

Summary
Patients with ALS show, in addition to the loss of motor neurons in the spinal cord, brainstem, and cerebral cortex, an abnormal depletion of energy stores alongside hypermetabolism. In this study, we show that bioenergetic defects and muscle remodeling occur in skeletal muscle of the SOD1G93A mouse model of ALS mice prior to disease onset and before the activation of muscle denervation markers, respectively. These changes in muscle physiology were followed by an increase in energy expenditure unrelated to physical activity. Finally, chronic treatment of SOD1G93A mice with Ranolazine, an FDA-approved inhibitor of fatty acid β-oxidation, led to a decrease in energy expenditure in symptomatic SOD1G93A mice, and this occurred in parallel with a robust, albeit temporary, recovery of the pathological phenotype.
Subject Areas: Drugs, Molecular Neuroscience, Cellular Neuroscience
Graphical Abstract
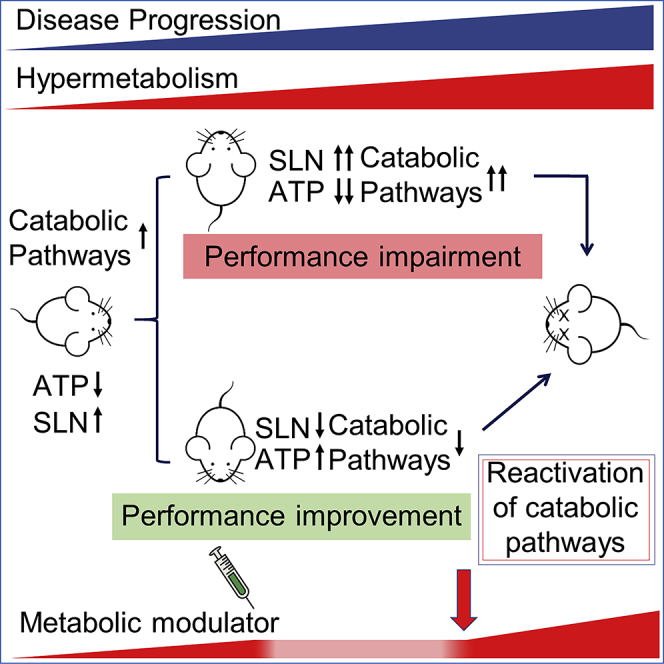
Highlights
-
•
Metabolic switch use occurs early in the skeletal muscle of SOD1G93A mice
-
•
Mitochondrial impairment precedes locomotor deficits and evokes catabolic pathways
-
•
Sarcolipin upregulation in presymptomatic SOD1G93A mice precedes hypermetabolism
-
•
Pharmacological modulation of hypermetabolism improves locomotor performance
Drugs; Molecular Neuroscience; Cellular Neuroscience
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive loss of upper and lower motor neurons. The loss of these neurons leads to the concomitant development of skeletal muscle atrophy (Rossi et al., 2015). ALS is a disease of obscure etiology, for which neither a cure nor a therapeutic option is available. ALS is a multifactorial disease and a number of mechanisms have been proposed to underpin disease pathology, including alterations in RNA metabolism, oxidative stress, axonal transport defects, neuroinflammation, impaired protein homeostasis, alterations in autophagic flux, aberrant cytoplasmic-nuclear shuttling, failure in DNA repair, excitotoxicity, mitochondrial dysfunction, and vesicular transport defects (Mejzini et al., 2019).
In spite of its elusive etiology, some well-described clinical signs in patients with ALS highlight promising lines of investigation to improve our understanding of disease. Notably, hypermetabolism (Steyn et al., 2018) and weight loss (Moglia et al., 2019) are associated with worse prognosis. However, weight loss, undernutrition, muscle atrophy, weakness, and reduced physical activity (Ioannides et al., 2016) appear incompatible with clinical studies reporting increased energy expenditure (EE) in patients with ALS (Bouteloup et al., 2009, Desport et al., 2005, Funalot et al., 2009, Kasarskis et al., 2014). The paradox may be reconciled by assuming that skeletal muscle plays a pathological role in ALS, contributing to defective energy metabolism and a derangement of basal metabolic rate. Indeed, skeletal muscle is a key determinant of whole-body metabolic rate (Zurlo et al., 1990).
In this context, the early events underlying defective muscle metabolism in ALS as well as the major molecular mechanisms that cause ALS-associated hypermetabolism are still unknown. We therefore undertook this investigation to provide insights into the early pathological events involved in ALS by examining the mechanisms responsible for maladaptive muscle oxidative metabolism and disease progression. Finally, in this study, we provided evidences to show that pharmacological modulation of metabolism can curtail excessive EE and improve ALS pathology, thereby demonstrating that limiting hypermetabolism could be a promising therapeutic target in ALS.
Results
An Increase in Fatty Acid Oxidation and Glucose Intolerance Precedes Hypermetabolism in SOD1G93A Mice
To investigate the whole-body metabolic status and fuel oxidation in SOD1G93A mice, we conducted indirect calorimetry over 2 or 3 continuous days. Measures were obtained from animals at the early presymptomatic (55 days of age) and symptomatic (120 days of age) stages of disease. Hypermetabolism, defined as an increase in total EE or resting energy expenditure (REE), was not detectable in early presymptomatic mice (Figures 1A–1C) but was strongly evident in symptomatic mice (Figures 1D–1F). Analysis of the respiratory exchange ratio (RER) revealed a significant genotype effect, with RER being reduced in both early presymptomatic and symptomatic SOD1G93A mice when compared with wild-type control mice (Figures 1G and 1H). These results are indicative of enhanced fatty acid oxidation and predominant use of fatty acids as fuel source in ALS mice.
Figure 1.

Energy Expenditure and Metabolic Profile of WT and SOD1G93A Mice
(A–L) (A) Energy expenditure profiles of early presymptomatic SOD1G93A mice (55 days) and their wild-type control littermates (WT). Black bar indicates dark cycle period. Mean (B) energy expenditure (EE) and (C) resting energy expenditure (REE) calculated from (A). (D) Energy expenditure profiles of symptomatic SOD1G93A mice (120 days) and their wild-type control littermates (WT). Mean (E) EE and (F) REE calculated from (D). (G and H) Mean RER from (A) and (D), respectively. (I) Gene expression changes in the tibialis anterior (TA) muscle from SOD1G93A mice and their wild-type control littermates. (J) Heatmap depicting expression of genes from the Reactome category “Glycolysis.” Shown genes are differentially expressed with adjusted p < 0.05. (K) Network plot of the "Pyruvate metabolism and Citric Acid (TCA) cycle" pathways. Nodes are genes colored based on log2FC, edges are knowledge-based interactions annotated by Reactome. (L) Glucose tolerance test performed in SOD1G93A and in their wild-type control littermates (WT) at 55, 90, and 120 days of age, respectively. Data in (B), (C), (E), (F), (G), and (H) are presented as mean ± SEM, ∗∗∗p < 0.0001, unpaired Student's t test.
After confirmed metabolic alteration in presymptomatic SOD1G93A mice and since skeletal muscle is the major factor affecting EE also at rest (Zurlo et al., 1990) we carried out RNA sequencing to assay gene expression changes in the tibialis anterior (TA) of SOD1G93A mice at the onset stage of disease, when compared with their wild-type littermate controls. Gene enrichment analysis, using Reactome categories, highlighted that the gene sets most significantly enriched among downregulated genes were associated with glucose metabolism (Figure 1I). In particular, we observed downregulation of key glycolysis pathway genes (Figures 1J and 1K). The decrease in the expression of glycolytic pathway genes was further supported through our assessment of glucose tolerance in SOD1G93A mice. At the early presymptomatic stage, SOD1G93A mice already show a marked decrease in blood glucose clearance with respect to wild-type control littermates (Figure 1L, upper panel). This metabolic alteration dramatically worsened with disease progression in SOD1G93A mice (Figure 1L, middle and lower panels).
