

Summary
Aryl halide (Br, Cl, I) is among the most important compounds in pharmaceutical industry, material science, and agrochemistry, broadly utilized in diverse transformations. Tremendous approaches have been established to prepare this scaffold; however, many of them suffer from atom economy, harsh condition, inability to be scaled up, or cost-unfriendly reagents and catalysts. Inspired by vanadium haloperoxidases herein we presented a biomimetic approach for halogenation (Br, Cl, I) of (hetero)arene catalyzed by tungstate under mild pH in a cost-efficient and environment- and operation-friendly manner. Broad substrates, diverse functional group tolerance, and good chemo- and regioselectivities were observed, even in late-stage halogenation of complex molecules. Moreover, this approach can be scaled up to over 100 g without time-consuming and costly column purification. Several drugs and key precursors for drugs bearing aryl halides (Br, Cl, I) have been conveniently prepared based on our approach.
Subject Areas: Pharmaceutical Engineering, Organic Chemistry, Green Chemistry
Graphical Abstract
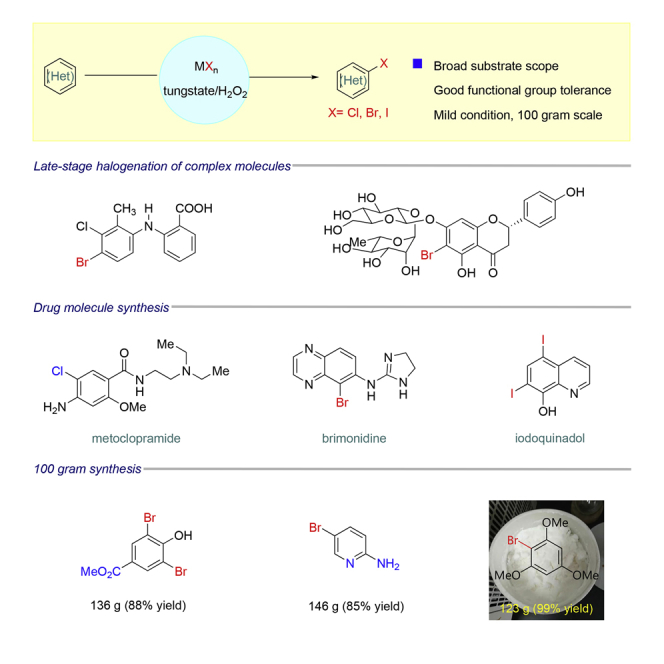
Highlights
-
•
Tungstate-catalyzed halogenation of (hetero)arenes under mild condition
-
•
Robust in 100-g-scale synthesis; good functional group tolerance
-
•
Late-stage halogenation of complex molecules; good application in drug synthesis
Pharmaceutical Engineering; Organic Chemistry; Green Chemistry
Introduction
(Hetero)aryl halides (Br, Cl, I) are embodied in many biologically important molecules, like natural products, drugs, and drug leads (Hernandes et al., 2010). In organic molecules, replacing a proton by a halide can significantly improve their properties, including solubility, polarity, melting point, stability, binding affinity, selectivity to biological targets, and metabolism. Recently, halogen bonding emerges as a useful tool in both catalyst and drug design (Wilcken et al., 2013). For example, chloride is normally used as bioisostere of methyl and hydroxyl groups, whereas bromide is frequently used as bioisostere of isopropyl and trifluoromethyl groups. The isotopes of iodide, like 123I and 131I, are widely used in medical setting for imaging, such as iopanoic acid. Meanwhile, as one of the most useful building blocks in organic synthesis, (hetero)aryl halides are broadly utilized in countless transformations like cross-couplings (e.g., Suzuki coupling, Buchwald-Hartwig coupling, Ullman coupling) (Johansson et al., 2012) (Figure 1A).
Figure 1.

Previous Approach for Halogenation (Br, Cl, I) and Our Strategy
(A) Typical examples of biologically important (hetero)aryl halides.
(B–D) (B) Previous reported approaches for halogenation developed in laboratories. (C) The reaction mechanism of vanadium haloperoxidases (V-HPO) and comparison of V-η2-peroxy and W-η2-peroxy intermediates. (D) Biomimetic halogenation catalyzed by tungstate. HMPT, hexamethylphos-phor triamide.
Conventional approach to introduce halides (Br, Cl, I) on (hetero)arenes heavily relies on electrophilic halogenation with various of reagents, like chlorine, bromine, iodine, NCS, NBS, tBuOCl, and Palau'chlor4 (Rodriguez et al., 2014). Such electrophilic halogenation process, unavoidably, generates another part of molecule as waste, like HBr and succinimide from bromine and NBS, respectively. Besides, they also suffer from being erosive, explosive, or toxic. The Sandmeyer reaction (Kumar et al., 2012) is another commonly used approach to prepare (hetero)aryl halides. However, multiple steps, lots of chemical wastes, and harsh reaction conditions are necessary. Oxidative halogenation serves as an important alternative (Podgoršek et al., 2009), like transition metal (TM)-catalyzed C-H bond functionalization (Petrone et al., 2016), photo-/electrocatalysis (Br, Cl) (Hering et al., 2016; Yuan et al., 2019, Liang et al., 2019), and HX (X = Br, Cl)/oxidant (Fosu et al., 2019, Srivastava et al., 1996, Ross and Burrows, 1987, Ben-Daniel et al., 2003). Albeit significant progress has been achieved, there is still large room to improve. For example, the TM-catalyzed oxidative halogenations normally require noble catalysts, directing groups, harsh conditions, or costly oxidants. Some frequently encountered functional groups cannot be tolerated in electrocatalysis, such as alkyl carboxylic acids (Kolbe electrolysis). Acid-sensitive groups (e.g., alkene, tert-butylcarbamate, alcohol, and basic N-atoms) and primary/secondary alcohols (Srivastava et al., 1996) cannot survive well in HX/oxidant as well. The large-scale synthesis (>100g) is also quite challenging for these methods, due to cost-unfriendly reagents, harsh conditions, or difficulties in purification. Halogenation in nature, on the other hand, can be achieved in high selectivity even with complex molecules under mild conditions via utilizing nucleophilic halides by enzymes, like metalloenzymes and flavoenzymes (Latham et al., 2018, Butler and Sandy, 2009, Gkotsi et al., 2018, Vailancourt et al., 2006). However, its industrial application still needs improvements in several aspects due to limited substrate scopes, high requirements of condition to maintain enzyme activity (solvent, temperature, pH, and so on), and inconvenient isolation process in large-scale synthesis (Figure 1B).
