

Abstract
Autophagy is a highly conserved multistep process and functions as passage for degrading and recycling protein aggregates and defective organelles in eukaryotic cells. Based on the nature of these materials, their size and degradation rate, four types of autophagy have been described, i.e. chaperone mediated autophagy, microautophagy, macroautophagy, and selective autophagy. One of the major regulators of this process is mTOR, which inhibits the downstream pathway of autophagy following the activation of its complex 1 (mTORC1). Alkylphosphocholine (APC) derivatives represent a novel class of antineoplastic agents that inhibit the serine–threonine kinase Akt (i.e. protein kinase B), which mediates cell survival and cause cell cycle arrest. They induce autophagy through inhibition of the Akt/mTOR cascade. They interfere with phospholipid turnover and thus modify signaling chains, which start from the cell membrane and modulate PI3K/Akt/mTOR, Ras-Raf-MAPK/ERK and SAPK/JNK pathways. APCs include miltefosine, perifosine, and erufosine, which represent the first-, second- and third generation of this class, respectively. In a high fraction of human cancers, constitutively active oncoprotein Akt1 suppresses autophagy in vitro and in vivo. mTOR is a down-stream target for Akt, the activation of which suppresses autophagy. However, treatment with APC derivatives will lead to dephosphorylation (hence deactivation) of mTOR and thus induces autophagy. Autophagy is a double-edged sword and may result in chemotherapeutic resistance as well as cancer cell death when apoptotic pathways are inactive. APCs display differential autophagy induction capabilities in different cancer cell types. Therefore, autophagy-dependent cellular responses need to be well understood in order to improve the chemotherapeutic outcome.
Keywords: types of autophagy, autophagy as drug target, alkylphosphocholines, Akt/mTOR pathway, miltefosine/perifosine/erufosine
Introduction
Autophagy is derived from a Greek word (αυτοϕαγϵἷν) meaning ‘self-eating’ and is a bulk degradation process, which includes the lysosomal-dependent degradation and recycling of components of eukaryotic cells. It has essential roles in keeping the cellular homeostasis and functions in cellular differentiation, control of cellular growth, cell defense, and promotes tissue remodeling and acclimatization. Autophagy either can have a protective function for cell survival or promote cell death. Alkylphosphocholines (APCs) are phospholipid-derived agents that cause changes in cell signaling by enriching in cell membranes including the lipid rafts. This physicochemical property and the resultant changes are basis for their anticancer, antiprotozoal, antibacterial, and antiviral activities. They induce autophagy as part of their mechanism of action. This review summarizes the current knowledge on alkylphosphocholines (APCs) regarding their influence on autophagy. It covers the following topics: Types of autophagy, role of autophagy in cancer, autophagy as a therapeutic target, APCs (miltefosine, perifosine, and erufosine) in general, as modulators of autophagy, and conclusion.
Types of Autophagy
Four types of autophagy have been recognized based on the nature of their cargo, their cargo size, and degradation rate, i.e. chaperone mediated autophagy (CMA), microautophagy, macroautophagy, and selective autophagy (Hayashi-Nishino et al., 2009; Bejarano and Cuervo, 2010; Li et al., 2012; Lippai and Szatmari, 2017; Bednarczyk et al., 2018). The lysosomal degradation of damaged proteins is common to all the previously mentioned types of autophagy, but the mechanism of delivering the substrate to the lysosome varies among the different types (Bednarczyk et al., 2018). The following part gives a short description of these four types.
Chaperone Mediated Autophagy
Chaperone proteins recognize substrate proteins for chaperone mediated autophagy (CMA, see Figure 1A) by the penta-peptide motif KFERQ (i.e. K: lysine–F: phenylalanine–E: glutamic or aspartic acid–R: arginine–Q: glutamine) (Jackson and Hewitt, 2016; Bednarczyk et al., 2018). Together with their chaperones, these proteins will be transported to lysosomes for breakdown in a receptor dependent manner (Bejarano and Cuervo, 2010). The native state of a substrate protein usually hides the recognition motif within the protein core, but it comes to be accessible by its respective chaperone regardless of its location within the protein. Examples for chaperones include heat shock cognate protein of 70 kD (Hsp70) and cochaperones as Hsp90, Hsp40, and Bcl-2-associated athanogene 1 (Bag-1). The latter will unfold the proteins before substrate–chaperone interactions can occur, eventually even without direct interaction. Then, the substrate is transferred to the lysosomal lumen after binding to the cytosolic tail of lysosome-associated membrane protein type 2A (LAMP-2A), which multimerizes to that purpose (Agarraberes and Dice, 2001; Bandyopadhyay et al., 2008; Bejarano and Cuervo, 2010; Jackson and Hewitt, 2016). CMA activity is directly proportional to the level of LAMP2A in the lysosomal membrane. CMA consists of four stages: recognition of the substrate, substrate binding, substrate translocation, which is an ATP dependent step, and finally substrate hydrolysis within the lysosome by proteolytic enzymes (Cuervo and Wong, 2014). Hsp90 maintains the stability of LAMP2A receptor during multimerization (Cuervo and Wong, 2014). Starvation of more than 10 h, oxidative stress, and exposure to toxic compounds will induce CMA. Under such conditions, the level of LAMP2A increases to meet the requirement of increasing CMA, and this occurs through degradation of the LAMP2A complex and transferal of its constituent proteins to the lysosomal membrane (Bednarczyk et al., 2018). The levels of LAMP2A protein are clearly organized by degradation at the lysosomal membrane, distribution between this structure and its lumen, or by de novo synthesis (Kaushik and Cuervo, 2006). The interaction of substrate and chaperone to form a complex is considered the rate-limiting step of the CMA process (Kaushik and Cuervo, 2006). However, the by age abridged stability of LAMP2A reduces CMA, which initially can be compensated by increased lysosome numbers (Kaushik and Cuervo, 2006; Bednarczyk et al., 2018). Reduced CMA activity as age-related effect results mainly in deficient binding and uptake of substrates into the lysosomal membrane, while kinetics of degradation is comparable to younger ages. The reduced CMA activity may have a role in the accumulation of altered products observed with aging (Kaushik and Cuervo, 2006).
Figure 1.

Types of autophagy: The four types of autophagy include chaperone mediated autophagy, microautophagy, selective autophagy, and macroautophagy. (A) Chaperone-mediated autophagy involves the recognition of a KFERQ penta-peptide motif in substrate proteins by corresponding chaperone proteins. The substrate is then transferred to the lysosomal lumen after binding to the LAMP protein. (B) Microautophagy is the process of sequestering minute parts of the cytoplasm and their engulfment by lysosomal invagination. (C) During selective autophagy, the respective cargo (e.g. invading pathogens, damaged mitochondria, or others) is specifically bound by autophagy receptors. The autophagy receptor has the ability to bind LC3 proteins through its LC3 interacting region (LIR) on the autophagosome beside binding molecular determinants, such as unfolded regions of a protein or conjugated ubiquitin (Ub) through its ubiquitin binding domain (UBD). (D) Macroautophagy consists of several steps of nucleation, elongation, maturation, and finally fusion and degradation. The process starts by the association of the ULK1 and BECN1 complexes that form the basis for recruiting other autophagy-related (ATG) proteins as well as the lipidated form of LC3 (LC3-II, i.e. LC3-I linked to phosphatidyl-ethanolamine). The ULK1 complex consists of the serine/threonine kinase UNC-51-like autophagy activating kinase (ULK1), focal adhesion kinase family interacting protein of 200 kDa (FIP200), ATG13, and ATG101. The PI3 kinase III nucleation complex (BECN1 complex) consists of Beclin-1, class III phosphoinositide 3-kinase [PI3K-III; also termed vacuolar protein sorting 34 (VPS34)] and its regulatory subunit VPS15. LC3-I protein is formed from its precursor protein, pro-LC3 with the contribution of ATG4. After attaching phosphatidyl-ethanolamine (PE) to LC3-I by ATG7 and ATG3, the lipophilic form (LC3-II) is created. The closed autophagosome fuses with a lysosome to form the autolysosome, where the proteins undergo degradation by different lysosomal enzymes.
Microautophagy
Microautophagy denotes the process of sequestering tiny parts of the cytoplasm and their subsequent engulfment through lysosomal invagination as shown in Figure 1B (Mijaljica et al., 2014).
Five phases of microautophagy have been identified: the first phase is the microautophagic invagination and formation of autophagic tubes, where the normal membrane bulges by lateral seclusion of lipids and local segregation of large transmembrane proteins toward the surface of lysosomes or vacuoles, and by an ATP dependent process then forms an autophagic tube. The second phase is vesicle formation, which is the equivalent of autophagosome formation in macroautophagy. It occurs because of the lateral organizing mechanism, where autophagic tubes invaginate because high-density lipids combine with low-density proteins. Vesicle expansion is the third step, which is characterized by the hanging of a prevesicular structure that dynamically moves back and forth in the lysosomal/vacuolar lumen. The formed vesicle does not contain proteins but lipids of high density because of the mechanism of lateral sorting. Finally, vesicle scission occurs because of the dynamic trend, where one or two vesicles bud into the lumen of the lysosome and move freely at high speed. Vesicle degradation happens because of the effect of some hydrolases that break down the freely moving vesicles. Recycling of the nutrients is done by a permease like action of ATG22p (Li et al., 2012).
Selective Autophagy
As compared to macroautophagy, which is considered nonselective, selective autophagy (Figure 1C) ensures recognition and elimination of specific cytosolic cargoes. Selective autophagy is specific for its substrates e.g., for damaged mitochondria (mitophagy), aggregated lipids (lipophagy), invading pathogens (xenophagy), or excess peroxisomes (pexophagy); these specific cargoes undergo degradation after being identified by autophagy receptors and are encircled into a double-membrane vesicle, the autophagosome, and transported to the lysosome for further breakdown (Rogov et al., 2014). The specificity is determined by identification of selective autophagy receptors (Rogov et al., 2014), which have the ability to bind LC3/GABARAP proteins on the forming autophagosome in addition to binding molecular elements, such as unfolded regions of a protein or conjugated ubiquitin (Ub). Then, through self-oligomerization, they contribute to the association of specific platforms on which autophagosomes form. Among the autophagy receptors are p62/sequestosome 1 (p62/SQSTM1), optineurin (OPTN), neighbor of BRCA1 (NBR1), and nuclear dot protein 52 kDa (NDP52). All of them possess a ubiqitin-binding domain (UBD) and LC3-interacting regions (LIRs) (Rogov et al., 2014; Stolz et al., 2014; Deng et al., 2017; Bednarczyk et al., 2018).
Macroautophagy
Macroautophagy (see Figure 1D, will be referred later as autophagy) involves the formation of isolation membranes (IMs), which extend as they ingest parts of the cytoplasm and organelles to produce autophagosomes (Hamasaki et al., 2013). This whole process is nonselective (Bednarczyk et al., 2018). Autophagosomes are double membrane structures, which function in delivering cargos to lysosomes or endosomes (Eskelinen, 2008). Autophagy is a complex mechanism with the involvement of several autophagy related proteins. Autophagy related proteins (ATG) responsible of the autophagy process were essentially discovered from yeast genome (Klionsky et al., 2003; Eskelinen, 2008). The source of the autophagosomal membrane is uncertain, as it is discussed to originate from endoplasmic reticulum (ER), mitochondria, or plasma membranes (Tooze and Yoshimori, 2010; Hamasaki et al., 2013; Nascimbeni et al., 2017; Wei et al., 2018). Hailey and coworkers found that the external membrane of the mitochondria adds to autophagosome production in fasting cells (Hailey et al., 2010). Both the endoplasmic reticulum (ER) and the early autophagic structures of IMs are interconnected. Electron tomography showed that the ER–IM complex is a subdomain of the ER that forms a frame surrounding the IM (Hayashi-Nishino et al., 2009). Other studies suggested’ that autophagosomes form at the ER–mitochondria contact site in mammalian cells (Hamasaki et al., 2013), or that the plasma membrane serves as a reservoir and participates openly in the development of positive autophagosome precursors during periods of increased autophagosome formation (Ravikumar et al., 2010). Furthermore, studying the intracellular dynamics of ATG9 in yeast showed that the Golgi apparatus derived ATG9 vesicles incorporate into the autophagosomal outer membrane at the initial stages of autophagosome generation (Yamamoto et al., 2012). Recycling endosomes represent membrane platforms that contribute to the formation of phagophores (Puri et al., 2018).
Mechanism and Regulation of Autophagy
Autophagy constitutes of several sequential steps, namely initiation or nucleation, phagophore elongation, autophagosome maturation, autophagosome fusion with the lysosome, and proteolytic degradation of the contents (Kardideh et al., 2019). The initial step usually begins with the association of the initiation complex (ULK1 complex). This complex consists of ULK1 (UNC-51-like autophagy activating kinase, a serine/threonine kinase), FIP200 (focal adhesion kinase family interacting protein of 200 kDa), ATG13 and ATG101, together with the nucleation complex (BECN1 complex) consisting of Beclin-1, class III phosphoinositide 3-kinase [PI3K-III or vacuolar protein sorting 34 (VPS34)] and its regulatory subunit VPS15. The latter represent the platform for recruiting other ATG proteins and for elongating the phagophore membrane (Lippai and Szatmari, 2017; Bednarczyk et al., 2018).
The three proteins VPS34, VPS15, and BECN1 make up the core, induce the elevated phosphatidylinositol 3-phosphate (PI3P) level of autophagic membranes, and can form, together with a fourth subunit, two different complexes. When this subunit is the UV radiation resistance associated (UVRAG) protein, the complex plays an essential role in endosomal maturation, but when the complex harbors ATG14L, it is required for autophagy. Engaging the BECN1 core complex to the phagophore assembly site (PAS) requires the function of ATG14L. Lipidation of ATG8/LC3 with phosphatidyl-ethanolamine (PE) is a crucial step in elongating the phagophore. Two ubiquitin-like conjugation systems act on this, namely: ATG7 with its E1 enzyme-like protein stimulating activity and ATG10 with its E2 conjugation enzyme like function. Their activities result into covalent bonding between ATG12 and ATG5. A multimeric complex, made of ATG5-ATG12 and ATG16L1, functions as a ubiquitin ligase like enzyme and facilitates binding of ATG8/LC3 to PE. Prerequisite to this step, LC3-I protein is formed by processing its precursor protein pro-LC3 with the protease ATG4. Then ATG7 and ATG3 activate ATG8 by E1 and E2 like enzymatic actions, and as a result, the lipophilic form (LC3-II) is created and bound to both membrane leaflets of the phagophore (Lippai and Szatmari, 2017). LC3-II can be considered as the best marker of autophagy, as its concentration is directly proportional to the number of autophagosomes formed (Burada et al., 2015) (Bednarczyk et al., 2018). All ATGs, with the exception of ATG8/LC3, dissociate from the membrane before closure and are recycled. Recycling of ATG8/LC3 occurs after closure of the autophagosome with the help of ATG4, while lysosomal enzymes in the autophagosome lumen cleave the proteins attached to the internal membrane (Lippai and Szatmari, 2017). Autophagosomes bind to late endosomes and lysosomes to form the autolysosomes and proceed for degradation (Lippai and Szatmari, 2017). LAMP proteins regulate the fusion and prevent degradation of the lysosomal membrane (Huynh et al., 2007; Lippai and Szatmari, 2017). The merger of the autophagosome with the lysosome to result in the autolysosome is facilitated by either the soluble NSF (N-ethylmaleimide sensitive factor) attachment protein (SNAP) receptor (SNARE) protein complex, or UVRAG, but many players can be involved in this process including cytoskeleton constituents and associated motor proteins, tethering factors, phospholipids, and specific SNARE complexes. This step is finished by degrading the interior autophagosomal membrane by lysosomal enzymes (Bednarczyk et al., 2018; Yu et al., 2018). Figure 1D shows the follow-up of steps described for macroautophagy.
