

Abstract
Recent advances in molecular phylogenetics provide us with information of Allium L. taxonomy and evolution, such as the subgenus Cyathophora, which is monophyletic and contains five species. However, previous studies detected distinct incongruence between the nrDNA and cpDNA phylogenies, and the interspecies relationships of this subgenus need to be furtherly resolved. In our study, we newly assembled the whole chloroplast genome of four species in subgenus Cyathophora and two allied Allium species. The complete cp genomes were found to possess a quadripartite structure, and the genome size ranged from 152,913 to 154,174 bp. Among these cp genomes, there were subtle differences in the gene order, gene content, and GC content. Seven hotspot regions (infA, rps16, rps15, ndhF, trnG-UCC, trnC-GCA, and trnK-UUU) with nucleotide diversity greater than 0.02 were discovered. The selection analysis showed that some genes have elevated Ka/Ks ratios. Phylogenetic analysis depended on the complete chloroplast genome (CCG), and the intergenic spacer regions (IGS) and coding DNA sequences (CDS) showed same topologies with high support, which revealed that subgenus Cyathophora was a monophyletic group, containing four species, and A. cyathophorum var. farreri was sister to A. spicatum with 100% bootstrap value. Our study revealed selective pressure may exert effect on several genes of the six Allium species, which may be useful for them to adapt to their specific living environment. We have well resolved the phylogenetic relationship of species in the subgenus Cyathophora, which will contribute to future evolutionary studies or phylogeographic analysis of Allium.
1. Introduction
Subgenus Cyathophora (R. M. Fritsch) R. M. Fritsch is a small group of Allium that has been put forward lately [1]. The special subgenus Cyathophora contains about six species and one variety according to Li et al. [2]; besides, A. spicatum (Prain) N. Friesen has a wild distribution range, extending from China to Nepal, while the rest of them are endemic species in China and mainly distributed in the southeastern margin of the Qinghai-Tibet Plateau (QTP): A. mairei Lév, A. kingdonii Stearn, A. rhynchogynum Diels, A. trifurcatum (F. T. Wang and Tang) J. M. Xu, and A. cyathophorum Bur. and Franch and its variety A. cyathophorum var. farreri (Stearn) Stearn. Although it contains a small number of species, the boundary of subgenus Cyathophora and the involved species have experienced some alterations with the development of molecular biology. In previous study, A. spicatum was classified at different taxonomic levels because of its idiographic spicate inflorescence based on morphological and molecular evidences [3]. Five species have been proposed by Huang et al. about subgenus Cyathophora [4]: A. mairei, A. rhynchogynum, A. cyathophorum, A. cyathophorum var. farreri, and A. spicatum, while A. kingdonii and A. trifurcatum did not belong to this group. Micromorphological and cytological features supported that the subgenus Cyathophora is a monophyly and contains five species [5, 6]. Among species of the subgenus Cyathophora, A. spicatum grows in the droughty western QTP with the extremely abnormal spicate inflorescence [3], while A. cyathophorum and A. cyathophorum var. farreri with the umbel inflorescence stretch to the moist HMR [7] (Figure 1). Furthermore, Li et al. [6] suggested that A. cyathophorum and A. farreri were independent species based on molecular phylogeny and the striking distinctiveness in micromorphology. A. rhynchogynum has never been sampled since it was published in 1912 [8]. We also performed a lot of field work to collect it but failed. The Flora of China recorded that A. rhynchogynum only distributed in northwest of Yunnan province in China. Therefore, we speculate that A. rhynchogynum might become extinct or there is an identification error in previous research studies. Li et al. [6] performed phylogenetic and biogeographic analyses for A. cyathophorum and A. spicatum based on chloroplast and nuclear ribosomal DNA and detected distinct different topologies between these two molecular methods, in which A. cyathophorum showed close relationship with A. spicatum in nuclear DNA tree but was sister to A. cyathophorum var. farreri in cpDNA tree [4, 6]. Other than this, the relationship between these species is not exactly determined, and phylogenetic analysis using single or several combined chloroplast fragments does not solve the problem effectively, and the complete cp genome can well resolve the relationship of subgenus Cyathophora. Hence, it is imperative to reconstruct the relationship of subgenus Cyathophora and clarify the contained species depending on the complete chloroplast genomes. To evaluate the subgenus Cyathophora resources comprehensively, we also need more efficient molecular markers.
Figure 1.

The morphological characters of flowers of subgenus Cyathophora species. (a) A. cyathophorum. (b) A. cyathophorum var. farreri. (c) A. mairei. (d) A. spicatum.
Chloroplast is one of the basic organelles in plant cells, which is in charge of photosynthesis of green plants [9]. The chloroplast genomes have a highly conserved structure and gene content, which have a quadripartite structure composed by large single-copy (LSC) and small single-copy (SSC) regions separated by two parts of inverted repeat (IR) [10, 11]. Previous studies suggested that genome size of angiosperms ranged from 120 kb to 170 kb with gene number changed from 120 to 130 [12]. Complete chloroplast genome has long been a core issue in plant molecular evolution and systematic studies because of its oversimplified structure, highly conservative sequence, and maternal hereditary traits [13]. Since the complete cp genome analysis can provide more genetic information contrasted with just single or few cpDNA fragments [14], by using cp genome sequences, many long existing phylogenetic problems of different angiosperms at various taxonomic levels have been successfully resolved [15–20].
In addition to exploring phylogenetic studies, the whole cp genome has important significance to reveal the photosynthesis mechanism, metabolic regulation, and adaptive evolution of plants. Research has shown that adaptive evolution is mainly promoted by evolutionary processes like natural selection, which affects genetic changes caused by genetic recombination and mutations [21]. Many recent studies have analyzed the selection pressures that undergo by species in the evolutionary processes based on complete chloroplast genome, for example, a positive selection for the atpF gene may suggest that it has made an important impact on the divergence in deciduous and evergreen oak tree [9], and there also existed positive selection on ycf2 in watercress chloroplasts [22]. With the development of sequencing technology, the number of cp genomic sequences has increased dramatically in recent years. However, a few plastid genomes of Allium were reported until now, and it is necessary to develop more complete chloroplast genome in Allium for future phylogenetic and evolutionary research studies.
In our report, we assembled and characterized the complete cp genome sequence of the six Allium species using next-generation sequencing technologies to (1) reveal common structural patterns and hotspot regions, (2) gain a better understanding of the relationship about subgenus Cyathophora based on complete chloroplast genome, and (3) investigate adaptive evolution in the cp genomes of the six Allium species. We hope our study will provide valuable genetic resources for further evolutionary studies about subgenus Cyathophora.
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequencing
Fresh leaves of A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. trifurcatum, and A. kingdonii were collected from different places (Table 1). Morphological characters were measured using karyotype [23]. The healthy leaves were immediately dried with silica gel to use for DNA extraction. The voucher specimens were stored in the Herbarium of Sichuan University (SZ Herbarium). Their total genomic DNA was extracted from the sampled leaves according to the manufacturer's instructions for the Plant Genomic DNA Kit (Tiangen Biotech, Beijing, China). Genomic DNA was indexed by tags and pooled together in one lane of Illumina HiSeq platform for sequencing (paired-end, 350 bp) at Novogene (Beijing, China).
Table 1.
Samples information.
| Species | Location | Geographical coordinate |
|---|---|---|
| A. cyathophorum | Mangkang, Tibet | 29°43′24″N, 98°31′51″E |
| A. cyathophorum var. farreri | Zhouqu, Gansu | 33°47′10″N, 104°22′7″E |
| A. spicatum | NaiDong, Tibet | 29°13′40″N, 91°45′36″E |
| A. mairei | Shangri-La, Yunnan | 27°25′19″N, 100°09′22″E |
| A. trifurcatum | Shangri-La, Yunnan | 27°52′22″N, 99°43′10″E |
| A. kingdonii | Nyingchi, Tibet | 29°37′27″N, 94°39′2″E |
2.2. Chloroplast Genome Sequence Assembly and Annotation
We firstly used FastQC v0.11.7 to assess the quality of all reads [24]. To select the best reference, we filtrated the chloroplast genome related reads by mapping all reads to the published chloroplast genome sequences in Allium. SOAPdenovo2 was used to assemble all relevant reads into contigs [25]. The clean reads were assembled using the program NOVOPlasty [26] with the complete chloroplast genome of its close relative A. cepa as the reference (GenBank accession no. KM088014). Geneious 11.0.4 was used to finish the annotation of the assembled chloroplast genome, and it was corrected manually after comparison with references [27]. The circular plastid genome maps were generated utilizing the OGDRAW program [28]. The GenBank accession numbers of A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. trifurcatum, and A. kingdonii are MK820611, MK931245, MK931246, MK820615, MK931247, and MK294559, respectively.
