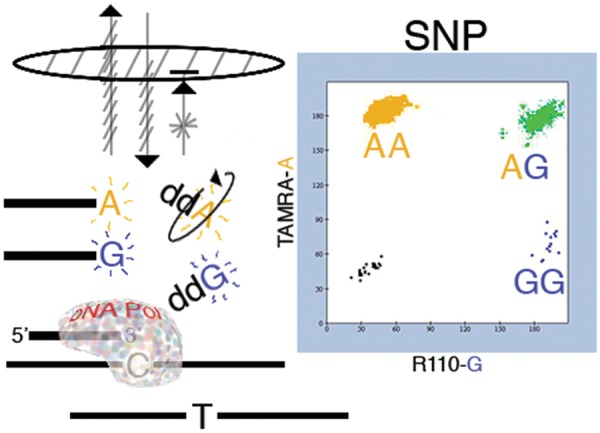
Graphic summary of the SNuPE assay. Allelic discrimination of a biallelic genetic variant (SNP or indel) relies upon the specificity of base incorporation by a thermostable polymerase as it extends a primer across a polymorphic variant position. For a subject who is heterozygous at a G/A SNP, e.g. a “blue” fluorescently labeled ddGTP is incorporated by extension across the complementary C template, while a “yellow” ddATP is incorporated by extension from the other T template. By contrast, assay of a homozygous subject would instead incorporate only a single corresponding reporter color. Detection of primer-incorporated reporter can capitalize upon its larger mass relative to residual unincorporated labeled ddNTPs. Excitation of the primer-incorporated reporter by polarized light will tend to emit back in the same plane, while relative movement of unincorporated reporter yields emission in other planes. Allelic detection thus does not require separation of unincorporated reagent from reaction product. The assay can be conducted in 96-well plate format; imaging by fluorescence polarization results in wells that are yellow, blue, both, or uncolored (e.g. negative controls). A scatterplot of the two-color measures for each well reveals corresponding subject genotype.
Materials and methods
The SNuPE assay entails three processing steps: (i) a PCR reaction; (ii) degradation of unincorporated PCR primer by exonuclease I (Exo I) and degradation of residual deoxynucleotide triphosphates (dNTPs) by calf intestinal alkaline phosphatase (CIAP), followed by heat inactivation of these enzymes; and (iii) extension of a primer annealed adjacent to the interrogated variant site by a thermostable DNA polymerase, adding either of two alternative fluorescently labeled dideoxynucleotide triphosphate (ddNTP) reporters.
Oligonucleotide design
We designed PCR primers with a preference for a 3′ G or C, to minimize primer-dimer and hairpin loop artifacts, to avoid annealing to nonunique sequence or polymorphic variant sites, and to target a melting temperature (Tm) of 55°C. We also designed a synthetic oligonucleotide template to evaluate reaction conditions and to provide a known genotype control. For a biallelic variant, two synthetic templates were used to represent genotypes AA, BB, or AB (mixed). Templates ranged from 68 to 100 nt in total length, artificially joining forward and reverse PCR primer sites to the flanks of ∼40 nt target variant site. From 5′ to 3′, a target oligonucleotide included forward PCR primer, forward extension primer, the position to be varied (SNP or indel site), reverse extension primer, and reverse PCR primer. Some synthetic targets were designed to represent four possible variant site bases. A synthetic oligonucleotide could optionally be directly used in quantity as an extension template (0.1 μM, without PCR amplification or cleanup). Oligonucleotide syntheses were desalted (Invitrogen/Thermo Fisher Scientific, Waltham, MA, Supplementary Tables S1–S3).
