

Tethered protein–protein complexes are presumed to offer functional advantages such as electron transfer due to constraints imposed on the conformational search. This work shows that tethering has a much more complex role.
Keywords: nitrogen cycle, denitrification, copper-containing nitrite reductase, electron transfer, catalysis, structural biology
Abstract
Copper-containing nitrite reductases (CuNiRs) are found in all three kingdoms of life and play a major role in the denitrification branch of the global nitrogen cycle where nitrate is used in place of dioxygen as an electron acceptor in respiratory energy metabolism. Several C- and N-terminal redox domain tethered CuNiRs have been identified and structurally characterized during the last decade. Our understanding of the role of tethered domains in these new classes of three-domain CuNiRs, where an extra cytochrome or cupredoxin domain is tethered to the catalytic two-domain CuNiRs, has remained limited. This is further compounded by a complete lack of substrate-bound structures for these tethered CuNiRs. There is still no substrate-bound structure for any of the as-isolated wild-type tethered enzymes. Here, structures of nitrite and product-bound states from a nitrite-soaked crystal of the N-terminal cupredoxin-tethered enzyme from the Hyphomicrobium denitrificans strain 1NES1 (Hd 1NES1NiR) are provided. These, together with the as-isolated structure of the same species, provide clear evidence for the role of the N-terminal peptide bearing the conserved His27 in water-mediated anchoring of the substrate at the catalytic T2Cu site. Our data indicate a more complex role of tethering than the intuitive advantage for a partner-protein electron-transfer complex by narrowing the conformational search in such a combined system.
1. Introduction
Denitrification is an important process in the global nitrogen cycle and has significant impacts on agronomy, the environment and health (Zumft, 1997 ▸). Copper-containing nitrite reductases (CuNiRs) are found in all three kingdoms of life and catalyze the reduction of nitrite to nitric oxide, which is the first committed step of denitrification; NO2
− + e− + 2 H+
 NO + H2O. The homotrimer structures are highly conserved among all the two-domain CuNiRs from organisms involved in agricultural denitrification such as Alcaligenes xylosoxidans (Ax) and Achromobacter cycloclastes (Ac) to bacterial pathogens Neisseria gonorrhoeae (Ng) and Neisseria meningitides (Nm). The catalytic type-2 copper center (T2Cu) is located at the interface of the adjacent monomers and is ‘hard-wired’ to the electron-donating type-1 copper center (T1Cu) via neighboring residues that form a Cys–His electron-transfer (ET) bridge. The T1Cu is close to the protein surface and functions as the electron acceptor from the physiological electron donor, cytochrome (Nojiri et al., 2009 ▸) or pseudoazurin (Nojiri, 2016 ▸). The two active-site residues, AspCAT and HisCAT around the T2Cu, are involved in substrate binding and catalysis with both residues starting in the deprotonated state prior to substrate-binding events (Godden et al., 1991 ▸; Dodd et al., 1998 ▸; Antonyuk et al., 2005 ▸; Boulanger et al., 2000 ▸; Tocheva et al., 2004 ▸; Kataoka et al., 2000 ▸; Fukuda et al., 2016 ▸; Halsted et al., 2019 ▸).
NO + H2O. The homotrimer structures are highly conserved among all the two-domain CuNiRs from organisms involved in agricultural denitrification such as Alcaligenes xylosoxidans (Ax) and Achromobacter cycloclastes (Ac) to bacterial pathogens Neisseria gonorrhoeae (Ng) and Neisseria meningitides (Nm). The catalytic type-2 copper center (T2Cu) is located at the interface of the adjacent monomers and is ‘hard-wired’ to the electron-donating type-1 copper center (T1Cu) via neighboring residues that form a Cys–His electron-transfer (ET) bridge. The T1Cu is close to the protein surface and functions as the electron acceptor from the physiological electron donor, cytochrome (Nojiri et al., 2009 ▸) or pseudoazurin (Nojiri, 2016 ▸). The two active-site residues, AspCAT and HisCAT around the T2Cu, are involved in substrate binding and catalysis with both residues starting in the deprotonated state prior to substrate-binding events (Godden et al., 1991 ▸; Dodd et al., 1998 ▸; Antonyuk et al., 2005 ▸; Boulanger et al., 2000 ▸; Tocheva et al., 2004 ▸; Kataoka et al., 2000 ▸; Fukuda et al., 2016 ▸; Halsted et al., 2019 ▸).
More recently, new classes of three-domain CuNiRs have been structurally characterized, where an extra cytochrome (Antonyuk et al., 2013 ▸; Tsuda et al., 2013 ▸) or cupredoxin (Opperman et al., 2019 ▸) domain is tethered to the C-terminus of the catalytic core domain corresponding to the two-domain CuNiR. Both of the C-terminal cytochrome-tethered CuNiRs from Ralstonia pickettii (Rp) (Antonyuk et al., 2013 ▸) and from Pseudoalteromonas haloplanktis (Ph) (Tsuda et al., 2013 ▸) show a trimeric structure but reveal different linking arrangements for the tethered cytochrome and T1Cu–T2Cu core domains via a long tethering linker. In both cases, however, these alternative arrangements place the heme of the cytochrome adjacent to the T1Cu at a distance of ∼10 Å for an effective ET from the heme to the T1Cu. The latest addition to the CuNiR family is the C-terminal cupredoxin-tethered CuNiR from Thermus scotoductus (Ts), the structure of which was very recently elucidated (Opperman et al., 2019 ▸). This enzyme, TsNiR, is trimeric with the T1CuC (T1Cu in the tethered cupredoxin domain) located near the core T1Cu with an ET compatible distance of ∼14 Å. In contrast to RpNiR and PhNiR, the tethered C-terminal domain interacts directly with the T1Cu–T2Cu core domain of the same subunit.
The only known structure of an N-terminal cupredoxin-tethered CuNiR is for the enzyme from Hyphomicrobium denitrificans strain A3151 (Hd A3151NiR) (Nojiri et al., 2007 ▸). Surprisingly its structure showed a prism-shaped homohexamer, whose monomers are organized into a tightly associated dimer of trimers with additional N-terminal cupredoxin (T1CuN) domains interacting head-to-head. Unlike the three representatives of the C-terminal tethered RpNiR (cytochrome), PhNiR (cytochrome) and TsNiR (cupredoxin), the tethered T1CuN domain is located far from the T1Cu–T2Cu catalytic core with a distance of ∼24 Å between the T1CuN and the T1Cu, thus questioning the role of N-terminal tethering in ET and catalysis. Pulse-radiolysis data (Nojiri et al., 2007 ▸) for Hd A3151NiR obtained in the presence of nitrite suggest that generated electrons attack the T1CuN, but not the T1Cucore. Subsequently, the reduced T1CuN gives up an electron to the type-2 Cu through the T1Cucore.
