

Abstract
Despite the ubiquity of reports describing titanium (Ti)-catalyzed [2 + 2 + 2] cyclotrimerization of alkynes, the incorporation of arynes into this potent manifold has never been reported. The in situ generation of arynes often requires fluoride, which instead will react with the highly fluorophilic Ti center, suppressing productive catalysis. Herein, we describe the use of group 4 diarylmetallocenes, CpR2MAr2 (CpR = C5H5, C5Me5; M = Ti, Zr), as aryne precursors for the Ti-catalyzed synthesis of substituted naphthalenes via coupling with 2 equiv of an alkyne. Fair-to-good yields of the desired naphthalene products could be obtained with 1% catalyst loadings, which is roughly an order of magnitude lower than similar reactions catalyzed by palladium or nickel. Additionally, naphthalenes find broad applications in the electronics, photovoltaics, and pharmaceutical industries, urging the discovery of more economic syntheses. These results indicate that aryne transfer from a CpR2M(η2-aryne) complex to another metal is a viable route for the introduction of aryne fragments into organometallic catalytic processes.
Graphical Abstract
INTRODUCTION
Titanium (Ti) is an attractive substitute to expensive noble metals owing to its natural abundance, low toxicity, and high activity in a number of synthetically relevant bond-forming reactions.1–3 Our laboratory has an ongoing interest in leveraging the reactivity of multiple valence states of Ti toward the modular assembly of complex small molecules.4–13 While TiII intermediates have been invoked in a multitude of catalytic and stoichiometric transformations, historically the chemistry of low-valent Ti species is weighted toward alkyne cycloaddition reactions such as alkyne cyclotrimerization.14 Alkyne cyclotrimerization is a powerful synthetic tool that allows the rapid construction of aromatic hydrocarbons through the formation of three bonds in one pot.
Naphthalenes and phenanthrenes have important applications in pharmaceuticals,15 photovoltaics,16 and the design of functional electronic materials.17 Moreover, substituted naphthalenes exhibit a myriad of biologically relevant activities including anticancer, antidepressant, antiinflammatory, antiviral, and antihypertensive activities.15 For example, Naproxen, marketed as Aleve, is a nonsteroidal antiinflammatory drug (NSAID) that is very commonly prescribed for the treatment of arthritis (Figure 1).
Figure 1.

Examples of naphthalene cores in drug molecules.
The synthesis of naphthalenes via formal [2 + 2 + 2] cycloaddition requires the engagement of key metallacyclic intermediates with an in situ generated reactive aryne substrate. Fast generation and capture of a stoichiometric aryne reagent is essential for selective trimerization. Modern techniques for the in situ generation of arynes have turned toward fluoride-based activators, which enjoy mild reaction conditions and easily accessible aryne precursors (Figure 2, top).18 These methods avoid the use of strong reductants such as magnesium(0) or organolithiums, synthetically elaborate aryne precursors such as those employed in the hexadehydro Diels–Alder (HDDA) reaction,19 or thermally activated substrates such as diazonium salts, which can pose serious operational hazards.20
Figure 2.

Modern methods of in situ aryne generation.
While catalytic synthesis of naphthalenes and phenanthrenes has been achieved using palladium (Pd),21–23 nickel (Ni),24 and rhodium25 (Figure 2, middle), analogous reactivity with Ti has never been reported. The prohibitive fluorophilicity of Ti [Ti–F bond dissociation energy (BDE) = 138 kcal/mol],26,27 which makes in situ aryne generation with fluoride incompatible with most potential Ti catalysts, likely contributes to this conspicuous absence. Considering the ubiquity of Ti-mediated trimerization reactions and the high catalytic activity reported therein, we were inspired to design aryne precursors that could be safely activated under conditions compatible with Ti.
The thermolysis and prolific ensuing insertion chemistry of group 4 diarylmetallocenes is well-established (Figure 3).28,29 This chemistry is proposed to proceed via a putative aryne–metallocene adduct, which is formed via β-abstraction under relatively mild conditions (∼80 °C). This aryne adduct can then be intercepted by a variety of unsaturated small molecules including alkynes, alkenes, nitriles, CO2, N2O, and N2.30–32
Figure 3.