Autophagy as a process is highly conserved and firmly controlled in mammals. The most important physiological regulator of autophagy is the availability of nutrients and amino acids. Other regulators include mTOR, especially its complex 1 (mTORC1), and inhibition of mTORC1 results in autophagy induction. In addition, starvation and amino acid depletion result into autophagy induction. This usually happens through activating adenosine monophosphate-activated protein kinase (AMPK), which transfers a phosphate group to ULK1, or by impeding mTORC1 activity, or through inhibitory phosphorylation of nonautophagic BECN1 complexes. Other autophagy regulators depend on an increase in cytosolic calcium, inhibition of inositol triphosphate or starvation induced autophagy (Petiot et al., 2000; Eskelinen, 2008; Jewell et al., 2013; Lippai and Szatmari, 2017). Furthermore, oxidative stress (Filomeni et al., 2015), DNA damage (Gomes et al., 2017) as well as hypoxia (Fang et al., 2015) can all induce autophagy. Activation of class I PI3-kinases inhibits autophagy, while class III PI3-kinase activity is required for autophagosome formation.
Autophagy breaks macromolecules and then provides nutrients and functions as a survival mechanism during short-term starvation (Eskelinen, 2008; Lippai and Szatmari, 2017). It eliminates damaged proteins and organelles and helps in the organelle turnover. It fights against attacking pathogens and in general preserves the cellular homeostasis and balance (Eskelinen, 2008; Lippai and Szatmari, 2017).
Autophagy also results into type II programmed cell death and is related to apoptosis in several ways. The tumor suppressor death-associated protein kinase (DAPk) may contribute to the signaling pathway linking autophagy to cell death. Certain types of cell death depend on autophagy proteins for the execution of cell death. Autophagy can protect cells from the apoptotic fate by providing nutrients, especially under conditions of starvation. The regulation of both processes is related to the pro-survival protein Bcl-2. Bcl-2 binds to Beclin 1 and prevents its interaction with VPS34. Thus, it inhibits the Beclin 1 dependent autophagy and maintains autophagy at levels harmonious with cell survival, rather than cell death (Eskelinen, 2008; Bednarczyk et al., 2018).
Different human diseases are associated with deregulation of autophagy, including neurodegenerative diseases and proteinopathies, lysosomal disorders, many cardiovascular diseases, cancer, diabetes and immune disorders. Infections and pathogens control autophagy based on their needs to assure their persistence in host cells (Lippai and Szatmari, 2017).
mTOR Regulation of Autophagy
Autophagy is controlled by a negative feedback mechanism of the mTOR pathway and modulators of this pathway have an impact on and can regulate autophagy (Jung et al., 2010; Kim and Guan, 2015). In normal cellular states and when amino acids are abundant, mTORC1 binds to the ULK complex by interaction of its component RAPTOR with ULK1. Then, mTOR transfers phosphate groups to ULK1 (specifically at its S757 and S637 residues) and ATG13 and thus hinders the ULK1 kinase activity. Under fasting conditions, the mTOR regulatory pathway is inhibited, and this leads to induction of autophagy because mTORC1 separates from the ULK complex and therefore its inhibitory control on ULK1 is gone, which allows autophagy to proceed (Rabanal-Ruiz et al., 2017). LKB1, AMPK, TSC1/TSC2 complex, and PTEN in mTOR signaling pathways can induce autophagy, while Akt and Rheb have an inhibitory effect. mTOR is inhibited at the beginning of autophagy and becomes activated later on, due to the release of breakdown molecules in the cytoplasm, which result into inhibition of the whole process. Enlarged mTOR activity then impedes autophagy and induces the formation of proto-lysosomal extensions (LAMP1+, LC3−) from autolysosomes (LAMP1+, LC3+). Finally, these proto-lysosomal extensions separate from the autolysosome and advance into functional lysosomes. Impediment of mTOR or its (auto-) lysosomal activity precludes autophagic lysosome restoration (Kapoor et al., 2014).
Role of Autophagy In Cancer
In carcinogenesis, the role of autophagy is controversial with many conflicting reports in the literature. Autophagy can either impede or favor cancer development and progression, which depends on the wild-type or transformed state of the cell, the underlying genetic lesion(s), the tumor type and stage, as well as the tumor microenvironment (Galluzzi et al., 2015; New et al., 2017; Bednarczyk et al., 2018; Kardideh et al., 2019).
Under normal conditions, autophagy acts as a protector against cancer development. In contrast, during stress situations, autophagy helps cells to adapt against hypoxia and nutrient deficiency and can save cancer cells from death (Galluzzi et al., 2015; New et al., 2017; Bednarczyk et al., 2018). Fighting mutagenic effects (e.g. DNA damage or instability of the genome) occurring from accumulation of reactive oxygen species (ROS) and degrading of oncogenic proteins are among the protective functions that autophagy can exert to inhibit cancer induction (Galluzzi et al., 2015; New et al., 2017; Bednarczyk et al., 2018). A reduced level of autophagy will hinder the ability of the cells to eliminate impaired proteins and damaged organelles and hence begin to mount up cytotoxic components that can cause damage to DNA and initiate carcinogenesis (Bednarczyk et al., 2018). Autophagy can suppress tumorigenesis through cell cycle and cell death regulation in conjunction with the ubiquitin–proteasome system (UPS), which can modify some key cell cycle components of CDK-Cyclin complexes (Kardideh et al., 2019).
Autophagy is required for immune activation as it plays a role in antigen presentation to T lymphocytes. It helps the maturation of some innate immune cells when activated and contributes to their antitumor activity. It can play a role in combatting cancer through activation of the immune system (Janji et al., 2018). Nevertheless, hypoxia-induced autophagy may result in the activation of immune escape mechanisms in the tumor microenvironment (Janji et al., 2018). Regarding the anticancer effects, some autophagic genes and inducers are mutated or deleted in certain cancers, as p53 and PTEN (phosphatase and tensin homolog), which are the most frequently altered tumor suppressor genes, and Beclin-1 (BECN1), which is deleted in breast and ovarian cancers (Bednarczyk et al., 2018).
Regarding the tumor supportive roles, autophagy provides tumor cells access to nutrients that are crucial to their metabolism, promotes DNA repair, reduces mitochondrial disorders, and increases drug resistance (Burada et al., 2015; Bednarczyk et al., 2018). Autophagy also helps tumor cells to resist stress and apoptotic signals; it provides cells with energy through increasing ATP concentrations that favor cell survival during hypoxia and starvation. Autophagy is linked in late cancer to poor prognosis and invasiveness (Galluzzi et al., 2015; New et al., 2017) and favors tumor growth through rendering cells more resistant to apoptotic signals and stress stimuli as well as resistance to therapy induced cell death. Furthermore, autophagy maintains the neoplastic stem cell compartment and epithelial-to-mesenchymal transition (EMT) (Galluzzi et al., 2015).
In metastasis, autophagy can also play a dual role, based on the stage. In early stages, autophagy may stimulate inflammatory responses and limit tumor necrosis. Further, autophagy may limit the development of dormant cancer cells into micrometastases, as well as prevent oncogene induced senescence. However, in advanced stages, autophagy tends to increase the life span of circulating metastatic cells, which lack an extracellular matrix, by inducing dormancy in the new environment until favorable conditions occur (Burada et al., 2015).
In this context, the autophagosome marker LC3B showed moderate to high expression in solid cancers, as breast cancer and melanomas, and its expression is linked to cell growth, invasion and metastasis, high tumor grade, and poor prognosis (Lazova et al., 2012).
To summarize, autophagy protects against malignant transformation under normal conditions by maintaining the cellular homeostasis, but increases tumor progression and invasiveness after tumor establishment (Galluzzi et al., 2015; New et al., 2017). The different roles of autophagy in cancer are shown in Table 1.
Table 1.
The dual role of autophagy in cancera).
| Onco-stimulatory roles | Onco-suppressive roles |
|---|---|
| Support of cancer stem cells | Clearance of intracellular pathogens |
| Providing cells with an alternative energy source | Favoring the genomic stability |
| Inhibition of cell death | Reduction of DNA damage and reactive oxygen species |
| Reduction of cellular sensitivity to stress stimuli | Reduction of inflammation |
| Maintaining the dormancy state of tumor cells until favorable conditions are met | Stimulation of autophagic cell death |
| Promotion of drug resistance | Removal of damaged organelles and proteins |
a)modified from (Burada et al., 2015; Bednarczyk et al., 2018).
Aspects of Autophagy in Disease Prevention
The physiologic functions of selective autophagy recently have been perceived as potentially preventive measures. Basis for this concept is e.g. the observation that aging mammalian cells accumulate dysfunctional mitochondria, which can be eliminated by selective autophagy (Hansen et al., 2018). Mitophagy, i.e. the elimination of aged mitochondria is therefore a concept of antiaging strategies as well as for diseases that are characterized by a pathophysiology, which is related to mitochondrial disorders, including heart disease, retinopathy, and a series of neurodegenerative sicknesses as e.g. Alzheimer’s disease. Tolerable induction of mitophagy could thus pave the way to neuroprotection and healthy longevity (Fang et al., 2014; Lou et al., 2020). Calorie restriction, low insulin/IGF1 levels, and the intake of NAD+ precursors are examples of a strategy aiming to increase the clearance of damaged mitochondria. NAD+ is a cofactor of the NAD-dependent deacetylase sirtuin-1, which can induce mitophagy (Lee et al., 2008). Sirtuin activating compounds include resveratrol, which together with NAD+ precursors (tryptophan, nicotinic acid, nicotinamide, nicotinamide mononucleotide, and nicotinamide riboside) could be basis of a dietary approach to healthy longevity (Bonkowski and Sinclair, 2016).
Autophagy as Therapeutic Target
Autophagy maintains the intracellular metabolic homeostasis, and its dysfunction is associated with numerous diseases including cancer, neurodegeneration, cardiac ischemia, metabolic dysfunctions, infections, autoimmune and pulmonary disorders. Aging and associated atherosclerotic cardiovascular diseases may be slowed down by increasing autophagic flux through calorie restriction, fasting, exercise, and nutritional support such as with spermidine-rich food (Maiuri and Kroemer, 2019). Autophagy is also being considered as a “druggable” process due to its fine regulation by diverse signaling pathways, hence involvement of multiple targets (Galluzzi et al., 2017; Morel et al., 2017). Whether pharmacological modulation of autophagy may be advantageous over life-style interventions remains an open question (Maiuri and Kroemer, 2019).
Autophagy as a therapeutic target in cancer is a double-edged sword, which can both enhance neoplastic growth or suppress cancer cell survival. Cancer cells can adapt to hyponutrient conditions and protect themselves against cancer chemotherapeutics by increasing autophagic flux (Yoshida, 2017). At the same time, induction of autophagy enables antigen cross-presentation, which stimulates antitumor immune response and may protect against relapses (Maiuri and Kroemer, 2019). On the other hand, autophagy contributes to multidrug resistance (MDR) development. Autophagy inhibitors or silencing of ATGs by microRNAs can thus sensitize cancer cells to chemotherapy, enable the use of lower dosage, and reduce adverse effects. However, autophagy inducers may also reverse MDR and sensitize apoptosis-resistant MDR cells to cancer chemotherapeutics (Li et al., 2017).
Deregulated autophagy can be modulated by either inhibiting or inducing identified targets in relevant pathways. Preclinical data followed by clinical trials demonstrate that some old drugs with already well-known safety profiles may be beneficial as autophagy modulators in certain diseases. Among these “conventional” drugs, metformin, the drug of choice in type 2 diabetes mellitus, is also being recognized as an antiaging agent because of its autophagy enhancing effect. In addition, antimalarial drugs have gained attention as autophagy inhibitors, including chloroquine (CQ) and hydroxychloroquine (HCQ). They are the mostly investigated drugs for this purpose in clinical studies, especially in cancer. Other autophagy inducers like mTOR inhibitors, BH3 mimetics, the receptor tyrosine kinase inhibitor sorafenib, the proteasome inhibitor bortezomib, the antigout agent colchicine, the nonreducing disaccharide trehalose, and 2-deoxyglucose have entered clinical phase trials against cancer and neurodegenerative diseases.
Many novel autophagy modulators were evaluated in preclinical trials, but only some of these molecules have succeeded to pass the clinical development phase, and most of the phase trials were based on repurposing of old drugs. Macroautophagy modulators, which have entered phase I–III trials and their mechanisms of action are illustrated in Figure 2. The key inhibitor of autophagy, mTORC, can be inhibited by AKT-inhibitors (alkylphosphocholines, see below) and mTOR-inhibitors (sirolimus, everolimus, temsirolimus, sorafenib, metformin, and vorinostat), and by adenosine monophosphate-activated protein kinase (AMPK) activation (devimistat, metformin and trehalose). The other two autophagy targets in the downstream pathway are ULK1 and BECLIN1, which can be activated by ULK1 activator/BECLIN1 releasers such as sorafenib, trehalose, gossypol and BH3 mimetics (BCL-2 inhibitors like obatoclax). Sorafenib and bortezomib can also induce ATG activation. Autophagic flux can be enhanced by endoplasmic reticulum stress and related unfolded protein response (UPR) signaling (alkylphosphocholines, sorafenib, 2-deoxyglucose), inhibition of glycolysis (2-deoxyglucose) and blockade of the cargo receptor p62/sequestome1 (p62/SQSTM1) degradation (bortezomib). The histone deacetylase 6 (HDAC6) inhibitor ricolinostat, lysosomal acidification inhibitors (CQ, HCQ), proton pump inhibitors (pantoprazole) and the autophagosome–lysosome fusion inhibitor trehalose can inhibit autophagosome maturation and autophagolysosomal degradation. The detailed mechanisms of actions of these autophagy modulators are given in Table 2.
Figure 2.
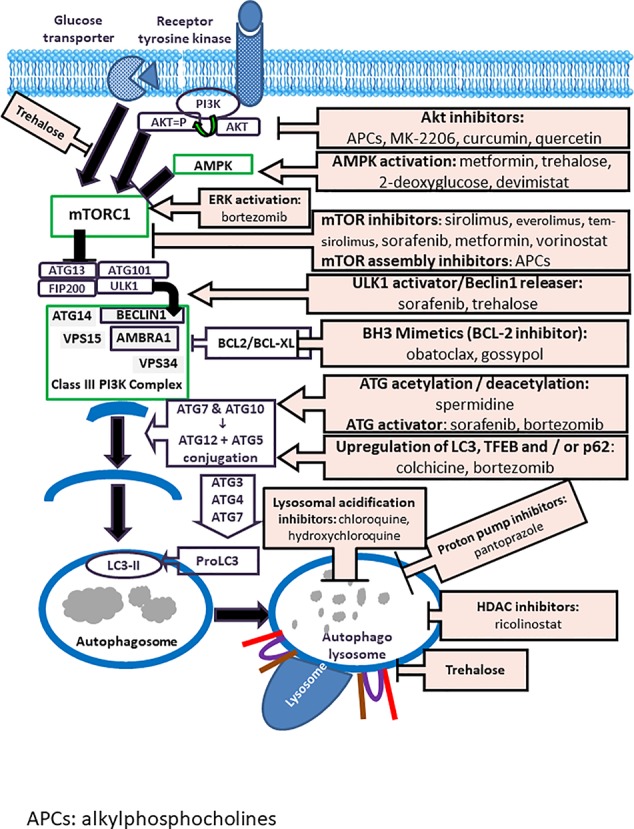
Modulators of autophagy Inhibitors of autophagy include Akt-inhibitors (alkylphosphocholines, MK-2206, curcumin, quercetin); AMPK activators (metformin, trehalose, 2-deoxyglucose, devimistat); ERK activators (bortezomib); mTOR-inhibitors (sirolimus, temsirolimus, sorafenib, metformin, vorinostat); mTOR-assembly inhibitors (alkylphosphocholines); ULK1 activators/Beclin releasers (sorafenib, trehalose); BH3-mimetics/BCL-2 inhibitors (obatoclax, gossypol); acetylators/deacetylators/activators of autophagy related (ATG) proteins (spermidine, sorafenib, bortezomib); upregulators of LC3; TFEB/p62 (colchicine, bortezomib); inhibitors of lysosomal acidification (chloroquine, hydroxychloroquine); proton pump inhibitors (pantoprazol), and inhibitors of the fusion of lysosomes with autophagosomes (trehalose).
Table 2.