PCR
Genomic DNA template or synthetic oligonucleotide targets were amplified in a 5 μl reaction in black 384-well plates. Each reaction included either 0.15 unit AmpliTaq Gold DNA polymerase (Applied Biosystems/Thermo Fisher Scientific, Waltham, MA) or 0.4× Titanium Taq (ClonTech Laboratories/Takara Bio Inc., Kyoto, Japan), 10 mM Tris-HCl pH 8.3, 50 mM KCl, 2.5 mM MgCl2, 0.25 mM dNTPs, 333 nM of each PCR primer, and 2 ng human genomic DNA or 0.1 nM synthetic target oligonucleotide. Where gel electrophoresis indicated weak or nonspecific amplification using the default condition of AmpliTaq Gold without betaine, a further test of each enzyme was done in both the presence and absence of 1 M betaine for selection of an optimal condition. The thermocycling protocol was 95°C for 12 min followed by 45 cycles of (94°C for 15 s, 55°C for 15 s, ramping to 72°C at 0.5°C/s, 72°C for 60 s, ramping to 94°C at 0.5°C/s), then held at 72°C for 10 min, followed by 10°C until further use (see Supplementary Tables S4 and S5).
Cleanup
After PCR, 4 μl of 2× cleanup reagent mix containing 0.95 units each of CIAP (Promega, Madison, WI) and Exo I (New England Biolabs, Ipswich, MA) was added, incubated at 37°C for 1 h, then 80°C for 15 min, and held at 10°C until further use.
SNuPE
The extension reagent mixture contained 3× buffer B [60 mM Tris-HCl pH 8.9, 15 mM (NH4)2SO4, 18 mM MgSO4, 0.15% Triton X-100, 15% glycerol], 1.5 uM extension primer and 0.21 units of Thermo Sequenase (GE Healthcare Life Sciences, Chicago, IL). It also included one TAMRA and one R110 labeled ddNTP (PerkinElmer, Waltham, MA) corresponding to the expected alleles. The possible biallelic extension combinations (A/C, A/G, A/T, C/G, C/T, and G/T) were each detected using a pair of terminators. The optimized 3× concentration for each terminator was 105 nM ddA-TAMRA, 60 nM ddC-TAMRA, 30 nM ddU-TAMRA, 12 nM ddC-R110, 12 nM ddG-R110, 12 nM ddU-R110, from which a given pair was included in the 3× extension reagent mix. For C/T SNPs, the ddU-TAMRA and ddC-R110 pair was used. Of 3× extension mix, 4 μl was added to a plate well of cleaned-up PCR products, then incubated at 93°C for 1 min followed by 26 cycles of (93°C for 10 s and 55°C for 30 s), and held at 10°C until final plate read.
Incorporation of R110- and TAMRA-labeled terminators was detected by measure of fluorescence polarization using a Molecular Devices plate reader (Molecular Devices, San Jose, CA). This approach does not require the separation of an extension primer with an incorporated fluorescent dideoxynucleotide from the residual unincorporated labeled ddNTPs [12]. Overall, a single black 384-well plate was carried forward with sequential reagent additions for the three reaction steps, with a final plate read. Use of a film seal facilitates reagent additions; we used Cycle Seal PCR plate sealers which can be cleaned for reuse (catalog # AB0580, Thermo Fisher Scientific, Waltham, MA). Excitation bandpass filters for TAMRA were 550-10 nm, and for R110 were 490-10 nm; emission bandpass filters for TAMRA were 580–10 nm, and for R110 were 520–10 nm. The dual-dichroic was 490/550 nm.
Performance measurement
The primer extension reaction for a given target template extends with a complimentary fluorescent ddNTP terminator. Nonspecific incorporation of a noncomplementary terminator can also be observed. As an index of specificity of incorporation, we subtracted the maximum noncomplementary terminator signal from the complementary terminator signal as the measure of specific incorporation. For development of the optimal reaction condition for a biallelic variant, we summed specific incorporation of the TAMRA and R110 terminators (fluorescence polarization (FP) sum) as an index of the overall performance (Supplementary Fig. S1).
Results
Assay development
We evaluated two thermostable polymerases designed for dideoxynucleotide incorporation to assess their capacity for allelic discrimination: Thermo Sequenase (Thermus aquaticus DNA polymerase F667Y) and Therminator (Thermococcus 9°N-7 DNA polymerase A485L). Figure 2 presents the specific terminator incorporation by these enzymes at a series of G/A SNPs, as a function of terminator concentration. PCR and cleanup of genomic DNA templates were as described under methods, with extension in the presence of supplied buffers and 10 mM ddNTP terminators. We assessed specific incorporation as the difference between the incorporation signals of the complementary and noncomplementary (incorrect) terminators. At high enzyme concentrations, nonspecific incorporation can be observed. With these initial tests, greatest specific incorporation was observed at extension enzyme concentrations of 0.004 units/μl for Therminator and, 0.02 units/μl for Thermo Sequenase.