ALS Skeletal Muscles Undergo Metabolic Reprogramming
Based on results obtained through indirect calorimetry and transcriptome analysis, we aimed to define the timing of the onset of metabolic changes in skeletal muscle of SOD1G93A mice by analyzing the oxidative capacity of the glycolytic TA at different time points. We found that in situ NADH-TR and SDH activities were dramatically increased in SOD1G93A mice at the early presymptomatic stage (55 days) and to a higher degree at the later stages of the disease (Figures 2A, 2B, S1A, and S1B).
Figure 2.

Muscle Metabolism Switches from Glycolytic to Oxidative in SOD1G93A Mice
(A) Representative NADH-tetrazolium reductase activity staining on tibialis anterior (TA) cross sections obtained from 55-, 90-, and 150-day-old SOD1G93A mice and their wild-type control littermates (WT). Scale bar, 100 μm, n = 3.
(B) Representative SDH activity staining on TA cross sections of 55-, 70-, 90-, and 120-day-old SOD1G93A mice and their wild-type control littermates (WT). The darker color indicates the site of SDH activity. Scale bar, 100 μm, n = 3.
(C–E) (C) Representative images of TA sections from 55-, 90-, and 150-day-old SOD1G93A and wild-type (WT) mice, immunostained with laminin (green) and the mitochondrial marker ATPB (red); nuclei were detected with Hoechst 33342 (blue). Scale bar, 100 μm, n = 3. Expression level of mRNAs coding for (D) Nor1 and Myoglobin and (E) MyHCI, MyHCIIa, MyHCIIIx, and MyHCIIb in TA sections obtained from SOD1G93A at the indicated ages (at least n = 4). Data are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, compared with wild-type control littermates at the same age (arbitrarily set at 1), unpaired Student's t test.
Also see Figures S1A–S1C.
In line with the rise in muscle oxidative capacity, we detected, by immunofluorescence analysis, a consistent increase in the intensity of ATPB, a known mitochondrial marker, in the TA muscle of SOD1G93A mice (Figures 2C and S1C). Accordingly, a striking increase in mitochondrial mass was observed, defined by an increase in the amount (μg) of mitochondria per milligram of tissue as well as an increase in the content of mitochondrial DNA, in different muscles of SOD1G93A mice during the course of the disease (Figures S2A–S2C). Interestingly, this phenomenon occurred mainly in glycolytic muscles and not in the oxidative soleus muscle.
The switching of skeletal muscle metabolism toward an oxidative phenotype in SOD1G93A mice was further corroborated by the upregulation in neuron-derived orphan nuclear receptor (Nor1) mRNA in the TA (Figure 2D). This transcription factor is known to promote muscle remodeling toward oxidative metabolism by activating pathways critical for adaptation to exercise or energy deficit (Goode et al., 2016). The upregulation of Nor1 in SOD1G93A mice occurred alongside an upregulation in mRNA encoding for myoglobin (Figure 2D) (Pearen et al., 2013). Moreover, we found significant alterations in the expression of mRNAs encoding myosin heavy chain isoforms. Specifically, we observed a reduction in the expression of the glycolytic isoform MyCHIIb and an increase in the expression of the oxidative/intermediate isoforms MyHCIIa and MyHCIIx (Figure 2E). No significant alterations were observed in the expression of the oxidative isoform MyHCI (Figure 2E). Overall, these data show that, in SOD1G93A mice, glycolytic muscles undergo a profound rearrangement in their physiology throughout the course of disease.
Mitochondrial Bioenergetics Are Perturbed in Skeletal Muscle of Early Presymptomatic SOD1G93A Mice
The metabolic modifications of skeletal muscle observed in SOD1G93A animals mirror adaptations to endurance training, wherein a switch from glycolytic to oxidative metabolism occurs (Granata et al., 2018, Widmann et al., 2019). During endurance training, this metabolic switch is accompanied by an increase in mitochondrial mass alongside significant remodeling of myofibers and a preferential use of fatty acids as fuel (Granata et al., 2018, Widmann et al., 2019). Although we found that mitochondrial mass in glycolytic TA muscle of SOD1G93A mice increased during the disease course, we also noted an early mitochondrial bioenergetic defect. In this regard, we found that total ATP was significantly lower in the TA of SOD1G93A mice at the early symptomatic through to the mid-stage of disease when compared with age-matched wild-type control mice (Figure 3A). Moreover, we observed a marked reduction in all mitochondrial functionalities in the TA of SOD1G93A mice at the early presymptomatic phase of the disease. Similarly, early reductions in mitochondrial function were also observed in other glycolytic muscles, gastrocnemius (GNM) and extensor digitorum longus (EDL) (Figures S3A and S3B). Interestingly, bioenergetic defects only become detectable in the spinal cord at onset of the pathology (Figures 3C and S3B).
Figure 3.

Mitochondrial Bioenergetic Failure Occurs in Skeletal Muscle of Early Presymptomatic SOD1G93A Mice
(A) Fluorometric measurements of total ATP in the tibialis anterior (TA) muscle from SOD1G93A and wild-type age-matched control mice (WT) at different ages. Data were analyzed by unpaired t test and presented as mean ± SEM, ∗∗∗p < 0.0001 compared with age-matched WT mice (n = 4 independent experiments).
(B) Coupling assay on isolated mitochondria purified from TA.
(C) Spinal Cord of SOD1G93A mice at the indicated ages. Mitochondrial respiration stages are reported as basal respiration (state 2), maximal coupled respiration (state 3), respiration due to proton leak (state 4o), and maximal uncoupled respiration (state 3u). Data are expressed as % Oxygen Consumption Rate (OCR), and 100% was arbitrarily assigned to values obtained from age-matched wild-type mice, ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, unpaired Student's t test (n = 5 independent experiments).
(D) Activity of electron transport chain complexes I, II\III, and IV of mitochondria obtained from TA.
(E) Spinal Cord of SOD1G93A and wild-type (WT) mice at the indicated ages. Values were normalized to citrate synthase activity and presented as mean ± SEM, ∗∗p < 0.001, ∗∗∗p < 0.0001 compared with age-matched WT mice (arbitrarily set at 100%), unpaired Student's t test (n = 5 independent experiments).
Also see Figures S2A–S2C and S3A–S3C.
To further investigate the mitochondrial bioenergetic deficit in SOD1G93A mice, we assessed respiration states of mitochondria isolated from skeletal muscle and spinal cord of SOD1G93A mice and wild-type litter matched controls. We observed a decrease in maximally coupled respiration (state 2), thereby confirming the impairment in ATP production. Moreover, maximal respiration (state 4), determined through the addition of the uncoupling agent FCCP, appeared to be significantly compromised in mitochondria obtained from glycolytic TA, GNM, and EDL of presymptomatic SOD1G93A mice (Figures 3B, S3A, and S3B). Interestingly, mitochondria purified from a purely oxidative muscle, the soleus, were not affected (Figures S3A and S3B). With the aim of characterizing the alterations in the electron transport chain, we performed a spectrophotometric analysis of the functional activities of the mitochondrial complexes and observed a specific impairment in Complex I in mitochondria purified from the TA (Figure 3D) and GNM (not shown) of early presymptomatic SOD1G93A mice. Again, this dysfunction was evident in skeletal muscle from early presymptomatic SOD1G93A mice, whereas defects in Complex I activity were only observed in the spinal cords of SOD1G93A mice at the symptomatic and end stage of disease (Figure 3E).