Our understanding of the role of tethered domains has been seriously hampered by the lack of substrate/product-bound structures of any tethered CuNiRs. This scarcity of substrate/product-bound structures extends to the C-terminal tethered CuNiRs, with the exception of RpNiR, where it required either mutation of the gate-keeper residue Tyr323 or alternatively AspCAT as well as pre-incubation of the crystals with nitric oxide (Dong et al., 2018 ▸; Hedison et al., 2019 ▸). Deconstruction of RpNiR into the NiR catalytic core and cytochrome domain showed the linker region that connects the two and harbors the gatekeeper tyrosine which unravels and results in a substantial conformational movement of the tethered cytochrome domain. This may place it far away from the catalytic core (T1Cu–T2Cu) suggesting that, in tethered domains, conformational dynamics may play an important role in substrate binding and regulation of catalysis (Hedison et al., 2019 ▸) in these tethered systems.
Here, we have identified, characterized and determined the crystallographic structure of the N-terminal cupredoxin tethered HdNiR from an alternative strain 1NES1 (Hd 1NES1NiR) and compared it with Hd A3151NiR. The chromatographic profile showed Hd 1NES1NiR to be primarily a hexamer but with enzymatic activity significantly lower than the classic two-domain CuNiRs. Hd 1NES1NiR crystallizes in a different space group, P6522, compared with P41 in the case of Hd A3151NiR, with a trimer of Hd 1NES1NiR in the asymmetric unit. It also forms a hexameric structure resulting from a dimer of trimers with the T1CuN located again too far away from the catalytic T1Cu–T2Cu NiR core for an effective ET. Despite a high sequence identity of 84% between Hd 1NES1NiR and Hd A3151NiR, significant structural differences were observed, which may account for more than an order of magnitude difference in specific NiR activity of these enzymes. Remarkably, in contrast to all other tethered CuNiR enzymes, we have been able to obtain a substrate-bound structure for Hd 1NES1NiR by simply soaking crystals with nitrite in a manner similar to the classic two-domain CuNiRs. In fact, structure determination of one nitrite-soaked Hd 1NES1NiR crystal revealed the trimeric assembly in the asymmetric unit with the catalytic T2Cu in both substrate- and product-bound states. In two molecules T2Cu bound the substrate at full occupancy and one molecule had NO bound to the T2Cu site, consistent with the functional asymmetry that has been noted recently for classic two-domain trimeric CuNiRs (Hedison et al., 2019 ▸). This is the first clear structural evidence for such asymmetry, suggesting a one-third reactivity of the T2Cu center of the core enzyme.
2. Materials and methods
2.1. Primary structure alignment
Primary sequence alignment was performed with ClustalW (Thompson et al., 1994 ▸) and amino-acid sequence identity was estimated with BLAST (Altschul et al., 1997 ▸) by performing one-to-one pairwise analysis. Primary sequence information was obtained from the Universal Protein Resource (UniProt) (http://www.uniprot.org).
2.2. Sample preparation
The Hd 1NES1NiR gene was ordered from GenScript with the NCBI reference code WP_015596837.1. The N-terminal signal peptide predicted by Signal-3L (version 2.0, Zhang & Shen, 2017 ▸) was removed from the ordered gene. The gene with a TEV cleavage site was cloned into pET-26b(+) (Novagen, Darmstadt, Germany) between the NdeI and XhoI sites. The resultant plasmid was verified by DNA sequencing.
An E. coli host strain BL21(DE3) cell (New England BioLabs Inc.) was transformed with the plasmid. A single colony was grown in 50 ml Luria–Bertani (LB) medium supplemented with 50 µg ml−1 kanamycin and incubated at 37°C for 16 h at 240 rev min−1. A 5 ml sample of culture was inoculated into 500 ml of LB medium supplemented with the same concentration of kanamycin and incubated at 37°C at 180 rev min−1 until OD600 nm reached ∼0.6. Subsequently, final concentrations of 1.0 mM CuSO4 and 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) were added and overexpression was induced at 18°C for 16 h at 180 rev min−1. The cells were harvested by centrifugation (4690g, 45 min, 4°C). The pellet was washed with 50 ml phosphate-buffered saline (PBS) pH 7.4 and harvested by centrifugation (3140g, 30 min, 4°C).
The cells were suspended in 50 ml of lysis buffer 100 mM Tris–HCl pH 8.0, 500 mM NaCl, 10 mM imidazole containing a protease inhibitor tablet (Roche) for 1 l culture. After lysozyme was added to a final concentration of 0.5 mg ml−1, the suspension solution was incubated on ice for 20 min. The cells were disrupted by sonication on ice. The cell debris was removed by centrifugation (29 900g, 45 min, 4°C). The supernatant was filtered and applied to a 5 ml of His-tag affinity column HisTrapTM HP (GE Healthcare, Buckinghamshire, UK) equilibrated with the lysis buffer. The resin was washed with the same buffer and the protein was eluted with 10 ml of elution buffer 100 mM Tris–HCl pH 8.0, 500 mM NaCl, 250 mM imidazole. The elution solution was dialyzed at 4°C for 24 h against size-exclusion chromatography (SEC) buffer 100 mM Tris–HCl pH 8.0, 500 mM NaCl, 10%(v/v) glycerol. After dialysis, a final concentration of 2 mM DTT was added and the protein was incubated with TEV protease (50:1) at 4°C for 16 h to remove the 6×His-tag. The protein solution was concentrated and applied on an SEC column HiLoad 16/600 Superdex 200 pg (GE Healthcare, Buckinghamshire, UK) equilibrium with SEC buffer. The protein was eluted at a flow rate of 1.0 ml min−1. The elution fractions were dialyzed at 4°C for 16 h against Cu-loading buffer, SEC buffer with 1.0 mM CuSO4, to reconstitute the T2Cu site. After dialysis, the protein solution was concentrated and applied again on the same SEC column equilibrated with SEC buffer. The protein was eluted at a flow rate of 1.0 ml min−1. The elution fractions were concentrated and stored at −80°C. All chromatography steps were performed at 4°C.