2.3. Repeat Sequences and Simple Sequence Repeat (SSR) Analysis
REPuter [29] was selected to investigate the location and size of repeat sequences, which included four types of repeats in the chloroplast genomes about the six Allium species. The sequence identity and minimum length of repeat size was set to >90% and 30 bp, with the hamming distance of 3. We used IMEx, ImperfectMicrosatelliteExtractor (http://43.227.129.132:8008/IMEX/imex_advanced.html) [30], to find chloroplast SSRs in six chloroplast genome sequences of Allium. Its specifications were set up as follows: the minimum number of repeats for mononucleotide, dinucleotides, trinucleotides, tetranucleotides, pentanucleotide and hexanucleotides was 10, 5, 5, 4, 3, and 3, respectively, the repeat type was imperfect, the imperfection % was Mono: 10%, Di: 10%, Tri: 15%, Tetra: 20%, Penta: 5%, and Hexa: 5%, mismatches allowed in pattern were Mono: 1, Di: 1, Tri: 2, Tetra: 4, Penta: 0, and Hexa: 3, and the level of standardization was level 1 standardization.
2.4. Codon Usage Analysis
Codon usage of the species in subgenus Cyathophora was analyzed by the software of CodonW [31]. Protein-coding genes (CDS) were selected with the following filter requirements: (1) each CDS was longer than 300 nucleotides [18, 32]; (2) repeat sequences were deleted. Totally, 53 CDS of each species in Allium were selected for further study.
2.5. Genome Comparison (IR Contraction and Expansion)
The mVISTA program was chosen to analyze the whole sequence similarity of all six Allium species with Shuffle-LAGAN model [33], using the chloroplast genomes to compare their difference in sequences at the chloroplast genome level and A. cyathophorum as the reference. The boundaries between single copy regions (LSC and SSC) and inverted repeats (IR) regions among the six chloroplast genome sequences were compared by using Geneious v11.0.4 software [27].
2.6. Hotspot Regions Identification in Subgenus Cyathophora
To analyze nucleotide diversity (Pi), we extracted the shared 112 genes of the six species in Allium after alignment. DnaSP 5.10 was employed to calculate the nucleotide variability [34].
2.7. Gene Selective Pressure Analysis of Six Allium Plastomes
To investigate selection pressures, nonsynonymous (Ka) and synonymous (Ks) substitution rates of 65 selected protein-coding genes between the cp genomes of subgenus Cyathophora and the other two Allium species were calculated by KaKs Calculator version 2.0 [35].
2.8. Subgenus Cyathophora Phylogenomic Analysis Based on Chloroplast Genome
Phylogenetic analysis of subgenus Cyathophora was totally depended on twenty-nine complete chloroplast genome sequences, which were twenty-one species of Allium (including 6 newly assembled species; 15 other species of Allium were collected from NCBI), six species of Lilium, and two species of Asparagus as the out groups (Table S1). Three different databases were used to build the phylogenetic tree, which include the complete genome sequences, the IGS sequences, and all CDS sequences, and three different methods, Bayesian-inference (MrBayes v3.2), maximum parsimony (PAUP-version4.0), and maximum likelihood (RAxmL8.0), were used to build the tree. The sequences were aligned using MAFFT [36] in Geneious 11.0.4 with the set parameters and manually trimmed. GTR + I + G was selected as the best model using software ModelTest v3.7 [37]. Maximum likelihood (ML) analyses were performed using RAxmL8.0 with 1000 bootstrap replications [38]. PAUP was used to conduct maximum parsimony (MP) analyses [39]. MP was run using a heuristic search with 1000 random addition sequence replicates with the tree-bisection-reconnection (TBR) branch-swapping tree search criterion. Bayesian inference (BI) was executed with Mrbayes v3.2 [40], and the Markov chain Monte Carlo (MCMC) analysis was run 1 × 108 generations. The trees were sampled every 1000 generations: the first 25% were discarded as burn in and the remaining trees were used to establish a 50% majority rule consensus tree. When the average standard deviation of the splitting frequency was kept below 0.001, it was considered that the stationarity is achieved.
3. Results
3.1. Chloroplast Genome Organization and Gene Content in Six Species
These six acquired Allium cp genomes were detected to have a circular DNA structure of angiosperm cp genomes that comprises LSC, SSC, and two IR regions (Figure 2). The sizes of the six CP genomes ranged from 152,913 bp for A. mairei to 154,174 bp for A. cyathophorum, which were similar with other Allium CP genomes [41]. The size varied from 82,493 bp (A. mairei) to 83,423 bp (A. kingdonii) in the LSC region, from 17,811 bp (A. kingdonii) to 21,706 bp (A. trifurcatum) in the SSC region, and from 24,561 bp (A. trifurcatum) to 26,467 bp (A. cyathophorum) in the IR region (Table 2). The entire GC content of the cp genome sequences was 36.8–36.9%, and the GC contents of the LSC, SSC, and IR regions were 34.6–34.8%, 29.5–31.2%, and 42.7–43.1%, respectively. A total of 132 genes were discovered from the complete cp genome: 8 ribosomal RNA (rRNA) genes, 86 protein-coding genes, and 38 transfer RNA (tRNA) genes (Table 3).
Figure 2.
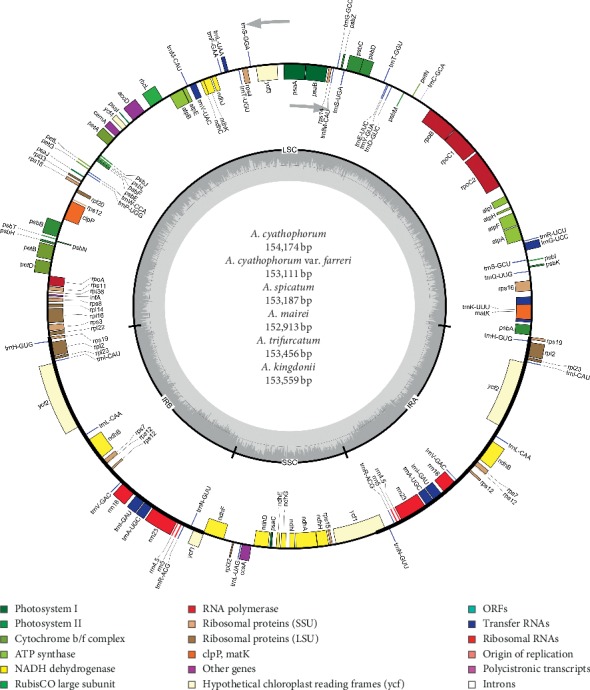
Gene map of A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. trifurcatum, and A. kingdonii complete chloroplast genomes. The circle inside genes is transcribed clockwise, and the outside genes are transcribed counterclockwise. Genes belonging to different functional groups are represented by distinct colors.
Table 2.
Details comparison of the complete chloroplast genomes of six species of Allium.
| A. cyathophorum | A. cyathophorum var. farreri | A. spicatum | A. mairei | A. trifurcatum | A. kingdonii | |
|---|---|---|---|---|---|---|
| Genome size (bp) | 154,174 | 153,111 | 153,187 | 152,913 | 153,456 | 153,559 |
| LSC length (bp) | 83,359 | 82,544 | 82,625 | 82,493 | 82,628 | 83,423 |
| SSC length (bp) | 17,881 | 17,811 | 17,920 | 18,762 | 21,706 | 17,810 |
| IR length (bp) | 26,467 | 26,378 | 26,321 | 25,829 | 24,561 | 26,163 |
| LSC GC content (%) | 34.6 | 34.7 | 34.7 | 34.8 | 34.8 | 34.8 |
| SSC GC content (%) | 29.5 | 29.7 | 29.7 | 29.9 | 31.2 | 29.9 |
| IR GC content (%) | 42.7 | 42.7 | 42.7 | 42.9 | 43.1 | 42.7 |
| Total GC content (%) | 36.8 | 36.9 | 36.9 | 36.9 | 36.9 | 36.9 |
| Total number of genes | 132 | 132 | 132 | 132 | 132 | 132 |
| Protein coding genes | 86 | 86 | 86 | 86 | 86 | 86 |
| rRNA | 8 | 8 | 8 | 8 | 8 | 8 |
| tRNA | 38 | 38 | 38 | 38 | 38 | 38 |
Table 3.