The biomimetic halogenation provides a possible access to such ideal halogenation. Inspired by vanadium haloperoxidases (V-HPO) (Latham et al., 2018, Winter and Moore, 2009), vanadium-catalyzed biomimetic halogenation (de la Rosa et al., 1992) has attracted lots of attention. According to the mechanism, a V-η2-peroxy intermediate (V-II) was formed first in the presence of H2O2 and then opened by halide assisted by H-bonding from proximate lysine, yielding an electrophilic hypohalite (OX−, HOX or V-OX, X = Br, Cl, I) captured by (hetero)arenes (Winter and Moore, 2009) (Figure 1C, I). However, vanadium is difficult for industrial production as critical hazard by Europea Union regulation (Assem and Levy, 2009). Theoretically, any transition metal can also fulfill such biomimetic halogenations if only they can form such η2-peroxy intermediates. Thus, many researches have focused on searching for other TM catalysts for this biomimetic halogenation (Herget et al., 2018). However, low pH condition was normally required to maintain the activity of catalysts, and many acid-sensitive functional groups cannot be tolerated. Actually, development of biomimetic halogenation under mild pH is a long-time challenge. Among them, tungsten (W) (Beinker et al., 1998, Sels et al., 1999, Badetti et al., 2015) has attracted great interest. Given that W-η2-peroxy intermediate has more charged metal ion-bearing larger-radii (W6+, 60 pm versus V5+ 46 pm) and longer O-O bonds than those of V-η2-peroxy intermediate (151, 153 pm for W, 143–146 nm for V) (Reynlds and Butler, 1996, Amato et al., 1986, Mimoun et al., 1983, Drew and Einstein, 1972, Stomberg, 1986, Djordjevic et al., 1985, Begin et al., 1975), it is supposed to be a better peroxy electrophile as observed in oxidation of (thiolato)-cobalt (III) complexes (Ghiron and Thompson, 1988). Meanwhile, the electrophilicity of W-η2-peroxy, we believe, can be further enhanced by Brønsted or Lewis acid (Kikushima et al., 2010), promoting the formation of hypohalous species (W-III, HOX) (Roy and Bhar, 2010). From this perspective, biomimetic W-catalyzed halogenation under mild pH could be possible (Figure 1C, II). Herein, we present such a tungstate-catalyzed, biomimetic oxidative halogenation (Br, Cl, I) of (hetero)arene in a scalable (>100 g), inexpensive, environment- and operation-friendly manner, along with broad substrate scope, diverse functional group tolerance, and good chemo- and regioselectivity (Figure 1D).
Results and Discussion
Condition Optimization for Tungstate-Catalyzed Oxidative Bromination of (Hetero)Arene
Initially, aniline 1-1 was selected as model substrate for our hypothesis. After lots of efforts in condition screening, ultimately the desired product 2-1 can be obtained in good yield in the presence of 5 mol % sodium tungstate, 1.1 equivalents of NaBr, and 6.0 equivalents of H2O2 (30% aq.) in EtOH assisted by adding 1.1 equivalents of HOAc (Table 1, entry 1). The reaction still moved on but at a much slower rate without adding HOAc (Table 1, entry 2). Only trace amount of the product can be detected by thin-layer chromatography without catalyst in background reaction (Table 1, entry 3). Polyoxotungstate also worked well in this reaction, with a little bit lower yield (Table 1, entry 4 and 5). Other bromides, like LiBr and KBr, afforded the product 2-1 as well in eroded efficiency (Table 1, entry 6 and 7). Sodium perborate failed to afford any product (Table 1, entry 8). Other protonic solvents, like MeOH, also worked smoothly (Table 1, entry 9). Notably, the reaction went on well even in H2O, although both starting material and product were not well dissolved (Table 1, entry 10). Decreasing the loading of either catalyst or H2O2 evidently reduced the reaction efficiency (Table 1, entry 11-12). Only trace ortho-bromination (2-1B), dibromination (2-1C), and nitroso products (2-1D) were observed in all condition screenings. In addition, no azo or azoxy products were observed (Ke et al., 2019), demonstrating good chemo- and regioselectivity of this transformation (Tables 1, see also S1 and S2 and Figure S6).
Table 1.
Condition Optimization
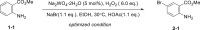 | |||
|---|---|---|---|
| Entry | Vary from Optimized Condition | Yielda | Major By-products |
| 1 | None | 78%–83% | 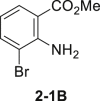 |
| 2 | Without HOAc | <50% conversion for 3 days | |
| 3 | Without Na2WO4-2H2O | Traceb | |
| 4 | H3O40PW12-xH2O instead of Na2WO4-2H2O | 64%c | |
| 5 | (NH4)10(H2W12O42)-xH2O instead of Na2WO4-2H2O | 77%c | 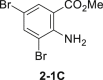 |
| 6 | LiBr instead of NaBr | 66%d | |
| 7 | KBr instead of NaBr | 75%d | |
| 8 | SPB instead of H2O2 | N.R | |
| 9 | MeOH instead of EtOH | 67% | 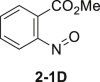 |
| 10 | H2O instead of EtOH | 67% | |
| 11 | Na2WO4-2H2O, 1 mol % instead of 5 mol % | 67% | |
| 12 | 2.0 equiv H2O2 instead of 6.0 equiv | 57% | |
N.R, no reaction
All the reactions were conducted in 1.0-mmol scale (1-1) for 12 h, isolated yield.