Autophagy modulators in clinical trials and their mechanisms of action.
| Modulator | Modulation | Mechanism of Action | Reference |
|---|---|---|---|
| HCQ and CQ | Inhibition | Inhibition of lysosomal acidification and thus the blockade of the terminal step of autophagic substrate degradation | (Perez-Hernandez et al., 2019) |
| Rapamycin (sirolimus) Everolimus Temsirolimus | Activation | mTOR inhibition. | (Mandrioli et al., 2018) |
| Pantoprazole | Inhibition | Inhibition of H+/K + ATPase proton pumps in membranes of intracellular endosomes and increase of endosomal pH, resulting in inhibition of autophagosome maturation. | (Marino et al., 2010) |
| Metformin | Activation | Activation of AMPK, a sensor of cellular energy levels (increased AMP/ATP ratio) in the cell | (Novita et al., 2019) |
| Ricolinostat (ACY-1215) | Inhibition | Selective inhibition of HDAC6, which mediates trafficking of ubiquitinated misfolded proteins to the aggresome/autophagy pathway. | (Vogl et al., 2017) |
| Vorinostat (suberoylanilide hydroxamic acid; SAHA) | Activation | Pan-HDAC inhibitor; inhibition of mTOR, which results in the dephosphorylation, and thus activation, of the autophagic protein kinase ULK1 and increases LC3 expression. | (Gammoh et al., 2012) |
| Devimistat (CPI-613) | Inhibition | Inhibition of pyruvate dehydrogenase and ketoglutarate dehydrogenase of TCA cycle, thus impairment of pancreatic cell mitochondrial metabolism. | (Philip et al., 2019) |
| Spermidine | Activation | Suppression of acetyltransferase activity of EP300, and inducing the acetylation or deacetylation of autophagy-related genes (Atgs). | (Yang et al., 2017) |
| Bortezomib | Inhibition; activation |
|
|
| Sorafenib | Activation | Activation of HP1-STAT3-Mcl-1-Beclin1 pathway and releasing Beclin1 from Mcl-1; mTORC1 inhibition; activation of IRE1 signaling pathway of ER stress, thus reduction of ER stress-induced cell death; activation of AMPK. | (Sun et al., 2017) |
| Colchicine | Activation | Upregulation of proteins involved in autophagy, including the master regulator transcription factor EB (TFEB), the TFEB regulated adaptor protein SQSTM1/p62 and autophagy player microtubule-associated protein 1A/1B-light chain 3 (LC3) | (Mandrioli et al., 2019) |
| 188Re-liposome | Inhibition | Microtubule-associated protein 1 light chain 3B (LC3) and lysosomal proteins, including Lamp-1 and cathepsin-B and p21WAF/Cip1 levels decline. 188Re-liposome is effective in the suppression of stemness markers’ expression. | (Chang et al., 2017) |
| Trehalose | Inhibition; activation |
|
|
| NAD+ (and its precursors) | Activation | Induction of autophagy/mitophagy | (Bonkowski and Sinclair, 2016) |
| 2-deoxyglucose | Activation | ER stress and unfolded protein response; inhibition of glycolysis. | (Stein et al., 2010) |
AMPK, 5′ AMP-activated protein kinase; HDAC, histone deacetylase; TCA cycle, tricarboxylic acid cycle; TFEB, master regulator transcription factor EB; SQSTM1/p62, the TFEB regulated adaptor protein; microtubule-associated protein 1A/1B-light chain 3; ER, endoplasmic reticulum, NAD+, nicotinamide adenine nucleotide.
Clinical Development
Today, published clinical data have demonstrated sufficient evidence for efficacy in various disorders. They also indicate the need for further studies, which is expected to open a new era of autophagy-based therapies. Published clinical trials with or without (only protocol) results are summarized in Tables 3 and 4. These studies were searched in the PubMed data base using the key words “phase I/II/III trial OR clinical study” AND “autophagy” (Tables 3 and 4) or they were included in the tables following searching in the NCT data base using the key words “autophagy” AND “drug/intervention”. Registered clinical trials listed in the NCT database are summarized in Table 5. Most of these clinical studies focus on cancer, but new fields such as infections, neurodegenerative diseases, antiaging, cognitive decline, venous endothelial function, and inflammation reduction in acute coronary syndrome are also under investigation.
Table 3.
Published clinical trials based on autophagy modulation in neurodegenerative, infectious, and other diseases.
| Study Design/Registry Number | Regimen | Indication/Autophagy Biomarker | Indication/Aims–Results/Status | Reference |
|---|---|---|---|---|
| Phase II R, DB, PC, MC NCT03693781 Eudract n.2017-004459-21 |
Colchicine 0.01 mg/kg/d; 0.005 mg/kg/d Riluzole: 100 mg/d Treatment: 30 wt Follow-up: 24w |
ALS/Quantification of mRNA and protein levels of p62, LC3, TFEB, ATGs, HSPB8, BAG3, BAG1, HSP70 and HSF1 in PBMCs, lymphoblasts and fibroblasts (transcriptome profile) | Efficacy of colchicine on disease progression as measured by ALS Functional Rating Scale - Revised (ALSFRS-R) at baseline and at treatment end. Results are not yet available. | (Mandrioli et al., 2019) |
| Phase II R, DB, PC, MC Eudract n.2016-002399-28 |
Sirolimus 1 mg/m2/d; 2 mg/m2/d |
ALS/mTOR downstream pathway (S6RP phosphorylation) | Efficacy of sirolimus in ALS patients on functional rating scale, survival, forced vital capacity, and quality of life. Results are not yet available. | (Mandrioli et al., 2018) |
| Case report, OL, R N (Test) = 22 N (control) = 20 |
1,000–1,500 mg metformin plus insulin and anti-TB treatment | Diabetes mellitus and tuberculosis/MAP1LC3B | MET has the potential to enhance the bactericidal effect of antituberculosis (sputum smear reversion after 2 months) via autophagy. MAP1LC3B level increased significantly by metformin treatment. | (Novita et al., 2019) |
| Phase IIa, R, PC, double blind NCT03094546 | Spermidine-based nutritional supplementation | Elderly with subjective cognitive decline/LC3 I/II, p62, EP300, proteomics, metabolomics, polyamine levels, metabolomics, proinflammatory biomarkers, and neurotrophin levels | Results are not yet available. | (Wirth et al., 2019) |
| Phase IIB, OL, RCT CTRI/2018/01/011176 |
İsoniazid: 150–300 mg/d Rifampicin. 300–600 mg/d Pyrazinamide: 800–1,600 mg/d Ethambutol: 550–1,100 mg/d Test group: plus metformin 1,000 mg/d |
Newly diagnosed smear positive pulmonary tuberculosis/immunological and autophagy biomarkers (T cell, monocyte and dendritic cell functions ESAT-6/CFP-10, Culture filtrate Protein, estimation of C reactive protein, tumor necrosis factor-alpha and other cytokines). | Autophagy response will be evaluated as a secondary endpoint. Results are not yet available. | (Padmapriyadarsini et al., 2019) |
| Phase IIa, R, PC, double blind NCT02755246 | Spermidine-rich plant extract supplement | Behavioral mnemonic similarity task/not assessed | Memory performance was moderately enhanced and mnemonic discrimination ability improved in the treatment group versus the placebo group/not assessed | (Wirth et al., 2018) |
| Phase IV, R, quadruple-blind N = 10 NCT02058173 |
CQ: 150 mg/d × 8 + 4 w Placebo 8 w |
HCV/HCV genotype, IL28 genetic polymorphism | A significant decrease in HCV-RNA after the treatments (week 8) was observed in all patients in the CQ-group. The IL28 polymorphism was not associated with 5 HCV RNA load in response to CQ. Preliminary evidence that CQ is possibly a safe treatment option for HCV nonresponders | (Peymani et al., 2016) |
R, randomized; DB, double-blind; OL, open label; PC, placebo-controlled; MC, multicenter; N, number of patients; d, day; w, week; m, month.
Table 4.
Published clinical trials based on autophagy modulation in cancer.
| Study Design/Registry Number | Regimen | Indication/Autophagy Biomarker | Results | Reference |
|---|---|---|---|---|
| Phase II, R, DB, PC N = 70 NCT02333890 |
CQ: 500 mg/d Treatment: 14–29 d, 2–6 weeks before surgery |
Newly diagnosed breast cancer/not assessed | No significant difference in Ki67 index (a proliferation-associated nuclear antigen). | (Arnaout et al., 2019) |
| Phase I/II, OL N = 33 NCT01510119 |
HCQ: 600 mg BID Everolimus:10 mg/d |
Clear-cell renal cell carcinoma (previously treated)/not assessed | PR + SD: 67%; PR: 6%; PFS ≥ 6 m: 45%. The primary endpoint (> 40% 6-month PFS rate) was met. HCQ is a tolerable inhibitor of autophagy. | (Haas et al., 2019) |
| Phase II, Simon’s two-stage design; N = 21 NCT01748500 |
Pantoprazole: 240 mg/3w Docetaxel: 75 mg/m2/3w |
mCRPC/not assessed | PR = 31%; mOS = 15.7 m; median PFS = 5.3 m. Tolerable but clinical activity is insufficient. | (Hansen et al., 2019) |
| Phase Ib/II, Single-arm, OL N = 40 NCT01649947 |
HCQ: 200 mg BID/1–21 Day 1: Paclitaxel: 200 mg/m2 Carboplatin: AUC = 6 Bevacizumab: 15 mg/kg |
mNSCLC/not assessed | ORR = 33% (44% in KRAS positive tumors); SD: 53%; PFS: 3.3 m (6.4 m in KRAS positive tumors). Addition of HCQ is safe and tolerable with a modest improvement in clinical responses. | (Malhotra et al., 2019) |
| Phase I, OL N = 35 NCT01480154 |
HCQ: 200–600 mg BID MK-2206 135 0r 200 mg/w |
Advanced solid tumors/pre-planned autophagy biomarkers are not assessed due to high attrition. | SD: 15%; combination increases HCQ plasma levels. Combination therapy is tolerable but antitumor activity is minimal. |
(Mehnert et al., 2019) |
| Phase 2, R, OL N = 112 NCT01506973 |
HCQ: 600 mg BID Gemcitabine hydrochloride +nab-paclitaxel (GA) with or without |
Pancreatic cancer/not assessed | HCQ: 12 m OS: non-HCQ: 41%; 49%, OS: 11.1 in HCQ; 12.1 in non-HCQ PFS: 5.7 m HCQ; 6.4 m non-HCQ, ORR: 38.2% in HCQ; 21.1% in non-HCQ HCQ did not improve the primary end point of OS but may play a role in the locally advanced setting, where tumor response may permit resection. |
(Karasic et al., 2019) |
| Phase III, OL, R N = 500 NCT03504423 (AVENGER 500 trial) |
CPI-613 + modified FOLFIRINOX (devimistat 500 mg/m2, oxaliplatin 65 mg/m2, irinotecan dose of 120 mg/m2; fluorouracil, 400 mg/m2 (bolus), then 2,400 mg/m2/2 w FOLFIRINOX (oxaliplatin, 85 mg/m2; irinotecan, 180 mg/m2; fluorouracil and leucovorin (same) | Metastatic adenocarcioma of pancreas/mitochondrial SOD2, PDK1-4, PDH, KGDH and CD79a and whole-exome sequencing | Objectives: evaluation of ORR and PFS; tumor response Results are not yet available. |
(Philip et al., 2019) |
| Phase I, OL, cohort N = 14 NCT01687179 |
HCQ: 100–200 mg BID Sirolimus: 2 mg (trough levels between 5 and 15 ng/ml) |
Lymphangioleiomyomatosis/metabolomic profiling of polyamine metabolism 5′-methylthioadenosine and arginine | Upregulation of 5′-methylthioadenosine and arginine in the plasma of patients with LAM | (Tang et al., 2019) |
| Phase I, OL, cohort N = 14 24 w treatment + 24 w observation NCT01687179 |
HCQ: 100–200 mg BID Sirolimus: 2 mg (trough levels between 5 and 15 ng/ml) |
Lymphangioleiomyomatosis/AXL receptor tyrosine kinase, brain-derived neurotrophic factor (BDNF), cathepsin D, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2, insulin, receptor tyrosine protein kinase erbB3, and soluble superoxide dismutase 1 | Only BDNF levels changed significantly. A consistent decrease of BDNF levels in comparison to baseline was observed which was not HCQ dosage-dependent. | (Lamattina et al., 2018) |
| NR, OL N = 30 (healthy) N = 43 (EM) |
None | Endometriosis/LC3B-II | The expression of LC3B-II in ectopic endometrium group was significantly lower than that of its eutopic endometrium group. Down-regulated autophagy of ectopic endometrium in secretory phase may be related to the progression of EMs. | (Li et al., 2018a) |
| Phase I, OL N = 20 NCT01835041 |
CPI-613 (devimistat): 500 mg/m2/d Modified FOLFIRINOX (oxaliplatin at 65 mg/m2, leucovorin at 400 mg/m2, irinotecan at 140 mg/m2, and fluorouracil 400 mg/m2 bolus followed by 2,400 mg/m2 over 46 h) |
Metastatic pancreatic cancer/not assessed | MTD of CPI-613 = 500 mg/m2 per day. 18 patients treated at MTD, ORR (CR + PR) = 61%. PFS = 9 m; mOS =19 m. Clinical activity requires Phase II validation. |
(Alistar et al., 2017) |
| Phase III, R 1. N = 219 2. N = 234 3. N = 204 NCT00719797 NCT00433927 |
1. TRIBE trial (discovery cohort) FOLFIRI (fluorouracil, leucovorin, and irinotecan) plus bevacizumab 2. FIRE-3 trial (validation cohort) FOLFIRI plus bevacizumab 3. FIRE-3 trial (negative control) FOLFIRI plus cetuximab |
mCRC/12 SNPs in eight autophagy-related genes were examined in this study (autophagy-related protein 13 [ATG13], ATG3, ATG5, ATG8, beclin 1, FIP200, ULK1 and UVRAG | G allele of the FIP200 rs1129660 SNP showed a significantly lower rate of grades 2–3 hypertension compared with the A/A genotype. Polymorphisms in autophagy-related FIP200 gene may be predictive of hypertension. |
(Berger et al., 2017) |
| OL, case report N = 2 NCT02271516 |
188Re-liposome (0.42 ± 0.04 mCi/kg) | Recurrent ovarian cancer/Cancer Antigen 125 (CA-125) as a marker of drug-resistance | 188Re-liposome reduces CA-125 levels and improves survival. | (Chang et al., 2017) |
| Phase I, OL, cohort N = 14; 24 w treatment + 24 w observation NCT01687179 |
HCQ: 100–200 mg BID Sirolimus: 2 mg (trough levels between 5 and 15 ng/ml) |
Lymphangioleiomyo-matosis/not assessed | Well tolerated; improvement in lung function at 24 weeks, with a decrease in lung function at the 48-week time point. | (El-Chemaly et al., 2017) |
| Phase I/II, NR, OL N = 12 NCT01023477 |
CQ: 250 mg/w and 500 mg/w × 4w | Breast ductal carcinoma in situ (DCIS)/LC3B positive puncta | Chloroquine reduces PCNA proliferation index in DCIS lesions and inhibits autophagic flux (LC3B positive puncta) | (Espina et al., 2017) |
| Phase I/II OL, R N (ricolinostat) = 15 N (combined) = 57 |
Ricolinostat: Phase I cohorts 1–6: 40, 80, 160, 240, and 360 mg on days 1–5 and 8–12 of each 21-day cycle. Bortezomib: 1.0 mg/m2/1.3 mg/m2. Dexamethasone | Relapsed or refractory multiple myeloma/not assessed | Ricolinostat of 160 mg daily, the combination with bortezomib and dexamethasone is safe, well tolerated, and active. | (Vogl et al., 2017) |
| OL, NR N = 65 |
5-FU based treatment (In vitro and in vivo clinical data with gossypol (AT-101) are linked to clinical data) |
Gastric cancer/APE1 expression | Expression of APE1 is associated with poor survival in gastric cancer patients. AT101, an APE1 inhibitor, may promote chemotherapeutic sensitivity. | (Wei et al., 2016) |
| Phase I, OL, NR N = 20 NCT01023737 |
HCQ: 400 mg/day Vorinostat: 600 mg/day |
mCRC/CTSD and LC3-II in on study-biopsies | SD > 16w: 26%; mPFS 2.8 m; mOS 6.7 m. Improved antitumor immunity (decreased exhausted and regulatory T cells and increased effector phenotype T cells) and reduced tumor autophagy | (Patel et al., 2016) |
| Phase Ib/OL, NR N = 38 NCT01583283 |
Ricolinostat: 40–320 mg/d Lenalidomide: 15–25 mg/d Dexamethasone: 40 mg/w |
Relapsed or refractory multiple myeloma/HDAC6 | DLT: ricolinostat ricolinostat 160 mg BID. The pharmacokinetics of ricolinostat and lenalidomide were not affected by coadministration. ORR:55% | (Yee et al., 2016) |
| Phase I, OL, NR N = 25 |
HCQ: 400 mg/d Sirolimus: 2 mg/d Metronomic chemotherapy |
Stage IV refractory metastatic solid tumors/not assessed | ORR=40%; SD:84% Tumor markers dropped >50%. Progression from PD to PR:2; SD to PR:8 Autophagy is a promising target and warrants further Phase II studies |
(Chi et al., 2015a) |
| Phase II, OL, single arm N = 10 NCT01842594 |
Sirolimus: 1 mg HCQ: 200 mg 2×1/d × 2 w |