Genes existing in the chloroplast genome of Allium.
| Category | Gene name | Number |
|---|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 |
| Cytochrome b6/f | petA, petB, petD, petG, petL, petN | 6 |
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | 6 |
| Rubisco | rbcL | 1 |
| NADH oxido reductase | ndhA, ndhB(×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 |
| Large subunit ribosomal proteins | rpl2(×2), rpl14, rpl16, rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 | 11 |
| Small subunit ribosomal proteins | rps3, rps4, rps7(×2), rps8, rps11, rps12(×2), rps14, rps15, rps16, rps18, rps19(×2) | 14 |
| RNAP | rpoA, rpoB, rpoC1, rpoC2 | 4 |
| Other proteins | accD, ccsA, matK, cemA, clpP, infA | 6 |
| Proteins of unknown function | ycf1(×2), ycf2(×2), ycf3, ycf4 | 6 |
| Ribosomal RNAs | rrn23(×2), rrn16(×2), rrn5(×2), rrn4.5(×2) | 8 |
| Transfer RNA | trnA-UGC(×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnH-GUG(×2), trnG-GCC, trnG-UCC, trnI-CAU(×2), trnI-GAU(×2), trnK-UUU, trnL-CAA(×2), trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC, trnW-CCA, trnY-GUA | 38 |
| Total | 132 |
3.2. Repeat and Simple Sequence Repeat (SSR) Analysis
Many research studies of cp genomes revealed that repeat sequences have been widely used in phylogeny, population genetics, and other studies [42]. Four types of repeats (forward repeats, reverse repeats, complement repeats, and palindromic repeats) were detected in the six Allium species. There were only 3 complement repeats in A. cyathophorum, while the other species did not have. The number of repeats varied from 37 to 77 in the six species; the A. cyathophorum showed the most abundant number of repeats, including 29, 40, 5 and 3 palindromic forward reverse and complement repeats, respectively. The number of forward repeats ranged from 15 to 40, the number of palindromic repeats ranged from 17 to 29, and the number of reverse repeats ranged from 1 to 5 (Figure 3). The lengths of forward, palindromic, and reverse repeats ranged from 30 to 267 bp, and most of them were concentrated in 30–50 bp (81.48%), while those of 50–70 bp (9.09%), >100 bp (6.40%), and 70–90 bp (3.03%) were less common (Figure S1). Earlier reports recommend that the appearance of the repeats indicates that this locus is a staple hots-pot for reconfiguration of the genome [43–45]. Nevertheless, these repeats are valuable for developing genetic markers in population genetics studies [46, 47].
Figure 3.
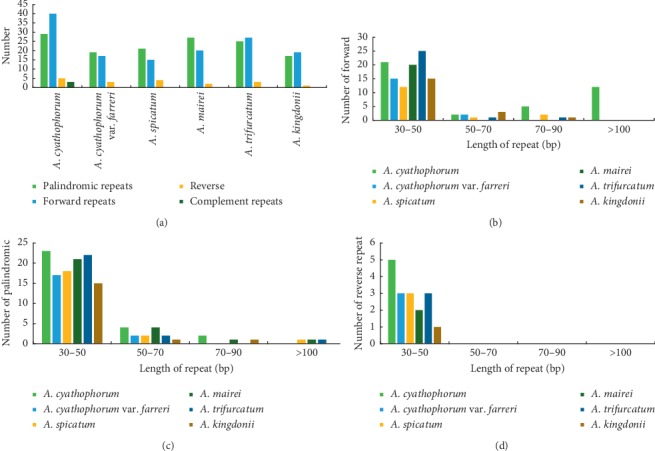
Investigation of repeated sequences in A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. trifurcatum, and A. kingdonii chloroplast genomes. (a) Four repeat types. (b) Number of the forward repeat by length. (c) Number of the palindromic repeat by length. (d) Number of the reverse repeat by length.
SSRs, also called as microsatellites, are 1- to 6-bp repeating sequences that are extensively distributed in the chloroplast genome. SSRs are highly polymorphic and codominant, which are valuable markers for study involving gene flow, population genetics, and gene mapping [48]. In this study, six classes of SSRs (mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats) were found in the cp genome of the six species, whereas the number of hexanucleotide repeats ranged from 1 to 3 in the six species, and the pentanucleotide repeats just existed in A. kingdonii and A. spicatum. The total number of SSRs in the genome of the six Allium species was 185 in A. cyathophorum, 158 in A. cyathophorum var. farreri, 159 in A. spicatum, 171 in A. mairei, 201 in A. trifurcatum, and 165 in A. kingdonii (Figure 4(a)). The highest number was mononucleotide repeat, which accounted for about 30.41% of the total SSRs (Figure 4(c)); the number ranged from 36 in A. kingdonii to 71 in A. trifurcatum, and all mononucleotide repeats are composed of A or T bases; these conclusions were unanimous in previous studies that SSRs in cp genomes usually contained short polyA or polyT repeats [49], while those of dinucleotide repeats (28.20%), trinucleotide repeats (20.79%), tetranucleotide repeats (19.15%), pentanucleotide repeats (0.38%), and hexanucleotide repeats were the least abundant (1.06%). In the whole SSR locus, the SSRs located in the LSC area are much more than those in the SSC and IR areas (Figure 4(b)), which is identical with previous research studies that SSRs are unevenly distributed in cp genomes [50].
Figure 4.
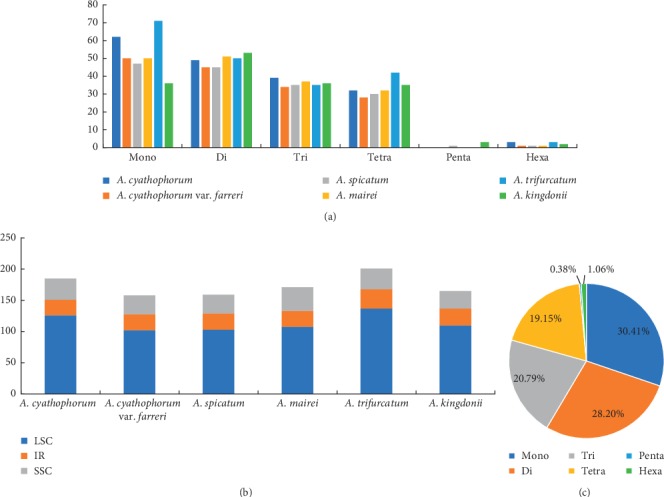
Analysis of simple sequence repeats (SSRs) in six Allium chloroplast genome sequences. (a) Number of six SSR types discovered in six Allium chloroplast genome sequences. (b) Number of SSRs in the LSC, IR, and SSC regions in six Allium chloroplast genome sequences. (c) Presence of different SSR types in total SSRs of six Allium chloroplast genome sequences.
3.3. Codon Usage Analysis
Codon usage bias is a phenomenon that the synonymous codons usually have different frequencies of use in plant genomes, which was caused by evolutionary factors that affect gene mutations and selections [51, 52]. The relative synonymous codon usage (RSCU) is a method that estimates nonuniform synonymous codon usage in coding sequences, in which RSCU less than 1 demonstrates lack of bias, whereas RSCU value greater than 1 stands for more frequent use of a codon. In view of the sequences of 53 protein-coding genes (CDS), the codon usage frequency was calculated for the six Allium cp genomes (Table 4). Altogether, the number of codons ranged from 33058 in A. kingdonii to 23791 in A. mairei. In addition, the result indicated that a total of 13218 codons encoding leucine in the cp genomes of the six species and 1453 codons encoding cysteine as the most common and least common universal amino acids, respectively. As recently discovered in other cp genomes of plants, our study revealed that except tryptophan and methionine, there was preference in the use of synonymous codons, and the RSCU value of 30 codons exceeded 1 for each species, and they were A or T-ending codons. The result is in accordance with other researches, which the codon usage preference for A/T ending in plants [53–55].
Table 4.
Codon usage in six Allium chloroplast genomes.