2.0 equivalents of HOAc was utilized.
1.5 equivalents NaBr and 2.0 equiv. HOAc were utilized.
1.5 equivalents of NaBr and 2.0 equivalents of HOAc were utilized, and the reactions were conducted at 50°C
Substrate Scope of Tungstate-Catalyzed Oxidative Bromination
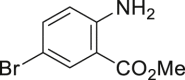
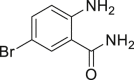
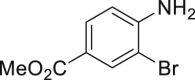
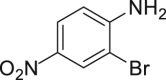
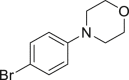
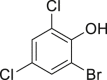
With the optimized reaction condition at hand, the substrate scope for bromination was investigated as summarized in Table 2. Overall, anilines, phenols, other electronically rich (hetero)arenes, and carbonyl compounds can all afford the bromination products smoothly in moderate to excellent yield. Diverse functional groups were well tolerated, including ester (2-1, 2-3, 2-11, 2-16, 2-20), amide (2-2, 2-12), hydroxy (2-7 to 2-12), nitrone (2-4, 2-10), halogens (2-7, 2-22), morpholine (2-6), methoxy (2-13, 2-14), carboxylic acid (2-9), and ketone (2-15). Good to excellent chemo- and regioselectivity were observed as well. The para-product was favored over ortho-product (e.g., 2-1, 2-2, and 2-6). The unprotected amine groups in anilines remained untouched, even though the azoxy formation could dominate the reaction (Ke et al., 2019). Unlike the dimerization and dearomatization frequently encountered under similar conditions (Dewar and Nakaya, 1968), the oxidative bromination of phenol still worked smoothly in this reaction. Adding 2.2 equiv NaBr and HOAc, the dibromination products can be easily prepared as well (2-10, 2-11, 2-12) (Figure 2B). Interestingly, only monobromination of 1,3,5-trimethoxy benzene (2-14) was observed without any dibromination product and 1,3-dicarbonyl compounds afforded the α-bromination product.
Table 2.
Substrate Scope of Tungstate-Catalyzed Oxidative Bromination of (Hetero)Arene
 |
 |
 |
 |
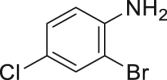 |
| 2-1, 91% (p: o: d = 21:1:1) | 2-2, 52% | 2-3, 85% (89%a in H2O) | 2-4, 75% (90% brsm) | 2-5, 69%b |
 |
 |
 |
 |
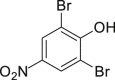 |
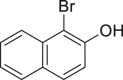
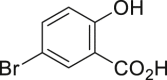
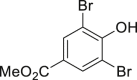
| 2-6, 57% | 2-7, 65% (76%a in H2O) | 2-8, 54% | 2-9, 70% | 2-10, 93%c |
 |
 |
 |
 |
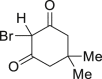 |
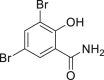
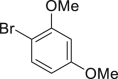
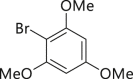
| 2-11, 86%c | 2-12, 83%c | 2-13, 86% | 2-14, 98% (92%a in H2O) | 2-15, 62% yield |
 |
 |
 |
 |
 |
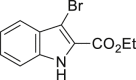
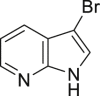
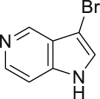
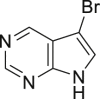
| 2-16, 81% | 2-17, 74% | 2-18, 73% | 2-19, 72% |
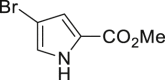
2-20 52% (m: d = 5:1) |
 |
 |
 |
 |
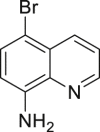 |
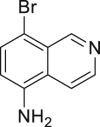
| 2-21, 38% (79% brsm) | 2-22, 92% | 2-23, 79% | 2-24, 61% (m: d = 2:1) | 2-25, 33% |
 |
 |
 |
 |
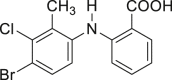
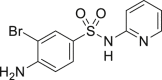
| 2-26, 97% (tolfenamic acid) | 2-27, 74% (sulfapyridine) |
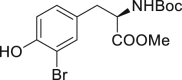
2-28, 34% (98% brsm) (derived from tyrosine) |
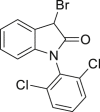
2-29, 31% (45% brsm) (indole-2-one) |
 |
 |
 |
|
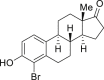
2-30, 48% (56% brsm, m: d = 11:1) (estrone) |
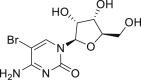
2-31, 83% (by HNMR) (cytidine) |
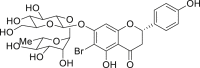
2-32, 75% (by NMR, 2:1 rr) (naringin) |
p, para-bromination product; o, ortho-bromination product; m, mono-bromination; d, dibromination product; rr, regioselective ration; brsm, based on recovered starting material.
Unless noted, all the reactions were conducted in 1.0-mmol scale (1) with Na2WO4-2H2O (5 mol %), NaBr (1.1 equivalents), H2O2 (30 % aq., 6.0 equivalents.), HOAc (1.1 equiv.) in EtOH (5.0 mL) at 30°C isolated yield (see also Figure S1).
The reactions were conducted in H2O with Na2WO4-2H2O (2.5 mol %), NaBr (1.1 equivalent), H2O2 (30 % aq., 1.1 equivalent) and HOAc (1.1 equivalent) (see also Figure S2).
5.0-mmol scale.
2.2 mmol NaBr (2.2 equivalents) and HOAc (2.2 equivalents) were utilized.
Figure 2.