Sarcoma/uptake of [18F]-fluorodeoxyglucose positron emission tomography (FDG PET) | An inhibition of glycolysis within the tumors without tumor growth was noted. PR: 6/10; SD:3/10, PD:1/10. |
(Chi et al., 2015b) |
| Phase II, OL N = 19 (evaluable) NCT01206530 |
12 cycles:HCQ: 600 mg BID+ FOLFOX (5-FU (400 mg/m2 bolus, then 2,400 mg/m2 over 46h) + leuco-vorin 200 mg/m2, oxaliplatin 85 mg/m2)/bevacizumab 5 mg/kg, all iv/2 w; after 12 cycles, no oxaliplatin. | Previously untreated mRCR/autophagy biomarkers in PBMC | Autophagy is inhibited in PBMCs. ORR: 52% (CR: 5%, PR: 47%) FOLFOX/bevacizumab + HCQ is an active regimen in mCRC. |
(Loaiza-Bonilla et al., 2015) |
| Phase I/II, OL, NR N = 35 NCT01128296 |
HCQ: 1200 mg/d for 31 d Gemcitabine: 1,500 mg/m2 on days 3 and 17 |
Pancreatic adenocarcinoma/LC3-II in PBMC | No dose-limiting toxicities and no Grade 4/5 events related to treatment. 61% had a decrease in CA19-9. Patients with a >51% increase of LC3-II in PBMC had improvement in PFS (15.03 vs. 6.9 months, p < 0.05) and OS (34.8 vs. 10.8 m, p < 0.05). Preoperative autophagy inhibition with HCQ plus gemcitabine is safe and well tolerated. Surrogate biomarker responses (CA 19-9) and surgical oncologic outcomes were encouraging. | (Boone et al., 2015) |
| Case series, OL, NR N = 5 |
CQ: 250 mg/d Reirradiation |
Recurrent glioblastoma | No CQ related toxicity. 2 PR, 1 SD, 1 PD. Encouraging responses were obtained. | (Bilger et al., 2014) |
| Phase I, OL N = 24 |
Pantoprazole: 80, 160, 240, and 360 mg iv prior to doxorubucin (60 mg/m2) | Solid tumors/not assessed | Pantoprazole 240 mg with doxorubicin 60 mg/m2 every 3 weeks: toxicity was predictable and manageable. | (Brana et al., 2014) |
| Phase I, OL, cohort N = 24 NCT01023737 |
HCQ: 400–800 mg/d (d2–d21) Vorinostat: 400 mg/d (d1–21) |
Advanced solid tumors/AV, lysosomal protease CTSD, CDKN1A | HCQ and VOR stimulate the expression of CTSD and CDKN1A and the accumulation of autophagic vacuoles in PBMC. HCQ addition had no significant impact on the pharmaco-kinetic profile of VOR. 46% had PR or SD for ≥2 cycles. Based on the safety and preliminary efficacy of this combination, additional clinical studies are currently being planned | (Mahalingam et al., 2014) |
| Phase Ib/II, OL N = 27 |
HCQ: 1200 mg/d Temsirolimus: 25 mg/w |
Advanced solid tumors and melanoma/AV accumulation in PBMC. | Significant AV accumulation with TEM + HCQ compared with baseline only with 1200 mg cohort. SD = 74%; further studies are warranted. TEM and HCQ: safe and tolerable, modulate autophagy in patients, and have significant antitumor activity. | (Rangwala et al., 2014a) |
| Phase Ib/II, OL N = 40 |
HCQ:1200 mg/d Temozolomide: 150 mg/m2 daily for 7/14 d |
Advanced solid tumors and melanoma/autophagic vacuoles peripheral blood mononuclear cells | PR = 14%; SD: 27% in 22 evaluable patients with advanced melanoma. Prolonged stable disease and responses suggest antitumor activity in melanoma patients. | (Rangwala et al., 2014b) |
| Phase I/II OL N (Phase I) = 16 N (Phase II) = 76 NCT00486603 |
HCQ: 200–800 mg/d Temozolomide: initially before RT-75 mg/m(2); maintenance-150 mg/m(2)/d; 5 d/m for 6 m. RT: 60 Gy in 30 fractions |
Newly diagnosed gliablastoma multiforme/AV and LC3-II | MTD of HCQ: 600 mg/d. 800 mg/d: Grades 3–4 neutropenia and thrombocytopenia; OS 12 m = 70%; OS 18 m = 36%; OS 24 m = 25%. HCQ-induced dose dependent increases in AV and LC3-II in PBMC. Autophagy was not consistently achieved. No significant improvement in overall survival. | (Rosenfeld et al., 2014) |
| Phase Ib/II, OL N = 25 |
HCQ: 1,200 mg/d Bortezomib: 1.3 mg/m2 |
Relapsed/refractory myeloma/AV accumulation and LC3-II in PBMC | Therapy-induced AV accumulation in bone marrow plasma cells. PR = 14%, MR = 14%, SD = 45%; further studies are warranted. | (Vogl et al., 2014) |
| Phase II OL N = 20 NCT01273805 |
HCQ: 800 and 1,200 mg/d Previously treated with other regimens |
Metastatic pancreatic adenocarcinoma (previously treated)/LC3-II in lymphocytes | Analysis of LC3-II showed inconsistent autophagy inhibition. SD: 10%; PFS: 46.5 d; OS: 69 d. Negligible therapeutic efficacy. | (Wolpin et al., 2014) |
| Phase II R, PC, DB N = 73 NCT01894633 |
CQ: 150 mg/d × 4 w WBI (30 Gy in 10 fractions/d over 2 weeks) |
Brain metastases from solid tumors/not assessed | ORR = CLQ-54%; PL-55%. PFS = CLQ-84%; PL-55% CLQ + WBI improved the control of brain metastasis with no increase in toxicity. CLQ did not improve the RR or OS. |
(Rojas-Puentes et al., 2013) |
| Pilot, single cohort N = 20 |
CQ: 250 mg/day × 5 w; started 1 w before WBRT | Newly diagnosed brain metastases from solid tumors | CR = 2; PR = 12; SD = 1 No treatment-related grade ≥ 3 toxicities or treatment interruption due to toxicity. Median/mean OS = 5.7 and 8.9 m |
(Eldredge et al., 2013) |
| Phase Ib/II, OL N = 12 |
2-deoxyglucose (2-DG) 45 mg/kg |
mCRPC/p62 as marker of 2-DG resistance | P62 decreased in 83% and fluorodeoxyglucose uptake decreased in 63% of patients. 2-DG alone or in combination can be used to target tumor metabolism. | (Stein et al., 2010) |
| Phase II, OL N = 30 |
Sorafenib: 400 mg × 2/d | Relapsed or refractory lymphoma/ LC3‐II in PBLs |
ORR: 13%; OS: 16 m. LC3-II levels at baseline were significantly higher in responsive patients than in nonresponsive patients. PBLs: responsive patients: reduction in LC3-II levels; nonresponsive patients: no change. | (Guidetti et al., 2012) |
| Phase I, OL, R N(HCQ) = 27 N(HCQ + Erlotinib) = 19 NCT01026844 |
HCQ: 400–1000 mg/d (escalated) Erlotinib: 150 mg/d |
Advanced NSCLC/not assessed | HCQ with or without erlotinib was safe and well-tolerated. The recommended phase 2 dose: HCQ 1000mg + erlotinib 150mg. | (Goldberg et al., 2012) |
| Phase II, R, DB, PC N = 30 NCT00224978 |
CQ: 150 mg/d Conventional chemotherapy + radiotherapy |
Glioblastoma multiforme | OS = 24m for CQ; 11m for PL Small sample size suggests larger, more definitive studies. |
(Sotelo et al., 2006) |
R, randomized; DB, double-blind; OL, open label; PC, placebo-controlled; MC, multicenter; N, number of patients; d, day; w, week; m, month; RT, Radiotherapy; PL, placebo; HCQ, hydroxychloroquine; CQ, chloroquine; OS, overall survival; ORR, objective response rate; CR, complete response; PR, partial response; MR, minor response; SD, stable disease; PFS, progression-free survival.
mCRPC, metastatic castration-resistant prostate cancer; mNSCLC, metastatic nonsmall cell lung cancer; mCRC, metastatic colorectal cancer; PBMC, peripheral blood mononuclear cells; AV, autophagic vacuole; UVRAG, UV radiation resistance-associated gene protein; FIP200, focal adhesion kinase family interacting protein of 200 kDa; ULK1, unc-51-like kinase 1; CDKN1A, cyclin-dependent kinase inhibitor 1A.
Table 5.
Registered clinical trials based on autophagy modulation in various diseases.
| NCT number | Study Design | Title | Regimen | Study Start/Study End | Result |
|---|---|---|---|---|---|
| NCT04160455 | Observational model: cohort | Study of autophagy and the effects of GALIG gene products in HIV-1 infected patients who are under antiretroviral therapy since primary-infection, chronic phase, or never treated. | HIV-1 infected patients under antiretroviral therapy since primary-infection, chronic phase, or never treated | November 7, 2019/November 7, 2029 | Recruiting |
| NCT04138134 | Observational model: cohort | Autophagy and venous endothelial function | Spermidine | December 1, 2019/June 1, 2020 | Not yet recruiting |
| NCT03979651 | NR, OL | MEK and autophagy inhibition in metastatic/locally advanced, unresectable neuroblastoma RAS (NRAS) melanoma | Trametinib plus HCQ | September 30, 2019/March 31, 2022 | Ongoing |
| NCT00786682 | Phase II, NR, OL | Docetaxel and hydroxychloroquine in treating patients with metastatic prostate cancer | Docetaxel and HCQ | December 2008–October 2012 | The study was stopped due to lack of improved efficacy compared to historical controls. |
| NCT00765765 | Phase I/II, NR, OL | Ixabepilone and hydroxychloro-quine in treating patients with metastatic breast cancer | Ixabepilone and HCQ | February 2009–December 2012 | The study was closed early due to slow accrual |
| NCT00969306 | Phase I, NR, OL | Chloroquine as an antiautophagy drug in stage IV small cell lung cancer (SCLC) patients | Chloroquine | September 2013–June 2016 | Terminated but no results posted |
| NCT03037437 | Phase II, NR, OL | Sorafenib induced autophagy using hydroxychloroquine in hepatocellular cancer | Sorafenib + HCQ | February 16, 2017–December 2020 | Ongoing |
| NCT01480154 | Phase I, NR, OL | Akt inhibitor MK2206 and hydroxychloroquine in treating patients with advanced solid tumors, melanoma, prostate or kidney cancer | MK2206 + HCQ | November 23, 2011–December 2019 | (Mehnert et al., 2019) |
| NCT01510119 | Phase I/II, NR, OL | Autophagy inhibition to augment mTOR Inhibition: A Phase I/II Trial of RAD001 and hydroxychloroquine in patients with previously treated renal cell carcinoma | RAD001 (everolimus) + HCQ | September 2011–January 2017 | (Haas et al., 2019) |
| NCT01023737 | Phase I, NR, OL | Hydroxychloroquine + Vorinostat in advanced solid tumors | HCQ + Vorinostat | November 2009–September 2020 | (Patel et al., 2016) |
| NCT03598595 | Phase I/II, NR, OL | Gemcitabine, docetaxel, and hydroxychloroquine in treating participants with recurrent or refractory osteosarcoma | Gemcitabine, Docetaxel, and HCQ | January 28, 2019–March 2, 2020 | Ongoing |
| NCT02631252 | Phase I, NR, OL | Phase I Study of mitoxantrone and etoposide combined with hydroxychloroquine, for relapsed acute myelogenous leukemia | Mitoxantrone and Etoposide and HCQ | August 18, 2016–October 2, 2017 | Terminated due to inability to accrue. |
| NCT03774472 | Phase I, NR, OL | Hydroxychloroquine, palbociclib, and letrozole before surgery in treating participants with estrogen receptor positive, HER2 negative breast cancer | HCQ, Palbociclib, and Letrozole | August 20, 2018–December 31, 2020 | Ongoing |
| NCT02339168 | Phase I, NR, OL | Enzalutamide and metformin-hydrochloride in treating patients with hormone-resistant prostate cancer | Enzalutamide and Metformin-Hydrochloride | June 2016–July 2020 | Ongoing |
| NCT01594242 | Phase I, NR, OL | Autophagy induction after bortezomib for myeloma | Bortezomib | July 10, 2012–March 2, 2015 | Completed but no results are available |
| NCT03700424 | Phase II, R, quadruple-blind | Inflammation reduction by Trehalose administration (in acute coronary syndrome) | Trehalose vs normal saline infusion weekly (15 g/week) for a period of 12 weeks | July 7, 2019–June 2020 | Ongoing |
| NCT01266057 | Phase I, NR, OL | Sirolimus or vorinostat and hydroxychloroquine in advanced cancer | Sirolimus or Vorinostat and HCQ |
April 28, 2011–February 2021 | Ongoing |
| NCT02378532 | Phase I, NR, OL | The addition of chloroquine to chemoradiation for glioblastoma | Chloroquine + temozolomide + chemoradiation | August 2016–June 2019 | Ongoing (still recruiting) |
| NCT01006369 | Phase II, NR, OL | Hydroxychloroquine, capecitabine, oxaliplatin, and bevacizumab in treating patients with metastatic colorectal cancer | HCQ, Capecitabine, Oxaliplatin, and Bevacizumab | May 2009–April 27, 2016 | Completed but results are not yet available |
| NCT03309007 | Phase III, R, quadruple-blind | A double-blind, placebo-controlled trial of anti-aging, pro-autophagy effects of metformin in adults with prediabetes | Metformin Placebo | September 1, 2017–July 31, 2021 | Active, not recruiting |
| NCT02042989 | Phase I, NR, OL | MLN9708 and Vorinostat in patients with advanced p53 mutant malignancies | MLN9708 and Vorinostat | June 27, 2014–June 2022 | Active, not recruiting |
| NCT00728845 | Phase I/II, NR, OL | Hydroxychloroquine, carboplatin, paclitaxel, and bevacizumab in recurrent advanced nonsmall cell lung cancer | HCQ, Carboplatin, Paclitaxel, and Bevacizumab | June 16, 2008–December 21, 2010 | Terminated due to slow accrual |
| NCT03754179 | Phase I/II, R, OL | Dabrafenib/trametinib/hydroxychloroquine for advanced pretreated BRAF V600 mutant melanoma | Dabrafenib/Trametinib/HCQ | January 23, 2018–December 2020 | Recruiting |
| NCT02316340 | Phase II, R, OL | Vorinostat Plus hydroxychloroquine vs regorafenib in colorectal cancer | Vorinostat + HCQ vs Regorafenib | February 2015–September 2020 | Active, not recruiting |
| NCT02432417 | Phase II, R, OL | The addition of chloroquine to chemoradiation for glioblastoma, | Chloroquine + Chemoradiation | January 2020–January 2024 | Not yet recruiting |
| NCT02257424 | Phase I/II, NR, OL | Dabrafenib, trametinib, and hydroxychloroquine in patients with advanced BRAF mutant melanoma | Dabrafenib, Trametinib and HCQ | October 2014–October 2026 | Active, recruiting |
| NCT02421575 | Phase I, NR, OL | Hydroxychloroquine in blocking autophagy in patients with prostate cancer undergoing surgery or active surveillance | HCQ | July 2012–February 26, 2016 | Terminated due to slow accrual |
| NCT01206530 | Phase I/II | FOLFOX/Bevacizumab/Hydroxychloroquine (HCQ) in Colorectal Cancer | HCQ, Oxaliplatin, Leuco-vorin, 5-fluorouracil, Bevacizumab | September 2010 –September 2017 | (Loaiza-Bonilla et al., 2015) |
| NCT00962845 | Phase I, NR, OL | Hydroxychloroquine in patients with stage III or stage IV melanoma that can be removed by surgery | HCQ | September 2010–May 2013 | Early Phase I is completed. Last update September, 2018. https://ichgcp.net/es/clinical-trials-registry/NCT00962845 |
| NCT01144169 | Phase I, NR, O | Study of hydroxychloroquine before surgery in patients with primary renal Cell carcinoma | HCQ | October 2010–September 2016 | Terminated due to barriers to accrual such as delay until surgery and additional preoperative visits |
| NCT03094546 | Phase III, R, triple-blind | Polyamine-enriched diet in elderly individuals with subjective cognitive decline | Polyamine-rich diet | June 2019 – June 2020 | (Wirth et al., 2019) |
| NCT01583283 | A Phase I/II, Open Label, Multi-center Study | Study of ACY-1215 in combination with lenalidomide, and dexamethasone in multiple myeloma | Ricolinostat (ACY-1215) Lenalidomide Dexamethasone | July 12, 2012–December 31, 2019 | (Yee et al., 2016) |
| NCT01997840 | Phase 1B/2 Multi-Center, Open Label, Dose-escalation Study | ACY-1215 (ricolinostat) in combination with pomalidomide and low-dose dex in relapsed-and-refractory multiple myeloma | ACY-1215 (ricolinostat) in combination with pomalidomide and dexamethasone | March 1, 2014–January 27, 2021 | Active, not recruiting |
| NCT02189343 | Phase 1b Multi-Center, Open Label, Dose-Escalation Study | Phase 1b Study evaluating ACY-1215 (ricolinostat) in combination with pomalidomide and dexamethasone in relapsed or relapsed-and-refractory multiple myeloma | ACY-1215 (Ricolinostat) Pomalidomide Dexamethasone | September 15, 2014–January 30, 2019 | Active, not recruiting |
R, randomized; DB, double-blind; OL, open label; PC, placebo-controlled; MC, multicenter; N, number of patients; d, day; w, week, m, month; RT, Radiotherapy; PL, placebo; HCQ, hydroxychloroquine; CQ, chloroquine; OS, overall survival; ORR, objective response rate; CR, complete response; PR, partial response; MR, minor response; SD, stable disease; PFS, progression-free survival.