| AA | Codon | Number | RSCU | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acy | Afa | Asp | Ama | Aki | Atr | Acy | Afa | Asp | Ama | Aki | Atr | ||
| Phe | UUU | 826 | 825 | 827 | 822 | 809 | 827 | 1.33 | 1.33 | 1.33 | 1.33 | 1.32 | 1.33 |
| UUC | 420 | 415 | 418 | 414 | 421 | 419 | 0.67 | 0.67 | 0.67 | 0.67 | 0.68 | 0.67 | |
| Leu | UUA | 758 | 758 | 756 | 751 | 744 | 746 | 2.05 | 2.05 | 2.05 | 2.05 | 2.04 | 2.04 |
| UUG | 447 | 450 | 447 | 441 | 446 | 444 | 1.21 | 1.22 | 1.21 | 1.21 | 1.22 | 1.21 | |
| CUU | 455 | 453 | 453 | 448 | 448 | 452 | 1.23 | 1.23 | 1.23 | 1.22 | 1.23 | 1.24 | |
| CUC | 138 | 137 | 138 | 139 | 136 | 138 | 0.37 | 0.37 | 0.37 | 0.38 | 0.37 | 0.38 | |
| CUA | 295 | 295 | 298 | 294 | 292 | 288 | 0.8 | 0.8 | 0.81 | 0.8 | 0.8 | 0.79 | |
| CUG | 122 | 121 | 124 | 122 | 119 | 125 | 0.33 | 0.33 | 0.34 | 0.33 | 0.33 | 0.34 | |
| Ile | AUU | 943 | 946 | 945 | 955 | 938 | 931 | 1.49 | 1.49 | 1.49 | 1.5 | 1.48 | 1.48 |
| AUC | 350 | 345 | 346 | 339 | 350 | 344 | 0.55 | 0.54 | 0.55 | 0.53 | 0.55 | 0.55 | |
| AUA | 611 | 610 | 613 | 615 | 616 | 616 | 0.96 | 0.96 | 0.97 | 0.97 | 0.97 | 0.98 | |
| Met | AUG | 497 | 500 | 498 | 495 | 497 | 495 | 1 | 1 | 1 | 1 | 1 | 1 |
| Val | GUU | 431 | 432 | 432 | 429 | 440 | 438 | 1.48 | 1.48 | 1.49 | 1.48 | 1.51 | 1.5 |
| GUC | 129 | 130 | 129 | 128 | 131 | 131 | 0.44 | 0.45 | 0.44 | 0.44 | 0.45 | 0.45 | |
| GUA | 432 | 435 | 434 | 434 | 430 | 433 | 1.49 | 1.49 | 1.5 | 1.5 | 1.47 | 1.48 | |
| GUG | 171 | 167 | 165 | 165 | 167 | 165 | 0.59 | 0.57 | 0.57 | 0.57 | 0.57 | 0.57 | |
| Ser | UCU | 467 | 468 | 473 | 461 | 473 | 476 | 1.73 | 1.73 | 1.75 | 1.72 | 1.76 | 1.76 |
| UCC | 246 | 254 | 249 | 252 | 249 | 244 | 0.91 | 0.94 | 0.92 | 0.94 | 0.93 | 0.9 | |
| UCA | 321 | 320 | 320 | 318 | 311 | 325 | 1.19 | 1.18 | 1.18 | 1.19 | 1.16 | 1.2 | |
| UCG | 151 | 151 | 150 | 151 | 159 | 154 | 0.56 | 0.56 | 0.55 | 0.56 | 0.59 | 0.57 | |
| Pro | CCU | 338 | 339 | 338 | 337 | 338 | 338 | 1.56 | 1.57 | 1.56 | 1.56 | 1.55 | 1.56 |
| CCC | 191 | 188 | 189 | 187 | 199 | 194 | 0.88 | 0.87 | 0.87 | 0.86 | 0.91 | 0.9 | |
| CCA | 247 | 244 | 247 | 243 | 246 | 238 | 1.14 | 1.13 | 1.14 | 1.12 | 1.13 | 1.1 | |
| CCG | 92 | 94 | 93 | 98 | 89 | 94 | 0.42 | 0.43 | 0.43 | 0.45 | 0.41 | 0.44 | |
| Thr | ACU | 448 | 449 | 449 | 446 | 438 | 440 | 1.65 | 1.67 | 1.67 | 1.65 | 1.63 | 1.63 |
| ACC | 186 | 184 | 184 | 185 | 191 | 193 | 0.69 | 0.68 | 0.68 | 0.69 | 0.71 | 0.72 | |
| ACA | 335 | 331 | 331 | 334 | 335 | 329 | 1.24 | 1.23 | 1.23 | 1.24 | 1.25 | 1.22 | |
| ACG | 116 | 114 | 114 | 114 | 111 | 115 | 0.43 | 0.42 | 0.42 | 0.42 | 0.41 | 0.43 | |
| Ala | GCU | 533 | 536 | 538 | 537 | 528 | 542 | 1.85 | 1.85 | 1.85 | 1.85 | 1.85 | 1.86 |
| GCC | 159 | 163 | 161 | 162 | 161 | 164 | 0.55 | 0.56 | 0.55 | 0.56 | 0.56 | 0.56 | |
| GCA | 343 | 345 | 348 | 341 | 335 | 348 | 1.19 | 1.19 | 1.2 | 1.18 | 1.18 | 1.19 | |
| GCG | 117 | 118 | 115 | 119 | 116 | 111 | 0.41 | 0.41 | 0.4 | 0.41 | 0.41 | 0.38 | |
| Tyr | UAU | 683 | 669 | 668 | 674 | 679 | 666 | 1.63 | 1.61 | 1.62 | 1.62 | 1.64 | 1.61 |
| UAC | 155 | 160 | 156 | 157 | 150 | 160 | 0.37 | 0.39 | 0.38 | 0.38 | 0.36 | 0.39 | |
| TER | UAA | 29 | 29 | 29 | 29 | 30 | 26 | 1.47 | 1.47 | 1.5 | 1.47 | 1.73 | 1.47 |
| UAG | 18 | 18 | 17 | 18 | 12 | 15 | 0.92 | 0.92 | 0.88 | 0.92 | 0.69 | 0.85 | |
| His | CAU | 415 | 416 | 415 | 412 | 419 | 420 | 1.56 | 1.57 | 1.56 | 1.57 | 1.56 | 1.56 |
| CAC | 118 | 113 | 116 | 113 | 118 | 119 | 0.44 | 0.43 | 0.44 | 0.43 | 0.44 | 0.44 | |
| Gln | CAA | 583 | 585 | 585 | 587 | 587 | 591 | 1.53 | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 |
| CAG | 178 | 176 | 177 | 177 | 175 | 177 | 0.47 | 0.46 | 0.46 | 0.46 | 0.46 | 0.46 | |
| Asn | AAU | 803 | 799 | 797 | 803 | 798 | 784 | 1.56 | 1.56 | 1.56 | 1.56 | 1.55 | 1.55 |
| AAC | 229 | 226 | 227 | 226 | 231 | 227 | 0.44 | 0.44 | 0.44 | 0.44 | 0.45 | 0.45 | |
| Lys | AAA | 876 | 878 | 884 | 866 | 838 | 863 | 1.55 | 1.55 | 1.55 | 1.53 | 1.51 | 1.52 |
| AAG | 255 | 258 | 254 | 264 | 269 | 269 | 0.45 | 0.45 | 0.45 | 0.47 | 0.49 | 0.48 | |
| Asp | GAU | 692 | 692 | 691 | 681 | 682 | 691 | 1.64 | 1.64 | 1.64 | 1.63 | 1.62 | 1.62 |
| GAC | 154 | 152 | 154 | 156 | 158 | 160 | 0.36 | 0.36 | 0.36 | 0.37 | 0.38 | 0.38 | |
| Glu | GAA | 861 | 864 | 857 | 861 | 869 | 852 | 1.49 | 1.49 | 1.48 | 1.49 | 1.5 | 1.49 |
| GAG | 296 | 299 | 299 | 291 | 291 | 289 | 0.51 | 0.51 | 0.52 | 0.51 | 0.5 | 0.51 | |
| Cys | UGU | 187 | 185 | 185 | 186 | 184 | 186 | 1.52 | 1.54 | 1.54 | 1.54 | 1.52 | 1.54 |
| UGC | 59 | 56 | 56 | 55 | 58 | 56 | 0.48 | 0.46 | 0.46 | 0.46 | 0.48 | 0.46 | |
| TER | UGA | 12 | 12 | 12 | 12 | 10 | 12 | 0.61 | 0.61 | 0.62 | 0.61 | 0.58 | 0.68 |
| Trp | UGG | 386 | 389 | 391 | 392 | 391 | 391 | 1 | 1 | 1 | 1 | 1 | 1 |
| Arg | CGU | 279 | 280 | 280 | 280 | 281 | 278 | 1.37 | 1.37 | 1.37 | 1.38 | 1.37 | 1.35 |
| CGC | 74 | 78 | 78 | 76 | 71 | 80 | 0.36 | 0.38 | 0.38 | 0.37 | 0.34 | 0.39 | |
| CGA | 269 | 270 | 269 | 269 | 272 | 274 | 1.32 | 1.32 | 1.31 | 1.32 | 1.32 | 1.33 | |
| CGG | 88 | 89 | 87 | 88 | 89 | 89 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | |
| Ser | AGU | 339 | 342 | 342 | 342 | 331 | 335 | 1.26 | 1.26 | 1.27 | 1.27 | 1.23 | 1.24 |
| AGC | 91 | 88 | 88 | 86 | 87 | 92 | 0.34 | 0.33 | 0.33 | 0.32 | 0.32 | 0.34 | |
| Arg | AGA | 390 | 392 | 395 | 388 | 398 | 400 | 1.91 | 1.91 | 1.93 | 1.91 | 1.93 | 1.94 |
| AGG | 125 | 121 | 119 | 119 | 124 | 119 | 0.61 | 0.59 | 0.58 | 0.59 | 0.6 | 0.58 | |
| Gly | GGU | 489 | 487 | 487 | 493 | 483 | 489 | 1.33 | 1.33 | 1.33 | 1.34 | 1.31 | 1.33 |
| GGC | 133 | 130 | 131 | 131 | 137 | 132 | 0.36 | 0.36 | 0.36 | 0.36 | 0.37 | 0.36 | |
| GGA | 620 | 621 | 625 | 622 | 623 | 616 | 1.69 | 1.7 | 1.7 | 1.69 | 1.69 | 1.67 | |
| GGG | 228 | 226 | 226 | 225 | 229 | 236 | 0.62 | 0.62 | 0.62 | 0.61 | 0.62 | 0.64 | |
RSCU represents relative synonymous codon usage. RSCU more than one is highlighted in bold. Acy, Afa, Asp, Ama, Aki, and Atr stand for A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. kingdonii, and A. trifurcatum, respectively.