2.3. UV–visible absorption spectrum measurement
UV–visible absorption spectra were recorded at room temperature on a Cary 300 Bio UV–visible spectrophotometer (Varian, Palo Alto, USA). Hd 1NES1NiR was prepared at 1.0 mg ml−1 in 100 mM Tris–HCl pH 8.0, 500 mM NaCl, 10%(v/v) glycerol for spectral measurements.
2.4. Oligomeric state analysis
The molecular mass of Hd 1NES1NiR was estimated by comparison with retention volumes of marker proteins (GE Healthcare, Buckinghamshire, UK). The marker proteins, blue dextran (2000 kDa), thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa) and ovalbumin (44 kDa), dissolved in SEC buffer 100 mM Tris–HCl pH 8.0, 500 mM NaCl, 10%(v/v) glycerol were applied on an SEC column HiLoad 16/600 Superdex 200 pg (GE Healthcare, Buckinghamshire, UK) equilibrium with SEC buffer and eluted at a flow of 1.0 ml min−1. A calibration curve [K av value versus log(Mw) where Mw = molecular weight] for these marker proteins was obtained. The K av value is defined with the equation K av = (V e − V o)/(V c − V o), where V e, V o and V c are the elution, column void and geometric column volumes, respectively. The molecular mass was estimated with their V e values obtained from the calibration curve.
2.5. NiR activity measurement
NiR activity was assessed under anaerobic conditions using an NO-detectable ISO-NOP electrode (World Precision Instruments, Serasota, USA). The 3 ml of assay mixture containing nitrogen saturated 50 mM HEPES buffer (pH 6.5), 8.0 mM sodium ascorbate, 80 µM phenazine methosulfate (PMS) and 8.0 mM sodium nitrite was prepared in the vessel under anaerobic conditions. The electrode was inserted into the mixture and the baseline voltage was confirmed to be constant for 1 min. The reaction was initiated by the addition of a tiny volume of the Hd 1NES1NiR sample at a final concentration of 300 nM and the time-course of NO production [voltage (V) versus time (s)] was monitored. The activity value [nmol s−1(nmol of protein)−1] for a linear slope was estimated using the experimentally determined calibration curve [voltage (V) versus NO production (nmol)].
2.6. Structure determination
For the as-isolated structure, the Hd 1NES1NiR sample in 20 mM Tris–HCl pH 7.5 was concentrated to 20 mg ml−1. The protein was crystallized by the hanging-drop vapor-diffusion method: 2 µl of sample solution was mixed with 1 µl of crystallization reagent 20%(w/v) PEG 1000, 0.1 M sodium citrate tribasic dihydrate pH 5.5, 0.1 M lithium sulfate monohydrate and equilibrated over 200 µl of the crystallization reagent at room temperature. The crystal was transferred in 20%(w/v) PEG 1000, 0.1 M sodium citrate tribasic dihydrate pH 5.5, 0.1 M lithium sulfate monohydrate, 20%(v/v) ethylene glycol and flash-cooled in liquid nitrogen.
For the substrate/product-bound structure, the Hd 1NES1NiR sample in 20 mM Tris–HCl pH 7.5 was concentrated to 20 mg ml−1. The protein was crystallized by the hanging-drop vapor-diffusion method: 1 µl of sample solution was mixed with 1 µl of crystallization reagent 22.5%(v/v) PEG Smear Low (Chaikuad et al., 2015 ▸), 0.1 M sodium cacodylate pH 5.3, 0.2 M ammonium nitrate and equilibrated over 200 µl of the crystallization reagent at room temperature. The crystal was transferred in 22.5%(v/v) PEG Smear Low, 0.1 M sodium cacodylate pH 5.3, 0.2 M ammonium nitrate, 100 mM NaNO2, 20%(v/v) glycerol and flash-cooled in liquid nitrogen.
Diffraction data were collected at the I04 beamline, Diamond Light Source, UK, at 100 K using an EIGER X 16M detector. The diffraction images were processed with DIALS (Winter et al., 2018 ▸) in XIA2 (Winter, 2010 ▸) and AIMLESS (Evans & Murshudov, 2013 ▸) for the as-isolated structure, and with a combination of autoPROC (Vonrhein et al., 2011 ▸) and STARANISO (Tickle et al., 2018 ▸) for the substrate/product-bound structure, both in space group P6522. For the substrate/product-bound crystal, an additional data set was collected at 1.33 Å wavelength to confirm the correct Cu incorporation. For the as-isolated structure, the initial model was obtained by molecular replacement with MOLREP (Vagin & Teplyakov, 2010 ▸) using the structure of the trimer Hd A3151NiR (PDB entry 2dv6). The substrate/product-bound structure was refined directly from the as-isolated structure. The models were refined with REFMAC5 (Murshudov et al., 2011 ▸) in CCP4 (Winn et al., 2011 ▸) and manually rebuilt with Coot (Emsley et al., 2010 ▸). The quality of the final models was assessed with MolProbity (Chen et al., 2010 ▸). Data collection and structure refinement statistics are summarized in Table 1 ▸. Sequence alignment was performed with TM-align (Zhang & Skolnick, 2005 ▸). Structural figures were prepared using PyMOL (v.1.4; Schrödinger).
Table 1. Data collection and refinement statistics.