Next, we performed Blue Native polyacrylamide gel electrophoresis (BN-PAGE) analysis of digitonin-extracted mitochondrial membranes obtained from the TA of SOD1G93A mice and their wild-type control littermates at different ages (Figure S3C). We found no difference in the assembly of electron transport chain complexes or supercomplexes in mitochondria obtained from SOD1G93A mice when compared with age-matched controls. Finally, bioenergetic assessment of primary cultures of skeletal muscle satellite cells isolated from early presymptomatic SOD1G93A mice (55 days of age) revealed significant decreases in basal respiration, ATP production, maximal respiration, and spare respiratory capacity when compared with those isolated from their wild-type control littermates (Figures 4A and 4B). The decline in maximal respiration and spare respiratory capacity suggests that ALS may inhibit the rapid adaptation of skeletal muscle satellite cells to metabolic changes. Thus, although the number of mitochondria in the whole muscle appears to increase in SOD1G93A mice, mitochondrial metabolism is compromised early in the course of the disease, leading to ATP deficit and impairments in OXPHOS functionality.
Figure 4.

Mitochondrial Bioenergetics Failure Occurs in Skeletal Muscle Satellite Cells Isolated from Early Presymptomatic SOD1G93A Mice
(A) Representative trace of oxygen consumption rate (OCR) in skeletal muscle satellite cells isolated from early presymptomatic SOD1G93A mice (55 days) and their wild-type control littermates (WT).
(B) Individual parameters obtained for basal respiration, ATP production, maximal respiration, and spare respiratory capacity. Each data point represents an OCR measurement. Data are presented as mean ± SEM, ∗∗p < 0.001, ∗∗∗p < 0.0001 relative to WT control, p values obtained from unpaired Student's t test. Data from four independent experiments, with each sample tested in quadruplicate.
Muscle Mitochondrial Deficit in SOD1G93A Mice Occurs in Parallel with an Activation of Molecular Pathways that Compensate for Energy Failure
In normal physiology, a decrease in ATP production (mitochondrial alterations) leads to an enhancement in AMPK activity to promote the binding of AMP (and ADP) to the kinase subunit. This in turn induces the phosphorylation of a wide range of downstream proteins that eventually lead to the activation of catabolic pathways (Hardie et al., 2016). In particular, pharmacological or genetic inhibition of Complex I is sufficient to activate AMPK (Hardie et al., 2016, Thomas et al., 2018). Therefore, in light of the bioenergetic defects observed in early presymptomatic mice and the specific impairment of Complex I activity, we analyzed the extent of AMPK activation in ALS mice.
Glycolytic skeletal muscles from SOD1G93A mice displayed a robust activation of AMPK, long before the appearance of locomotor symptoms (Figures 5A and S4). AMPK activation occurred in parallel with the phosphorylation/inactivation of the AMPK downstream target Acetyl-CoA Carboxylase (ACC), which catalyzes the first step of fatty acid synthesis (Figures 5A and S4). Moreover, we found an upregulation in carnitine palmitoyltransferase 1 (CPT1) in the skeletal muscle of presymptomatic SOD1G93A mice (Figures 5A and S4). Finally, in support of a metabolic shift toward lipid use, we observed a decrease in the expression of Glucose Transporter type 4 (Glut4) mRNA and the concomitant increase in pyruvate dehydrogenase kinase 4 (Pdk4) mRNA in skeletal muscle of presymptomatic mice (Figure 5B).
Figure 5.

Alterations in Expression of Metabolic Targets Occurs Early in the tibialis anterior (TA) of SOD1G93A Mice
(A) Representative western blot images of pAMPK, AMPK, pACC, ACC, and CPT1 (upper panel) in the TA of SOD1G93A and wild-type mice at 55 days of age. GAPDH was used as loading control. Densitometric analysis of pAMPK/AMPK and pACC/ACC ratios and CPT1 expression (lower panel) from n = 4 independent experiments.
(B) Expression of mRNAs coding for Pdk4 and Glut4 in the TA of SOD1G93A mice relative to wild-type litter-matched controls. Data presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001 when compared with control littermates at the same age (arbitrarily set at 1), unpaired Student's t test.
Also see Figures S4 and S5A–S5C.
Sarcolipin Is an Early Player in ALS Metabolic Alterations
Recent studies have outlined the pivotal role of sarcolipin (SLN) in orchestrating the transition toward oxidative metabolism in skeletal muscle (Maurya et al., 2018). In line with the literature (Babu et al., 2007), we found that SLN is highly expressed in the TA of wild-type control mice until 14 days of age, after which it gradually declines (Figure 6A). By contrast, in SOD1G93A mice, the expression level of SLN remained detectable in TA beyond 14 days of age, increasing during the course of the disease (Figure 6A). Indeed, in SOD1G93A mice, SLN overexpression was strongly upregulated through to the end stage of disease in all glycolytic and oxidative muscles assessed. This is in contrast to the expression profile of SLN in age-matched wild-type control mice, where SLN was only detectable in the oxidative soleus (Figure 6B). Of note, we did not observe any significant variation in mRNA expression of phospholamban (PLN) or myoregulin (MLN) (Figure S5A), muscular micropeptides that function similarly to SLN. Thus, our data suggest that SLN is the only SERCA-regulatory micropeptide involved in ALS.
Figure 6.

Upregulation of Sarcolipin in ALS Occurs before Denervation in SOD1G93A Mice
(A and B) Representative western blots of sarcolipin (SLN) protein expression in the (A) tibialis anterior (TA) or (B) Extensor digitorum longus (EDL), gastrocnemius (GNM), and soleus muscle of SOD1G93A (+) and wild-type mice (−) at the indicated ages.
(C) Western blot of pCaMKII and CaMKII in the TA of SOD1G93A (+) and wild type mice (−) at the indicated ages; GAPDH was used as loading control in (A–C).
(D) SLN protein expression in different muscle biopsies derived from MELAS, neuropathy (N), not Limb Girdle Muscle Dystrophy (LGMD) and patients with ALS (1 = SOD1D90A mutation, soleus; 2 = sporadic, vastus lateralis; 3 = Valosin Containing Protein (VCP) mutation, vastus lateralis; 4 = C9ORF72 repeat expansions, soleus; 5 = sporadic, vastus lateralis; 6 = sporadic, rectus femoralis).
(E) Densitometric analysis of (D) normalized to GAPDH protein expression. Data are presented as a percentage, where an arbitrary value of 100% was assigned to patients with ALS, ∗∗∗p < 0.0001, one-way parametric ANOVA and Tukey's post hoc test.
(F) Expression of mRNAs coding for Sarcolipin (Sln), Acetylcholine Receptor α subunit (Achrα), Acetylcholine Receptor ϵ subunit (Achrϵ), Voltage-gated Na channels 1.5 (Nav1.5), Histone Deacetylase 4 (Hdac4), Myogenin, MuscleRING Finger-1 (Murf1), and Myostatin in the TA of SOD1G93A mice relative to their wild-type control littermates, ∗p < 0.05, ∗∗p < 0.001 ∗∗∗p < 0.0001 when compared with age control littermates (arbitrarily set at 1).
(G) Expression of mRNAs coding for Sln and denervation markers (Achrα, Nav1.5, Hdac4, and Myogenin) in the TA of wild-type mice 3 and 15 days after sciatic nerve axotomy. Data are expressed as the ratio between the average of values from not axotomized and axotomized mice, ∗p < 0.05, ∗∗p < 0.001 when compared with control not axotomized mice (arbitrarily set on at 1), and ##p < 0.001 between mice after 3 and 15 days from axotomy. All data are presented as mean ± SEM, unpaired Student's t test (E) and parametric ANOVA and a Bonferroni post hoc test (F), n = 3 or 4 independent experiments.
See also Table 1.
An upregulation in SLN is known to trigger an increase in cytosolic calcium concentration in skeletal muscle (Maurya and Periasamy, 2015). To this end, we found a significant activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) in skeletal muscle of SOD1G93A at the presymptomatic stage of the disease, when the glycolytic muscle was highly oxidative (Figure 6C). Moreover, we observed a significant increase in PGC-1α protein expression in the TA of SOD1G93A mice at the presymptomatic phase of disease (70 days of age) as well as throughout the course of the disease (Figure S5B).