Application in Synthesis of Selected Drugs and Key Precursors
(A) Synthesis of quinoline drugs or key precursors, including clioquinol, iodoquinado, broxyquinoline, and precursor for broxaldine.
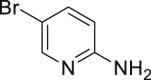
(B) Synthesis of key precursor for brimonidine, a drug to treat ocular hypertension, rosacea, and open-angle glaucoma.
(C) Synthesis of key precursor bromopride and metoclopramide, antiemetic drugs.
(D) Synthesis for benzofuran drugs: benzbromaron, benziodarone, and amiodarone.
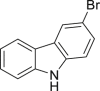
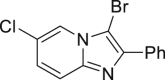
As it is well known, the Lewis basicity of nitrogen (N) could hamper the reaction by coordinating to transition metals as observed in halogenation catalyzed by Pd, Cu, Rh, and Ru (Petrone et al., 2016, Wan et al., 2006). In addition, heteroarenes bearing strong basic nitrogen (e.g., pyridine, isoquinoline, and quinolone) can form salts with HOAc, leading to a decrease in their nucleophilicity and enhancement of the pH value in the reaction. Surprisingly, the bromination of N-containing heteroarenes still proceeded smoothly in our reaction, including indole (2-16), indole analogs (2-17, 2-18, 2-19), pyrroles (2-20, see also Figure S7), carbazone (2-21), imidazo[1,2-a]pyridine (2-22), pyridine (2-23), isoquinoline (2-24, see also Figure S8), and quinoline (2-25). Actually, the reactions were conducted in neutral condition to some extent for those basic heteroarenes. Of note, no N-oxide products were observed in all tested heteroarenes, indicating excellent chemoselectivity in this reaction.
Late-stage bromination of complex molecules, like drug leads and bioactive natural products, is highly appealing, facilitating quick structure-activity relationship studies given the diverse transformations based on aryl bromides. It is reasonable to hypothesize that the selective late-stage bromination of complex molecules can be achieved, considering the good functional group tolerance as observed in previous studies. However, it can be challenging to obtain good chemo- and regioselectivities for substrates bearing multiple reaction sites with slightly different chemical surroundings. Nonetheless, the late-stage bromination of complex molecules was proved to be successful in our reaction. For instance, the slight difference of multiple reactive positions in olfenamic acid (2-26) and sulfapyridine (2-27) could be distinguished, affording the single monobromination products in high yield. Tyrosine (2-28), indole-2-one (2-29), and estrone (2-30, see also Figure S9) also yielded the monobromination products in good chemo- and regioselectivity. It is worth pointing out that the acid-sensitive tert-butylcarbamate group was well tolerated in our reaction, demonstrating that our approach has better functional group tolerance than that of reported HX/oxidant system. Notably, saccharide scaffolds are maintained untouched, as shown in cytidine (2-31, see also Figure S10) and naringin (2–32, see also Figure S11), albeit many oxidant transformations could occur with such multiple unprotected hydroxyl groups. Water is the ideal reaction solvent, from the perspective of cost and environmental concerns. Delightfully, our reaction worked in water as well (see also Table S3), giving comparable results as those in EtOH (2-2, 2-3, 2-7, 2-14).
Substrate Scope of Tungstate-Catalyzed Oxidative Chlorination and Iodination
The oxidative chlorination and iodination were also investigated as summarized in Table 3. Noticing that the redox potential of chloride is higher than that of bromide, oxidative chlorination was more challenging. Indeed, unlike the fact that the bromination worked smoothly with NaBr and KBr, no chlorination product was observed in the presence of NaCl or KCl. After tedious efforts in condition optimization, ultimately it was found that BaCl2 could afford the chlorination products in best yield with model substrate 1-1 (see also Tables S4 and S5). Aniline, phenol, other electronically rich (hetero)arenes, and carbonyl compounds all worked well. Good functional group tolerance was observed as well, including ester (2-33, see also Figure S12; 2-40), free aniline (2-33, 2-34), morpholine (2-35, see also Figure S13), halide (2-36, 2-42), alkoxyl (2-37), carboxylic acid (2-38), and ketone (2-39). Compared with bromination, chlorination generally required longer reaction time and higher reaction temperature. Although iodide is easier to be oxidized than bromide, such biomimetic oxidative iodination of (hetero)arenes is scarcely reported (Sels et al., 2005). Moreover, the aryl iodide product can potentially be further oxidized into hyperiodide species (Banik et al., 2016), leading to undesired by-products. To our delight, such overoxidation was not observed in this reaction (Emmanuvel et al., 2006). Selected substrates were investigated, and all afforded the products in moderate to excellent yield, including aniline (2-43, 2-45), phenol (2-44), and heteroarene (2-46, 2-47). It should be pointed out that the background of iodination worked as well without catalyst. However, the catalyst acceleration was also evident as observed in some substrates, and longer reaction time is required without catalyst (2-45, 2-47).
Table 3.