Numbers in parentheses represent the value for the lowest/highest resolution shell (innermost/outermost shells).
| Hd 1NES1NiR (as-isolated) | Hd 1NES1NiR (nitrite-soaked) | |
|---|---|---|
| Ligands | W1 | NO2/NO |
| Data collection | ||
| Space group | P6522 | P6522 |
| Wavelength (Å) | 0.9795 | 0.9795 |
| a, b, c (Å) | 77.07, 77.07, 754.55 | 77.72, 77.72, 758.20 |
| Resolution (Å) | 66.75–2.05 (66.75–10.85/2.09–2.05) | 126.37–2.10 (126.37–5.7/2.14–2.10) |
| No. of reflections, total/unique | 363227/85026 | 987567/81889 |
| R merge † (%) | 12.1 (2.9/92.6) | 21.6 (6.2/284.5) |
| R p.i.m. ‡ (%) | 8.4 (1.9/67.7) | 6.4 (2.0/82.8) |
| I/σ(I) | 3.5 (9.5/0.8) | 7.7 (24.0/0.9) |
| CC1/2 | 0.995 (0.998/0.610) | 0.998 (0.998/0.426) |
| Completeness (%) | 98.7 (99.8/98.2) | 100 (100/100) |
| Multiplicity | 4.3 (3.4/4.4) | 12.1 (10.6/12.3) |
| Refinement | ||
| R work §/R free ¶ | 0.227/0.279 | 0.172/0.227 |
| Resolution (Å) | 66.75–2.05 | 126.37–2.25 |
| No. of atoms | ||
| Protein | 9630 | 9625 |
| Ligand/ion | 9 | 17 |
| Water | 528 | 639 |
| Average B factor (Å2) | ||
| Protein | 51.8 | 54.9 |
| Ligand/ion | 47.1 | 52.3 |
| Water | 50.5 | 52.6 |
| R.m.s. deviations | ||
| ond lengths (Å) | 0.007 | 0.007 |
| Bond angles (°) | 1.511 | 1.601 |
| Ramachandran plot | ||
| Favored (%) | 96.6 | 97.2 |
| Allowed (%) | 99.8 | 99.7 |
| PDB entry | 6tfo | 6tfd |
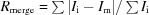 , where I
i is the intensity of the measured reflection and I
m is the mean intensity of all symmetry related reflections.
, where I
i is the intensity of the measured reflection and I
m is the mean intensity of all symmetry related reflections.
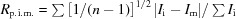 where I
i is the intensity of the measured reflection, I
m is the mean intensity of all symmetry related reflections and n is the redundancy.
where I
i is the intensity of the measured reflection, I
m is the mean intensity of all symmetry related reflections and n is the redundancy.
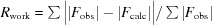 , where F
obs and F
calc are the observed and calculated structure factors, respectively.
, where F
obs and F
calc are the observed and calculated structure factors, respectively.
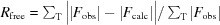 , where T is a test data set of 5% of the total reflections randomly chosen and set aside prior to refinement.
, where T is a test data set of 5% of the total reflections randomly chosen and set aside prior to refinement.
3. Results
3.1. Spectroscopic and functional characterization of Hd 1NES1NiR
We have purified the N-terminal cupredoxin-tethered three-domain CuNiR from Hyphomicrobium denitrificans strain 1NES1 (Hd 1NES1NiR), which is the same class of enzyme as the structurally characterized CuNiR from Hyphomicrobium denitrificans strain A3151 (Hd A3151NiR) (Nojiri et al., 2007 ▸). The primary structure alignment shows 84% sequence identity between these with complete conservation of the ligand residues to the catalytic core T1Cu and T2Cu centers and the active-site residues AspCAT and HisCAT involved in substrate-anchoring and catalysis (Fig. S1 of the supporting information). The different amino-acid residues are predominantly distributed on the N-terminal tethered cupredoxin domain and the N-terminal and signal peptides. The UV–visible absorption spectrum revealed Hd 1NES1NiR to have an A 600/A 460 ratio of ∼1.6 [Fig. S2(a)] compared with ∼1.9 for Hd A3151NiR (Deligeer et al., 2002 ▸). These spectral features have been assigned to S(Cys)-to-Cull charge transfer transitions, the intensity of which depends on the detailed geometry of the T1Cu site. In the case of HdNiRs, the difference in this ratio results in a stronger greenish appearance of Hd 1NES1NiR and is the result of the structural differences in the T1CuN sites of the two Hyphomicobium species. The MW of Hd 1NES1NiR determined by size-exclusion chromatography is consistent with it being a hexamer [Fig. S2(b)]. The broad elution peak may indicate the presence of additional unresolved lower oligomeric states. The NiR activity assayed by direct measurements of NO formation demonstrated that the specific NiR activity of Hd 1NES1NiR is 8.3 ± 0.8 nmol s−1 (nmol of protein)−1 [Fig. S2(c)], which is ∼40-fold less than that reported for Hd A3151NiR (Deligeer et al., 2002 ▸). Unexpectedly, the time course assay profile for NO production by Hd 1NES1NiR exhibited a lag period of ∼200 s before the rate became linear when the reaction was initiated by the addition of the enzyme to the assay mixture, but was eliminated when Hd 1NES1NiR was pre-incubated with substrate (nitrite) [Fig. S2(d)] or reductant (ascorbate) [Fig. S2(e)]. The specific activity without pre-incubation (1.7 ± 0.2) was increased by approximately fivefold more when pre-incubated with substrate (8.3 ± 0.8), whereas with reductant showed it a marginal increase (2.6 ± 0.3).
3.2. Crystallographic structure of as-isolated Hd 1NES1NiR
The crystallographic structure of Hd A3151NiR isolated from the native source was reported more than a decade ago as the first and only structure of an N-terminal tethered three-domain CuNiR (Nojiri et al., 2007 ▸). It remained the only hexameric structure for a CuNiR where the tethered domain T1Cu was placed too far away from the T1Cu–T2Cu NiR core, hence, casting doubt on its role in electron transfer. It has thus remained imperative to discover another representative of this class of NiR and determine its high-resolution structure in order to resolve the details of structure–function relationships in this class of CuNiRs. With this goal in mind we have determined the crystallographic structure of HdNiR from a different strain, 1NES1 (Hd 1NES1NiR), at 2.05 Å resolution using protein heterologously expressed in E. coli (Fig. 1 ▸, Table 1 ▸).
Figure 1.

Overall structures of Hd 1NES1NiR in trimeric and hexameric forms. (a) Top (upper) and side (lower) views of a trimeric Hd 1NES1NiR, colored green, magenta and cyan for each monomer. Threefold axis symmetry is indicated by a black closed triangle (upper) and black line (lower). The T1Cu and T2Cu ions in the core domain and T1CuN ion in the extra cupredoxin domain are shown by deep-blue spheres. (b) Side view of a hexameric Hd 1NES1NiR coloured green, magenta and cyan for each monomer generated by crystallographic symmetry. Threefold axis symmetry for each trimer is indicated by a black line. The interaction interface between the two trimers through extra cupredoxin domains is indicated by a black broken line. The T1Cu and T2Cu ions in the core domain and T1CuN ion in the extra cupredoxin domain are represented by deep-blue spheres.