Group 4 diarylmetallocenes undergo β-hydrogen abstraction, leading to aryne adducts that can be intercepted via insertion.
The aryne intermediate can be stabilized via the addition of L-type ligands33 or C–H activation of the cyclopentadienyl ligands.32,34 Given this rich chemistry, we hypothesized that this putative benzyne adduct could serve as an aryne coupling partner via transfer from the metallocene fragment to a catalytic metal center. Unlike other aryne generation methods, we speculated that this simple and safe thermal generation of arynes would be compatible with Ti-mediated catalysis.
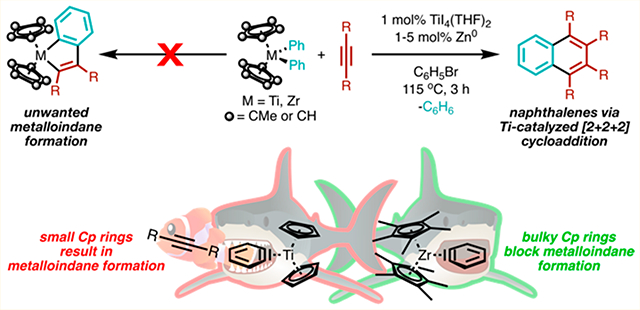
Herein, we report the investigation of group 4 diarylmetallocenes as aryne precursors for the catalytic synthesis of substituted naphthalenes (Figure 2, bottom). Good yields of naphthalene products could be obtained with catalyst loadings as low as 1%, which is an order of magnitude lower than similar reactions catalyzed by Pd21–23 or Ni.24 Overall, our experiments demonstrate that aryne transfer from a CpR2M(η2-aryne) complex to another metal is a viable route for the introduction of aryne fragments into organometallic catalytic processes.
RESULTS AND DISCUSSION
Model Reactions
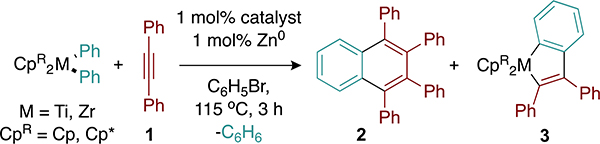
We began our investigation by choosing a suitable model reaction (Table 1). Diphenylacetylene was chosen as the alkyne coupling partner owing to the low proclivity toward self-cyclotrimerization and the high symmetry expected for the desired naphthalene products. Diphenyltitanocene was chosen as an initial benzyne precursor owing to its ease of synthesis. All reactions were carried out in the presence of stoichiometric or catalytic amounts of titanium tetrahalide catalyst precursors and zinc(0) (Zn0), along with equimolar amounts of diphenylacetylene and a metallocene benzyne precursor. Access to the active TiII catalytic species is proposed to occur via the initial reduction of titanium tetrahalide to TiIII by Zn0 followed by disproportionation to TiII and TiIV.35 The product yields were determined by quantitative gas chromatography flame ionization detection (GC-FID) with respect to an internal standard (trimethoxybenzene).
Table 1.
Screening of Reaction Conditionsa
 | ||||
|---|---|---|---|---|
| entry | CpR2Mb | catalyst | % yield 2c | % yield 3d |
| 1 | Cp2Ti | TiI4(THF)2e | >95 | 0 |
| 2 | Cp2Ti | TiI4(THF)2 | 0 | >95 |
| 3 | Cp*2Ti | TiI4(THF)2 | 0 | 0 |
| 4 | Cp*2Zr | TiI4(THF)2 | 71 | 0 |
| 5 | Cp*2Zr | TiI4(THF)2f | 55 | 0 |
| 6 | Cp*2Zr | TiBr4(THF)2 | trace | 0 |
| 7 | Cp*2Zr | TiCl4(THF)2 | trace | 0 |
| 8 | Cp*2Zr | TiI4(THF)2g | 0 | 0 |
| 9 | Cp*2Zr | none | 0 | 0 |
Reaction conditions: metallocene (0.05 mmol), diphenylacetylene (0.05 mmol), catalyst (0.05–0.0005 mmol of Ti), Zn0 (0.05–0.0005 mmol), 0.5 mL of C6H5Br, 115 °C, 3 h.