The expression pattern of mRNAs coding for different isoforms of SERCA pumps reflects the metabolic profile of muscle. SERCA2a and SERCA2b isoforms that are normally associated with oxidative muscle, were more highly expressed in the TA from SOD1G93A mice, whereas the glycolytic muscle-associated SERCA1 isoform was downregulated at the presymptomatic stage (Figure S5C). It is noteworthy that SLN selectively binds SERCA2 isoforms, interacting with these to trigger the transition of skeletal muscle from glycolytic to oxidative metabolism (Anderson et al., 2016). Finally, highly relevant was the finding that SLN expression was upregulated in muscle biopsies obtained from six patients affected by different forms of ALS (for details see Table 1). Indeed, all ALS biopsies showed higher expression levels of SLN when compared with control patients affected by other muscular disorders (Figures 6D and 6E).
Table 1.
Demographics of Patients with ALS
| Patient | Age (years) | Sex | ALS Mutation | Months from Diagnosis | Muscle (Biopsies Were Done on Limb Muscle where the Disease Began) | Patient Information |
|---|---|---|---|---|---|---|
| 1 | 47 | F | SOD1D90A | 12 | soleus dx | Spinal onset |
| 2 | 72 | F | Sporadic | 42 | vastus lateralis dx | Spinal onset |
| 3 | 50 | M | VCP mutation | 54 | vastus lateralis dx | Spinal onset with IBM (inclusion body myositis) and Paget's disease |
| 4 | 62 | F | C9ORF72 (58 repeats) | 33 | soleus dx | Spinal onset with progressive muscle atrophy |
| 5 | 77 | F | Sporadic | 19 | vastus lateralis dx | Spinal onset |
| 6 | 71 | F | Sporadic | 14 | rectus femoris dx | N/A |
Sarcolipin Overexpression in SOD1G93A Mice Is Not Related to Muscle Denervation
Several pathological conditions that lead to skeletal muscle disuse and subsequent atrophy affect not only muscle mass but also muscle metabolism (Tintignac et al., 2015). SLN has been described to be a key mediator of metabolic modifications occurring in unloaded/disused muscle (Fajardo et al., 2017). Thus, to discriminate whether SLN upregulation is causally linked to the altered metabolic state observed in ALS mice, or whether it is merely a consequence of early denervation, we analyzed the expression levels of several denervation/atrophy markers. Our results demonstrate that, in TA from SOD1G93A mice, SLN is upregulated at the early presymptomatic stage, whereas mRNAs coding for Hdac4, Nav1.5, Myogenin, AchRε, AchRα, Murf1, and Myostatin, all associated with the denervation process, are only increased at the later stages of disease (Figure 6F).
In order to further decipher the relationship between denervation and metabolic alterations in skeletal muscle from ALS mice, we used an experimental paradigm of acute denervation. As shown in Figure 6G, sciatic nerve axotomy in control mice evoked an upregulation in denervation markers after 3 days, whereas SLN was only found to increase after 15 days. Overall, our data strongly suggest that, in ALS, muscle metabolic reprogramming is orchestrated by SLN and this occurs independent of denervation.
Altered Muscle Performance in SOD1G93A Mice Is Related to Hypermetabolism
Given the altered bioenergetic profiles observed in SOD1G93A mice, we next tested whether pharmacological targeting of altered energy metabolism could be a beneficial therapeutic approach in ALS. To address this aim, we chronically administered ranolazine (RAN) to SOD1G93A mice. RAN is a US Food and Drug Administration (FDA)-approved drug that is used to treat ventricular hypertrophy, and it has been shown to exert cardioprotective effects by facilitating higher energy production under conditions of lower oxygen supply (Stanley, 2002). This drug decreases β-oxidation, thereby restoring the use of glucose as fuel (McCormack et al., 1998).
In a preliminary dose-response study, a small cohort of SOD1G93A mice and age-matched wild-type control mice received daily intraperitoneal (i.p.) injections, for seven consecutive days, of three different doses of RAN (25, 50, 100 mg/kg). Treatment commenced at an age that corresponds to the symptomatic stage of the disease in SOD1G93A mice (110 days). Bioavailability of RAN in plasma was assessed by high-performance liquid chromatography (HPLC), and results confirmed a dose-dependent increase (Table 2). RAN concentration values were consistent with those registered in more extended pharmacokinetics and pharmacodynamics studies reported by others (Patel and Hasumati, 2015). Analysis of grip strength and SLN expression were used as readouts to determine the efficacy of RAN treatment. Strikingly, SOD1G93A mice receiving 50 and 100 mg/kg of RAN showed a marked increase in muscular strength after 2 days of treatment. This improvement was maintained throughout the 7-day treatment period (Figure S6A). Similar results were obtained with a lower dose of RAN (25 mg/kg), which showed a significant, although delayed, efficacy. Furthermore, in the TA muscle of SOD1G93A mice, SLN expression was reduced after 7 days of treatment. This was most evident at a RAN dose of 50 mg/kg (Figure S6B). As such, 50 mg/kg of RAN was selected as the most effective dose.
Table 2.
HPLC Determination of Ranolazine (RAN) Plasma Concentrations 12 h after i.p. Administration of Ranolazine at 25, 50, or 100 mg/kg
| RAN Dose (mg/kg) | WT (μM) | SOD1G93A (μM) |
|---|---|---|
| 25 | 0.63 ± 0.16 | 0.79 ± 0.11 |
| 50 | 1.22 ± 0.20 | 1.30 ± 0.18 |
| 100 | 3.33 ± 0.21 | 2.87 ± 0.17 |
Data are expressed as mean ± SD, n = 5 per group.
A second, larger cohort of SOD1G93A mice received 50 mg/kg RAN from the onset stage of disease (90 days) through the end stage of disease. As shown in Figure 7A, RAN treatment improved hanging grid performance in SOD1G93A mice up until the fully symptomatic stage of the disease (132 days). However, this improvement was not sustained; hanging grid performance in RAN-treated SOD1G93A mice was comparable with that of non-treated SOD1G93A mice by the end stage of disease (Figure 7A). Interestingly, RAN exerted similar effects on wild-type mice (Figure S7). Despite improving hanging grid performance in SOD1G93A mice, RAN failed to improve survival (Figure 7B) and it did not provide weight changes (data not shown). Interestingly, RAN treatment improves glycemia and reverses glucose intolerance in murine models of metabolic alterations without modifying body weight development (Batran et al., 2019, Ning et al., 2011).
Figure 7.

Ranolazine Improves Locomotor Abilities but Does Not Affect Survival
(A) Locomotor abilities of SOD1G93A mice receiving daily intraperitoneal injections of Ranolazine 50 mg/kg (SOD1G93A RAN) or physiological solution (SOD1G93A) evaluated by hanging grid test at the indicated days. Performance was evaluated from 90 days of age (start of treatment) to the end stage of disease. Data are presented as mean ± SEM, ∗p < 0.05 ∗∗p < 0.001 ∗∗∗p < 0.0001 when compared with age-matched SOD1G93A untreated mice, and #p < 0.05 ##p < 0.01 ###p < 0.0001 when compared with SOD1G93A mice at the beginning of treatment. (n = 12 SOD1G93A, n = 12 SOD1G93A + RAN).
(B) Kaplan-Meier survival curve of SOD1G93A mice and their wild-type control littermates (WT) receiving daily intraperitoneal injections of Ranolazine 50 mg/kg (SOD1G93A RAN and WT RAN) or physiological solution (SOD1G93A and WT). p Values were obtained using parametric two-way ANOVA with Bonferroni post hoc test (A) or one-way ANOVA with Bonferroni post hoc test (B).
Also Figure S7.