The overall structure of a monomer is quite similar to that of Hd A3151NiR with an r.m.s.d. value of 0.64 (Cαs) for 422 amino-acid residues. The asymmetric unit of the crystal (space group P6522) contains a trimer, which creates a hexamer with a second symmetry related trimer [Fig. 1 ▸(b)], also observed for Hd A3151NiR (PDB entry 2dv6; Nojiri et al., 2007 ▸). In both of these HdNiRs, the T1CuN in the N-terminal tethered cupredoxin domain is placed at a distance of ∼24 Å from the catalytic core T1Cu, which may be considered too far away for an effective electron transfer. Nevertheless, pulse radiolysis data for Hd A3151NiR, which is also hexameric in solution, has shown that in the presence of nitrite, type-1 CuN receives the electron first and then passes it onto the type-1 Cucore of the adjacent monomer ready for catalysis (Nojiri et al., 2007 ▸).
A close examination of the structural differences between the two HdNiRs was made with particular emphasis on the non-conserved amino-acid residues between the two enzymes. These residues are predominantly located on the surface of the N-terminal tethered cupredoxin domain, which is exposed to solvent [Fig. S3(c)], though some are also in the inter- and/or intra monomer interface (Fig. S4). The substitutions in this domain in Hd 1NES1NiR result in a much less negative and water-inaccessible surface compared with Hd A3151NiR [Figs. S3(a) and S3(b)]. Differences are also observed in the water networks near T1Cucore and T1CuN in the catalytic core and N-terminal tethered cupredoxin domains [Figs. S3(d) and S4(d)], respectively, together with a number of non-conserved amino-acid residues between the two HdNiRs. In both T1Cu sites in Hd 1NES1NiR, a bridging water molecule expected to be hydrogen-bonded to the ligand histidine His271 and His122 (for T1Cucore and T1CuN, respectively) is missing [Figs. S3(d) and S4(d)], which may also contribute to the UV-spectral difference between the two HdNiRs together with subtle changes in the T1CuN geometry (Deligeer et al., 2002 ▸).
4. Structure of the tethering linker, N-terminal peptide and catalytic T2Cu site of Hd 1NES1NiR
Structural differences are also observed in the central part of the tethering linker between the catalytic core domain and N-terminal tethered cupredoxin domain in the two HdNiRs (Fig. 2 ▸). Lys139 and Gly142 of Hd A3151NiR are replaced by Pro142 and Ala145 in Hd 1NES1NiR, increasing the rigidity of the main chain. This results in a different orientation of the side chains of Glu140 and Met141 in Hd 1NES1NiR [Fig. 2 ▸(b)]. These residues are located in the middle part of the tethering linker, which is between the outside flexible loop exposed to solvent and the inner region that interacts with the core domain [Fig. 2 ▸(a)]. The N-terminal tethered cupredoxin domain of Hd 1NES1NiR is positioned ∼1.0 Å away from the core domain compared with Hd A3151NiR, probably arising from combinatorial structural differences in this region.
Figure 2.

Structural differences in the tethering linker and N-terminal peptide between Hd 1NES1NiR and Hd A3151NiR. (a) Monomer of Hd 1NES1NiR colored green superimposed on the core domain of Hd A3151NiR colored yellow. The monomer is constructed with the core domain, extra cupredoxin domain and tethering linker between them. The 2F o F c electron-density map at the 1.0σ level is shown for the tethering linker. The main-chain structural difference between the two HdNiRs is indicated by a black arrow. (b) The middle part of the tethering linker and (c) the N-terminal peptide near the T2Cu of Hd 1NES1NiR colored green superimposed on the core domain of Hd A3151NiR coloured yellow. The T2Cu ion in the core domain is represented by a deep-blue sphere. The ligand water (W1) molecules for Hd 1NES1NiR and Hd A3151NiR are represented by red and yellow spheres, respectively. Coordination to the T2Cu ion is shown by a red broken line. The 2F o F c electron density map at the 1.0σ level is shown for the His27 of Hd 1NES1NiR. The distances between His27 and His24 and T2Cu are indicated (∼10 and ∼20 Å for 1NES1 and A3151, respectively).
Despite the fact that HdNiR from two different strains of bacteria crystallize in different space groups, neither structure shows electron density for the N-terminal peptide, reflecting the intrinsic high flexibility of the region. The two structures show visible electron density starting from an equivalent residue at the N-terminus, His27 and His24 for Hd 1NES1NiR and Hd A3151NiR, respectively. However, they differ significantly in conformation consistent with the flexibility of the region [Fig. 2 ▸(c)]. The His27 of Hd 1NES1NiR is positioned closer to the T2Cu at a distance of ∼10 Å, compared with ∼20 Å in Hd A3151NiR. This difference likely represents a different conformational state of the tethering linker [Fig. 2 ▸(a)] of the HdNiR enzymes. The different position of the histidine could be derived from the substitution of Ile412 in Hd A3151NiR by the less bulky valine in Hd 1NES1NiR. Like the C-terminal tethered RpNiR, the dynamic features of the linker may be important for communication between the redox center of the tethered domain and the core NiR (Hedison et al., 2019 ▸).
5. Structures of substrate- and product-bound forms of Hd 1NES1NiR trapped in the same nitrite-soaked crystal
None of the C- or N-terminal cytochrome or cupredoxin tethered CuNiRs have demonstrated successful soaking of substrate into the crystals. Using simple soaking of Hd 1NES1NiR crystals, the substrate-bound structure has been determined to 2.1 Å resolution. Intriguingly, three T2Cu sites belonging to the trimer in the asymmetric unit are occupied by different ligands: two by the substrate nitrite and one by a diatomic molecule consistent with it being the product nitric oxide. Each of these independent sites have full occupancy of copper and the ligand (Fig. 3 ▸).
Figure 3.