Cp = cyclopentadiene; Cp* = pentamethylcyclopentadiene.
Determined by quantitative GC-FID with respect to an internal standard (1,3,5-trimethoxybenzene).
Determined by 1H NMR with respect to an internal standard (1,3,5-trimethoxybenzene).
Reaction conducted with 100 mol % TiI4(THF)2.
Reaction conducted with 0.1 mmol of diphenylacetylene.
Reaction conducted in the absence of Zn0.
Under stoichiometric conditions, diphenyltitanocene served as a suitable benzyne precursor and generated 1,2,3,4-tetraphenylnaphthalene (2) in near-quantitative yields (Table 1, entry 1). However, under catalytic conditions, the quantitative formation of metalloindane species 3 was observed (Table 1, entry 2). This suggests that interception of the putative metallocene–benzyne adduct with diphenylacetylene is kinetically competitive to productive catalysis. Prolonged thermolysis of metalloindane 3 in the presence of additional diphenylacetylene does not afford naphthalene 2, demonstrating that 3 is likely an off-cycle thermodynamic sink. We considered that increasing the steric bulk of the Cp rings (Cp = cyclopentadiene) on the titanocene might suppress the formation of parasitic metalloindane species such as 3. However, the substitution of Cp2TiPh2 for Cp*2TiPh2 (Cp* = pentamethylcyclopentadiene) led to no product formation (Table 1, entry 3). Cp*2TiPh2 is known to favor Ti–C bond homolysis to form phenyl radical rather than β-abstraction to afford the desired benzyne adduct and benzene.36
Bearing in mind that the zirconium (Zr)–phenyl (Ph) bonds in Cp*2ZrPh2 (BDE ∼ 75 kcal/mol) have a higher BDE than analogous Ti–Ph bonds (BDE ∼ 67 kcal/mol),37 we were prompted to consider Cp*2ZrPh2 as a benzyne precursor in the hopes of promoting β-abstraction over extrusion of the phenyl radical. Indeed, using Cp*2ZrPh2 as the benzyne precursor, 2 was delivered in 71% yield with no detectable amount of 3 (Table 1, entry 4). Reactions using a 1:2 stoichiometry of Cp*2ZrPh2 and diphenylacetylene afforded 2 in slightly lower yields, likely owing to decomposition of the incipient Zr–benzyne complex (entry 5). Control reactions conducted in the absence of catalytic Ti or Zn (Table 1, entries 8 and 9) showed no formation of 2. Other TiX4 precatalysts (Table 1, entries 6 and 7) only afforded trace amounts of 2, demonstrating the importance of iodide ligands to catalysis. We have previously observed that TiI4-based precatalysts are significantly more productive for alkyne trimerization than TiCl4-based precatalysts,7 likely due to the decreased donor ability of iodide (I−) compared to chloride (Cl−).38 Reactions performed at lower temperatures (80 or 100 °C) afforded no naphthalene product; however, evolution of stoichiometric amounts of benzene signaled that β-hydrogen abstraction of Cp*2ZrPh2 did occur. Moreover, low-valent Ti is competent for trimerization under mild conditions.39 Thus, the high reaction temperature may be more readily associated with the barrier for the generation of an active TiII species. Notably, 2 is furnished in good yield at 1 mol % catalyst loading, while similar reactions employing Ni or Pd catalysts require 10 mol % catalyst loading to achieve comparable yields. This highlights the inherent advantage of utilizing Ti-based catalyst systems.
1H NMR spectra of catalytic reactions show decomposition of Cp*2ZrII to multiple ill-defined species. In order to ascertain whether any of these species are catalytically competent, the cycloaddition of tetraphenylnaphthalene was attempted using catalytic amounts of Cp*2ZrCl2/Zn. This system did not afford tractable amounts of naphthalene product. Similarly, attempts to catalyze the cyclotrimerization of 3-hexyne with Cp*2ZrCl2/Zn were also unsuccessful. These observations suggest that Cp*2ZrII-borne species are likely not involved in promoting cycloaddition catalysis.