It is noteworthy that treatment efficacy followed the trend of expression/activation of muscle metabolic markers. Indeed, after 30 days of treatment with RAN, SLN was markedly downregulated and CaMKII and AMPK were inactivated by dephosphorylation, whereas the AMPK target, ACC, was dephosphorylated and hence reactivated (Figures 8A and 8B). Similarly, expression levels of mRNAs encoding myosin subtypes and SERCA pumps were decreased in response to RAN (Figure 8C), whereas mRNAs encoding Nor1 and Glut4 returned to levels similar to that observed in wild-type control mice (Figure 8C). In line with RAN's known mechanism of action in improving glucose metabolism, fluorometric determination of total ATP content showed a net increase in ATP concentration in the TA from symptomatic, 120-day-old SOD1G93A mice when compared with their wild-type control littermates (Figure 8D).
Figure 8.

Chronic Ranolazine (RAN) Administration Restores Metabolic Homeostasis
Mid-term evaluation (120 days) of SOD1G93A mice receiving daily RAN (50 mg/kg i.p.) from 90 days of age.
(A) Western blot analysis of SLN, pCaMKII, CaMKII, pAMPK, AMPK, pACC, and ACC in the tibialis anterior (TA) of SOD1G93A mice (+) and wild-type control littermates (−) receiving daily RAN (+) or vehicle (−) at 120 days of age. GAPDH was used as loading control.
(B) Densitometric analysis of data obtained in (A). Data are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.001 when compared with wild-type untreated mice (arbitrarily set on at 1) and #p < 0.05 ##p < 0.001 when compared with SOD1G93A untreated mice (n = 6 independent experiments), parametric one-way ANOVA and a Bonferroni post hoc test.
(C) Expression of mRNAs coding for Nor1, Glut4, Serca1, Serca2a, Serca2b, MyHCI, MyHCIIa, MyHCIIx, MyHCIIb in the TA of SOD1G93A mice and wild-type control littermates treated with RAN (SOD1G93A, RAN and WT RAN) or not treated with RAN (SOD1G93A and WT) at 120 days of age (n = 4 for each genotype/treatment). Data are presented as mean ± SEM, ∗∗p < 0.001 when compared with wild-type untreated mice (arbitrarily set on at 1) and #p < 0.05 when compared with SOD1G93A untreated mice (n = 6 independent experiments), parametric one-way ANOVA and a Bonferroni post hoc test.
(D) Fluorometric measurement of total ATP in the TA of SOD1G93A mice and wild-type control littermates receiving daily RAN (+) or vehicle (−) at 120 days of age. Data were analyzed by one-way ANOVA with multiple comparisons using Dunnet's t test and Tukey's HSD (Honestly Significantly Different) post hoc test. Data presented as mean ± SEM, ∗∗∗p < 0.0001 when compared with WT untreated mice and ##p < 0.001 when compared with SOD1G93A untreated mice (n = 4 independent experiments).
(E) Energy expenditure profile (EE) of WT and SOD1G93A treated with RAN or vehicle at 120 days of age. Black bar indicates the dark cycle period.
(F–H) (F) Percentage of increase in EE between 55 and 120days of age, and effects of RAN treatment (initiation of treatment marked with an arrow) from day 90 onward. Mean of (G) EE and (H) Resting EE (REE) obtained from WT and SOD1G93A mice in (E). Data are presented as mean ± SEM, ∗∗∗p < 0.0001 when compared with WT mice; ###p < 0.0001 when compared with SOD1G93A mice (at least n = 4 animals), two-way ANOVA with Tukey post hoc test.
(I–K) (I) Respiratory exchange ratio (RER) demonstrating the extent of lipid versus carbohydrate oxidation. Data are presented as mean ± SEM, ∗∗∗p < 0.0001 when compared with WT mice; (at least n = 4 animals), two-way ANOVA with Tukey post hoc test. Long-term evaluation (150 days) of RAN treatment started at 90 days of age: (J) western blot analysis of SLN, pCaMKII, CaMKII, pAMPK, AMPK, pACC, and ACC in the TA of SOD1G93A (+) and wild-type (−) RAN-treated (+) or untreated (−) mice. GAPDH was used as loading control. (K) Densitometric analysis of data in (I). Data are presented as mean ± SEM, ∗∗p < 0.001 when compared with WT untreated mice (arbitrarily set on at 1) and #p < 0.05 when compared with SOD1G93A untreated (n = 6 independent experiments), parametric one-way ANOVA and a Bonferroni post hoc test.
Given that progressive changes in muscle metabolic markers in SOD1G93A mice occurred alongside the presentation of hypermetabolism, we next aimed to determine whether RAN would attenuate hypermetabolism in SOD1G93A mice. Indeed, indirect calorimetry analysis showed that hypermetabolism was partially abrogated after treatment with RAN (Figures 8E and 8F). RAN restored EE (Figure 8G) and REE (Figure 8H) but had no effect on RER (Figure 8I). Surprisingly, the weakening of RAN effects during the end stage of the disease was paralleled with an increase in the expression of SLN, the reactivation of CaMKII and AMPK, and thus ACC inactivation (Figures 8, 8J, and 8K). Overall, these results show that decreasing EE is linked to an improvement in grip strength in SOD1G93A mice. Our data highlight the potential relevance of pharmacological interventions aimed at counteracting hypermetabolism in ALS.
Discussion
In this study, we detail the existence of early events responsible for the remodeling of myofiber-type composition and defective energy metabolism in the SOD1G93A mouse model of ALS. Our observations are in agreement with previous work demonstrating a shift in fast-to-slow muscle fiber type composition (Peggion et al., 2017), as well as aberrant mitochondrial metabolism (Salvatori et al., 2017) and alterations in muscle glucose metabolism in response to muscle-restricted overexpression of SOD1G93A (Dobrowolny et al., 2008, Dobrowolny et al., 2018). Moreover, in line with the derangement in whole-body EE that we identified in symptomatic SOD1G93A mice, hypermetabolism has been described in many patients with ALS (Bouteloup et al., 2009, Desport et al., 2005, Funalot et al., 2009, Jésus et al., 2018, Steyn et al., 2018) and often associated with dyslipidemia (Dupuis et al., 2008). Notably, prognosis is less favorable in hypermetabolic patients (Steyn et al., 2018), whereas dyslipidemia/hyperlipidemia may confer protection and increase survival (Dupuis et al., 2008).
Given that skeletal muscle is a major determinant of whole-body energy metabolism (Zurlo et al., 1990), we focused our investigation on the contribution of skeletal muscle to SOD1G93A metabolic dysfunction. Indeed, we found some prodromal molecular signatures of oxidative metabolism at 55 days of age, including an initial increase in NADH-TR and SDH activities and an increase in mitochondrial mass in fast-twitch glycolytic muscles. Thus, we identified a fast-to-slow skeletal muscle fiber transition in the early presymptomatic stage; this pathological rearrangement of muscle phenotype might partially account for the marked increase of fatty acid β-oxidation observed before disease onset. However, according to previous evidence (Doshi et al., 2017), hypermetabolism occurs in SOD1 G93A mice only at a symptomatic stage of the disease, whereas it is undetectable at 55 days of age. It should be considered that, around 55 days of postnatal life, the skeletal muscle of SOD1 G93A mice are not yet fully oxidative.
Skeletal muscle is a dynamic tissue that is highly adaptable to environmental changes and to alterations in energy need. Physical exercise is a key modulator of muscle plasticity, as enduring contractile activity, it is a powerful stimulus for enhancing mitochondrial biogenesis (Hood, 2009). This condition upregulates mitochondrial enzymes involved in fatty acid β-oxidation (Lundsgaard et al., 2018) to promote myofiber switching from fast (glycolytic) to slow (oxidative) fiber types (Flück, 2006). In this view, the muscle alterations that we have observed in SOD1G93A mice appear to mimic a state of long-lasting endurance exercise. Indeed, Palamiuc et al. (2015) have previously reported increased endurance capacity in SOD1G86R mice at an advanced phase of disease. Although it remains unknown as to why there is a shift in muscle fiber type, and enhanced fatty acid utilization in the absence of environmental pressure and/or physical exercise in SOD1G93A mice, this exercise-mimetic phenotype may be associated with aberrant energy needs that may be associated with early mitochondrial dysfunction and/or SLN overexpression.