3.4. Comparative Analysis of the Chloroplast Genomes among Six Species in Allium
mVISTA online software in the Shuffle-LAGAN mode was employed to analyze the comprehensive sequence discrepancy of the six chloroplast genomes of Allium with the annotation of A. cyathophorum as a reference. In this study, the whole chloroplast genome alignment showed great sequence consistency of the six cp genomes, indicating that Allium cp genomes are very conservative (Figure 5). We found that among the six cp genomes, their IR region is more conserved compared to the LSC and SSC regions, which is similar with other plants [56, 57]. Furthermore, as we have found in other angiosperms, the coding areas were more conserved than the noncoding areas, and there were more variations in the intergenic spacers of the LSC and SSC areas, whereas the IR areas presented a lower sequence divergence [58, 59]. A. cyathophorum var. farreri had the highest sequence similarity to A. cyathophorum in sequence identity analysis. Noncoding regions displayed varying degrees of sequence differences in these six Allium cp genomes, including trnK-rps16, trnS-trnG, atpH-atpI, petN-psbM, trnT-psbD, trnF-ndhJ, accD-psaI, and petA-psbL. The coding areas with significant diversity contain matK, rps16, rpoC2, infA, ycf1, ndhF, and rps15 genes. The highly diverse regions found in this study may be used to develop molecular markers that can improve efficiency to study phylogenetic relationships within the Allium species.
Figure 5.
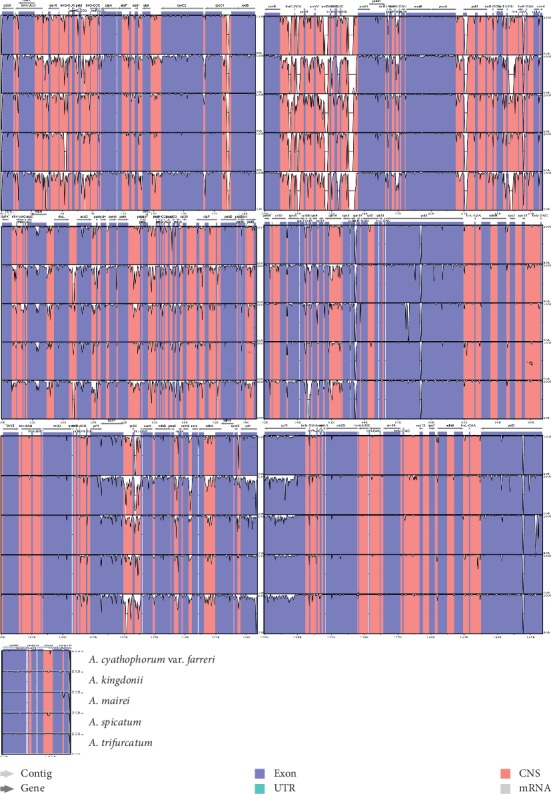
Sequence alignment of six Allium chloroplast genomes (A. cyathophorum as the reference). The y-axis represents the percent similarity between 50% and 100%. Different colors represent different genetic regions.
Though the cp genome is usually well conserved, having typical quadripartite structure, gene number, and order, a phenomenon recognized as ebb and flow exists, and this is where the IR area often expands or contracts [60]. Expansion and contraction of IR region is related to the size variations in the cp genome and has great differences in its evolution [61, 62]. We compared the IR/SC boundary areas of the six Allium cp genomes, and we found that there are obvious differences in the IR/LSC and IR/SSC connections (Figure 6). At the boundary of LSC/IRa junction, rps19 gene of different species distance the boundary were from 1 to 81 bp, while the rpl22 genes distance the border were from 29 to 273 bp. At the boundary of LSC/IRb connections, the psbA genes distance the border were reached from 108 to 605 bp. The inverted repeat b (IRb)/SSC border located in the coding region, and the ycf1 genes of the six species with a region ranged from 4193 to 5223 bp located in the SSC regions, which the ycf1 gene of A. trifurcatum all located in the SSC region. The shorter ycf1 gene crossed the inverted repeat (IRa)/SSC boundary, with 56–919 bp locating in the SSC regions. And the ndhF genes were situated in the SSC regions, which distance from the IRa/SSC boundary ranged from 1 to 1962 bp. Undoubtedly, the full-length differences in the sequence of the six cp genomes are caused by changes in the IR/SC boundaries.
Figure 6.
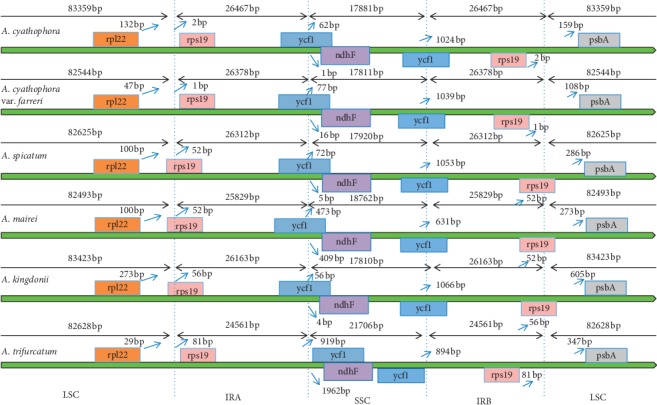
Comparison of the boundaries of the LSC, SSC, and IR areas of the whole chloroplast genomes of the six species.
3.5. Hotspot Regions Identification in Subgenus Cyathophora
We totally extracted the shared 112 genes of the six species in chloroplast genomes; the nucleotide variability (Pi) ranged from 0.00041 (rrn16) to 0.08125 (infA) among these shared genes (Figure 7; Table S2). Seven genes (infA, rps16, rps15, ndhF, trnG-UCC, trnC-GCA, and trnK-UUU) were considered to be hotspot regions with a nucleotide diversity greater than 0.02. These regions can be used to develop useful markers for phylogenetic analysis and distinguish the species in Allium.
Figure 7.

The nucleotide diversity of the shared 112 genes of the six species in chloroplast genomes.
3.6. Synonymous (Ks) and Nonsynonymous (Ka) Substitution Rate Analysis
The Ka/Ks ratio is a significant index for understanding the evolution of protein-coding genes to assess gene differentiation rates and to determine whether positive, purified, or neutral selections have been performed; a Ka/Ks ratio >1 illustrates positive selection and Ka/Ks < 1 illustrates purifying selection, while the ratio of Ka/Ks close to 1 illustrates neutral selection [63]. In our study, the Ka/Ks ratio was calculated for 65 shared protein-coding genes in all six chloroplast genomes (Table S3), and the results are shown in Figure 8. The conservative genes with Ka/Ks ratio of 0.01, indicating powerful purifying selection pressure, were rpl2, rpl32, psaC, psbA, rpoC2, petN, psbZ, psaB, psaJ, and psbT, when the averaging Ka/Ks method showed ycf1 and ycf2 genes with Ka/Ks > 1, which shows that they may undergo some selective pressure among the six Allium species. The Ka/Ks ratios ranging from 0.5 to 1 were found for matK, rps16, psaI, cemA, petA, and rpl20, representing relaxed selection. The majority (56 of 65 genes) had an average Ka/Ks ratio ranging from 0 to 0.49 for the six compared groups, indicating that most genes were under purifying selection. Other than this, four genes (matK, rpoB, petA, and rpoA) with Ka/Ks > 1 in one or more pairwise comparisons (Figure 8) suggest that these genes may undergo selective pressure which is unknown, which is very important for researching the evolution of species.
Figure 8.

KA/KS analysis of 65 common protein coding genes in six Allium species. Acy, Afa Asp, Ama, Aki, and Atr stand for A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, A. kingdonii, and A. trifurcatum, respectively. KA: nonsynonymous; KS: synonymous.
3.7. Phylogenetic Analysis of Subgenus Cyathophora Depends on Chloroplast Genome
The cp genome of sequence is significant and helpful to construct phylogenetic relationships and explore the evolutionary history in many previous reports [64, 65]. To explore the phylogenetic relationship of the six Allium species, we constructed the phylogenetic tree using three different methods and databases containing twenty-one Allium species, six Lilium species, and two Asparagus species as the out groups (Figure 9). Three databases of the complete genome sequences, the IGS sequences, and all CDS sequences using MP, BI, and ML methods all showed the same topologies with high support (Figures S2 and S3). The results strongly supported that subgenus Cyathophora is a monophyletic group, comprising A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, and A. mairei in this study with 100% bootstrap value; subgenus Cyathophora does not contain A. kingdonii and A. trifurcatum, and the phylogenetic tree indicates that A. cyathophorum var. farreri is a direct sister to A. spicatum, which is in accordance with the results of previous molecular research studies [4, 6]. The sister relationship of A. cyathophorum var. farreri and A. spicatum strongly suggests that A. spicatum is closely related to subgenus Cyathophora though it is a special species with the significant abnormal spicate inflorescence compared to other species with capitate or umbellate inflorescence. Furthermore, Allium kingdonii was the closest relative of Allium paradoxum and Allium ursinum.