Substrate Scope of Tungstate-Catalyzed Oxidative Chlorination and Iodination
 |
 |
 |
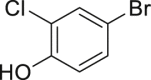 |
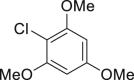 |
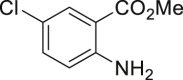
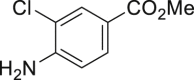
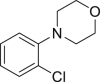
| 2-33a, 81% (p: o = 2.2: 1) | 2-34a, 49% (64% brsm) | 2-35a, 58% (p: o = 1: 3.5) | 2-36a, 82%b | 2-37a, 51% (74% brsm) |
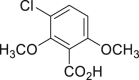 |
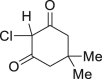 |
 |
 |
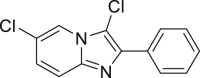 |
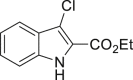
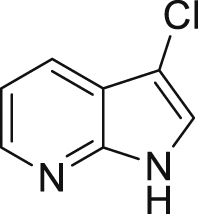
| 2-38a, 44% (56% brsm) | 2-39a, 43% | 2-40a, 70% (91% brsm) | 2-41a, 56% (62% brsm) | 2-42a, 35% (78% brsm) |
 |
 |
 |
 |
 |
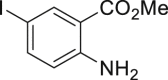
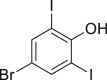
| 2-43c, 39% (48% brsm) |
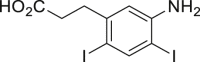
2-44c,d,e, 97%d (13 h) (93%e, w/o. cat.,17 h) |
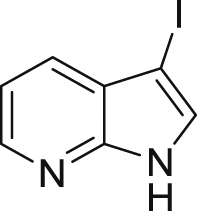
2-45c,d, 65%d (40 min) (66%e, w/o. cat., 6 h) |
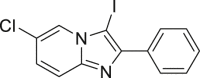
2-46c, 89% (15 min) (96%e, w/o. cat., 15 min) |
2-47c, 97% (20 min) (99%e, w/o. cat., 1 h) |
brsm, based on recovered starting material; p, para-chlorination product; o, ortho-chlorination product.
Unless noted, the chlorination reactions were carried out in 1.0-mmol scale (1) in MeCN (5.0 mL) at 50°C with Na2WO4-2H2O (5 mol %), H2O2 (30% aq. 4.0 equiv.), BaCl2·2H2O (1.2 equiv), and HOAc (1.0 equiv) (see also Figure S3).
Trifluoro acetic acid was used instead of HOAc.
The iodination reaction was conducted in 1.0-mmol scale (1) in EtOH at room temperature (20–25°C) with Na2WO4-2H2O (5 mol %), H2O2 (30% aq. 6.0 equiv), KI (1.1 equiv), and HOAc (1.1 equiv) (see also Figure S4).
2.2 equivalents of KI and HOAc were used.
No catalyst was added as control experiments.
100-g-Scale Preparation of (Hetero)Aryl Bormide
The reaction can be scaled up to over 100 g without any erosion of efficiency, including aniline (Table 4, 2-1 and 2-3), phenol (Table 4, 2-11), and other electronically rich (hetero)arene (Table 4, 2-14 and 2-23). Of note, the reaction can be conducted in beakers in the open air, and the products precipitated upon completion during gram-scale reaction, obtained in high purity only by filtration and washing (see also Figure S5). These results suggested the good potential of our reaction in industrial preparation of (hetero)aryl halide.
Table 4.
100-g-Scale Bromination
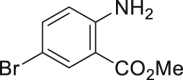 |
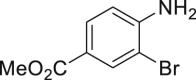 |
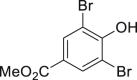 |
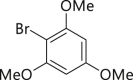 |
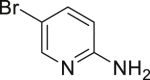 |
| 2-1, 93%, 221g (p: o: d = 20: 3: 3) | 2-3, 94%, 107 g | 2-11, 88%,136 g | 2-14, 99%, 123 g | 2-23, 85%, 146 g |
Synthetic Application in Drugs and Key Precursors for Drugs
Many drugs and drug leads contain aryl halide (Br, Cl, I) motifs, thus we would like to explore the utility of our approach in their preparation. The quinoline halides were embodied in several antibiotic drugs and bioactive compounds, like chlorquinaldol, iodoquinadol, cloxiquine, clioquinol, and broxyquinoline. Although they were reported to be good ligands for tungsten (Archer and Bonds, 1967), the oxidative halogenation of 8-hydoxyquinolines was achieved successfully. Clioquinol (2-48), iodoquinadol (2-49), broxyquinoline (2-50), and precursor for broxaldine (2-51) were all obtained in moderate to good yield. Of note, all these reactions were conducted in multiple gram scale without silica gel column purification, typically like the precursor for broxalide (2-51) (Swain et al., 1986) (Figure 2A). The key intermediate for brimonidine (2-52) (Jeon et al., 1995), a medicine to treat ocular hypertension, rosacea, and open-angle glaucoma, was conveniently prepared (Figure 2B). As dopamine antagonists are used as antiemetic drugs, both bromopride and metoclopramide bear an aryl halide motif derived from salicylic acid (Kato et al., 1991), which were conveniently prepared from methyl 4-amino-2-methoxybenzoate according to our approach (2-53 and 2-54) (Figure 2C). Benzbromaron is a broadly utilized drug to treat gout. However, the dibromination of its precursor, compound 1-43, with bromine, only afford 35% yield of benzbromarone along with the monobromination impurities (Wempe et al., 2011). By contrast, pure benzbromarone (2-55) was obtained in excellent yield under mild condition even without column purification by utilizing our approach. Benziodarone (2–56), a vasodilator, was obtained in excellent yield as well. Compared with the poor yield and chemoselectivity by utilizing NaClO/KI/NaOH (Snead et al., 2008) the iodination of compound 1-50 worked quite well in our reaction, giving the key precursor for amiodarone (2-57), a drug to treat arrhythmias (Figure 2D).