As seen in Tables 3 and 4, the role of autophagy in the prognosis of various diseases received increasing interest, especially in the last decade. Various biomarkers were utilized in these clinical trials to measure autophagy response. In the early studies, autophagic vacuoles were evaluated by transmission electron microscopy, but this method was unreliable (Levy et al., 2017). Selected biomarkers of autophagy include microtubule associated protein light chain 3 (LC3), p62, ATGs in peripheral blood mononuclear cells (PBMCs), lymphoblasts, and fibroblasts of patients. Proteomics, metabolomics, lysosomal protease cathepsin D (CTSD), cyclin-dependent kinase inhibitor 1 (CDKN1A, p21/cip1/waf1), and S6RP phosphorylation are among the potential biomarkers considered.
Cancer
Cancer is the extensively studied field in autophagy (Table 4). CQ and HCQ are mostly included in treatment regimens to increase the sensitivity of chemotherapeutics. CQ treatment at a relatively high daily dose of 500 mg resulted in no significant difference in the classic cellular proliferation marker, Ki67 index, in newly diagnosed breast cancer patients in a Phase II, double-blind, randomized trial (Arnaout et al., 2019). CQ at a daily dose of 150 mg was able to reduce the proliferating cell nuclear antigen (PCNA) index in breast ductal carcinoma (Espina et al., 2017). CQ at 150 mg/day together with conventional chemotherapy plus radiotherapy improved overall survival when compared to placebo in glioblastoma multiforme (GBM), but the small sample size of 30 patients was not sufficient for reaching a conclusion (Sotelo et al., 2006). A higher dose of CQ (250 mg/day) added to radiotherapy gave encouraging results in a pilot study of five patients with GBM (Bilger et al., 2014). A small size study, which enrolled newly diagnosed brain metastasis patients, showed that CQ at 250 mg/day, starting 1 week before radiotherapy and continuing for 5 weeks, was well tolerated with a PR rate of 60% (Eldredge et al., 2013). In comparison, CQ at a daily dose of 150 mg for four weeks plus radiotherapy, when compared to placebo, also improved brain metastasis control and prolonged progression free survival (PFS),but achieved no benefit in terms of response rate or overall survival (Rojas-Puentes et al., 2013).
Combination of HCQ with mTOR inhibitors, which enhance cytoprotective autophagy, was tested in Phase I/IIa trials. HCQ in combination with everolimus was tolerable and the primary endpoint of a 6-month progression free survival (PFS) rate over 40% was met in clear cell renal carcinoma (Haas et al., 2019). HCQ in combination with sirolimus significantly increased the “forced expiratory volume” but not the “forced vital capacity” in lymphangioleiomyomatosis (El-Chemaly et al., 2017). Metabolomic profiling of polyamine metabolism (upregulation of 5′-methylthioadenosine) and brain derived neurotrophic factor were suggested as candidate markers associated with autophagy in lymphangioleiomyomatosis (Lamattina et al., 2018; Tang et al., 2019). HCQ combined with temsirolimus showed significant antitumor activity in advanced solid tumors and melanoma (Rangwala et al., 2014a). HCQ and sirolimus decreased glycolysis mainly in cancer associated fibroblasts, hence attenuated their metabolic-parasite relationship with sarcoma cells; this double autophagy modulation resulted in an overall response rate (ORR) of 90% (Chi et al., 2015b). Addition of sirolimus and HCQ to the ongoing metronomic chemotherapy, to which patients were previously unresponsive, resulted in high salvage rates in stage IV refractory metastatic tumors (Chi et al., 2015a).
HCQ alone or in combination provided minimal to modest benefit with the EGFR inhibitor erlotinib in metastatic nonsmall cell lung cancer (Goldberg et al., 2012; Malhotra et al., 2019); with FOLFOX/bevacizumab in metastatic colorectal cancer (Loaiza-Bonilla et al., 2015); with temozolomide in advanced solid tumors and melanoma (Rangwala et al., 2014b); with vorinostat (Mahalingam et al., 2014; Patel et al., 2016) and MK-2206 (Mehnert et al., 2019) in advanced solid tumors; as a single agent (Wolpin et al., 2014), with gemcitabine (Boone et al., 2015) and with gemcitabine plus nab-paclitaxel (Karasic et al., 2019) in advanced pancreatic cancer; with temozolomide plus radiotherapy in newly diagnosed glioblastoma multiforme (Rosenfeld et al., 2014) and with bortezomib in refractory myeloma (Vogl et al., 2014).
Ricolinostat, a selective HDAC6 inhibitor, was well tolerated and moderately effective in combination with bortezomib (Vogl et al., 2017) and the immunomodulator lenalidomide plus dexamethasone (Yee et al., 2016) in refractory multiple myeloma.
The proton pump inhibitor, pantoprazole, was tolerable when combined with doxorubicin in solid tumors (Brana et al., 2014) but showed insufficient clinical activity when combined with docetaxel in metastatic castration resistant prostate cancer (mCRPC) (Hansen et al., 2019).
Two novel drugs seem to be promising as autophagy inhibitors. Devimistat (CPI-613) inhibits two key enzymes of the tricarboxcylic acid cycle, i.e. pyruvate dehydrogenase and ketoglutarate dehydrogenase, and thus impairs pancreatic cell mitochondrial metabolism. Devimistat, in combination with the FOLFIRINOX regimen, exhibited an ORR of 61% (Alistar et al., 2017) in a Phase I trial and is also under evaluation in a large scale Phase III trial (AVENGAR 500) in metastatic pancreas adenocacinoma (Philip et al., 2019). 188Re-liposome treatment was successful in inducing mitochondrial autophagy and improved survival in two cases with recurrent ovarian cancer (Chang et al., 2017).
Natural products were also investigated as autophagy modulators. Spermidine, a natural polyamine, triggers autophagy by inducing the acetylation or deacetylation of autophagy-related genes (Yang et al., 2017). Spermidine-based nutritional supplementation, leading to the acetylation or deacetylation of autophagy related genes, is considered as an autophagy-based strategy against memory under-performance, mnemonic discrimination ability and cognitive decline in the elderly (Wirth et al., 2018; Wirth et al., 2019). Gossypol (AT-101) is a polyphenolic compound extracted from cotton. In addition to its contraceptive and anti-infective effects, its roles as an anticancer agent by targeting apoptotic and autophagic pathways have also been studied in the last decade. It is a pan-Bcl-2 inhibitor and induces autophagy by liberating Beclin-1 from Bcl2 (Perez-Hernandez et al., 2019). Most of the clinical studies in PubMed focused on its apoptotic effect; only Wei et al. reported that the expression of apurinic/apyrimidinic endonuclease (APE1), which regulates the DNA base excision repair process, reduces survival in gastric cancer patients and the APE1 inhibitor AT-101 may be a potential sensitizer for 5-fluorouracil (Wei et al., 2016). Curcumin, the major constituent of Curcuma longa (turmeric), and quercetin, a plant flavonol, are two other autophagy inducers, which inhibit the Akt/mTOR pathway. No phase trial on autophagy is yet registered with the above-mentioned natural compounds in the NCT database, except for spermidine (Kondapuram et al., 2019; Perez-Hernandez et al., 2019).
Autophagy related polymorphisms might be predictive of anticancer treatment associated adverse effects. Berger et al. reported that FIP200 gene polymorphisms might predict bevacizumab-induced hypertension (Berger et al., 2017).
In summary, the application of autophagy inducers/inhibitors in cancer treatment is a quickly growing area of research, which has the aim of balancing the potentially good and bad effects. The success in keeping this balance will determine the future development and use of such compounds.
Neurodegenerative Disorders
In amyotrophic lateral sclerosis (ALS), degradation of mutant protein aggregates and damaged organelles is disrupted. Two autophagy modulators, colchicine, which upregulates autophagy-related proteins in motor neurons in combination with riluzole (Mandrioli et al., 2019), and the mTOR inhibitor rapamycin (sirolimus) (Mandrioli et al., 2018) are being evaluated for their therapeutic efficacy against ALS in two separate double-blind, randomized, placebo-controlled phase-II trials.
Infectious Diseases
The mycobacterium tuberculosis virulence is associated with perturbations in the autophagy process and AMPK signaling. Novita et al. showed that metformin enhances the bactericidal effect of anti-tuberculosis treatment via autophagy in patients with tuberculosis and diabetes (Novita et al., 2019). Another study was designed to assess the effect of metformin by means of a detailed autophagy biomarker panel in tuberculosis (Padmapriyadarsini et al., 2019). Chloroquine significantly decreased HCV-RNA in Hepatitis C in a quadruple-blind, randomized Phase IV study (Peymani et al., 2016).
Alkylphosphocholines
Synthetic APCs include miltefosine, perifosine, and erufosine, which correspond to the first, second, and third generations of APCs (see Figure 3). They are derived from alkylphospholipids (ALPs) or ‘synthetic antitumor lipids’, which include edelfosine and ilmofosine. The two related groups of antitumor agents target cell membranes rather than DNA (Van Blitterswijk and Verheij, 2013). ALPs are metabolically stable, nonmutagenic and versatile drugs derived from lysophosphatidylcholine (Kaleagasioglu et al., 2019; Zaremberg et al., 2019). All agents possess long hydrocarbon chains that slow their catabolism (Van Blitterswijk and Verheij, 2008).
Figure 3.

Structures and names of alkylphosphocholines.
Cancer researchers became interested in APCs during the 80s of the last century for their selective and high anticancer efficacy in autochthonous, methylnitrosourea-induced rat mammary carcinoma (Berger et al., 1987; Muschiol et al., 1987), which was later confirmed in 7,12-dimethylbenzanthracene-induced rat mammary carcinoma (Hilgard et al., 1988). This unusual spectrum of activity pointed to an influence of APCs on the ras signaling chain, as both chemically induced models exhibit point mutations in the ras gene as driving force for tumor development. In subsequent years, this assumption was confirmed (Berger et al., 1992; Berger et al., 2003; Dineva et al., 2012). Apart from breast cancer models, tumor cell lines from other cancer types were sensitive to ALPs, as well (Erdlenbruch et al., 1998; Jendrossek et al., 1999; Konstantinov and Berger, 1999; Georgieva et al., 2002).
The first generation of APCs, the prototype of which is miltefosine, possesses a typical phosphocholine polar moiety and a saturated alkyl chain of varying length (Berger et al., 1987; Berger et al., 1993). The second generation includes perifosine, which has a cyclic polar head and saturated alkyl chain (Hilgard et al., 1997). Finally, the third generation possesses a classic or modified phosphocholine polar moiety and an unsaturated alkyl chain. The best-known representative of this group is erufosine (Kaleagasioglu et al., 2019).
An ether linkage to its glycerol backbone characterizes the prototype of ALPs, edelfosine (Kaleagasioglu et al., 2019; Zaremberg et al., 2019). The first APC generation is represented by miltefosine (hexadecylphosphocholine, HPC), which lacks the glycerol backbone and thus shows a less complex metabolism (Kaleagasioglu et al., 2019; Zaremberg et al., 2019). In order to reduce the acetylcholine-like side effects observed for derivatives with a phosphocholine headgroup, as miltefosine, perifosine contained a piperidine moiety instead of phosphocholine and thus showed less side effects in clinical applications. Erufosine represents the most novel generation of APCs, which are characterized by a chain length of 22 carbons and the presence of a single double bond (Fiegl et al., 2008; Konigs et al., 2010; Kaleagasioglu and Berger, 2014; Kaleagasioglu et al., 2019; Zaremberg et al., 2019). At clinical concentrations, APCs disturb cell membranes and hamper their phospholipid turnover as well as lipid-based signal and transduction pathways. They may interfere with signaling by hindering the contact of proteins with other cell membrane proteins, or with distinct membrane lipids or lipid microdomains. At high concentrations, they may cause cell lysis owing to their detergent properties (Van Blitterswijk and Verheij, 2013). APCs hinder the normal lipid metabolism and lipid dependent signal transduction by their accumulation in cell membranes. Therefore, they can induce apoptosis in rapidly growing cells, such as tumor cells. They can lead to selective reduction of tumor metastases, impairment of angiogenesis, differentiation of tumor cells, impediment of cytokinesis, cell cycle halt, immune stimulation and intensification of immune reactions against tumors (Van Blitterswijk and Verheij, 2008; Kostadinova et al., 2015; Kaleagasioglu et al., 2019; Zaremberg et al., 2019). Among their mechanism of actions are interference with phospholipid metabolism, the inhibition of survival pathways and alteration of signal transduction (e.g. SAPK/JNK AKT-mTOR Ras/Raf, PKC), activation of pro-apoptotic signaling through ALP-triggered stress, and ALP-stimulated Fas/CD95 assembly in lipid rafts (Van Blitterswijk and Verheij, 2013; Kostadinova et al., 2015). This group of drugs interferes with cell division without inhibiting nuclear division, causing increased percentages of cells in the G2/M phase of the cell cycle, with multinucleate cell formation, and ensuing apoptotic cell death (Kostadinova et al., 2015).