Ligand-bound structures of Hd 1NES1NiR compared with those of AcNiR. (a) Ligand water (W1)-, (b) nitrite (NO2 −)- and (c) nitric oxide (NO)-bound T2Cu of Hd 1NES1NiR colored green, magenta and cyan for each monomer. The T2Cu ion is represented by a deep-blue sphere. The water molecules are represented by red spheres and the bridging water is indicated by a black arrow. Coordination to the T2Cu ion is shown by a red broken line and the interaction is shown by a black broken line. The F o F c electron density map at the 5.0σ level is shown for nitrite (NO2 −) and nitric oxide (NO). The 2F o F c electron-density map at the 1.0σ level is shown for the His27, the ligand-water (W1) and the other waters (W2, W3, W). (d) The ligand water (W1)-, (e) nitrite (NO2 −)- and (f) nitric oxide (NO)-bound T2Cu of the AcNiR are colored green and cyan for each monomer. The T2Cu ion is represented by a deep-blue sphere. The water molecules are represented by red spheres and the bridging water is indicated by a black arrow. Coordination to the T2Cu ion is shown by a red broken line and the interaction is shown by a black broken line. The structural coordinates for (d), (e) and (f) are from the PDB entries 6gsq and 6gto (Halsted et al., 2019 ▸), and 5of8 (Horrell et al., 2018 ▸), respectively.
Thus, we are able to compare for the first time a tethered CuNiR with two-domain NiRs in three different catalytically important states, namely the as-isolated water (W1)-bound structure and the substrate (NO2 −)- and product (NO)-bound structures. Interestingly, comparison among these shows different structural arrangements around the T2Cu including the positioning of His27 in the flexible N-terminal peptide. For the ligand water (W1)-bound structure [Fig. 3 ▸(a)], the N-terminal His27 assumes an outward open conformation, where a single water molecule is positioned near the His27. The relative position of the W1 to the T2Cu with a W1–T2Cu–His419 angle of ∼90° is very similar to that observed in equivalent structures from other classical two-domain CuNiRs such as AcNiR [Fig. 3 ▸(d)], suggesting that the displacement by substrate should be similarly favorable. The NO2 −-bound structure [Figs. 3 ▸(b) and S5(a)] shows a remarkable inward pointing of His27 towards the T2Cu and its closed conformation involving W2–W3 mediation. This suggests it plays a role in anchoring the substrate. The nitrogen atom of His27 is linked to bound NO2 −, mediated by the second water (W2), as well as to Asp225 (AspCAT), mediated by the third water (W3). The NO2 − is bound to the T2Cu in a side-on conformation via a single nitrogen atom and a single proximal oxygen atom with distances of 1.9 and 2.0 Å, respectively. The distal oxygen atom of the NO2 − with a longer distance of 3.0 Å to the T2Cu is linked to the nitrogen atom of His27. The AspCAT forms a hydrogen bond to the proximal oxygen atom of the NO2 − with a distance of 2.5 Å. The His368 (HisCAT) residue also forms a hydrogen bond to the bridging water [Figs. 3 ▸(b) and S5(a)], which is located at the terminus of the proton channel extending from the solvent region (see below). This is not the case in the water (W1)- and NO-bound structures [Figs. 3 ▸(a), 3 ▸(c) and S5(b)]. In the case of the NO2 −-bound structure, W2 and W3 form a hydrogen bond to each other with a distance of 2.7 Å [Figs. 3 ▸(b) and S5(a)].
For the NO-bound structure [Figs. 3 ▸(c) and S5(b)], again, the N-terminal His27 assumes an outward opened conformation directed towards the solvent and consequently loses water-mediated linkages to the ligand and AspCAT. In this case both the water and His27 itself show poorer electron density compared with the other two structures, suggesting higher flexibility of the N-terminal peptide that may facilitate release of the NO from the T2Cu with consequent return to the resting state. The corresponding histidine in Hd A3151NiR (His24 of Hd A3151NiR) has a more open conformation placing it quite far from the T2Cu [Fig. 2 ▸(c)]. The NO is bound to the T2Cu in a side-on manner similar to that observed for two-domain AcNiR [Fig. 3 ▸(f)] and with N and O at 2.0 and 2.5 Å with a tilt angle of 30°. The nitrogen atom is located 3.1 Å from the closest side-chain oxygen atom of Asp225. Unlike the NO2 −-bound structure [Figs. 3 ▸(b) and S5(a)], Asp225 forms a hydrogen bond with water in the proton channel with a distance of 2.8 Å [Figs. 3 ▸(c) and S5(b)]. We note that the unprecedented crystallographic observations of side-on NO binding geometry in CuNiR (Antonyuk et al., 2005 ▸; Tocheva et al., 2004 ▸) have been treated with scepticism by the chemical biology and synthetic chemistry communities until very recently (Ghosh et al., 2007 ▸; Bower et al., 2019 ▸). The observation of side-on NO binding geometry seen here adds to the gathering evidence that this geometry is energetically stable at the T2Cu site of CuNiR and is an intrinsic part of the catalytic turnover.
6. Structure of the proton channel and the alternative electron transfer pathway of Hd 1NES1NiR
The proton channel, which extends from the solvent to the catalytic T2Cu at the inter-monomer interface, is different in the two HdNiRs [Fig. 4 ▸(a)]. Val381 of Hd A3151NiR, which is located at the entrance to the channel, is replaced with the more hydrophilic threonine (Thr384), whilst Tyr256 is replaced with the more hydrophobic phenylalanine (Phe259 of Hd 1NES1NiR). Ile252 of Hd A3151NiR is replaced with the less bulky valine (Val255 of Hd 1NES1NiR). More importantly, the three water molecules which form a proton pathway in Hd A3151NiR are missing in Hd 1NES1NiR. These structural differences may contribute to the lower NiR activity of Hd 1NES1NiR.
Figure 4.

Structural differences in the proton channel and structural preservation of the alternative electron transfer route between Hd 1NES1NiR and Hd A3151NiR. (a) Proton channel of Hd 1NES1NiR colored green and cyan for each monomer superimposed on Hd A3151NiR colored yellow for all monomers for simplicity. The T2Cu ion in the core domain is represented by a deep-blue sphere. The water molecules for Hd 1NES1NiR and Hd A3151NiR are represented by red and yellow spheres, respectively, and the bridging water is indicated by a black arrow. Coordination to the T2Cu ion is shown by a red broken line and the interaction is shown by a black broken line. (b) Alternative electron transfer route of Hd 1NES1NiR colored green and cyan for each monomer superimposed on the extra cupredoxin domain of Hd A3151NiR colored yellow for all monomers for simplicity. The T1CuN ion in the extra cupredoxin domain and T2Cu ion in the core domain are represented by deep-blue spheres. The mediation water molecules (W2 and W3) and ligand water molecule (W1) for Hd 1NES1NiR and Hd A3151NiR are shown by red and yellow spheres, respectively. Coordination to the T1CuN and T2Cu ions is shown by a red broken line and the interaction is shown by a black broken line. The distance between Val30 and Thr36 is indicated (∼20 Å).