Alkyne Scope
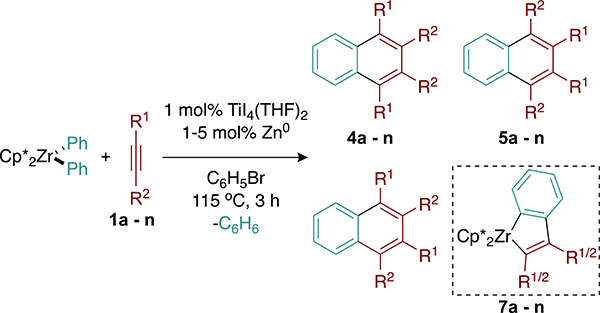
With optimized reaction conditions in hand, we set out to explore the scope of the alkyne coupling partner. We hypothesized that only alkynes with sufficient steric bulk could engage in productive catalysis by avoiding the formation of parasitic metalloindane species. A simple structure–activity relationship was developed in order to qualitatively correlate the alkyne steric size to reactivity. While several models for substituent size have been developed,40–45 A values proved sufficient. The A values for both alkyne substituents were summed to give a “composite A value” and plotted against the total yield of naphthalene products formed under catalytic conditions (Figure S20).
Alkynes with composite A values of below 4 delivered naphthalene products in zero or very poor yield (Table 2, alkynes 1a–1f). These reactions were dominated by formation of the corresponding metalloindanes 7a–7f. In the cases of 3-hexyne (Table 1, alkyne 1a) and 4-octyne (Table 1, alkyne 2a), alkylated benzenes formed via alkyne trimerization were observed in low yields (∼10%).
Table 2.
Alkyne Partner Substrate Scopea
 | |||||
|---|---|---|---|---|---|
| R1, R2 | % yield 4:5:6b | % yield 7c | % yield trimerd | A valuee | |
| 1a | Et, Et | 5 | 86 | 8 | 3.5 |
| 1b | nPr, nPr | 4 | 88 | 9 | 3.6 |
| 1c | TMS, H | 0 | >95 | 0 | 2.5 |
| 1d | Ph, H | 0 | >95 | 0 | 3.0 |
| 1e | iPr, Me | 0 | >95 | 0 | 3.9 |
| 1f | Ph, Me | 0 | >95 | 0 | 4.7 |
| 1g | Ph, nBu | 0:4:10 | 19 | 16 | 4.8 |
| 1h | Ph, Ph | 71 | 0 | 24 | 6.0 |
| 1i | p-CF3Ph,p-CF3Ph | 64 | 0 | 19 | 6.0 |
| 1j | p-OMe-Ph,p-OMe-Ph | 78 | 0 | 15 | 6.0 |
| 1k | p-tBuPh,p-tBuPh | 76 | 0 | 22 | 6.0 |
| 1l | Ph, TMS | 8:14:26 | 0 | 0 | 5.5 |
| 1m | nBu, TMS | 0:34:31 | 0 | 0 | 4.3 |
| 1n | Me, tBu | 0 | 0 | 0 | 6.6 |
Reaction conditions: metallocene (0.05 mmol), alkyne (0.05 mmol), catalyst (0.0005 mmol of Ti), Zn0 (0.0005 mmol), 0.5 mL of C6H5Br, 115 °C, 3 h. Cp* = pentamethylcyclopentadiene.
Determined by quantitative GC-FID with respect to an internal standard (1,3,5-trimethoxybenzene).
Determined by 1H NMR with respect to an internal standard (1,3,5-trimethoxybenzene).
% yield of all alkyne cyclotrimer regioisomers; determined by 1H NMR with respect to an internal standard (1,3,5-trimethoxybenzene).
Sum of both alkyne substituent A values.
Composite A values between 4 and 6 appear necessary but are not sufficient to furnish naphthalene products 4–6 in acceptable yields (Table 2, alkynes 1f–1m). The product selectivity for asymmetric alkynes appears entirely substratebiased in accordance with literature precedent.21–24 Notably, slight changes to the steric profile of the alkyne coupling partner can substantially affect the catalytic performance. While phenylpropyne affords the metalloindane complex 7f as the exclusive product (Table 2, alkyne 1f), phenylhexyne furnished the metalloindane complex 7g in a substantially lower yield and produced both naphthalene and trimer products in comparable yields (∼15%; Table 2, alkyne 1g). The difference in the A values between Me and nBu is only 0.1 kcal/mol, illustrating how even small steric perturbations can manifest in observable differences in the catalytic performance.