Modulation of Phosphatidylinositol 3-Kinase/Akt/mTOR Signaling by APCs
The PI3K/Akt/mTOR pathway is an important regulator of cell survival, which is activated by receptor tyrosine kinases (RTKs). Following stimulation by growth factors, dimerization of the RTKs triggers an autophosphorylation process and activates PI3K. PIP2 (phosphatidylinositol-3,4-bisphosphate) is then converted to PIP3 (phosphatidylinositol-3,4,5-triphosphate) by PI3K (Zhang, 2004). PI3K signaling is inhibited through the de-phosphorylation of PIP3 by PTEN (tumor suppressor phosphatase and Tensin homolog deleted on chromosome 10) (Liu et al., 2009). PIP3 binds to the pleckstrin homology (PH) domain of the serine/threonine kinase Akt, leading to its allosterical activation. At the next step, phosphorylated Akt (p-Akt) activates mTOR, the key inhibitor of autophagy. Therefore, APCs, by inhibiting Akt, induce autophagy via Akt/mTOR signaling pathway (Figure 2) (Kaleagasıoglu et al., 2019). Additionally, perifosine inhibits mTOR signaling by promoting degradation of mTOR, raptor, and rictor (Sun, 2010).
Miltefosine inhibits phosphorylation of Akt at its Ser473 residue in A431 epidermoid carcinoma cells, which were stimulated by insulin (Ruiter et al., 2003). The related PI3K/Akt signaling has also been implicated in various viral infections. Miltefosine inhibits Chikungunya virus replication by inhibiting Akt phosphorylation in human primary dermal fibroblasts (Sharma et al., 2018). Primary effusion lymphoma (PEL) displays activated PI3K/Akt/mTOR signaling. Both perifosine and miltefosine inhibit proliferation of PEL cell lines and retarded PEL tumor progression in vivo (Bhatt et al., 2010).
Perifosine prevents membrane recruitment of Akt and displaces PIP2 and PIP3 from this enzyme. As a result, Akt can no longer proceed through the required conformational change, for its dual phosphorylation, hence activation (Van Blitterswijk and Verheij, 2013). Perifosine blocks phosphorylation of constitutively active Akt within 3 h and also S6 kinase within 6 h in PTEN-mutant human glioma cell lines T98G and U373MG (Momota et al., 2005). Perifosine decreases phosphorylated Akt levels in a dose- and time-dependent manner in the aggressive thyroid cancer cell line FTC133 that has genetic alterations in the PI3K/Akt pathway (Liu et al., 2009). Inhibition of aberrant PI3K/Akt signaling by perifosine in various cancer types including thyroid cancer (Liu et al., 2009), renal cell carcinoma (Porta and Figlin, 2009), neuroblastoma (Sun and Modak, 2012), neuroendocrine tumors (Zitzmann et al., 2012), mantle cell lymphoma (Reis-Sobreiro et al., 2013), and multiple myeloma (Cirstea et al., 2010) seems to be a promising therapeutic strategy. Perifosine also plays a protective role against kainic acid-induced epileptogenesis, which involves abnormal activity of the mTOR pathway. Perifosine pretreatment significantly decreases p-Akt/Akt and p-S6/S6 ratios which results in marked decrease of spontaneous seizure frequency and neuronal death (Zhu et al., 2018)
Erufosine can be infused systemically and shows penetration through the blood brain barrier (Erdlenbruch et al., 1999; Martelli et al., 2010). Erufosine inhibits the aberrant PI3K/Akt/mTOR signaling pathway in various cancer types including leukemia, oral squamous cell carcinoma, glioma and prostate cancer (Erdlenbruch et al., 1998; Rudner et al., 2010; Kapoor et al., 2012; Awde et al., 2013; Kapoor et al., 2014; Ansari et al., 2017; Avsar Abdik et al., 2019). Remarkably, erufosine was less toxic to normal bone marrow than to cancer cells (Bagley et al., 2011; Yosifov et al., 2011). Phosphorylation of PKB/Akt is linked to several proliferation pathways, which are inhibited, correspondingly [for overview see (Kaleagasioglu et al., 2019)]. Erufosine prevents the Akt–mTOR pathway reactivation and development of drug resistance because it targets this pathway at multiple levels. Firstly, it induces dephosphorylation of PKB/Akt at Ser473, thus inhibiting the mTORC2 complex. Furthermore, it reduces the phosphorylation of PKB/Akt at its Thr308 residue, the upstream kinase activator of the mTORC1 complex. In addition, erufosine causes a dose-dependent decrease in p-PTEN levels, with total PTEN levels remaining unaffected, dephosphorylation of p-mTOR at Ser2448, with total mTOR expression levels remaining unaffected. Erufosine also dephosphorylates other constituents of the mTORC1 complex, such as p-PRAS40 and p-Raptor in a concentration-dependent manner, as well as the downstream substrates of mTORC1, i.e. p70S6K and p-4EBP1. Knockdown of mTOR by siRNA enhances the cytotoxic potential of erufosine (Kapoor et al., 2014). The effects of erufosine on mTOR explain the dose-dependent induction of autophagy observed with this drug. In addition to these effects, erufosine induces cell cycle arrest in combination with its activity on the Rb gene (Zaharieva et al., 2014; Ansari et al., 2018b).
Autophagy and Alkylphosphocholines
Miltefosine
Miltefosine, the first generation APC, is the only oral drug used for the treatment of leishmaniasis. In trypanosomatids, the shift in balance between posttranslational modifications, polyglutamylation, and deglutamylation may determine cell death and autophagic survival, respectively. Overexpression of polyglutamylases increases miltefosine-induced cell-death, whereas overexpression of deglutamylases, CCP5A and CCP5B, increases miltefosine-induced cell survival process of autophagy (Basmaciyan et al., 2019).
Atherosclerosis is driven by deposition of LDL in the arterial intima, which triggers endothelial cell activation and inflammation followed by the activation of toll-like receptor (TLR) pathways and the assembly of the NLRP3 inflammasome. Autophagy and cholesterol efflux, which deteriorate with age, are important protective mechanisms against atherosclerosis. Miltefosine activates AMPK and ULK1, inhibits TLR signaling pathway, and reduces NLRP3 inflammasome assembly. In this regard, autophagy enhancement by APCs may have a therapeutic potential in atherosclerosis (Iacano et al., 2019).
Miltefosine enhances autophagy in cutaneous T-cell lymphoma as shown by increased levels of LC3B in MJ (from patients with mycosis fungoides) and Hut78 (from a patient with Sézary syndrome) cells (Yosifov et al., 2014).
Perifosine
The second generation APC, perifosine, activates autophagy by inhibiting Akt and the assembly of mTOR raptor and mTOR/rictor complexes and also by increasing degradation of the major components (mTOR, rictor, raptor, p70S6K, and 4E-BP1) in the mTOR axis through an ubiquitin/proteasome-mediated mechanism in human lung cancer cells (Fu et al., 2009). Combination of perifosine with other autophagy modulators may provide benefits in cancer regimens as shown by preclinical studies (Cirstea et al., 2010; Tong et al., 2012). Perifosine-induced autophagy may be protective and induces resistance against its cytotoxic and apoptotic effects which can be restored by addition of an autophagy inhibitor CQ in human chronic myelogenous leukemia cells (Tong et al., 2012). Perifosine can augment the autophagy induced by the mTOR inhibitor sirolimus and cause synergistic cytotoxicity in multiple myeloma cells; this mechanism may present a rationale for designing clinical trials in relapsed/refractory multiple myeloma (Cirstea et al., 2010). Perifosine and also the ether lipid edelfosine can induce autophagy by evoking an ER stress response, as demonstrated by a two- to threefold increased expression of the apoptotic transcription factor CHOP (C/EBP homologous protein)/GADD153 (growth arrest and DNA damage inducible gene 153) in human hepatoblastoma HepG2 cells, but not in human glioblastoma U-87 MG cells. It should be noted that miltefosine was devoid of this ER stress inducing effect (Rios-Marco et al., 2015), but erufosine was able to strongly cause ER stress ((Ansari et al., 2018a) and see below).
A recent study revealed the potential role of APCs against neurodegenerative diseases. Enhancement of autophagy by perifosine may combat against TNF-α induced Akt/mTOR signaling, which promotes microglia polarization toward the neurotoxic M1 phenotype and inflammation (Jin et al., 2018).
Erufosine
APC induced autophagy is indicated by increased levels of the autophagic marker LC3B. Kapoor et al. (Kapoor et al., 2012) were the first to demonstrate that erufosine can cause both, apoptosis and autophagy, by affecting the Akt–mTOR signaling pathway (Kapoor et al., 2014). Exposure of oral squamous carcinoma cells to erufosine induced formation of acidic vesicular organelles as tested by acridine orange stain. Erufosine increased the ratio of lipidated LC3B-II to LC3B-I, denoting autophagosome formation in oral squamous carcinoma cells when compared to untreated controls. This effect probably results from the capability of the drug to inhibit the PI3K/Akt/mTOR pathway as a regulator of the autophagy process (Kapoor et al., 2012). The treated tumor cells showed reduced size, detached growth, and membrane blebbing as main characteristics of apoptosis (Kapoor et al., 2012; Kapoor et al., 2014).
In pancreatic ductal adenocarcinoma cells, erufosine showed also a dose-dependent increase in LC3B expression at mRNA and protein levels. Moreover, it caused an increased expression of the autophagy receptor optineurin in Suit2-007 cells (Ali et al., 2019).
Furthermore, erufosine induces endoplasmic reticulum and mitochondrial stress, which is linked to its effects on autophagy, apoptosis, and ROS induction. The association between erufosine and ER stress was shown by silencing and drug-induced inhibition of the ER stress sensors PERK and XBP1, which diminished the cellular effects of erufosine towards e.g. inhibition of proliferation, induction of apoptosis and autophagy (Ansari et al., 2018a).
Conclusion
In conclusion, autophagy is a multistep process, which can either suppress or favor cancer development. There are four subtypes, which have been named chaperone mediated autophagy, microautophagy, macroautophagy, and selective autophagy. Among other stimuli, autophagy is triggered by the complex 1 of mTOR, which, following its activation, inhibits autophagy induction. Alkylphosphocholine derivatives represent a membrane seeking class of antineoplastic agents that induce autophagy by inhibiting the Akt/mTOR cascade. They primarily interfere with phospholipid turnover and thus modify signaling chains, which start from the cell membrane and modulate PI3K/Akt/mTOR, Ras-Raf-MAPK/ERK and SAPK/JNK pathways. APCs include miltefosine, perifosine and erufosine, which represent the first-, second- and third generations of this class, respectively.
APCs inhibit Akt which acts as the main regulator of cell survival. In human cancers, the autophagic process is usually suppressed in vitro and in vivo by the constitutively active Akt and its downstream target mTOR. Therefore, exposure to APC derivatives, administered as monotherapy or in combination with other drugs, increases autophagy by reducing Akt/mTOR phosphorylation. Autophagy is a double-edged sword and may result in chemotherapeutic resistance as well as cancer cell death when apoptotic pathways are inactive. APCs display differential autophagy induction capabilities in different cancer cell types. Therefore, autophagy-dependent cellular responses need to be well understood in order to improve the chemotherapeutic outcome.
Author Contributions
FK, DA, and MB wrote the manuscript. MB composed and edited the review. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Agarraberes F. A., Dice J. F. (2001). A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 114, 2491–2499. [DOI] [PubMed] [Google Scholar]
- Ali D. M., Ansari S. S., Zepp M., Knapp-Mohammady M., Berger M. R. (2019). Optineurin downregulation induces endoplasmic reticulum stress, chaperone-mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discovery 5, 128. 10.1038/s41420-019-0206-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alistar A., Morris B. B., Desnoyer R., Klepin H. D., Hosseinzadeh K., Clark C., et al. (2017). Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 18, 770–778. 10.1016/S1470-2045(17)30314-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari S. S., Akgun N., Berger M. R. (2017). Erufosine increases RhoB expression in oral squamous carcinoma cells independent of its tumor suppressive mode of action - a short report. Cell Oncol. (Dordr) 40, 89–96. 10.1007/s13402-016-0302-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari S. S., Sharma A. K., Soni H., Ali D. M., Tews B., Konig R., et al. (2018. a). Induction of ER and mitochondrial stress by the alkylphosphocholine erufosine in oral squamous cell carcinoma cells. Cell Death Dis. 9, 296. 10.1038/s41419-018-0342-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari S. S., Sharma A. K., Zepp M., Ivanova E., Bergmann F., Konig R., et al. (2018. b). Upregulation of cell cycle genes in head and neck cancer patients may be antagonized by erufosine’s down regulation of cell cycle processes in OSCC cells. Oncotarget 9, 5797–5810. 10.18632/oncotarget.23537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout A., Robertson S. J., Pond G. R., Lee H., Jeong A., Ianni L., et al. (2019). A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res. Treat 178, 327–335. 10.1007/s10549-019-05381-y [DOI] [PubMed] [Google Scholar]
- Avsar Abdik E., Kaleagasioglu F., Abdik H., Sahin F., Berger M. R. (2019). ABT-737 and erufosine combination against castration-resistant prostate cancer: a promising but cell-type specific response associated with the modulation of anti-apoptotic signaling. Anticancer Drugs 30, 383–393. 10.1097/CAD.0000000000000736 [DOI] [PubMed] [Google Scholar]
- Awde A. R., Boisgard R., Theze B., Dubois A., Zheng J., Dolle F., et al. (2013). The translocator protein radioligand 18F-DPA-714 monitors antitumor effect of erufosine in a rat 9L intracranial glioma model. J. Nucl. Med. 54, 2125–2131. 10.2967/jnumed.112.118794 [DOI] [PubMed] [Google Scholar]
- Bagley R. G., Kurtzberg L., Rouleau C., Yao M., Teicher B. A. (2011). Erufosine, an alkylphosphocholine, with differential toxicity to human cancer cells and bone marrow cells. Cancer Chemother. Pharmacol. 68, 1537–1546. 10.1007/s00280-011-1658-0 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U., Kaushik S., Varticovski L., Cuervo A. M. (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 28, 5747–5763. 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmaciyan L., Robinson D. R., Azas N., Casanova M. (2019). (De)glutamylation and cell death in Leishmania parasites. PloS Negl. Trop. Dis. 13, e0007264. 10.1371/journal.pntd.0007264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk M., Zmarzly N., Grabarek B., Mazurek U., Muc-Wierzgon M. (2018). Genes involved in the regulation of different types of autophagy and their participation in cancer pathogenesis. Oncotarget 9, 34413–34428. 10.18632/oncotarget.26126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano E., Cuervo A. M. (2010). Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 7, 29–39. 10.1513/pats.200909-102JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M. R., Muschiol C., Schmahl D., Eibl H. J. (1987). New cytostatics with experimentally different toxic profiles. Cancer Treat Rev. 14, 307–317. 10.1016/0305-7372(87)90023-5 [DOI] [PubMed] [Google Scholar]
- Berger M. R., Yanapirut P., Reinhardt M., Klenner T., Scherf H. R., Schmeiser H. H., et al. (1992). Antitumor activity of alkylphosphocholines and analogues in methylnitrosourea-induced rat mammary carcinomas. Prog. Exp. Tumor Res. 34, 98–115. 10.1159/000420836 [DOI] [PubMed] [Google Scholar]
- Berger M. R., Betsch B., Gebelein M., Amtmann E., Heyl P., Scherf H. R. (1993). Hexadecylphosphocholine differs from conventional cytostatic agents. J. Cancer Res. Clin. Oncol. 119, 541–548. 10.1007/BF01686464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M. R., Tsoneva I., Konstantinov S. M., Eibl H. (2003). Induction of apoptosis by erucylphospho-N,N,N-trimethylammonium is associated with changes in signal molecule expressionand location. Ann. N. Y. Acad. Sci. 1010, 307–310. 10.1196/annals.1299.054 [DOI] [PubMed] [Google Scholar]