Figure 2:

Extension polymerase concentration curves. (A) Specific incorporation as a function of Therminator concentration. (B) Specific incorporation as a function of Thermo Sequenase concentration.
Both Thermo Sequenase and Therminator showed expected assay performance variation across different SNPs (e.g. Supplementary Fig. S2). As an aid to assay optimization, we devised a synthetic target system to provide control over template and variant site context. These targets encompassed flanking PCR priming site sequences as well as sufficient sequence surrounding a variant position for subsequent hybridization of an extension primer, facilitating terminator incorporation at the variant position. Examples of synthetic target template assay performance are shown in Fig. 3.
Figure 3:

Example assay of a synthetic target template (SynFP_C and SynFP_T) as an engineered SNP using Therminator (A) or Thermo Sequenase (B).
We investigated the impact of extension buffer composition upon terminator incorporation by Therminator. We employed synthetic targets SynFP_C and SynFP_T (incorporating ddG-R110 and ddA-TAMRA) and an initial buffer of 2 mM, 10 mM KCl, 10 mM (NH4)2SO4, 0.1% Triton X-100, and 20 mM Tris-HCl pH 8.8. Our approach was to test a range of concentrations of one component, each in the presence of a range of concentrations of a second component, while holding other variables constant. We selected the optimum for each of the two tested components, adopting the new condition as a change to the initial buffer. We followed this approach until optima for each variable had been selected. The optimized reaction was 0.5 μM extension primer and 1× buffer A: 2 mM MgSO4, 5 mM (NH4)2SO4, and 0.1% Triton X-100, and 20 mM Tris-HCl pH 9.3. Thermo Sequenase also performed well in these conditions, though we later analogously optimized buffer B for it (described further below).
We next evaluated the efficiency of ddU-TAMRA, ddG-R110, ddC-R110, and ddA-TAMRA terminator incorporation by both Therminator and by Thermo Sequenase in buffer A using four corresponding synthetic targets (Fig. 4). Only Thermo Sequenase incorporated all four of these terminators efficiently, and so was chosen for all subsequent experiments. For Thermo Sequenase, optimal 1× terminator concentrations of these four terminators were 35 nM ddA-TAMRA, 4 nM ddC-R110, 4 nM ddG-R110, and 10 nM ddU-TAMRA (Fig. 5). We further optimized an extension buffer specifically for Thermo Sequenase using synthetic targets and the general approach outlined above (Fig. 6). The resulting 1× buffer B contained: 6 mM MgSO4, 5 mM (NH4)2SO4, 0.05% Triton X-100, 20 mM Tris-HCl pH 8.9, and the addition of 5% glycerol. The Thermo Sequenase concentration at which terminator incorporation was most specific was 0.0175 units/μl. An additional two terminators were also evaluated, choosing optimal concentrations of 4 nM ddU-R110 and 20 nM ddC-TAMRA for Thermo Sequenase (Fig. 7). Even with efficient and specific terminator incorporation, the accumulation of labeled extension primer is a function of the number of linear thermal cycles. The sum of the specific signals of both possible extension products (FP sum) of an assayed SNP plateaued at roughly 26 extension cycles (illustrated in Supplementary Fig. S3).
Figure 4:

Specificity of fluorescently labeled ddNTP terminator incorporation by Therminator and Thermo Sequenase, evaluated using synthetic targets SynFP_A, SynFP_C, SynFP_G, and SynFP_T.
Figure 5:

Specificity of terminator incorporation by Thermo Sequenase as a function of ddNTP concentration, evaluated using synthetic targets SynFPz_A, SynFPz_C, SynFPz_G, and SynFPz_T.
Figure 6:

Specificity of terminator incorporation by Thermo Sequenase as a function of buffer components and enzyme concentration. Each panel presents an evaluated reaction component curve for the selection of an optimum.