Mechanism Study
To figure out whether the bromine radical is involved in this reaction, substrate 1-45 was selected as a probe. If the bromine radical existed as reaction intermediate, product 2-59 should be observed. However, only product 2-58 was isolated in moderate yield, excluding that the bromination went through radical reaction pathway (Figures 3A and see also S14). HOAc was used only in 1.1 equivalents as reagent in this reaction, rather than solvent, to stabilize the Br+ species. We suppose the HOAc played dual key roles in this halogenation (Maheswari et al., 2006): (1) neutralizing the hydroxyl anion from H2O2 and (2) providing H-bonding to the key W-η2-peroxy intermediate W-II. For chlorination, the chloride salts (MCln) also played key dual role: (1) providing chloride and (2) being a Lewis acid to the W-η2-peroxy intermediate (W-II) similar to V-catalyzed bromination (Kikushima et al., 2010). Both the HOAc and metal ion (Mn+) can increase the electrophilicity of W-η2-peroxy intermediate W-II by H-bonding or Lewis acid-base interaction (Figure 3B). From this perspective, weakening such H-boding effect will decrease the reaction efficiency. Indeed, the reaction proceeded much slower without HOAc or in aprotic solvent, such as THF (Figure 3C, see also Table S2). On the other side, the strength of the Lewis acidity of metal cations can control the chlorination as well, in consist with evidently different reactionefficiency when utilizing different chloride salts. For example, no chlorination product was detected in the presence of LiCl, NaCl, and KCl; however, dichlorination product 2-34C was isolated as major product in the presence of CaCl2 or SrCl2 even reducing their loading. By contrast, monochlorination products (2-34, 2-34B) were obtained as major products with BaCl2 even extending reaction time. In addition, the major hypohalous species in chlorination should be W-III rather than HOCl; otherwise, the chloride salts should not have significant impact on products' distribution on monochlorination and dichlorination (Figure 3D, see also Table S4).
Figure 3.

Hypothesis of Mechanism of Bromination/Iodination and Control Experiments
(A) Radical trapping experiment.
(B) Hypothesis of H-bonding effect with Brønsted acid.
(C) Control experiments to probe the importance of H-bonding.
(D) Effects of metal ion in chloride salts in chlorination.
Proposed Reaction Mechanism
Other polyoxotungstate catalyst intermediates might also be involved (Corsini and Subramanian, 1978), like dinuclear peroxotungstate W-VI (Kamata et al., 2007), thus we used W-IV as a model to illustrate the reaction mechanism given that the reaction center is the peroxyl moiety. Based on all the control experiments, we proposed the reaction mechanism as following. Initially, the tungstate W-V would form W-η2-peroxy intermediate W-V and then W-I in the presence of H2O2 and HOAc. The peroxy moiety in W-I was opened by halide (Roy and Bhar, 2010) in the assistance of Brønsted acid (bromination, iodination) or Lewis acid (chlorination) to yield hypohalous species W-III or HOX, which was trapped by (hetero)arene via electrophilic halogenation to afford the desired products. The HOAc and EtOH can also serve as a ligand to tungstate catalyst (Figure 4).
Figure 4.

Proposed Reaction Mechanism
In summary, we have developed a tungstate-catalyzed, biomimetic, cost-efficient, environment- and operation-friendly approach for halogenation (Br, I, Cl) of (hetero)arene under mild pH. Broad substrate scope, diverse functional group tolerance, and late-stage bromination of bioactive complex molecule were achieved in this reaction. Besides, water can be utilized as solvent and >100-g scale reaction was conveniently accessed without column purification. Furthermore, several drugs and key precursors for drugs have been conveniently prepared. Primary mechanism studies suggested that Brønsted or Lewis acid can accelerate the reaction and control the products' distribution.
Limitations of the Study
Our reaction provides a green and robust access to (hetero)aryl halides and works well for most tested substrates. However, limitation still exists. For example, the chlorination normally takes longer reaction time and has lower efficiency than bromination and iodination. Some too electronically rich substrates like indole involve other undesired oxidative transformations along with halogenation. In addition, more than 1 equivalent of H2O2 is required for the completion of this reaction under current optimized conditions. Further improvement to enhance the reaction efficiency with broader substrate scope is under investigation in our laboratory.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Financial support was provided by the National Natural Science Foundation of China (21602011) and the talent recruitment fund of Huazhong University of Science and Technology (HUST). The authors also would like to thank the Analytical and Testing Center in HUST for assistance in NMR and HRMS data collection.
Author Contributions
Z.C. conceived and directed the project; Z.C., Z.M., H.L., and K.L. designed the experiments; Z.M. and K.L. performed the condition optimization for bromination/chlorination; Z.M. performed the condition optimization of iodination; H.L. performed the reactions in water; Z.M. performed the substrates scope investigation; Z.C. and Z.M. collected and analyzed the data; Z.C. prepared the manuscript.
Declaration of Interests
The authors declare no conflicts of interest.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101072.
Data and Code Availability
Procedure for experiments and characterization data for products are available in Supplemental Informations. (Figures S15–S149) Any other data are available from the corresponding author upon reasonable request.