A substrate screen utilizing para-substituted diarylalkynes shows a slight yield dependence on the electronic character of the alkyne (Table 2, alkynes 1i–1k). More electron-rich alkyne coupling partners deliver naphthalene products in higher yields. Alkynes with composite A values exceeding 6 did not convert under the reaction conditions (Table 2, alkyne 1n) likely owing to the steric strain associated with the formation of metallacyclic intermediates. While limited in overall scope, the reactivity of the alkyne coupling partners appears to be more closely governed by the steric rather than electronic properties of the alkyne. Although the reaction yields delivered by productive alkynes are moderate, the relatively high turnover numbers are encouraging, and current work is directed toward developing more robust catalytic systems.
Only trace amounts of benzyne homocoupled products could be detected in the reactions with any alkynes. Thermolysis of Cp*2ZrPh2 in the absence of alkyne or catalyst also only affords trace yields of benzyne homocoupling products. This stands in contrast to other synthetically relevant metal–benzyne adducts of ruthenium,46 Pd,21–23 and Ni,24 where benzyne coupling products such as biphenylene or triphenylene are furnished in more substantial yields. Additionally, Ti species generated during catalysis do not appear to promote the formation of metalloindane byproducts. The propensity of alkynes to form metalloindanes is identical in the presence or absence of catalytic Ti.
Proposed Mechanism
Synthetic success with a fairly unusual benzyne precursor prompted us to consider the mechanistic aspects of the ensuing catalytic reaction. The most likely pathway for naphthalene synthesis is as follows (Figure 4): after generation of an active TiII species (I) by the Zn reductant, 2 equiv of alkyne undergo oxidative coupling to yield a titanacyclopentadiene (II). Next, the titanacyclopentadiene reacts with the in situ generated Zr–benzyne adduct, either via transmetalation of the benzyne to Ti or direct [4 + 2] cycloaddition of Zr–benzyne to the diene fragment. The resulting bound arene can either then dissociate to regenerate the reduced TiII species I or undergo associative displacement by alkyne to generate II. We favor II over titanaindane (Figure 4a) formation owing to the observed product and byproduct selectivity: catalysis results in the formation of only naphthalenes and a small amount of hexasubstituted benzene as a consequence of alkyne cyclotrimerization. In principle, a titanaindane intermediate (or a Ti–benzyne adduct) could result in the formation of phenanthrene or triphenylene byproducts, neither of which are observed.
Figure 4.

Plausible mechanism for the formation of naphthalene products from aryne/alkyne cyclization.
We were prompted to interrogate the role of Zn during catalysis because the ZnX2 byproduct of Ti reduction could facilitate either transmetalation or act as a Lewis acid—in either case possibly impacting how naphthalenes are formed. We previously reported the generation of TiII trimerization catalysts through the alkyne-triggered reduction of titanium imidos7 instead of Zn reduction of titanium halides. Conducting the coupling reaction with equimolar amounts of Cp*2ZrPh2 and PhCCPh in the presence of 10% py3TiI2(NPh) and 3-hexyne afforded naphthalene 2 in 60% yield (Figure 5). 3-Hexyne was chosen as a sacrificial alkyne owing to its high reactivity in the formation of the requisite pyrrole catalyst activation product. The formation of 2 in in the absence of Zn demonstrates that Zn is “innocent”—not likely participating as a transmetalation agent or Lewis acid during the catalytic synthesis of naphthalenes.
Figure 5.

Zn-free reactions yield significant amounts of 2, indicating that Zn is not critically important for catalysis.