- Berger M. D., Yamauchi S., Cao S., Hanna D. L., Sunakawa Y., Schirripa M., et al. (2017). Autophagy-related polymorphisms predict hypertension in patients with metastatic colorectal cancer treated with FOLFIRI and bevacizumab: Results from TRIBE and FIRE-3 trials. Eur. J. Cancer 77, 13–20. 10.1016/j.ejca.2017.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt A. P., Bhende P. M., Sin S. H., Roy D., Dittmer D. P., Damania B. (2010). Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 115, 4455–4463. 10.1182/blood-2009-10-251082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilger A., Bittner M. I., Grosu A. L., Wiedenmann N., Meyer P. T., Firat E., et al. (2014). FET-PET-based reirradiation and chloroquine in patients with recurrent glioblastoma: first tolerability and feasibility results. Strahlenther. Onkol. 190, 957–961. 10.1007/s00066-014-0693-2 [DOI] [PubMed] [Google Scholar]
- Bonkowski M. S., Sinclair D. A. (2016). Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690. 10.1038/nrm.2016.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone B. A., Bahary N., Zureikat A. H., Moser A. J., Normolle D. P., Wu W. C., et al. (2015). Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 22, 4402–4410. 10.1245/s10434-015-4566-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brana I., Ocana A., Chen E. X., Razak A. R., Haines C., Lee C., et al. (2014). A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and tissue penetration of doxorubicin. Invest. New Drugs 32, 1269–1277. 10.1007/s10637-014-0159-5 [DOI] [PubMed] [Google Scholar]
- Burada F., Nicoli E. R., Ciurea M. E., Uscatu D. C., Ioana M., Gheonea D. I. (2015). Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest Oncol. 7, 271–284. 10.4251/wjgo.v7.i11.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. M., Lan K. L., Huang W. S., Lee Y. J., Lee T. W., Chang C. H., et al. (2017). (188)Re-Liposome Can Induce Mitochondrial Autophagy and Reverse Drug Resistance for Ovarian Cancer: From Bench Evidence to Preliminary Clinical Proof-of-Concept. Int. J. Mol. Sci. 18, E903. 10.3390/ijms18050903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi K. H., Ko H. L., Yang K. L., Lee C. Y., Chi M. S., Kao S. J. (2015. a). Addition of rapamycin and hydroxychloroquine to metronomic chemotherapy as a second line treatment results in high salvage rates for refractory metastatic solid tumors: a pilot safety and effectiveness analysis in a small patient cohort. Oncotarget 6, 16735–16745. 10.18632/oncotarget.3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi M. S., Lee C. Y., Huang S. C., Yang K. L., Ko H. L., Chen Y. K., et al. (2015. b). Double autophagy modulators reduce 2-deoxyglucose uptake in sarcoma patients. Oncotarget 6, 29808–29817. 10.18632/oncotarget.5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirstea D., Hideshima T., Rodig S., Santo L., Pozzi S., Vallet S., et al. (2010). Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol. Cancer Ther. 9, 963–975. 10.1158/1535-7163.MCT-09-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A. M., Wong E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z., Purtell K., Lachance V., Wold M. S., Chen S., Yue Z. (2017). Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 27, 491–504. [DOI] [PubMed] [Google Scholar]
- Dineva I. K., Zaharieva M. M., Konstantinov S. M., Eibl H., Berger M. R. (2012). Erufosine suppresses breast cancer in vitro and in vivo for its activity on PI3K, c-Raf and Akt proteins. J. Cancer Res. Clin. Oncol. 138, 1909–1917. 10.1007/s00432-012-1271-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Chemaly S., Taveira-Dasilva A., Goldberg H. J., Peters E., Haughey M., Bienfang D., et al. (2017). Sirolimus and Autophagy Inhibition in Lymphangioleiomyomatosis: Results of a Phase I Clinical Trial. Chest 151, 1302–1310. 10.1016/j.chest.2017.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldredge H. B., Denittis A., Duhadaway J. B., Chernick M., Metz R., Prendergast G. C. (2013). Concurrent Whole Brain Radiotherapy and Short-Course Chloroquine in Patients with Brain Metastases: A Pilot Trial. J. Radiat. Oncol. 2, 315–321. 10.1007/s13566-013-0111-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdlenbruch B., Jendrossek V., Marx M., Hunold A., Eibl H., Lakomek M. (1998). Antitumor effects of erucylphosphocholine on brain tumor cells in vitro and in vivo. Anticancer Res. 18, 2551–2557. [PubMed] [Google Scholar]
- Erdlenbruch B., Jendrossek V., Gerriets A., Vetterlein F., Eibl H., Lakomek M. (1999). Erucylphosphocholine: pharmacokinetics, biodistribution and CNS-accumulation in the rat after intravenous administration. Cancer Chemother. Pharmacol. 44, 484–490. 10.1007/s002800051122 [DOI] [PubMed] [Google Scholar]
- Eskelinen E.-L. (2008). New insights into the mechanisms of macroautophagy in nammalian cells. Int. Rev. Cell Mol. Biol. 266 207–247. 10.1016/S1937-6448(07)66005-5 [DOI] [PubMed] [Google Scholar]
- Espina V. A., Liotta L., Rassulova S., Gallimore H., Grant-Wisdom T., Menezes G., et al. (2017). PINC trial: Preventing invasive breast neoplasia with chloroquine. Cancer Res. 77, Abstract nr CT140. 10.1158/1538-7445.am2017-ct140 [DOI] [Google Scholar]
- Fang E. F., Scheibye-Knudsen M., Brace L. E., Kassahun H., Sengupta T., Nilsen H., et al. (2014). Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD(+)/SIRT1 Reduction. Cell 157, 882–896. 10.1016/j.cell.2014.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Tan J., Zhang Q. (2015). Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol. Int. 39, 891–898. 10.1002/cbin.10463 [DOI] [PubMed] [Google Scholar]
- Fiegl M., Lindner L. H., Juergens M., Eibl H., Hiddemann W., Braess J. (2008). Erufosine, a novel alkylphosphocholine, in acute myeloid leukemia: single activity and combination with other antileukemic drugs. Cancer Chemother. Pharmacol. 62, 321–329. 10.1007/s00280-007-0612-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomeni G., De Zio D., Cecconi F. (2015). Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388. 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Kim Y. A., Wang X., Wu X., Yue P., Lonial S., et al. (2009). Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 69, 8967–8976. 10.1158/0008-5472.CAN-09-2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Pietrocola F., Bravo-San Pedro J. M., Amaravadi R. K., Baehrecke E. H., Cecconi F., et al. (2015). Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880. 10.15252/embj.201490784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Bravo-San Pedro J. M., Levine B., Green D. R., Kroemer G. (2017). Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discovery 16, 487–511. 10.1038/nrd.2017.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N., Lam D., Puente C., Ganley I., Marks P. A., Jiang X. (2012). Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. U. S. A. 109, 6561–6565. 10.1073/pnas.1204429109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva M. C., Konstantinov S. M., Topashka-Ancheva M., Berger M. R. (2002). Combination effects of alkylphosphocholines and gemcitabine in malignant and normal hematopoietic cells. Cancer Lett. 182, 163–174. 10.1016/S0304-3835(02)00088-5 [DOI] [PubMed] [Google Scholar]
- Goldberg S. B., Supko J. G., Neal J. W., Muzikansky A., Digumarthy S., Fidias P., et al. (2012). A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 7, 1602–1608. 10.1097/JTO.0b013e318262de4a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes L. R., Menck C. F. M., Leandro G. S. (2017). Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 18 (11), 2351. 10.3390/ijms18112351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti A., Carlo-Stella C., Locatelli S. L., Malorni W., Pierdominici M., Barbati C., et al. (2012). Phase II study of sorafenib in patients with relapsed or refractory lymphoma. Br. J. Haematol. 158, 108–119. 10.1111/j.1365-2141.2012.09139.x [DOI] [PubMed] [Google Scholar]
- Haas N. B., Appleman L. J., Stein M., Redlinger M., Wilks M., Xu X., et al. (2019). Autophagy Inhibition to Augment mTOR Inhibition: a Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 25, 2080–2087. 10.1158/1078-0432.CCR-18-2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey D. W., Rambold A. S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P. K., et al. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667. 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. 10.1038/nature11910 [DOI] [PubMed] [Google Scholar]
- Hansen M., Rubinsztein D. C., Walker D. W. (2018). Autophagy as a promoter of longevity: insights from model organisms (vol 19, pg 579, 2018). Nat. Rev. Mol. Cell Biol. 19, 611–611. 10.1634/theoncologist.2018-0621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A. R., Tannock I. F., Templeton A., Chen E., Evans A., Knox J., et al. (2019). Pantoprazole Affecting Docetaxel Resistance Pathways via Autophagy (PANDORA): Phase II Trial of High Dose Pantoprazole (Autophagy Inhibitor) with Docetaxel in Metastatic Castration-Resistant Prostate Cancer (mCRPC). Oncologist 24, 1188–1194. 10.1634/theoncologist.2018-0621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Nishino M., Fujita N., Noda T., Yamaguchi A., Yoshimori T., Yamamoto A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437. 10.1038/ncb1991 [DOI] [PubMed] [Google Scholar]
- Hilgard P., Stekar J., Voegeli R., Engel J., Schumacher W., Eibl H., et al. (1988). Characterization of the antitumor activity of hexadecylphosphocholine (D 18506). Eur. J. Cancer Clin. Oncol. 24, 1457–1461. 10.1016/0277-5379(88)90336-7 [DOI] [PubMed] [Google Scholar]
- Hilgard P., Klenner T., Stekar J., Nossner G., Kutscher B., Engel J. (1997). D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur. J. Cancer 33, 442–446. 10.1016/S0959-8049(97)89020-X [DOI] [PubMed] [Google Scholar]
- Huynh K. K., Eskelinen E. L., Scott C. C., Malevanets A., Saftig P., Grinstein S. (2007). LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 26, 313–324. 10.1038/sj.emboj.7601511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacano A. J., Lewis H., Hazen J. E., Andro H., Smith J. D., Gulshan K. (2019). Miltefosine increases macrophage cholesterol release and inhibits NLRP3-inflammasome assembly and IL-1beta release. Sci. Rep. 9, 11128. 10.1038/s41598-019-47610-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. P., Hewitt E. W. (2016). Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 60, 173–180. 10.1042/EBC20160005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janji B., Berchem G., Chouaib S. (2018). Targeting Autophagy in the Tumor Microenvironment: New Challenges and Opportunities for Regulating Tumor Immunity. Front. Immunol. 9, 887. 10.3389/fimmu.2018.00887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendrossek V., Erdlenbruch B., Hunold A., Kugler W., Eibl H., Lakomek M. (1999). Erucylphosphocholine, a novel antineoplastic ether lipid, blocks growth and induces apoptosis in brain tumor cell lines in vitro. Int. J. Oncol. 14, 15–22. 10.3892/ijo.14.1.15 [DOI] [PubMed] [Google Scholar]
- Jewell J. L., Russell R. C., Guan K. L. (2013). Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 14, 133–139. 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M. M., Wang F., Qi D., Liu W. W., Gu C., Mao C. J., et al. (2018). A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 10, 378. 10.3389/fnagi.2018.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C. H., Ro S. H., Cao J., Otto N. M., Kim D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaleagasioglu F., Berger M. R. (2014). Differential effects of erufosine on proliferation, wound healing and apoptosis in colorectal cancer cell lines. Oncol. Rep. 31, 1407–1416. 10.3892/or.2013.2942 [DOI] [PubMed] [Google Scholar]
- Kaleagasioglu F., Zaharieva M. M., Konstantinov S. M., Berger M. R. (2019). Alkylphospholipids are Signal Transduction Modulators with Potential for Anticancer Therapy. Anticancer Agents Med. Chem. 19, 66–91. 10.2174/1871520618666181012093056 [DOI] [PubMed] [Google Scholar]
- Kao C., Chao A., Tsai C. L., Chuang W. C., Huang W. P., Chen G. C., et al. (2014). Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis. 5, e1510. 10.1038/cddis.2014.468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor V., Zaharieva M. M., Das S. N., Berger M. R. (2012). Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt-mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett. 319, 39–48. 10.1016/j.canlet.2011.12.032 [DOI] [PubMed] [Google Scholar]
- Kapoor V., Zaharieva M. M., Berger M. R. (2014). Erufosine Induces Autophagy and Apoptosis in Oral Squamous Cell Carcinoma: Role of the Akt–mTOR Signaling Pathway, in Autophagy: Cancer, other pathologies, inflammation, immunity, infection, and aging . Ed. Hayat M., (San Diego, London, Waltham: Academic Press/Elsevier; ), 3, 229–245. [Google Scholar]
- Karasic T. B., O’hara M. H., Loaiza-Bonilla A., Reiss K. A., Teitelbaum U. R., Borazanci E., et al. (2019). Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 5, 993–998. 10.1001/jamaoncol.2019.0684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardideh B., Samimi Z., Norooznezhad F., Kiani S., Mansouri K. (2019). Autophagy, cancer and angiogenesis: where is the link? Cell Biosci. 9, 65. 10.1186/s13578-019-0327-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A. M. (2006). Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol. Aspects Med. 27, 444–454. 10.1016/j.mam.2006.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. C., Guan K. L. (2015). mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25–32. 10.1172/JCI73939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Cregg J. M., Dunn W. A., Jr., Emr S. D., Sakai Y., Sandoval I. V., et al. (2003). A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539–545. 10.1016/S1534-5807(03)00296-X [DOI] [PubMed] [Google Scholar]
- Kondapuram S. K., Sarvagalla S., Coumar M. S. (2019). Targeting autophagy with small molecules for cancer therapy. J. Cancer Metastasis Treat. 5, 32. 10.20517/2394-4722.2018.105 [DOI] [Google Scholar]
- Konigs S. K., Pallasch C. P., Lindner L. H., Schwamb J., Schulz A., Brinker R., et al. (2010). Erufosine, a novel alkylphosphocholine, induces apoptosis in CLL through a caspase-dependent pathway. Leuk Res. 34, 1064–1069. 10.1016/j.leukres.2009.12.003 [DOI] [PubMed] [Google Scholar]
- Konstantinov S. M., Berger M. R. (1999). Human urinary bladder carcinoma cell lines respond to treatment with alkylphosphocholines. Cancer Lett. 144, 153–160. 10.1016/S0304-3835(99)00219-0 [DOI] [PubMed] [Google Scholar]
- Kostadinova A., Topouzova-Hristova T., Momchilova A., Tzoneva R., Berger M. R. (2015). Antitumor Lipids–Structure, Functions, and Medical Applications. Adv. Protein Chem. Struct. Biol. 101, 27–66. 10.1016/bs.apcsb.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Lamattina A. M., Taveira-Dasilva A., Goldberg H. J., Bagwe S., Cui Y., Rosas I. O., et al. (2018). Circulating Biomarkers From the Phase 1 Trial of Sirolimus and Autophagy Inhibition for Patients With Lymphangioleiomyomatosis. Chest 154, 1070–1082. 10.1016/j.chest.2018.08.1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazova R., Camp R. L., Klump V., Siddiqui S. F., Amaravadi R. K., Pawelek J. M. (2012). Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 18, 370–379. 10.1158/1078-0432.CCR-11-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J., Yoon Y. S., Lee S. J. (2018). Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 9, 712. 10.1038/s41419-018-0749-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. H., Cao L., Mostoslavsky R., Lombard D. B., Liu J., Bruns N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U. States America 105, 3374–3379. 10.1073/pnas.0712145105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J. M. M., Towers C. G., Thorburn A. (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542. 10.1038/nrc.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Lu M., Liu M., Deng S., Tang X., Han C., et al. (2018. a). An observation of the role of rutophagy in patients with endometriosis of different stages during secretory phase and proliferative phase. Curr. Gene Ther. 18, 286–295. 10.2174/1566523218666181008155039 [DOI] [PubMed] [Google Scholar]
- Li W. W., Li J., Bao J. K. (2012). Microautophagy: lesser-known self-eating. Cell Mol. Life Sci. 69, 1125–1136. 10.1007/s00018-011-0865-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Liang M., Jiang J., He R., Wang M., Guo X., et al. (2018. b). Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 14, 1291–1305. 10.7150/ijbs.26776b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. J., Lei Y. H., Yao N., Wang C. R., Hu N., Ye W. C., et al. (2017). Autophagy and multidrug resistance in cancer. Chin. J. Cancer 36, 52. 10.1186/s40880-017-0219-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippai M., Szatmari Z. (2017). Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 33, 145–168. 10.1007/s10565-016-9374-5 [DOI] [PubMed] [Google Scholar]