Figure 9.

Phylogenetic relationship of subgenus Cyathophora with relational species. The whole chloroplast genomes dataset was analyzed using three different methods: maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI). Numbers on the branches stand for bootstrap values in the ML, MP, and posterior probabilities in the BI trees. Six species of Lilium and two species of Asparagus were considered as the out groups. Purple shows the branch of the subgenus Cyathophora.
4. Discussion
4.1. Variations among the Six Allium Species
In this research, we assembled the complete cp genome of the six species in Allium. They were very conservative in genome structure and size; it showed a typical circular DNA structure and similar cp genome sequence length, ranging from 152,913 bp in A. mairei to 154,174 bp in A. cyathophorum. The six species had the identical numbers of protein-coding, tRNA, and rRNA genes. There were some expansion or contraction of IRs among these species (Figure 6); the expansion and contraction of IR regions are related to the divergences in chloroplast genome size [66]. To some extent, it is contributed to the cp genome variation and evolution. Other than this, variations in the IR/SC boundaries in the six cp genomes lead to the distinction in the whole length of sequence [61]. Previous research studies showed that SSRs have been widely known as important resources of molecular markers and have been broadly applied in phylogenetic and biogeographic studies [67, 68]. We surveyed and analyzed the quantities and distributions of SSRs with the six species in Allium, the largest number of SSR type was mononucleotide repeats, and the SSRs in the LSC area are much higher than those in the SSC and IR areas (Figure 4), showing that SSRs have a unevenly distribution in cp genome [50]. Additionally, we also explored seven common genes (infA, rps16, rps15, ndhF, trnG-UCC, trnC-GCA, and trnK-UUU) with nucleotide diversity more than 0.02 in the six cp genome sequences of Allium; among them, trnK-UUU, trnG-UCC, ndhF, and rps15 have been previously known as hypervariable regions in Allium [17], and we consider that these SSRs and genes with greater nucleotide diversity can be used as helpful DNA barcodes to identify the species in Allium.
4.2. Phylogenetic Relationships
The results of phylogenetic analysis clearly show that Allium subgenus Cyathophora is a monophyletic group, and comprise four species (A. cyathophorum, A. farreri, A. spicatum and A. mairei), A. cyathophorum var. farreri has been upgraded to the level of the species as A. farreri in a recent study [69]. Besides A. farreri is a direct sister to A. spicatum with 100% strong bootstrap value, while the previous study showed low bootstrap value by the combined plastid dataset (trnL-F + rpl32-trnL) [4]. Currently, most phylogenetic relationships are obtained with chloroplast fragments, while single ITS, chloroplast fragment, or chloroplast combined fragment does not have a better effect in phylogenetic analysis compared to the whole cp genome. We convinced that the complete chloroplast genomes have more advantages to solve the phylogenetic issues about the subgenus Cyathophora. In previous studies, many phylogenetic problems in many plants have been successfully resolved by using complete cp genome sequences [18, 19, 70]; the lately published article about Allium also well resolved the phylogenetic relationship [17, 71]. Although the morphological characteristics of the A. farreri and A. spicatum are obviously different, in which A. spicatum has distinctive spicate inflorescence compared to A. farreri with umbel hemispheric inflorescence, our results undoubtedly showed A. farreri is a direct sister to A. spicatum, which is in accordance with Li et al. [6]. According to previous study, different inflorescence may imply that the umbel inflorescence was replaced by spicate inflorescence to adapt the harsh environment [6]. The phylogenetic tree revealed A. cyathophorum had a closer relationship with A. spicatum and A. farreri compared to A. mairei. Furthermore, the members of subgenus Cyathophora do not contain A. kingdonii and A. trifurcatum; A. kingdonii was the closest relative of A. paradoxum and A. ursinum, which is consistence with previous studies [4, 6]. Certainly, our study persuasively constructed reliable phylogeny relationship of subgenus Cyathophora by using the complete cp genome data.
4.3. Selection Events in Protein Coding Genes
DNA base mutations can be divided into two categories based on their effects to the encoded amino acids: synonymous mutations (Ks) and nonsynonymous mutations (Ka). Synonymous mutations do not result in amino acid changes, the frequency of which is represented by Ks; nonsynonymous mutations resulted in a change of amino acid, the frequency of which is indicated by Ka [72]. The ratio (Ka/Ks) is an important indicator to reveal evolutionary rate and natural selection pressure [73]. Interestingly, the synonymous nucleotide substitutions occurred at a higher frequency than nonsynonymous substitutions, and thus Ka/Ks ratios are constantly <1 in most genes [9, 74], and our study is similar with this. Between different regions and genes, the Ka/Ks ratios were usually specific (Figure 8). Most conserved genes (56 of 65 genes) had an average Ka/Ks value ranging from 0 to 0.49 for the fifteen comparison groups, indicating that most genes were under purifying selection. On the contrary, the average Ka/Ks values of the ycf1 and ycf2 genes were >1 in the fifteen comparison groups, revealing that some selective pressure may execute on them in six Allium species. Previous studies have shown that ycf1 and ycf2 genes were two large open reading frames; they were important to tobacco, and the gene knockout experiments showed that ycf1 and ycf2 played important role in a healthy cell [75]. Hu et al. [76] suggested that plants have a variety of adaptation strategies in response to unforeseen environmental conditions. Recent studies about Allium species also suggested that the selective pressure in chloroplast genomes play an important role in Allium species adaptation and evolution [17, 71]. In our field investigations, the species of subgenus Cyathophora grows in slopes or grasslands with altitude ranging from 2700 m to 4800 m. The elevated Ka/Ks ratios observed about some genes in the six Allium species may suggest that it is relate to their specific living environment. What is more, there were four genes (matK, rpoB, petA, and rpoA) with Ka/Ks > 1 in at one or more pairwise comparisons (Figure 8, Table S3), and among these genes, rpoA was also undergone positive selection in species of Annonaceae [77]. Previous study demonstrated that rpoA encodes the α subunit of plastid RNA polymerase (PEP), which is in charge of the expression of most photosynthesis-related genes [78]. It is generally believed that low temperature and strong ultraviolet radiation are not conducive to effective photosynthesis of plants; therefore, plants that survive and reproduce at high altitudes need a special photosynthetic protection strategy [79, 80]. In this study, the population of subgenus Cyathophora is mainly distributed in the Qinghai-Tibet Plateau and its adjacent high-altitude regions [2]. Therefore, we speculated that the positive selection of these genes may be related to the difference between their optimal growth environment.
5. Conclusions
Here, we sequenced, assembled, and annotated six chloroplast genomes of Allium with high-throughput sequencing technology. The gene contents and orders of the cp genomes were extremely conservative, and their cp genomes are also quadripartite structure. Repeated sequence and SSRs are helpful sources for developing new molecular markers. Codon usage analyses detected that some amino acids of the six species showed distinct codon usage preferences, and we should comprehend codon usage bias to learn evolution process. We also discovered seven highly variable common genes which can be used to develop useful markers for phylogenetic analysis and distinguish species in Allium. The Ka/Ks analysis indicated that some selective pressure may exert on several genes in the chloroplast genomes of six Allium L. species. The maximum likelihood (ML), BI, and MP phylogenetic results clearly showed that subgenus Cyathophora comprised the four assembled species: A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, and A. mairei, and A. cyathophorum var. farreri has a closer relationship with A. spicatum. This study will not only provide insights into the cp genome characteristics of species in subgenus Cyathophora but also supply useful genetic resources for phylogenetic analysis of genus Allium.
Acknowledgments
The authors appreciate the open cp genome data in NCBI and acknowledge Min Jie Li for the help in collection of materials. The authors also thank Xian Lin Guo, Fu Ming Xie, Dan Mei Su, Hao Li, and Sheng Bin Jia for their help in the use of software. This work was supported by the National Natural Science Foundation of China (grant nos. 31872647 and 31570198), National Specimen Information Infrastructure, Educational Specimen Sub-Platform (http://mnh.scu.edu.cn/), and Science and Technology Basic Work (grant no. 2013FY112100).
Data Availability
The complete chloroplast genome sequences of A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, Allium trifurcatum, and Allium kingdonii are saved in the GenBank of NCBI, and the accession numbers are MK820611, MK931245, MK931246, MK820615, MK931247, and MK294559, respectively.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
Table S1: all used accession numbers of cp genome sequences from GenBank in this article.