Thus, benzyne incorporation into titanacyclopentadiene II likely occurs via direct engagement of the Zr–benzyne adduct with Ti, through either transmetalation (Figure 4c) or direct [4 + 2] cycloaddition with II (Figure 4d). Thermolysis of Cp*2ZrPh2 in the presence of furan and cyclohexadiene as benzyne traps yielded no products from free benzyne capture (Figure 4c). Instead, only the tuck-in complex V (Figure 6) was observed. Identical benzyne trap experiments were conducted with Cp2TiPh2, which cannot form a tuck-in complex, but still no products assignable to free benzyne capture were detected. Notably, the Zr tuck-in complex V does not exhibit evidence of interconversion to the benzyne adduct IV up to 90 °C;32 however, a small yet reactive population of the benzyne adduct may be responsible for direct transmetalation from Zr to Ti. How the bulky zironocene fragment may hinder formation of the requisite transition state(s) needed for transmetalation remains a lingering question.
Figure 6.

Free benzyne trap experiments led only to formation of the tuck-in complex V.
In order to determine whether the Cp*2Zr benzyne adduct IV is indeed a relevant species during catalysis, we examined substituted aryne coupling partner 8 as an isomerization probe to detect the intermediacy of an aryne complex (Figure 7). Metallocene aryne complexes that can be stabilized via C–H activation of the Cp* ring (as in V) are expected to “proton walk” via the successive protonation of an incipient aryne intermediate (Figure 7).34 By measurement of the product distribution, the aryne adduct isomer of Cp*2Zr(C6H3Me) can be implicated as an on-cycle species in catalysis if isomerization occurs. Indeed, after complex 8 was subjected to the catalytic reaction conditions, a ∼2:1 mixture of 6-methyl-1,2,3,4-tetraphenylnaphthalene (9) and 5-methyl-1,2,3,4-tetraphenylnaphthalene (10) was afforded in 64% yield. The formation of naphthalene 10 can only arise following isomerization via an aryne adduct and thus implicates the intermediacy of a complex such as IV during catalysis (Figure 6). Notably, thermolysis of 8 shows almost immediate scrambling of the tolyl group to an equimolar mixture of ortho-, meta-, and para-substituted analogues of V (Figure S21), demonstrating that isomerization is energetically facile and not dictated by Cp* ring sterics. This isomerization behavior is consistent with the observed reactivity of the analogous Cp2Zr(tolyl)2 complex.47
Figure 7.

Formation of 10 implicating isomerization through a Zr−aryne adduct.
Overall, the mechanistic data are most consistent with the direct engagement of a Zr–aryne adduct with a titanocyclopentadiene to afford the observed naphthalene products. Future work is focused on elucidating the fine details of this transmetalation event and how it may affect or effect product selectivity.
CONCLUSIONS
Cp*2ZrPh2 was employed as a benzyne precursor in a Ti-catalyzed formal [2 + 2 + 2] cycloaddition of arynes and alkynes to form highly substituted naphthalene products. Bulky Cp* rings were required to suppress the formation of parasitic metalloindane byproducts. A screen of potential alkyne coupling partners showed that formation of the desired naphthalene products is best correlated to alkyne steric rather than electronic properties. The product selectivity toward naphthalenes over other potential coupling products including triphenylenes or phenanthrenes suggests a mechanism in which a titanacyclopentadiene is intercepted by a Zr–benzyne adduct. An isomerization probe substrate, 8, was designed to interrogate this possibility. The product distribution arising from this substrate implicates isomerization through a Zr–aryne adduct during productive catalysis.
While the yields of naphthalene products are modest, the relatively low catalyst loading—nearly an order of magnitude lower than comparable systems with Ni or Pd—is encouraging. The high oxo- and fluorophilicity of Ti could be circumvented with an unusual aryne precursor, demonstrating a novel way to introduce aryne fragments into organometallic catalytic processes. Future work is directed toward expanding the scope of reactions that incorporate these metallocene-born arynes in the hopes of assembling more complex small molecules and understanding the mechanistic implications of aryne transmetalation in catalysis.