Mitochondrial Dysfunction
In light of the critical role that mitochondria play in muscle metabolism and plasticity, it is conceivable that early alterations in muscle mitochondria might contribute to altered energy homeostasis in ALS. In agreement with this hypothesis, we report mitochondrial impairments in skeletal muscle in early presymptomatic SOD1G93A mice, which occur prior to the presentation of motor symptoms. Recently, an association between myopathy and defective mitochondrial function has been described in a different ALS mouse model (Genin et al., 2019), thus underlining the importance of muscle bioenergetics in neuron motor disease. In line with premature atrophy and loss of function of fast-twitch fibers (Hegedus et al., 2008, Palamiuc et al., 2015, Peggion et al., 2017), we detected early mitochondrial metabolic alterations in glycolytic EDL, TA, and GNM but not in oxidative soleus. Interestingly, skeletal muscle satellite cells isolated from early presymptomatic SOD1G93A mice also exhibited defective bioenergetic profiles. The drop in energy production in these skeletal muscle stem cells and their inability to respond to metabolic changes might affect muscle regeneration in response to denervation as previously described in ALS pathology (Tsitkanou et al., 2016, Yin et al., 2013).
We found an early impairment of Complex I activity in skeletal muscle of SOD1G93A mice. An impairment of Complex I activity has previously been described in muscle biopsies from patients with sporadic ALS (Wiedemann et al., 1998). Moreover, functional alterations of the Complex I has been observed in GNM isolated from SOD1G93A mice at 7 weeks of age (Capitanio et al., 2012). Collectively, these observations corroborate the hypothesis that, in patients with ALS (Ghiasi et al., 2012) and in preclinical models (Salvatori et al., 2018, Wang et al., 2016), Complex I activity is a selective target of different ALS-related proteins. Complex I can be considered as an energy-transducing enzyme whose activity regulates cell energy request and activates catabolic pathways following the failure of its activity (Hunter et al., 2018, Leonard et al., 2017). Accordingly, pharmacological inhibition or a failure in Complex I activity triggers the activation of the AMPK (Hu et al., 2017, Hunter et al., 2018). AMPK activation promotes catabolic pathways, such as lipid oxidation in skeletal muscle (Merrill et al., 1997, O’Neill et al., 2013). This known action of AMPK is in line with the increase in lipid oxidation and inhibition of glucose utilization that we observed here in early presymptomatic mice. In the context of ALS, mitochondrial failure could drive an increase in the use of lipids as primary fuel substrate to compensate for energy deficit and a decline in ATP synthesis. Subsequent to this, excess ROS generation as a result of excessive mitochondrial lipid oxidation may further aggravate mitochondrial dysfunction (Quijano et al., 2016, Rosca et al., 2012).
SLN Overexpression
SLN is a micropeptide inhibitor of SERCA activity that uncouples Ca2+ transport from ATP hydrolysis (Bal et al., 2012, Smith et al., 2002). Thus, SLN has a critical role in determining metabolic rate, EE, and muscle-derived thermogenesis (Bal et al., 2012, Maurya and Periasamy, 2015, Maurya et al., 2018). Notably, selective SLN overexpression in fast-twitch glycolytic muscle has been shown to reprogram mitochondria and induce an oxidative phenotype (Maurya et al., 2018). The expression of SLN is tightly regulated during development, and in rodents, its expression is detectable in skeletal muscle during late embryonic development and early neonatal life (Babu et al., 2007). Accordingly, in control mice SLN expression was downregulated in glycolytic muscle by postnatal day 15, whereas its expression was highly upregulated in the same muscle during the entire life course of SOD1G93A mice. More interestingly, we found marked SLN overexpression in muscle biopsies from patients affected by genetic and sporadic forms of ALS. Although SLN has already been viewed as a feasible prognostic or early diagnostic biomarker in ALS, its upregulation has been associated with disease-specific denervation (Calvo et al., 2012) rather than specific metabolic signatures. In contrast to this, our results indicate that SLN is overexpressed before the upregulation of any denervation markers. Although our data do not pinpoint SLN upregulation as being the primary “source” of hypermetabolism in ALS, early mitochondrial failure and energy deficit could underlie SLN overexpression and activate compensatory oxidative metabolism. Indeed, SLN plays a central role in the adaptive mechanisms that couple increased energy request (e.g., diet-induced obesity), cold acclimatization, and exercise with mitochondrial ATP and energy production (Maurya et al., 2018). Thus, the increase of SLN expression in ALS might represent a classical adaptation to increased energy demand, leading to a shift in substrate utilization, mitochondrial biogenesis, and oxidative metabolism in glycolytic muscle.
RAN Treatment
To determine whether decreasing energy demand and re-balancing substrate utilization might be of benefit in ALS, we aimed to assess the potential therapeutic role of RAN, an FDA-approved fatty acid oxidation inhibitor that is used for the management of cardiac dysfunction (Bhandari and Subramanian, 2007, Stanley, 2002). We demonstrate that chronic RAN treatment in SOD1G93A mice significantly improves motor function in SOD1G93A by slowing the decline in muscle strength over a 6-week treatment period. Moreover, during mid-term treatment evaluation (120 days of age), we found that RAN attenuated excessive whole-body EE in SOD1G93A mice, and although it did not offset increased fatty acid β-oxidation, RAN treatment improved energy metabolism by increasing muscle ATP content.
Interestingly, we found that, by the end stage of disease (at 150 days), RAN had lost its efficacy and there was a re-emergence of hypermetabolism. It should be noted that, in patients with stable angina pectoris, the recommended posology includes the escalation of drug dosage during therapy (www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000805/human_med_001009.jsp&mid=WC0b01ac058001d124). This suggests that the re-emergence of pathology at the end stage of disease in SOD1G93A mice may be attributed to the loss of RAN efficacy rather than disease exacerbation. Of interest, RAN also acts as an inhibitor of late inward sodium currents (IpNa) during cardiac repolarization (Belardinelli et al., 2013). Hence, we cannot rule out that sodium channel facilitation of membrane repolarization might decrease cell energy demand to compensate for mitochondrial failure in skeletal muscle and neurons of SOD1G93A mice.
Conclusions
Our study provides compelling evidence to demonstrate specific metabolic dysfunction and mitochondrial failure as early pathophysiological events in ALS. Here, we may consider that a slight drop in ATP production via mitochondrial impairment or SLN overexpression may become pathologically relevant when energy demand increases as a result of lifestyle changes or during aging. In this scenario, a timely pharmacological intervention impinging on hypermetabolism may be a promising therapeutic option to improve quality of life.
Limitations of the Study
In this study we use an FDA-approved drug, Ranolazine, to counteract hypermetabolism in an ALS mouse model. In our hands, Ranolazine ameliorates locomotor symptoms of fully symptomatic ALS mice, but its effect is temporary. In this regard, RAN does not improve life span in SOD1G93A mice. Given that we have observed temporary restoration and recovery of metabolic indicators, the loss of effectiveness of Ranolazine requires further investigation through extended pharmacokinetics and pharmacodynamics studies. From our study, we provide proof of concept that hypermetabolism appears to be considered a viable therapeutic target in ALS. As such, alternate drugs that are known to modulate metabolism should also be considered.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by AriSLA through HyperALS project (A.F.); AFM-Telethon project n. 2018; AFM-Telethon project n. 21021 (J.P.L. and A.F.); the National Health and Medical Research Council (1101085 and 1185427 to F.J.S. and S.T.N.); Scott Sullivan MND Research Fellowship (Queensland Brain Institute, the Royal Brisbane & Women's Hospital Foundation, The MND and Me Foundation) (S.T.N.); and Australian Institute for Bioengineering and Nanotechnology (S.T.N.).