Table S2: the nucleotide variability (Pi) of 112 shared genes.
Table S3: the Ka/Ks ratio of 65 shared protein-coding genes in all six chloroplast genomes.
Figure S1: the proportion of the different lengths of the four dispersed repeats.
Figure S2: phylogenetic tree of the subgenus Cyathophora built by the CDS sequences using maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI).
Figure S3: phylogenetic tree of the subgenus Cyathophora built by the IGS sequences using maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI).
References
- 1.Friesen N., Fritsch R., Blattner F. Phylogeny and new intrageneric classification of allium (alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso. 2006;22(1):372–395. doi: 10.5642/aliso.20062201.31. [DOI] [Google Scholar]
- 2.Li Q.-Q., Zhou S.-D., He X.-J., Yu Y., Zhang Y.-C., Wei X.-Q. Phylogeny and biogeography of allium (amaryllidaceae: allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Annals of Botany. 2010;106(5):709–733. doi: 10.1093/aob/mcq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friesen N., Fritsch R. M., Pollner S., Blattner F. R. Molecular and morphological evidence for an origin of the aberrant genus Milula within himalayan species of Allium (Alliacae) Molecular Phylogenetics and Evolution. 2000;17(2):209–218. doi: 10.1006/mpev.2000.0844. [DOI] [PubMed] [Google Scholar]
- 4.Huang D.-Q., Yang J.-T., Zhou C.-J., Zhou S.-D., He X.-J. Phylogenetic reappraisal of allium subgenus cyathophora (amaryllidaceae) and related taxa, with a proposal of two new sections. Journal of Plant Research. 2014;127(2):275–286. doi: 10.1007/s10265-013-0617-8. [DOI] [PubMed] [Google Scholar]
- 5.Li M.-J., Guo X.-L., Li J., Zhou S.-D., Liu Q., He X.-J. Cytotaxonomy of allium (amaryllidaceae) subgenera cyathophora and amerallium sect. bromatorrhiza. Phytotaxa. 2017;331(2):185–198. doi: 10.11646/phytotaxa.331.2.3. [DOI] [Google Scholar]
- 6.Li M.-J., Tan J.-B., Xie D.-F., Huang D.-Q., Gao Y.-D., He X.-J. Revisiting the evolutionary events in Allium subgenus Cyathophora (amaryllidaceae): insights into the effect of the Hengduan mountains region (HMR) uplift and quaternary climatic fluctuations to the environmental changes in the Qinghai-Tibet Plateau. Molecular Phylogenetics and Evolution. 2016;94(Pt B):802–813. doi: 10.1016/j.ympev.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Li B. On the boundaries of the Hengduan mountains. Journal of Mountain Research. 1987;5:74–82. [Google Scholar]
- 8.Diels L. Plantae chinenses forrestianae: new and imperfectly known species. Notes of the Royal Botanic Gardens Edinburgh. 1912;5(25):161–304. [Google Scholar]
- 9.Yin K., Zhang Y., Li Y., Du F. Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: evidence from comparative analysis of the complete chloroplast genome of quercus aquifolioides with other oak species. International Journal of Molecular Sciences. 2018;19(4):p. 1042. doi: 10.3390/ijms19041042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olejniczak S. A., Łojewska E., Kowalczyk T., Sakowicz T. Chloroplasts: state of research and practical applications of plastome sequencing. Planta. 2016;244(3):517–527. doi: 10.1007/s00425-016-2551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu A., Guo W., Gupta S., Fan W., Mower J. P. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytologist. 2016;209(4):1747–1756. doi: 10.1111/nph.13743. [DOI] [PubMed] [Google Scholar]
- 12.Green B. R. Chloroplast genomes of photosynthetic eukaryotes. The Plant Journal. 2011;66(1):34–44. doi: 10.1111/j.1365-313x.2011.04541.x. [DOI] [PubMed] [Google Scholar]
- 13.Jansen R. K., Raubeson L. A., Boore J. L., et al. Methods in Enzymology. Amsterdam, Netherlands: Elsevier; 2005. Methods for obtaining and analyzing whole chloroplast genome sequences; pp. 348–384. [DOI] [PubMed] [Google Scholar]
- 14.Burke S. V., Grennan C. P., Duvall M. R. Plastome sequences of two new world bamboos-Arundinaria gigantea and Cryptochloa strictiflora (poaceae)-extend phylogenomic understanding of bambusoideae. American Journal of Botany. 2012;99(12):1951–1961. doi: 10.3732/ajb.1200365. [DOI] [PubMed] [Google Scholar]
- 15.Ma P.-F., Zhang Y.-X., Zeng C.-X., Guo Z.-H., Li D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe arundinarieae (poaceae) Systematic Biology. 2014;63(6):933–950. doi: 10.1093/sysbio/syu054. [DOI] [PubMed] [Google Scholar]
- 16.Xi Z., Ruhfel B. R., Schaefer H., et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proceedings of the National Academy of Sciences. 2012;109(43):17519–17524. doi: 10.1073/pnas.1205818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie D. F., Yu H. X., Price M., et al. Phylogeny of Chinese allium species in section daghestanica and adaptive evolution of allium (amaryllidaceae, allioideae) species revealed by the chloroplast complete genome. Frontiers in Plant Science. 2019;10:p. 460. doi: 10.3389/fpls.2019.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Zhu J., Feng L., et al. Plastid genome comparative and phylogenetic analyses of the key genera in fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Frontiers in Plant Science. 2018;9:p. 82. doi: 10.3389/fpls.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong H., Liu W., Yao G., Gong W. A comparison of chloroplast genome sequences in aconitum (Ranunculaceae): a traditional herbal medicinal genus. PeerJ. 2017;5 doi: 10.7717/peerj.4018.e4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gitzendanner M. A., Soltis P. S., Wong G. K.-S., Ruhfel B. R., Soltis D. E. Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. American Journal of Botany. 2018;105(3):291–301. doi: 10.1002/ajb2.1048. [DOI] [PubMed] [Google Scholar]
- 21.Scott-Phillips T. C., Laland K. N., Shuker D. M., Dickins T. E., West S. A. The niche construction perspective: a critical appraisal. Evolution. 2014;68(5):1231–1243. doi: 10.1111/evo.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan C., Du J., Gao L., Li Y., Hou X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): genome organization, adaptive evolution and phylogenetic relationships in cardamineae. Gene. 2019;699:24–36. doi: 10.1016/j.gene.2019.02.075. [DOI] [PubMed] [Google Scholar]
- 23.Altınordu F., Peruzzi L., Yu Y., He X. A tool for the analysis of chromosomes: KaryoType. Taxon. 2016;65(3):586–592. doi: 10.12705/653.9. [DOI] [Google Scholar]
- 24.Andrews S., FastQC A. A quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 25.Luo R., Liu B., Xie Y., et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):p. 18. doi: 10.1186/2047-217x-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dierckxsens N., Mardulyn P., Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research. 2016;45(4):p. e18. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kearse M., Moir R., Wilson A., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohse M., Drechsel O., Kahlau S., Bock R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research. 2013;41(W1):W575–W581. doi: 10.1093/nar/gkt289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurtz S., Choudhuri J. V., Ohlebusch E., Schleiermacher C., Stoye J., Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research. 2001;29(22):4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mudunuri S. B., Nagarajaram H. A. IMEx: imperfect microsatellite extractor. Bioinformatics. 2007;23(10):1181–1187. doi: 10.1093/bioinformatics/btm097. [DOI] [PubMed] [Google Scholar]
- 31.Peden J. CodonW. Dublin, Ireland: Trinity College; 1997. [Google Scholar]
- 32.Wright F. The “effective number of codons” used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 33.Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Research. 2004;32(2):W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Librado P., Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Wang D., Zhang Y., Zhang Z., Zhu J., Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics, Proteomics & Bioinformatics. 2010;8(1):77–80. doi: 10.1016/s1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katoh K., Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Posada D., Crandall K. A. Selecting the best-fit model of nucleotide substitution. Systematic Biology. 2001;50(4):580–601. doi: 10.1080/106351501750435121. [DOI] [PubMed] [Google Scholar]
- 38.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 39.Swofford D. L. Phylogenetic Analysis Using Parsimony (∗ and Other Methods) Sunderland, MA, USA: Sinauer Associates; 2002. [Google Scholar]
- 40.Ronquist F., Teslenko M., van der Mark P., et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J., Chon J., Lim J., Kim E.-K., Nah G. Characterization of complete chloroplast genome of Allium victorialis and its application for barcode markers. Plant Breeding and Biotechnology. 2017;5(3):221–227. doi: 10.9787/pbb.2017.5.3.221. [DOI] [Google Scholar]
- 42.Bull L. N., Pabon-Pena C. R., Freimer N. B. Compound microsatellite repeats: practical and theoretical features. Genome Research. 1999;9(9):830–838. doi: 10.1101/gr.9.9.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asano T., Tsudzuki T., Takahashi S., Shimada H., Kadowaki K.-i. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Research. 2004;11(2):93–99. doi: 10.1093/dnares/11.2.93. [DOI] [PubMed] [Google Scholar]