Supplemental Information
References
- Amato G., Arcoria A., Ballistreri F.P., Tomaselli G.A., Bortolini O., Conte V., Di Furia F., Modena G., Valle G. Oxidations with peroxotungsten complexes: rates and mechanism of stoichiometric olefin epoxidations. J. Mol. Catal. 1986;37:165–175. [Google Scholar]
- Archer R.D., Bonds W.D., Jr. A completely cheated spin-paired eight-coordinate tungsten(IV) complex. J. Am. Chem. Soc. 1967;89:2236–2237. [Google Scholar]
- Assem F.L., Levy L.S. A review of current toxicological concerns on vanadium pentoxide and other vanadium compounds: gaps in knowledge and directions for future research. J. Toxicol. Environ. Health B Crit. Rev. 2009;12:289. doi: 10.1080/10937400903094166. [DOI] [PubMed] [Google Scholar]
- Badetti E., Romano F., Marchiò L., Taşkesenlioğlu S., Daştan A., Zonta C., Licini G. Effective bromo and chloro peroxidation catalyzed by tungsten(VI) amino triphenolate complexes. Dalton. Trans. 2015;45:14603. doi: 10.1039/c6dt01780k. [DOI] [PubMed] [Google Scholar]
- Banik S.M., Medley J.W., Jacobsen E.N. Catalytic, asymmetric difluorination of alkenes to generate difluoromethylated stereocenters. Science. 2016;353:51–54. doi: 10.1126/science.aaf8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begin D., Einstein F.W.B., Field J. Asymmetrically coordinated diperoxo compound. Crystal structure of tripotassium oxodiperoxooxalatovanadate(V) monohydrate. Inorg. Chem. 1975;14:1785–1790. [Google Scholar]
- Beinker P., Hanson J.R., Meindl N., Medina I.C.R. Oxidative iodination of aromatic amides using sodium perborate or hydrogen peroxide with sodium tungstate. J. Chem. Res. 1998;1998:204–205. [Google Scholar]
- Ben-Daniel R., De Visser S.P., Shaik S., Neumann R. Electrophilic aromatic chlorination and haloperoxidation of chloride catalyzed by polyfluorinated alcohols: a new manifestation of template catalysis. J. Am. Chem. Soc. 2003;125:12116–12117. doi: 10.1021/ja0364524. [DOI] [PubMed] [Google Scholar]
- Butler A., Sandy M. Mechanistic considerations of halogenating enzymes. Nature. 2009;460:848–854. doi: 10.1038/nature08303. [DOI] [PubMed] [Google Scholar]
- Corsini A., Subramanian K.S. Studies on the tungstate solution in acid medium. J. Inorg. Nucl. Chem. 1978;40:1777–1779. [Google Scholar]
- Dewar M.J.S., Nakaya T. Oxidaive coupling of phenols. J. Am. Chem. Soc. 1968;90:7134–7135. [Google Scholar]
- Djordjevic C., Craig S.A., Sinn E. A polymeric peroxo heteroligand vanadate(V). Synthesis, spectra, and structure of MI[VO(O2)(C4H5O4N)] Inorg. Chem. 1985;24:1281–1283. [Google Scholar]
- Drew R.E., Einstein F.W.B. Crystal structure of ammonium oxodiperoxoamminevanadate(V) Inorg. Chem. 1972;11:1079–1083. [Google Scholar]
- Emmanuvel L., Shukla R.K., Sudalai A., Gurunath S., Sivaram S. NaIO4/KI/NaCl: a new reagent system for iodination of activated aromatics through in situ generation of iodine monochloride. Tetra. Lett. 2006;47:4793–4796. [Google Scholar]
- Fosu S.C., Hambira C.M., Chen A.D., Fuchs J.R., Nagib N.D. Site-selective C–H functionalization of (hetero)arenes via transient, non-symmetric iodanes. Chem. 2019;5:417–428. doi: 10.1016/j.chempr.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiron A.F., Thompson R.C. Kinetic study of the oxygen-transfer reactions from the oxo diperoxo complexes of molybdenum (VI) and tungsten(VI) to (thiolato)- and (sulfenato) cobalt (III) complexes. Inorg. Chem. 1988;26:4766–4771. [Google Scholar]
- Gkotsi D.S., Dhaliwal J., McLachlan M.M., Mulholand K.R., Goss R.J.M. Halogenases: powerful tools for biocatalysis (mechanisms application and scope) Curr. Open Chem. Biol. 2018;43:119–126. doi: 10.1016/j.cbpa.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Herget K., Freichs H., Pfitzner F., Tahir M.N., Tremel W. Functional enzyme mimics for oxidative halogenation reactions that combat biofilm formation. Adv. Mater. 2018;30:1707073. doi: 10.1002/adma.201707073. [DOI] [PubMed] [Google Scholar]
- Hering T., Mühldorf B., Wolf R., König B. Halogenase-inspired oxidative chlorination using flavin photocatalysis. Angew. Chem. Int. Ed. 2016;55:5342–5345. doi: 10.1002/anie.201600783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandes Z.M., Cavalcanti S.M.T., Moreira D.R.M., Azevedo, Lima W.F.D. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr. Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Jeon Y.T., Luo C., Forray C., Vaysse P.J.J., Branchek T.A., Gluchowski C. Pharmacological evaluation of UK-14,304 analogs at cloned human α adrenergic receptors. Bio. Med. Chem. Lett. 1995;5:2255–2258. [Google Scholar]
- Johansson S.C., Kitching M.O.T.J., Colacot S.V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- Kamata K., Kuzuya S., Uehara K., Yamaguchi S., Mizuno N. μ-η1: η1-peroxo-bridged dinuclear peroxotungstate catalytically active for epoxidation of olefins. Inorg. Chem. 2007;46:3768–3773. doi: 10.1021/ic0701211. [DOI] [PubMed] [Google Scholar]
- Kato S., Morie T., Kon T., Yoshida N., Karasawa T., Matsumoto J. Novel benzamides as selective and potent gastrokinetic agents. 2. Synthesis and structure-activity relationships of 4-amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)-2-morpholinyl]methyl]benzamide citrate (AS-4370) and related compounds. J. Med. Chem. 1991;34:616–624. doi: 10.1021/jm00106a023. [DOI] [PubMed] [Google Scholar]
- Ke L., Zhu G., Qian H., Xiang G., Chen Q., Chen Z. Catalytic selective oxidative coupling of secondary N-alkylanilines: an approach to azoxyarene. Org. Lett. 2019;21:4008–4013. doi: 10.1021/acs.orglett.9b01200. [DOI] [PubMed] [Google Scholar]