- Liu P., Cheng H., Roberts T. M., Zhao J. J. (2009). Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discovery 8, 627–644. 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loaiza-Bonilla A., O’hara M. H., Redlinger M., Damjanov N., Teitelbaum U. R., Vasilevskaya I., et al. (2015). Phase II trial of autophagy inhibition using hydroxychloroquine (HCQ) with FOLFOX/bevacizumab in the first-line treatment of advanced colorectal cancer. J. Clin. Oncol. 33, 3614–3614. 10.1200/jco.2015.33.15_suppl.3614 [DOI] [Google Scholar]
- Lou G. F., Palikaras K., Lautrup S., Scheibye-Knudsen M., Tavernarakis N., Fang E. F. (2020). Mitophagy and Neuroprotection. Trends Mol. Med. 26, 8–20. 10.1016/j.molmed.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Mahalingam D., Mita M., Sarantopoulos J., Wood L., Amaravadi R. K., Davis L. E., et al. (2014). Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10, 1403–1414. 10.4161/auto.29231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri M. C., Kroemer G. (2019). Therapeutic modulation of autophagy: which disease comes first? Cell Death Differ. 26, 680–689. 10.1038/s41418-019-0290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J., Jabbour S., Orlick M., Riedlinger G., Guo Y., White E., et al. (2019). Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat Res. Commun. 21, 100158. 10.1016/j.ctarc.2019.100158 [DOI] [PubMed] [Google Scholar]
- Mandrioli J., D’amico R., Zucchi E., Gessani A., Fini N., Fasano A., et al. (2018). Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Med. (Baltimore) 97, e11119. 10.1097/MD.0000000000011119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrioli J., Crippa V., Cereda C., Bonetto V., Zucchi E., Gessani A., et al. (2019). Proteostasis and ALS: protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 9, e028486. 10.1136/bmjopen-2018-028486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A. M., Papa V., Tazzari P. L., Ricci F., Evangelisti C., Chiarini F., et al. (2010). Erucylphosphohomocholine, the first intravenously applicable alkylphosphocholine, is cytotoxic to acute myelogenous leukemia cells through JNK- and PP2A-dependent mechanisms. Leukemia 24, 687–698. 10.1038/leu.2010.32 [DOI] [PubMed] [Google Scholar]
- Mardones P., Rubinsztein D. C., Hetz C. (2016). Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 9, fs2. 10.1126/scisignal.aaf1937 [DOI] [PubMed] [Google Scholar]
- Marino M. L., Fais S., Djavaheri-Mergny M., Villa A., Meschini S., Lozupone F., et al. (2010). Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 1, e87. 10.1038/cddis.2010.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert J. M., Kaveney A. D., Malhotra J., Spencer K., Portal D., Goodin S., et al. (2019). A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 84, 899–907. 10.1007/s00280-019-03919-x [DOI] [PubMed] [Google Scholar]
- Mijaljica D., Prescott M., Devenish R. J. (2014). Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 7, 673–682. 10.4161/auto.7.7.14733 [DOI] [PubMed] [Google Scholar]
- Momota H., Nerio E., Holland E. C. (2005). Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 65, 742. 10.1158/0008-5472.CAN-05-1042 [DOI] [PubMed] [Google Scholar]
- Morel E., Mehrpour M., Botti J., Dupont N., Hamai A., Nascimbeni A. C., et al. (2017). Autophagy: A Druggable Process. Annu. Rev. Pharmacol. Toxicol. 57, 375–398. 10.1146/annurev-pharmtox-010716-104936 [DOI] [PubMed] [Google Scholar]
- Muschiol C., Berger M. R., Schuler B., Scherf H. R., Garzon F. T., Zeller W. J., et al. (1987). Alkyl phosphocholines: toxicity and anticancer properties. Lipids 22, 930–934. 10.1007/BF02535558 [DOI] [PubMed] [Google Scholar]
- Nascimbeni A. C., Giordano F., Dupont N., Grasso D., Vaccaro M. I., Codogno P., et al. (2017). ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 36, 2018–2033. 10.15252/embj.201797006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- New M., Van Acker T., Long J. S., Sakamaki J. I., Ryan K. M., Tooze S. A. (2017). Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 7, 28. 10.3389/fonc.2017.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novita B. D., Ali M., Pranoto A., Soediono E. I., Mertaniasih N. M. (2019). Metformin induced autophagy in diabetes mellitus - Tuberculosis co-infection patients: A case study. Indian J. Tuberc. 66, 64–69. 10.1016/j.ijtb.2018.04.003 [DOI] [PubMed] [Google Scholar]
- Padmapriyadarsini C., Bhavani P. K., Natrajan M., Ponnuraja C., Kumar H., Gomathy S. N., et al. (2019). Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF): study protocol for a randomised clinical trial. BMJ Open 9, e024363. 10.1136/bmjopen-2018-024363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S., Hurez V., Nawrocki S. T., Goros M., Michalek J., Sarantopoulos J., et al. (2016). Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 7, 59087–59097. 10.18632/oncotarget.10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Hernandez M., Arias A., Martinez-Garcia D., Perez-Tomas R., Quesada R., Soto-Cerrato V. (2019). Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers (Basel) 11, 1599. 10.3390/cancers11101599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A., Ogier-Denis E., Blommaart E. F., Meijer A. J., Codogno P. (2000). Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992–998. 10.1074/jbc.275.2.992 [DOI] [PubMed] [Google Scholar]
- Peymani P., Yeganeh B., Sabour S., Geramizadeh B., Fattahi M. R., Keyvani H., et al. (2016). New use of an old drug: chloroquine reduces viral and ALT levels in HCV non-responders (a randomized, triple-blind, placebo-controlled pilot trial). Can. J. Physiol. Pharmacol. 94, 613–619. 10.1139/cjpp-2015-0507 [DOI] [PubMed] [Google Scholar]
- Philip P. A., Buyse M. E., Alistar A. T., Rocha Lima C. M., Luther S., Pardee T. S., et al. (2019). A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 15, 3189–3196. 10.2217/fon-2019-0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C., Figlin R. A. (2009). Phosphatidylinositol-3-Kinase/Akt Signaling Pathway and Kidney Cancer, and the Therapeutic Potential of Phosphatidylinositol-3-Kinase/Akt Inhibitors. J. Urol. 182, 2569–77. 10.1016/j.juro.2009.08.085 [DOI] [PubMed] [Google Scholar]
- Puri C., Vicinanza M., Rubinsztein D. C. (2018). Phagophores evolve from recycling endosomes. Autophagy 14, 1475–1477. 10.1080/15548627.2018.1482148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal-Ruiz Y., Otten E. G., Korolchuk V. I. (2017). mTORC1 as the main gateway to autophagy. Signal. Mech. Autophagy 61, 565–584. 10.1042/EBC20170027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala R., Chang Y. C., Hu J., Algazy K. M., Evans T. L., Fecher L. A., et al. (2014. a). Combined mTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 10, 1391–1402. 10.4161/auto.29119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala R., Leone R., Chang Y. C., Fecher L. A., Schuchter L. M., Kramer A., et al. (2014. b). Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 10, 1369–1379. 10.4161/auto.29118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Moreau K., Jahreiss L., Puri C., Rubinsztein D. C. (2010). Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12, 747–757. 10.1038/ncb2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis-Sobreiro M., Roue G., Moros A., Gajate C., De La Iglesia-Vicente J., Colomer D., et al. (2013). Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J. 3, e118. 10.1038/bcj.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios-Marco P., Rios A., Jimenez-Lopez J. M., Carrasco M. P., Marco C. (2015). Cholesterol homeostasis and autophagic flux in perifosine-treated human hepatoblastoma HepG2 and glioblastoma U-87 MG cell lines. Biochem. Pharmacol. 96, 10–19. 10.1016/j.bcp.2015.04.015 [DOI] [PubMed] [Google Scholar]
- Rogov V., Dotsch V., Johansen T., Kirkin V. (2014). Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178. 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- Rojas-Puentes L. L., Gonzalez-Pinedo M., Crismatt A., Ortega-Gomez A., Gamboa-Vignolle C., Nunez-Gomez R., et al. (2013). Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 8, 209.’ 10.1186/1748-717X-8-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M. R., Ye X. B., Supko J. G., Desideri S., Grossman S. A., Brem S., et al. (2014). A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10, 1359–1368. 10.4161/auto.28984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner J., Ruiner C. E., Handrick R., Eibl H. J., Belka C., Jendrossek V. (2010). The Akt-inhibitor Erufosine induces apoptotic cell death in prostate cancer cells and increases the short term effects of ionizing radiation. Radiat. Oncol. 5, 108. 10.1186/1748-717X-5-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter G. A., Zerp S. F., Bartelink H., Van Blitterswijk W. J., Verheij M. (2003). Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anticancer Drugs 14, 167–173. 10.1097/00001813-200302000-00011 [DOI] [PubMed] [Google Scholar]
- Sharma A., Bhomia M., Yeh T. J, Singh J., Maheshwari R. K. (2018). Miltefosine inhibits Chikungunya virus replication in human primary dermal fibroblasts. F1000Research 7, 9. 10.12688/f1000research.13242.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J., Briceno E., Lopez-Gonzalez M. A. (2006). Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern Med. 144, 337–343. 10.7326/0003-4819-144-5-200603070-00008 [DOI] [PubMed] [Google Scholar]
- Stein M., Lin H., Jeyamohan C., Dvorzhinski D., Gounder M., Bray K., et al. (2010). Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 70, 1388–1394. 10.1002/pros.21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A., Ernst A., Dikic I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. 10.1038/ncb2979 [DOI] [PubMed] [Google Scholar]
- Sun S. Y. (2010). Enhancing perifosine’s anticancer efficacy by preventing autophagy. Autophagy 6, 184–5. 10.4161/auto.6.1.10816 [DOI] [PubMed] [Google Scholar]
- Sun W., Modak S. (2012). Emerging treatment options for the treatment of neuroblastoma: Potential role of perifosine. Onco. Targets. Ther. 5, 21–9. 10.2147/OTT.S14578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T., Liu H., Ming L. (2017). Multiple Roles of Autophagy in the Sorafenib Resistance of Hepatocellular Carcinoma. Cell. Physiol. Biochem. 44, 716–727. 10.1159/000485285 [DOI] [PubMed] [Google Scholar]
- Tang Y., El-Chemaly S., Taveira-Dasilva A., Goldberg H. J., Bagwe S., Rosas I. O., et al. (2019). Alterations in Polyamine Metabolism in Patients With Lymphangioleiomyomatosis and Tuberous Sclerosis Complex 2-Deficient Cells. Chest 156, 1137–1148. 10.1016/j.chest.2019.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y., Liu Y. Y., You L. S., Qian W. B. (2012). Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol. Sin. 33, 542–550. 10.1038/aps.2011.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze S. A., Yoshimori T. (2010). The origin of the autophagosomal membrane. Nat. Cell Biol. 12, 831–835. 10.1038/ncb0910-831 [DOI] [PubMed] [Google Scholar]
- Van Blitterswijk W. J., Verheij M. (2008). Anticancer alkylphospholipids: mechanisms of action, cellular sensitivity and resistance, and clinical prospects. Curr. Pharm. Des. 14, 2061–2074. 10.2174/138161208785294636 [DOI] [PubMed] [Google Scholar]
- Van Blitterswijk W. J., Verheij M. (2013). Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 1831, 663–674. 10.1016/j.bbalip.2012.10.008 [DOI] [PubMed] [Google Scholar]
- Vogl D. T., Stadtmauer E. A., Tan K. S., Heitjan D. F., Davis L. E., Pontiggia L., et al. (2014). Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10, 1380–1390. 10.4161/auto.29264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl D. T., Raje N., Jagannath S., Richardson P., Hari P., Orlowski R., et al. (2017). Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 23, 3307–3315. 10.1158/1078-0432.CCR-16-2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Duan W., Li Y., Zhang S., Xin X., Sun L., et al. (2016). AT101 exerts a synergetic efficacy in gastric cancer patients with 5-FU based treatment through promoting apoptosis and autophagy. Oncotarget 7, 34430–34441. 10.18632/oncotarget.9119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y., Liu M., Li X., Liu J., Li H. (2018). Origin of the Autophagosome Membrane in Mammals. BioMed. Res. Int. 2018, 1012789. 10.1155/2018/1012789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M., Benson G., Schwarz C., Kobe T., Grittner U., Schmitz D., et al. (2018). The effect of spermidine on memory performance in older adults at risk for dementia: A randomized controlled trial. Cortex 109, 181–188. 10.1016/j.cortex.2018.09.014 [DOI] [PubMed] [Google Scholar]
- Wirth M., Schwarz C., Benson G., Horn N., Buchert R., Lange C., et al. (2019). Effects of spermidine supplementation on cognition and biomarkers in older adults with subjective cognitive decline (SmartAge)-study protocol for a randomized controlled trial. Alzheimers Res. Ther. 11, 36. 10.1186/s13195-019-0484-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin B. M., Rubinson D. A., Wang X., Chan J. A., Cleary J. M., Enzinger P. C., et al. (2014). Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19, 637–638. 10.1634/theoncologist.2014-0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H., Kakuta S., Watanabe T. M., Kitamura A., Sekito T., Kondo-Kakuta C., et al. (2012). Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 198, 219–233. 10.1083/jcb.201202061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Chen S. C., Zhang Y. Q., Lin X. X., Song Y. Y., Xue Z. L., et al. (2017). Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. 8, e2738. 10.1038/cddis.2017.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee A. J., Bensinger W. I., Supko J. G., Voorhees P. M., Berdeja J. G., Richardson P. G., et al. (2016). Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 17, 1569–1578. 10.1016/S1470-2045(16)30375-8 [DOI] [PubMed] [Google Scholar]
- Yoshida G. J. (2017). Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J. Hematol. Oncol. 10, 67. 10.1186/s13045-017-0436-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosifov D. Y., Todorov P. T., Zaharieva M. M., Georgiev K. D., Pilicheva B. A., Konstantinov S. M., et al. (2011). Erucylphospho-N,N,N-trimethylpropylammonium (erufosine) is a potential antimyeloma drug devoid of myelotoxicity. Cancer Chemother. Pharmacol. 67, 13–25. 10.1007/s00280-010-1273-5 [DOI] [PubMed] [Google Scholar]
- Yosifov D. Y., Kaloyanov K. A., Guenova M. L., Prisadashka K., Balabanova M. B., Berger M. R., et al. (2014). Alkylphosphocholines and curcumin induce programmed cell death in cutaneous T-cell lymphoma cell lines. Leuk Res. 38, 49–56. 10.1016/j.leukres.2013.10.011 [DOI] [PubMed] [Google Scholar]
- Yu L., Chen Y., Tooze S. A. (2018). Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215. 10.1080/15548627.2017.1378838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaharieva M. M., Kirilov M., Chai M., Berger S. M., Konstantinov S., Berger M. R. (2014). Reduced expression of the retinoblastoma protein shows that the related signaling pathway is essential for mediating the antineoplastic activity of erufosine. PloS One 9, e100950. 10.1371/journal.pone.0100950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremberg V., Ganesan S., Mahadeo M. (2019). Lipids and Membrane Microdomains: The Glycerolipid and Alkylphosphocholine Class of Cancer Chemotherapeutic Drugs. Handb. Exp. Pharmacol. 1–28. 10.1007/164_2019_222 [DOI] [PubMed]
- Zhang H. (2004). “NFluc-FHA2-Aktpep-CFluc,” in Molecular Imaging and Contrast Agent Database ((Bethesda (MD)): (MICAD)). [Google Scholar]
- Zhu F., Kai J., Chen L., Wu M., Dong J., Wang Q., et al. (2018). Akt Inhibitor Perifosine Prevents Epileptogenesis in a Rat Model of Temporal Lobe Epilepsy. Neurosci. Bull. 34, 283–290. 10.1007/s12264-017-0165-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitzmann K., Vlotides G., Brand S., Lahm H., Spöttl G., Göke B., et al. (2012). Perifosine-mediated Akt inhibition in neuroendocrine tumor cells: Role of specific Akt isoforms. Endocr. Relat. Cancer 19, 423–34. 10.1530/ERC-12-0074 [DOI] [PubMed] [Google Scholar]