Figure 7:

Specificity of ddC-TAMRA and ddU-R110 incorporation by Thermo Sequenase as a function of ddNTP concentration, evaluated using synthetic targets SynFPz_A, SynFPz_C, SynFPz_G, and SynFPz_T.
Incorrect terminator incorporation becomes problematic with the use of decreasing concentration of either Exo I or CIAP for post-PCR cleanup (Fig. 8). Residual PCR primers and residual dNTPs can allow incorporation of a labeled terminator at a position other than the intended, interrogated variant site. The optimal concentration of each enzyme for specific terminator incorporation was 0.95 units per reaction, with no difference between heat inactivation at 80°C for 15 min versus 95°C for 30 min.
Figure 8:

Effect of CIAP and Exo I concentration on terminator incorporation specificity. Incorporation of specific and nonspecific fluorescently labeled ddNTP terminators are illustrated for synthetic template targets SynFP_G (A) and for SynFP_A (B).
Assay performance with production genotyping
We applied the iteratively optimized SNuPE assay to a set of 98 SNPs and indels, designing assays for each to genotype 2202 DNA samples. We selected PCR conditions for each using the approach described under Methods; 87 used AmpliTaq Gold/no betaine, 9 used AmpliTaq Gold/betaine, and 2 used Titanium Taq/betaine. For each desired variant assay, we then amplified synthetic targets designed to represent AA, AB (mixed), and BB genotypes for comparison of forward and reverse extension assay performance. The version with greatest FP sum was selected for a subsequent test of a sample of study genomic DNAs that had been extracted from whole blood. This screen of two 96-well plates evaluated 151 subjects (3 present in triplicate), 5 negative controls, and 30 synthetic targets (10 of each homozygote and 10 of the mixed/heterozygote). The latter were especially helpful for establishing AA, AB, and BB cluster positions and assay performance of rare SNPs (versus testing a novel assay of uncertain performance on genomic DNAs of unknown genotype). Of the 98 designed and tested assays, 85 yielded clean genotypes in study DNAs (an 87% assay conversion rate).
We proceeded to production genotyping with 77 of these SNP and indel assays (the subset that proved necessary for our work) on 2202 DNA samples to generate ∼170 000 total genotypes. We included as controls 67 duplicate genomic DNA pairs, observing two mismatched genotype calls (estimated error rate 0.0004). Independent of the SNuPE assays, we also genotyped the same DNA samples by Illumina Infinium MEGAEX array. Note that an array survey is more appropriate for assessment of genetic ancestry than customized assay of specific, required variants. Although not by our design, genotypes of 12 of the 77 variants assayed by SNuPE were also generated by the array, enabling comparison of genotype calls from an orthogonal method. One was errantly monomorphic by array. The remaining 11 SNPs yielded 17 346 duplicate genotypes with 43 discrepancies, a discrepancy rate of 0.003. These data support an accuracy for the SNuPE assay in line with that of other production genotyping approaches.
Six of the SNuPE assays that were genotyped in production had FP sums in assay development stages that we recognized could be improved by altering extension primer Tm. In the course of production genotyping, we evaluated extension primer Tm as an additional assay variable with potential to improve specific incorporation. We optimized six assays by evaluating and choosing higher Tm extension primers (Fig. 9). Overall, extension primer Tm’s of successful assays ranged from 52.6°C to 73.1°C, averaging 58.1°C. We estimate the optimal extension primer Tm design goal to be between 60°C and 65°C.
Figure 9:

Effect of extension primer Tm on FP sum. For each variant, the FP sum of the initial and of the optimized Tm is presented as an extended line.
A significant proportion of specific required variants typically fail to successfully convert for assay by any given alternative method. More than one approach is often necessary. As an independent example, among a set of 26 SNPs for which we had previously sought TaqMan assays, half were available predesigned and half required custom design. Among the custom set, six failed design, one passed design but failed actual assay, and the remaining six had good performance. Thus, 19 of 26 (73%) successfully converted for TaqMan assay, with an estimated error rate of 0.004 (four genotype mismatches among 1,139 duplicate genotype pairs).