- Kikushima K., Moriuchi T., Hirao T. Vanadium-catalyzed oxidative bromination promoted by brønsted acid or Lewis acid. Tetrahedron. 2010;66:6906–6911. [Google Scholar]
- Kumar L., Mahajan T., Agarwal D.D. Aqueous bromination method for the synthesis of industrially important intermediates catalyzed by micellar solution of sodium dodecyl sulfate (SDS) Ind. Eng. Chem. Res. 2012;51:2227–2234. [Google Scholar]
- Latham J., Brandenburger E., Shepherd S.A., Menon B.R.K., Micklefield J. Development of halogenase enzymes for use in synthesis. Chem. Rev. 2018;118:232–236. doi: 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]
- Liang Y., Lin F., Adeli Y., Jin R., Jiao N. Efficient electrocatalysis for the preparation of (hetero)aryl chlorides and vinyl chloride with 1,2-dichloroethane. Angew. Chem. Int. Ed. 2019;58:4566–4570. doi: 10.1002/anie.201814570. [DOI] [PubMed] [Google Scholar]
- Maheswari P.U., Tang X., Hage R., Gamez P., Reedijk J. The role of carboxylic acids on a Na2WO4/H2WO4-based biphasic homogeneous alkene epoxidation, using H2O2 as oxidant. J. Mol. Cat. A. 2006;258:295–301. [Google Scholar]
- Mimoun H., Saussine L., Daire E., Postel M., Fischer J., Weiss R. Vanadium(V) peroxy complexes. New versatile biomimetic reagents for epoxidation of olefins and hydroxylation of alkanes and aromatic hydrocarbons. J. Am. Chem. Soc. 1983;105:3101–3110. [Google Scholar]
- Petrone D.A., Ye J., Lautens M. Modern transition-metal-catalyzed carbon–halogen bond formation. Chem. Rev. 2016;116:8003–8104. doi: 10.1021/acs.chemrev.6b00089. [DOI] [PubMed] [Google Scholar]
- Podgoršek A., Zupan M., Iskra J. Oxidative halogenation with “green” oxidants: oxygen and hydrogen peroxide. Angew. Chem. Int. Ed. 2009;48:8424–8450. doi: 10.1002/anie.200901223. [DOI] [PubMed] [Google Scholar]
- Reynlds M.S., Butler A. Oxygen-17 NMR, electronic and vibrational spectroscopy of transition metal peroxo complexes: correlation with reactivity. Inorg. Chem. 1996;35:2378–2383. doi: 10.1021/ic951212d. [DOI] [PubMed] [Google Scholar]
- Rodriguez R.A., Pan C.-M., Yabe Y., Kawamata Y., Eastgate M.D., Baran P.S. Palaúchlor: a practical and reactive chlorinating reagent. J. Am. Chem. Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa R.I., Clague M.J., Butler A. A functional mimic of vanadium bromoperoxidase. J. Am. Chem. Soc. 1992;114:760–761. [Google Scholar]
- Ross S.A., Burrows C.J. Bromination of pyrimidines using bromide and monoperoxysulfate: a competition study between cytidine, uridine and thymidine. Tetra. Lett. 1987;38:2805–2808. [Google Scholar]
- Roy S., Bhar S. Sodium tungstate-catalyzed “On-water” synthesis of β-arylvinyl bromides. Green. Chem. Lett. Rev. 2010;3:341–347. [Google Scholar]
- Sels B., Levecque P., Brosius R., De Vos D., Jacobs P., Gammon D.W., Kinfe H.H. A new catalytic route for the oxidative halogenation of cyclic enol ethers using tungstate exchanged on takovite. Adv. Synth. Catal. 2005;347:93–104. [Google Scholar]
- Sels B., Vos D.D., Buntinx M., Pierard F., Mesmaeker K.-D., Jacobs P. Layered double hydroxides exchanged with tungstate as biomimetic catalysts for mild oxidative bromination. Nature. 1999;400:855–857. [Google Scholar]
- Snead A.N., Miyakawa M., Tan E.S., Scanlan T.S. Trace amine-associated receptor 1 (TAAR1) is activated by amiodarone metabolites. Bio. Med. Chem. Lett. 2008;18:5920–5922. doi: 10.1016/j.bmcl.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S.K., Chauhan P.M.S., Bhaduri A.P. Novel site-specific one-step bromination of substituted benzene. Chem. Commun. (Camb.) 1996;23:2679–2680. [Google Scholar]
- Stomberg R. The crystal structures of potassium bis(oxalato)oxoperoxovanadate(V) hemihydrate, K3[VO(O2)(C2O4)2].½H2O, and potassium bis(oxalato)dioxovanadate(V) trihydrate, K3[VO2(C2O4)2].3H2O. Acta Chem. Scand. 1986;A40:168–176. [Google Scholar]
- Swain R., Bapna J.S., Das A.K., Chandrasekar S., Swaminathan R.P., Bosco B., Veliath S., Thombre D.P. A study on the neurotoxicity of broxyquinoline and brobenzoxaldine combination in therapeutic doses, Human. Toxicol. 1986;5:35–41. doi: 10.1177/096032718600500107. [DOI] [PubMed] [Google Scholar]
- Vailancourt F.H., Yeh E., Vosburg D.A., Garneau-Tsodikova S., Walsh C.T. Nature’s inventory of halogenation catalysts: oxidative strategies predominate. Chem. Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- Wan X., Ma Z., Li B., Zhang K., Cao S., Zhang S., Shi Z. Highly selective C-H functionalization/halogenation of acetanilide. J. Am. Chem. Soc. 2006;128:7416–7417. doi: 10.1021/ja060232j. [DOI] [PubMed] [Google Scholar]
- Wempe M.F., Jutabha P., Quade B., Iwen T.J., Frick M.M., Ross I.R., Rice P.J., Anzai N., Endou H. Developing potent human uric acid transporter 1 (hURAT1) inhibitors. J. Med. Chem. 2011;54:2701–2713. doi: 10.1021/jm1015022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R., Zimmermann M.O., Lange A., Joerger A.C., Boeckler F.M. Principles and applications of halogen Bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Winter J.M., Moore B.S. Exploring the chemistry and biology of vanadium-dependent haloperoxidases. J. Bio. Chem. 2009;284:18577–18581. doi: 10.1074/jbc.R109.001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Yao A., Zheng Y., Gao M., Zhou Z., Qiao J., Hu J., Ye B., Zhao J., Wen H., Lei A. Electrochemical oxidative clean halogenation using HX/NaX with hydrogen evolution. iScience. 2019;12:293–303. doi: 10.1016/j.isci.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Procedure for experiments and characterization data for products are available in Supplemental Informations. (Figures S15–S149) Any other data are available from the corresponding author upon reasonable request.