We have investigated an alternative ET route from the T1CuN in the tethered cupredoxin domain to the T2Cu in the core domain via long-range electron tunneling through a β-strand polypeptide over a distance of ∼16 to 26 Å, as has been reported for Ru-labeled cupredoxin by Gray & Winkler (2005 ▸). The completely conserved hydrophobic β-strand is present in the structures of the two HdNiRs, starting from Val30 to Thr36 (for Hd 1NES1NiR) with a distance of ∼20 Å within this region, suggesting the possibility for electron transfer through this strand. The structurally conserved glutamic acid (Glu76 and Glu73 of Hd 1NES1NiR and Hd A3151NiR, respectively) forms a hydrogen bond with threonine (Thr36 and Thr33) with a distance of ∼2.7 Å as well as with the ligand histidine to the T1CuN (His80 and His77) with a distance of ∼2.6 Å. We suggest a role for this residue in making an electron-transfer bridge between the T2CuN and Thr36, the starting residue of the probable electron tunneling hydrophobic β-strand. Val30, the terminal residue of this β-strand, connects to the T2Cu through the N-terminal peptide including the NO2 −-capturing His27 and the W2–W3 water mediation system in the NO2 −-bound structure of Hd 1NES1NiR. This suggests that electron transfer from the T1CuN in the tethered cupredoxin domain to the T2Cu in the core domain is dependent on both the presence of substrate and the dynamics of His27 movement. Pulse radiolysis data for Hd A3151NiR indicate that ET only occurs in the presence of NO2 −and that the electron preferentially goes to the T1CuN. The mutational studies of the T1Cucore of Hd A3151NiR revealed that the T1Cu-deficient enzyme still exhibited a significant NiR activity (Yamaguchi et al., 2004 ▸). This can now be rationalized neatly by the proposed long-range ET through the hydrophobic β-strand to the catalytic T2Cu site.
7. Discussion
Among nearly a couple of hundred structures of CuNiRs in the Protein Data Bank (PDB) we provide the second example of an enzyme with a hexameric rather than trimeric structure. The hexameric structure observed for the N-terminal cupredoxin-tethered three-domain CuNiR from two strains of Hyphomicrobium denitrificans suggests that it is likely to offer an advantage for these organisms. In both of the hexameric structures of Hd 1NES1NiR and Hd A3151NiR, the T1CuN in the N-terminal tethered cupredoxin domain is placed too far away from the catalytic core T1Cu for effective electron transfer (Fig. 1 ▸) (Nojiri et al., 2007 ▸), suggesting that conformational changes may be required to place the tethered cupredoxin domain close to the core domain or an alternative mechanism is at work. Differences in the proton channel between the two HdNiRs are also likely to contribute to these differences.
Though several C-terminal and N-terminal tethered CuNiRs have been identified and structurally characterized, the scarcity of substrate/product-bound structures has hampered understanding of the role of tethered partner electron donor proteins. When these enzymes were discovered it was simply assumed that tethering would provide functional advantage by narrowing the range of conformational searches that are generally required in encounter complexes. We have successfully determined the substrate-bound structure Hd 1NES1NiR, obtained by simple soaking of an as-isolated enzyme crystal. This represents the first substrate-bound structure of the wild-type tethered CuNiR.
The nitrite-soaked crystal trapped the substrate in two of the monomers while the third monomer showed the product (NO) with full occupancy. The product may have formed from enzyme turnover catalyzed by solvated electrons generated during X-ray data collection, but the full occupancy of a single species is surprising. This suggests a very slow off-rate for dissociation of the product which is consistent with the lower NiR activity in these enzymes. Our data provides clear evidence for the role of the N-terminal peptide that carries His27 (Fig. 3 ▸) in water-mediated anchoring of the substrate at the T2Cu site. The conformational flexibility of this N-terminal peptide and His27 may be critical for the substrate entry, anchoring and product formation stages of the catalytic reaction, requiring it to move to the outward conformation for the eventual release of the product. Our data also provide an explanation for the significant activity for the core T1Cu mutant for Hd A3151NiR by identifying a long-range electron tunneling route via a hydrophobic β-strand, thereby bypassing the T1Cucore and delivering electrons directly to the catalytic T2Cu center.
8. Data availability
The atomic coordinates and structure factors of Hd 1NES1NiR have been deposited in the Protein Data Bank (http://www.rcsb.org/) under the accession codes 6tfo and 6tfd for the as-isolated and substrate/product-bound structures, respectively.
Supplementary Material
Supporting information file. DOI: 10.1107/S2052252520005230/jt5044sup1.pdf
PDB reference: Hd1NES1NiR (as-isolated), 6tfo
PDB reference: Hd1NES1NiR (nitrite-soaked), 6tfd
Acknowledgments
RRE, RCG, SVA and SSH conceived and designed the project. DS and TFW performed the experiments. DS, TFW, RRE, RCG, SVA and SSH analyzed the data. DS, RRE, RCG, SVA and SSH wrote the paper. The authors would like to thank Diamond Light Source, UK (MX15991 BAG). The authors declare no competing financial interests.
Funding Statement
This work was funded by Biotechnology and Biological Sciences Research Council grant BB/N013972/1 to S. Samar Hasnain and Svetlana V. Antonyuk. Conselho Nacional de Desenvolvimento Científico e Tecnológico grant 407438/2013-0 to S. Samar Hasnain and Tatiana F. Watanabe.