Discussion
The application space of genotyping few required genetic variants in many DNA samples is not well served by current array or sequencing technologies, given the relatively high cost that would be incurred to generate the needed data set. Surprisingly few methods have been advanced to practical use for this application. For any given method, a specific required variant site can fail to successfully convert for assay; no single approach will successfully genotype all desired genetic variants. This motivated our adaptation of the SNuPE assay an additional practical alternative using relatively basic reagents and instrumentation. We present our implementation of this assay, advancing it from concept to practical usage with demonstrated performance. Our assay had a high conversion rate, able to accurately genotype an estimated 87% of SNPs and indels. Where an alternative method may fail to capture a given variant, this assay has a reasonable probability of success. A synthetic template system can be used for assay development and to generate reference genotypes, designating expected scatterplot cluster positions. Synthetic templates proved particularly valuable for rare variant assays. We employed the assay to generate 170,000 genotypes in routine production genotyping, with error rate estimates under 0.003 from duplicates as well as by comparison to orthogonal methods.
The application space for which this assay is particularly suited is genotyping a small set of genetic variants that are specifically required and without ready substitute, where ability to customize a nonproprietary assay may be at a premium to ensure performance. The ability to customize this assay is a highly desirable aspect because a specific genetic variant of particular interest can warrant effort to capture it. The SNuPE assay can be further adapted, e.g., to genotype variants within nonunique regions of a genome using nested PCR strategies [11, 13]. Scalability is also an advantage; the plate format of the SNuPE assay is amenable to automation. Ability to employ a plate reader that is nonproprietary and can be multipurposed as a general laboratory instrument is also an advantage. Relative disadvantages include the processing requirement of sequential reagent additions, and potentially also cost (48 cents per genotype in our application, more than half enzyme cost). Cost is an important consideration. Perspective of cost will vary with the application; e.g., the cost of targeted genotyping of a required variant may be minor relative to the cost of a genome-wide association study that led to its identification. The adapted SNuPE assay is a viable practical alternative for the application space of genotyping large numbers of samples and few SNP or indel variants.
Funding
This work was supported by awards from the V Foundation and the Veteran’s Administration. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding sources.
Conflict of interest statement. None declared.
Supplementary Material
References
- 1. Livak KJ. Allelic discrimination using fluorogenic probes and the 5’ nuclease assay. Genet Anal 1999;14:143–9. [DOI] [PubMed] [Google Scholar]
- 2. Orita M, Iwahana H, Kanazawa H. et al. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 1989;86:2766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kan YW, Dozy AM.. Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation. Proc Natl Acad Sci USA 1978;75:5631–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu DY, Ugozzoli L, Pal BK. et al. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc Natl Acad Sci USA 1989;86:2757–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greenwood AD, Southard-Smith EM, Galecki AT. et al. Coordinate control and variation in X-linked gene expression among female mice. Mamm Genome 1997;8:818–22. [DOI] [PubMed] [Google Scholar]
- 6. Kuppuswamy MN, Hoffmann JW, Kasper CK. et al. Single nucleotide primer extension to detect genetic diseases: experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proc Natl Acad Sci USA 1991;88:1143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haff LA, Smirnov IP.. Single-nucleotide polymorphism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry. Genome Res 1997;7:378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shumaker JM, Metspalu A, Caskey CT.. Mutation detection by solid phase primer extension. Hum Mutat 1996;7:346–54. [DOI] [PubMed] [Google Scholar]
- 9. Kwok PY. SNP genotyping with fluorescence polarization detection. Hum Mutat 2002;19:315–23. [DOI] [PubMed] [Google Scholar]
- 10. Bradley KM, Elmore JB, Breyer JP. et al. A major zebrafish polymorphism resource for genetic mapping. Genome Biol 2007;8:R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yaspan BL, McReynolds KM, Elmore JB. et al. A haplotype at chromosome Xq27.2 confers susceptibility to prostate cancer. Hum Genet 2008;123:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen X, Levine L, Kwok PY.. Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res 1999;9:492–8. [PMC free article] [PubMed] [Google Scholar]
- 13. Breyer JP, Dorset DC, Clark TA. et al. An expressed retrogene of the master embryonic stem cell gene POU5F1 is associated with prostate cancer susceptibility. Am J Hum Genet 2014;94:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.