This work is dedicated to the memory of our dear Mentor and Friend Maria Teresa Carrì.
Author Contributions
S.S. and I.S. designed and performed experiments. R.C. designed and G.G. performed the indirect calorimetry experiments. E.F., L.M., C.Q., and D.P. performed histological analysis. C.H. performed bioinformatics analyses, M.R. performed experiments on cells. S.R., E.F., and L.M. expanded and completed the study. S.B., N.V., and F.G. provided clinical samples. J.P.L., F.R., F.J.S., R.C., E.F., L.M., and S.T.N. designed the experiments and critically revised the manuscript. A.F. and C.V. designed the study and wrote the paper.
Declaration of Interests
The authors declare that they have no conflict of interest.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101087.
Contributor Information
Cristiana Valle, Email: c.valle@hsantalucia.it.
Alberto Ferri, Email: alberto.ferri@cnr.it.
Supplemental Information
References
- Anderson D.M., Makarewich C.A., Anderson K.M., Shelton J.M., Bezprozvannaya S., Bassel-Duby R., Olson E.N. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci. Signal. 2016;9:ra119. doi: 10.1126/scisignal.aaj1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu G.J., Bhupathy P., Carnes C.A., Billman G.E., Periasamy M. Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J. Mol. Cell. Cardiol. 2007;43:215–222. doi: 10.1016/j.yjmcc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal N.C., Maurya S.K., Sopariwala D.H., Sahoo S.K., Gupta S.C., Shaikh S.A., Pant M., Rowland L.A., Goonasekera S.A., Molkentin J.D. Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med. 2012;18:1575–1579. doi: 10.1038/nm.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batran R. Al, Gopal K., Aburasayn H., Eshreif A., Almutairi M., Greenwell A.A., Campbell S.A., Saleme B., Court E.A., Eaton F. The antianginal ranolazine mitigates obesity-induced nonalcoholic fatty liver disease and increases hepatic pyruvate dehydrogenase activity. JCI Insight. 2019;4:e124643. doi: 10.1172/jci.insight.124643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli, Liu, Smith-Maxwell, Wang, El-Bizri, Hirakawa, Karpinski, Li, Hu, Li, Crumb, Wu, Koltun, Zablocki, Yao, Dhalla, Rajamani, Shryock J. Pharmacol. Exp Ther. 2013 doi: 10.1124/jpet.112.198887. [DOI] [PubMed] [Google Scholar]
- Bhandari B., Subramanian L. Ranolazine, a partial fatty acid oxidation inhibitor, its potential benefit in angina and other cardiovascular disorders. Recent Pat. Cardiovasc. Drug Discov. 2007;2:35–39. doi: 10.2174/157489007779606095. [DOI] [PubMed] [Google Scholar]
- Bouteloup C., Desport J.-C., Clavelou P., Guy N., Derumeaux-Burel H., Ferrier A., Couratier P. Hypermetabolism in ALS patients: an early and persistent phenomenon. J. Neurol. 2009;256:1236–1242. doi: 10.1007/s00415-009-5100-z. [DOI] [PubMed] [Google Scholar]
- Calvo A.C., Manzano R., Atencia-Cibreiro G., Oliván S., Muñoz M.J., Zaragoza P., Cordero-Vázquez P., Esteban-Pérez J., García-Redondo A., Osta R. Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS One. 2012;7:e32632. doi: 10.1371/journal.pone.0032632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitanio D., Vasso M., Ratti A., Grignaschi G., Volta M., Moriggi M., Daleno C., Bendotti C., Silani V., Gelfi C. Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1G93A mouse model. Antioxid. Redox Signal. 2012;17:1333–1350. doi: 10.1089/ars.2012.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desport J.-C., Torny F., Lacoste M., Preux P.-M., Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener. Dis. 2005;2:202–207. doi: 10.1159/000089626. [DOI] [PubMed] [Google Scholar]
- Dobrowolny G., Aucello M., Rizzuto E., Beccafico S., Mammucari C., Boncompagni S., Bonconpagni S., Belia S., Wannenes F., Nicoletti C. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–436. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Dobrowolny G., Lepore E., Martini M., Barberi L., Nunn A., Scicchitano B.M., Musarò A. Metabolic changes associated with muscle expression of SOD1 (G93A) Front. Physiol. 2018;9:831. doi: 10.3389/fphys.2018.00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi S., Gupta P., Kalb R.G. Genetic induction of hypometabolism by ablation of MC4R does not suppress ALS-like phenotypes in the G93A mutant SOD1 mouse model. Sci. Rep. 2017;7:13150. doi: 10.1038/s41598-017-13304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L., Corcia P., Fergani A., Gonzalez De Aguilar J.L., Bonnefont-Rousselot D., Bittar R., Seilhean D., Hauw J.J., Lacomblez L., Loeffler J.P. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis symbol. Neurology. 2008;70:1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27. [DOI] [PubMed] [Google Scholar]
- Fajardo V.A., Rietze B.A., Chambers P.J., Bellissimo C., Bombardier E., Quadrilatero J., Tupling A.R. Effects of sarcolipin deletion on skeletal muscle adaptive responses to functional overload and unload. Am. J. Physiol. Cell Physiol. 2017;313:C154–C161. doi: 10.1152/ajpcell.00291.2016. [DOI] [PubMed] [Google Scholar]
- Flück M. Functional, structural and molecular plasticity of mammalian skeletal muscle in response to exercise stimuli. J. Exp. Biol. 2006;209:2239–2248. doi: 10.1242/jeb.02149. [DOI] [PubMed] [Google Scholar]
- Funalot B., Desport J.-C., Sturtz F., Camu W., Couratier P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009;10:113–117. doi: 10.1080/17482960802295192. [DOI] [PubMed] [Google Scholar]
- Genin E.C., Madji Hounoum B., Bannwarth S., Fragaki K., Lacas-Gervais S., Mauri-Crouzet A., Lespinasse F., Neveu J., Ropert B., Augé G. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10 S59L/+ mouse. Acta Neuropathol. 2019;138:123–145. doi: 10.1007/s00401-019-01988-z. [DOI] [PubMed] [Google Scholar]
- Ghiasi P., Hosseinkhani S., Noori A., Nafissi S., Khajeh K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012;34:297–303. doi: 10.1179/1743132812Y.0000000012. [DOI] [PubMed] [Google Scholar]
- Goode J.M., Pearen M.A., Tuong Z.K., Wang S.C.M., Oh T.G., Shao E.X., Muscat G.E.O. The nuclear receptor, Nor-1, induces the physiological responses associated with exercise. Mol. Endocrinol. 2016;30:660–676. doi: 10.1210/me.2015-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata C., Jamnick N.A., Bishop D.J. Training-induced changes in mitochondrial content and respiratory function in human skeletal muscle. Sports Med. 2018;48:1809–1828. doi: 10.1007/s40279-018-0936-y. [DOI] [PubMed] [Google Scholar]
- Hardie D.G., Schaffer B.E., Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J., Putman C.T., Tyreman N., Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood D.A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab. 2009;34:465–472. doi: 10.1139/H09-045. [DOI] [PubMed] [Google Scholar]
- Hu R., Yan H., Fei X., Liu H., Wu J. Modulation of glucose metabolism by a natural compound from Chloranthus japonicus via activation of AMP-activated protein kinase. Sci. Rep. 2017;7:778. doi: 10.1038/s41598-017-00925-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter R.W., Hughey C.C., Lantier L., Sundelin E.I., Peggie M., Zeqiraj E., Sicheri F., Jessen N., Wasserman D.H., Sakamoto K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018;24:1395–1406. doi: 10.1038/s41591-018-0159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides Z.A., Ngo T., Henderson D. Altered metabolic homeostasis in amyotrophic lateral sclerosis: mechanisms of energy imbalance and contribution to disease progression. Neurodegener. Dis. 2016;16:382–397. doi: 10.1159/000446502. [DOI] [PubMed] [Google Scholar]