References
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Antonyuk, S. V., Han, C., Eady, R. R. & Hasnain, S. S. (2013). Nature, 496, 123–126. [DOI] [PMC free article] [PubMed]
- Antonyuk, S. V., Strange, R. W., Sawers, G., Eady, R. R. & Hasnain, S. S. (2005). Proc. Natl Acad. Sci. 102, 12041–12046. [DOI] [PMC free article] [PubMed]
- Boulanger, M. J., Kukimoto, M., Nishiyama, M., Horinouchi, S. & Murphy, M. E. (2000). J. Biol. Chem. 275, 23957–23964. [DOI] [PubMed]
- Bower, J. K., Sokolov, A. Y. & Zhang, S. (2019). Angew. Chem. Int. Ed. 58, 10225–10229. [DOI] [PubMed]
- Chaikuad, A., Knapp, S. & von Delft, F. (2015). Acta Cryst. D71, 1627–1639. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Deligeer, Fukunaga, R., Kataoka, K., Yamaguchi, K., Kobayashi, K., Tagawa, S. & Suzuki, S. (2002). J. Inorg. Biochem. 91, 132–138. [DOI] [PubMed]
- Dodd, F. E., Van Beeumen, J., Eady, R. R. & Hasnain, S. S. (1998). J. Mol. Biol. 282, 369–382. [DOI] [PubMed]
- Dong, J., Sasaki, D., Eady, R. R., Antonyuk, S. V. & Hasnain, S. S. (2018). IUCrJ, 5, 510–518. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Fukuda, Y., Tse, K. M., Nakane, T., Nakatsu, T., Suzuki, M., Sugahara, M., Inoue, S., Masuda, T., Yumoto, F., Matsugaki, N., Nango, E., Tono, K., Joti, Y., Kameshima, T., Song, C., Hatsui, T., Yabashi, M., Nureki, O., Murphy, M. E., Inoue, T., Iwata, S. & Mizohata, E. (2016). Proc. Natl Acad. Sci. USA, 113, 2928–2933. [DOI] [PMC free article] [PubMed]
- Ghosh, S., Dey, A., Usov, O. M., Sun, Y., Grigoryants, V. M., Scholes, C. P. & Solomon, E. I. (2007). J. Am. Chem. Soc. 129, 10310–10311. [DOI] [PMC free article] [PubMed]
- Godden, J. W., Turley, S., Teller, D. C., Adman, E. T., Liu, M. Y., Payne, W. J. & LeGall, J. (1991). Science, 253, 438–442. [DOI] [PubMed]
- Gray, H. B. & Winkler, J. R. (2005). Proc. Natl Acad. Sci. USA, 102, 3534–3539. [DOI] [PMC free article] [PubMed]
- Halsted, T. P., Yamashita, K., Gopalasingam, C. C., Shenoy, R. T., Hirata, K., Ago, H., Ueno, G., Blakeley, M. P., Eady, R. R., Antonyuk, S. V., Yamamoto, M. & Hasnain, S. S. (2019). IUCrJ, 6, 761–772. [DOI] [PMC free article] [PubMed]
- Hedison, T. M., Shenoy, R. T., Iorgu, A. I., Heyes, D. J., Fisher, K., Wright, G. S. A., Hay, S., Eady, R. R., Antonyuk, S. V., Hasnain, S. S. & Scrutton, N. S. (2019). ACS Catal. 9, 6087–6099. [DOI] [PMC free article] [PubMed]
- Horrell, S., Kekilli, D., Sen, K., Owen, R. L., Dworkowski, F. S. N., Antonyuk, S. V., Keal, T. W., Yong, C. W., Eady, R. R., Hasnain, S. S., Strange, R. W. & Hough, M. A. (2018). IUCrJ 5, 283–292. [DOI] [PMC free article] [PubMed]
- Kataoka, K., Furusawa, H., Takagi, K., Yamaguchi, K. & Suzuki, S. (2000). J. Biochem. 127, 345–350. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nojiri, M. (2016). Metalloenzymes in Denitrification: Applications and Environmental Impacts, edited by I. Moura, J. J. G. Moura, S. R. Pauleta & L. B. Maia, pp. 91–113. Cambridge: The Royal Society of Chemistry.
- Nojiri, M., Koteishi, H., Nakagami, T., Kobayashi, K., Inoue, T., Yamaguchi, K. & Suzuki, S. (2009). Nature, 462, 117–120. [DOI] [PubMed]
- Nojiri, M., Xie, Y., Inoue, T., Yamamoto, T., Matsumura, H., Kataoka, K., Deligeer, Yamaguchi, K., Kai, Y. & Suzuki, S. (2007). Proc. Natl Acad. Sci. USA, 104, 4315–4320. [DOI] [PMC free article] [PubMed]
- Opperman, D. J., Murgida, D. H., Dalosto, S. D., Brondino, C. D. & Ferroni, F. M. (2019). IUCrJ, 6, 248–258. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Tickle, I. J., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2018). STARANISO. Global Phasing Ltd, Cambridge, UK.
- Tocheva, E. I., Rosell, F. I., Mauk, A. G. & Murphy, M. E. (2004). Science, 304, 867–870. [DOI] [PubMed]
- Tsuda, A., Ishikawa, R., Koteishi, H., Tange, K., Fukuda, Y., Kobayashi, K., Inoue, T. & Nojiri, M. (2013). J. Biochem. 154, 51–60. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., Womack, T. & Bricogne, G. (2011). Acta Cryst. D67, 293–302. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Winter, G. (2010). J. Appl. Cryst. 43, 186–190.
- Winter, G., Waterman, D. G., Parkhurst, J. M., Brewster, A. S., Gildea, R. J., Gerstel, M., Fuentes-Montero, L., Vollmar, M., Michels-Clark, T., Young, I. D., Sauter, N. K. & Evans, G. (2018). Acta Cryst. D74, 85–97. [DOI] [PMC free article] [PubMed]
- Yamaguchi, K., Kataoka, K., Kobayashi, M., Itoh, K., Fukui, A. & Suzuki, S. (2004). Biochemistry, 43, 14180–14188. [DOI] [PubMed]
- Zhang, Y. & Skolnick, J. (2005). Nucleic Acids Res. 33, 2302–2309. [DOI] [PMC free article] [PubMed]
- Zhang, Y. Z. & Shen, H. B. (2017). J. Chem. Inf. Model. 57, 988–999. [DOI] [PubMed]
- Zumft, W. G. (1997). Microbiol. Mol. Biol. Rev. 61, 533–616. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information file. DOI: 10.1107/S2052252520005230/jt5044sup1.pdf
PDB reference: Hd1NES1NiR (as-isolated), 6tfo
PDB reference: Hd1NES1NiR (nitrite-soaked), 6tfd
Data Availability Statement
The atomic coordinates and structure factors of Hd 1NES1NiR have been deposited in the Protein Data Bank (http://www.rcsb.org/) under the accession codes 6tfo and 6tfd for the as-isolated and substrate/product-bound structures, respectively.