- 44.Gao L., Yi X., Yang Y.-X., Su Y.-J., Wang T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evolutionary Biology. 2009;9(1):p. 130. doi: 10.1186/1471-2148-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C., Zheng Y., Liu S., et al. The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ. 2017;5 doi: 10.7717/peerj.3820.e3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie D.-F., Yu Y., Deng Y.-Q., et al. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. International Journal of Molecular Sciences. 2018;19(7):p. 1847. doi: 10.3390/ijms19071847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nie X., Lv S., Zhang Y., et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora) PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0036869.e36869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Hao Z., Xu H., et al. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Frontiers in Plant Science. 2015;6:p. 447. doi: 10.3389/fpls.2015.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuang D.-Y., Wu H., Wang Y.-L., Gao L.-M., Zhang S.-Z., Lu L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–673. doi: 10.1139/g11-026. [DOI] [PubMed] [Google Scholar]
- 50.Qian J., Song J., Gao H., et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One. 2013;8(2) doi: 10.1371/journal.pone.0057607.e57607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong G. K.-S., Wang J., Tao L., et al. Compositional gradients in Gramineae genes. Genome Research. 2002;12(6):851–856. doi: 10.1101/gr.189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ermolaeva M. D. Synonymous codon usage in bacteria. Current Issues in Molecular Biology. 2001;3(4):91–97. doi: 10.21775/cimb.003.091. [DOI] [PubMed] [Google Scholar]
- 53.Wang W., Yu H., Wang J., et al. The complete chloroplast genome sequences of the medicinal plant forsythia suspensa (oleaceae) International Journal of Molecular Sciences. 2017;18(11):p. 2288. doi: 10.3390/ijms18112288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yi D.-K., Kim K.-J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PloS One. 2012;7(5) doi: 10.1371/journal.pone.0035872.e35872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu X., Zuo L., Lu D., Lu B., Yang M., Wang J. Comparative analysis of chloroplast genomes of five Robinia species: genome comparative and evolution analysis. Gene. 2019;689:141–151. doi: 10.1016/j.gene.2018.12.023. [DOI] [PubMed] [Google Scholar]
- 56.Dong W., Xu C., Cheng T., Zhou S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0077965.e77965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y. J., Du L. W., Liu A., et al. The complete chloroplast genome sequences of five epimedium species: lights into phylogenetic and taxonomic analyses. Frontiers in Plant Science. 2016;7:p. 306. doi: 10.3389/fpls.2016.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu L. X., Wang Y. W., He P. Z., et al. Chloroplast genome analyses and genomic resource development for epilithic sister genera oresitrophe and mukdenia (saxifragaceae), using genome skimming data. BMC Genomics. 2018;19(1):p. 235. doi: 10.1186/s12864-018-4633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menezes A. P. A., Resende-Moreira L. C., Buzatti R. S. O., et al. Chloroplast genomes of byrsonima species (malpighiaceae): comparative analysis and screening of high divergence sequences. Scientific Reports. 2018;8(1):p. 2210. doi: 10.1038/s41598-018-20189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goulding S. E., Olmstead R. G., Morden C. W., Wolfe K. H. Ebb and flow of the chloroplast inverted repeat. Molecular and General Genetics MGG. 1996;252(1-2):195–206. doi: 10.1007/s004389670022. [DOI] [PubMed] [Google Scholar]
- 61.Yang M., Zhang X., Liu G., et al. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.) PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012762.e12762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang R.-J., Cheng C.-L., Chang C.-C., Wu C.-L., Su T.-M., Chaw S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biology. 2008;8(1):p. 36. doi: 10.1186/1471-2148-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kimura M. The neutral theory of molecular evolution and the world view of the neutralists. Genome. 1989;31(1):24–31. doi: 10.1139/g89-009. [DOI] [PubMed] [Google Scholar]
- 64.Hong S. Y., Cheon K. S., Yoo K. O., et al. Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Frontiers in Plant Science. 2017;8:p. 1696. doi: 10.3389/fpls.2017.01696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaney L., Mangelson R., Ramaraj T., Jellen E. N., Maughan P. J. The complete chloroplast genome sequences for four amaranthus species (amaranthaceae) Applications in Plant Sciences. 2016;4(9) doi: 10.3732/apps.1600063.1600063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravi V., Khurana J. P., Tyagi A. K., Khurana P. An update on chloroplast genomes. Plant Systematics and Evolution. 2008;271(1-2):101–122. doi: 10.1007/s00606-007-0608-0. [DOI] [Google Scholar]
- 67.Pauwels M., Vekemans X., Godé C., Frérot H., Castric V., Saumitou-Laprade P. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (brassicaceae) New Phytologist. 2012;193(4):916–928. doi: 10.1111/j.1469-8137.2011.04003.x. [DOI] [PubMed] [Google Scholar]
- 68.Powell W., Morgante M., McDevitt R., Vendramin G. G., Rafalski J. A. Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proceedings of the National Academy of Sciences. 1995;92(17):7759–7763. doi: 10.1073/pnas.92.17.7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li M. J., Liu J. Q., Guo X. L., Xiao Q. Y., He X. J. Taxonomic revision of Allium cyathophorum (amaryllidaceae) Phytotaxa. 2019;415(4):240–246. doi: 10.11646/phytotaxa.415.4.9. [DOI] [Google Scholar]
- 70.Jansen R. K., Cai Z., Raubeson L. A., et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences. 2007;104(49):19369–19374. doi: 10.1073/pnas.0709121104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huo Y., Gao L., Liu B., et al. Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Scientific Reports. 2019;9(1):1–14. doi: 10.1038/s41598-019-48708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hurst L. D. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genetics. 2002;18(9):p. 486. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- 73.Yang Z., Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution. 2000;17(1):32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 74.Makałowski W., Boguski M. S. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proceedings of the National Academy of Sciences. 1998;95(16):9407–9412. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drescher A., Ruf S., Calsa T., Jr., Carrer H., Bock R. The two largest chloroplast genome‐encoded open reading frames of higher plants are essential genes. The Plant Journal. 2000;22(2):97–104. doi: 10.1046/j.1365-313x.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- 76.Hu H., Al-Shehbaz I. A., Sun Y. S., Hao G. Q., Wang Q., Liu J. Q. Species delimitation in orychophragmus (brassicaceae) based on chloroplast and nuclear DNA barcodes. Taxon. 2015;64(4):714–726. doi: 10.12705/644.4. [DOI] [Google Scholar]
- 77.Blazier J. C., Ruhlman T. A., Weng M.-L., Rehman S. K., Sabir J. S., Jansen R. K. Divergence of RNA polymerase α subunits in angiosperm plastid genomes is mediated by genomic rearrangement. Scientific Reports. 2016;6(1) doi: 10.1038/srep24595.24595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hajdukiewicz P. T., Allison L. A., Maliga P. The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. The EMBO Journal. 1997;16(13):4041–4048. doi: 10.1093/emboj/16.13.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Streb P., Aubert S., Gout E., Bligny R. Cold‐and light‐induced changes of metabolite and antioxidant levels in two high mountain plant species Soldanella alpina and Ranunculus glacialis and a lowland species Pisum sativum. Physiologia Plantarum. 2003;118(1):96–104. doi: 10.1034/j.1399-3054.2003.00099.x. [DOI] [PubMed] [Google Scholar]
- 80.Ikeda H., Fujii N., Setoguchi H. Molecular evolution of phytochromes in Cardamine nipponica (brassicaceae) suggests the involvement of PHYE in local adaptation. Genetics. 2009;182(2):603–614. doi: 10.1534/genetics.109.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: all used accession numbers of cp genome sequences from GenBank in this article.
Table S2: the nucleotide variability (Pi) of 112 shared genes.
Table S3: the Ka/Ks ratio of 65 shared protein-coding genes in all six chloroplast genomes.
Figure S1: the proportion of the different lengths of the four dispersed repeats.
Figure S2: phylogenetic tree of the subgenus Cyathophora built by the CDS sequences using maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI).
Figure S3: phylogenetic tree of the subgenus Cyathophora built by the IGS sequences using maximum likelihood (ML), maximum parsimony (MP), and Bayesian inference (BI).
Data Availability Statement
The complete chloroplast genome sequences of A. cyathophorum, A. cyathophorum var. farreri, A. spicatum, A. mairei, Allium trifurcatum, and Allium kingdonii are saved in the GenBank of NCBI, and the accession numbers are MK820611, MK931245, MK931246, MK820615, MK931247, and MK294559, respectively.