- Jésus P., Fayemendy P., Nicol M., Lautrette G., Sourisseau H., Preux P.-M., Desport J.-C., Marin B., Couratier P. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2018;25:97–104. doi: 10.1111/ene.13468. [DOI] [PubMed] [Google Scholar]
- Kasarskis E.J., Mendiondo M.S., Matthews D.E., Mitsumoto H., Tandan R., Simmons Z., Bromberg M.B., Kryscio R.J. Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2014;99:792–803. doi: 10.3945/ajcn.113.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard S., Tobin L.M., Findlay J.B.C. The signalling mechanisms of a novel mitochondrial complex I inhibitor prevent lipid accumulation and attenuate TNF-α-induced insulin resistance in vitro. Eur. J. Pharmacol. 2017;800:1–8. doi: 10.1016/j.ejphar.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Lundsgaard A.-M., Fritzen A.M., Kiens B. Molecular regulation of fatty acid oxidation in skeletal muscle during aerobic exercise. Trends Endocrinol. Metab. 2018;29:18–30. doi: 10.1016/j.tem.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Maurya S.K., Periasamy M. Sarcolipin is a novel regulator of muscle metabolism and obesity. Pharmacol. Res. 2015;102:270–275. doi: 10.1016/j.phrs.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya S.K., Herrera J.L., Sahoo S.K., Reis F.C.G., Vega R.B., Kelly D.P., Periasamy M. Sarcolipin signaling promotes mitochondrial biogenesis and oxidative metabolism in skeletal muscle. Cell Rep. 2018;24:2919–2931. doi: 10.1016/j.celrep.2018.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack J.G., Stanley W.C., Wolff A.A. Ranolazine: a novel metabolic modulator for the treatment of angina. Gen. Pharmacol. 1998;30:639–645. doi: 10.1016/s0306-3623(97)00301-7. [DOI] [PubMed] [Google Scholar]
- Mejzini R., Flynn L.L., Pitout I.L., Fletcher S., Wilton S.D., Akkari P.A. ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 2019;13:1310. doi: 10.3389/fnins.2019.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill G.F., Kurth E.J., Hardie D.G., Winder W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. Endocrinol. Metab. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Moglia C., Calvo A., Grassano M., Canosa A., Manera U., D’ovidio F., Bombaci A., Bersano E., Mazzini L., Mora G. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J. Neurol. Neurosurg. Psychiatry. 2019;90:666–673. doi: 10.1136/jnnp-2018-319611. [DOI] [PubMed] [Google Scholar]
- Ning Y., Zhen W., Fu Z., Jiang J., Liu D., Belardinelli L., Dhalla A.K. Ranolazine increases β-cell survival and improves glucose homeostasis in low-dose streptozotocin-induced diabetes in mice. J. Pharmacol. Exp. Ther. 2011;337:50–58. doi: 10.1124/jpet.110.176396. [DOI] [PubMed] [Google Scholar]
- O’Neill H.M., Holloway G.P., Steinberg G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol. Cell. Endocrinol. 2013;366:135–151. doi: 10.1016/j.mce.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Palamiuc L., Schlagowski A., Ngo S.T., Vernay A., Dirrig-Grosch S., Henriques A., Boutillier A.-L., Zoll J., Echaniz-Laguna A., Loeffler J.-P. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015;7:526–546. doi: 10.15252/emmm.201404433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.D., Hasumati R. Ranolazine: a review on analytical method and its determination in synthetic mixture. Asian J. Pharm. Anal. 2015;5:214. [Google Scholar]
- Pearen M.A., Goode J.M., Fitzsimmons R.L., Eriksson N.A., Thomas G.P., Cowin G.J., Mary Wang S.C., Tuong Z.K., Muscat G.E.O. Transgenic muscle-specific Nor-1 expression regulates multiple pathways that effect adiposity, metabolism, and endurance. Mol. Endocrinol. 2013;27:1897–1917. doi: 10.1210/me.2013-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggion C., Massimino M.L., Biancotto G., Angeletti R., Reggiani C., Sorgato M.C., Bertoli A., Stella R., Stella R. Absolute quantification of myosin heavy chain isoforms by selected reaction monitoring can underscore skeletal muscle changes in a mouse model of amyotrophic lateral sclerosis. Anal. Bioanal. Chem. 2017;409:2143–2153. doi: 10.1007/s00216-016-0160-2. [DOI] [PubMed] [Google Scholar]
- Quijano C., Trujillo M., Castro L., Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016;8:28–42. doi: 10.1016/j.redox.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca M.G., Vazquez E.J., Chen Q., Kerner J., Kern T.S., Hoppel C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61:2074–2083. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S., Serrano A., Gerbino V., Giorgi A., Di Francesco L., Nencini M., Bozzo F., Schininà M.E., Bagni C., Cestra G. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015;128:1787–1799. doi: 10.1242/jcs.165332. [DOI] [PubMed] [Google Scholar]
- Salvatori I., Valle C., Ferri A., Carrì M.T. SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochem. Int. 2017;109:184–192. doi: 10.1016/j.neuint.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Salvatori I., Ferri A., Scaricamazza S., Giovannelli I., Serrano A., Rossi S., D’Ambrosi N., Cozzolino M., Di Giulio A., Moreno S. Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 2018;146:585–597. doi: 10.1111/jnc.14465. [DOI] [PubMed] [Google Scholar]
- Smith W.S., Broadbridge R., East J.M., Lee A.G. Sarcolipin uncouples hydrolysis of ATP from accumulation of Ca2+ by the Ca2+-ATPase of skeletal-muscle sarcoplasmic reticulum. Biochem. J. 2002;361:277–286. doi: 10.1042/0264-6021:3610277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley W.C. Partial fatty acid oxidation inhibitors for stable angina. Expert Opin. Investig. Drugs. 2002;11:615–629. doi: 10.1517/13543784.11.5.615. [DOI] [PubMed] [Google Scholar]
- Steyn F.J., Ioannides Z.A., Van Eijk R.P.A., Heggie S., Thorpe K.A., Ceslis A., Heshmat S., Henders A.K., Wray N.R., Van Den Berg L.H. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry. 2018;89:1016–1023. doi: 10.1136/jnnp-2017-317887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas H.E., Zhang Y., Stefely J.A., Veiga S.R., Thomas G., Kozma S.C., Mercer C.A. Mitochondrial complex I activity is required for maximal autophagy. Cell Rep. 2018;24:2404–2417.e8. doi: 10.1016/j.celrep.2018.07.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintignac L.A., Brenner H.R., Rüegg M.A. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 2015;95:809–852. doi: 10.1152/physrev.00033.2014. [DOI] [PubMed] [Google Scholar]
- Tsitkanou S., Gatta P.A.D., Russell A.P. Skeletal muscle satellite cells, mitochondria, and MicroRNAs: their involvement in the pathogenesis of ALS. Front. Physiol. 2016;7:403. doi: 10.3389/fphys.2016.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wang L., Lu J., Siedlak S.L., Fujioka H., Liang J., Jiang S., Ma X., Jiang Z., Da Rocha E.L. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016;22:869–878. doi: 10.1038/nm.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann M., Nieß A.M., Munz B. Physical exercise and epigenetic modifications in skeletal muscle. Sports Med. 2019;49:509–523. doi: 10.1007/s40279-019-01070-4. [DOI] [PubMed] [Google Scholar]
- Wiedemann F.R., Winkler K., Kuznetsov A.V., Bartels C., Vielhaber S., Feistner H., Kunz W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998;156:65–72. doi: 10.1016/s0022-510x(98)00008-2. [DOI] [PubMed] [Google Scholar]
- Yin H., Price F., Rudnicki M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurlo F., Larson K., Bogardus C., Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Invest. 1990;86:1423–1427. doi: 10.1172/JCI114857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.