EXPERIMENTAL SECTION
General Considerations
All air- and moisture-sensitive compounds were manipulated in a glovebox under a N2 atmosphere. Benzene, pentane, diethyl ether (Et2O), tetrahydrofuran (THF), and toluene were sparged with ultrahigh-purity argon and dried via passage through drying columns using a solvent purification system from Pure Process Technologies. All solvents were stored over 4 Å molecular sieves. C6D5Br was dried over CaH2 and distilled before use. All high-boiling liquid reagents were freeze−pump−thawed three times, brought into the glovebox, diluted in hexanes or Et2O, passed through activated basic alumina, pumped dry in vacuo, and checked by 1H NMR in CDCl3 to ensure the dryness. 1,2-Bis(4-tert-butylphenyl)ethyne,48 bis(4-methoxyphenyl)ethyne,49 bis(4-trifluoromethylphenyl)ethyne,50 Cp2TiPh2,36 Cp*2TiPh2,36 Cp*2ZrPh2,32 and p-tolyllithium51 were prepared according to reported procedures. 1H and 13C NMR spectra were collected on a Bruker Avance III HD NanoBay 400 MHz spectrometer. Chemical shifts are reported with references of the residual protiosolvent impurity: 1H (s, 7.26 ppm for CHCl3), 13C (t, 77.16 ppm for CDCl3). GC-FID was performed using an Agilent 7890 series gas chromatograph system (HP-5 column, 30 m length, 0.32 mm inside diameter, 0.25 μm film thickness; temperature program = 50 °C for 1.5 min, 20 °C/min to 290 °C, and held for 6.5 min) equipped with a Polyarc reactor for quantitative C detection.
General Procedure for Catalytic Reactions
In an N2-filled glovebox, a vial was charged with Cp*2ZrPh2 (25.8 mg, 0.05 mmol, 1 equiv), alkyne (0.05 mmol, 1 equiv), TiI4(THF)2 (0.350 mg, 0.0005 mmol, 0.01 equiv), C6D5Br (0.5 mL), and trimethoxybenzene (8.4 mg, 0.05 mmol, 1 equiv) as an internal standard. The reaction mixture was transferred into an NMR tube charged with Zn (0.1−0.3 mg, 0.0015−0.0030 mmol, 0.03−0.05 equiv). The reaction was heated at 115 °C for 3 h in a preheated oil bath outside the glovebox. After being cooled to ambient temperature, the reaction was concentrated in vacuo, and the resulting residue was dissolved in ethyl acetate (EtOAc) and passed over a plug of silica. Reactions using tetramethylsilane−alkynes that afforded naphthalene products were concentrated in vacuo and mixed with 10 mL of 2 M HCl in methanol. After the mixture was stirred for 2 h, it was diluted by 20 mL of deionized water and extracted by EtOAc (3 × 15 mL). The organic layer was washed with brine and dried by MgSO4. All reactions were analyzed by 1H NMR in a CDCl3 solvent, and spectra containing naphthalene products were compared to literature examples. 24,52−58 NMR samples were then diluted with dichloromethane (CH2Cl2) and analyzed by GC-FID. Product yields were determined by quantitative GC-FID.
Synthesis of 8
A solution of p-tolyllithium (68 mg, 0.69 mmol, 3 equiv) in THF (2 mL) was added dropwise to a stirring solution of Cp*2ZrCl2 (100 mg, 0.23 mmol, 1 equiv) in Et2O (5 mL) at −30 °C. The reaction was allowed to warm to room temperature and stirred for 6 h, affording a hazy orange solution. CH2Cl2 (0.5 mL) was added dropwise to quench the remaining organolithium species. The reaction solvent was removed in vacuo, and the resulting residue was extracted into Et2O and filtered over Celite. The solution was concentrated to quarter volume and left to stand in the freezer overnight. The resulting white precipitate was collected and washed with hexanes (3 × 2 mL). 8 was isolated as a white microcrystalline powder (yield: 78 mg, 63%). 1H NMR (400 MHz, CDCl3): δ 7.11 (d, 2H, t, 3JH−H = 7.4 Hz, tolyl H), 6.97−6.91 (m, 6H), 2.26 (s, 6H, tolyl Me), 1.64 (s, 30H, Cp*-Me). 13C{1H} NMR (101 MHz, CDCl3): δ 189.1 (s, 2C, Zr C), 136.7 (s, 2C, tolyl Me), 134.11 (s, 2C, o-tolyl C), 133.0 (s, 2C, o-tolyl C), 126.9 (s, 1C, m-tolyl C), 126.6 (s, 1C, m-tolyl C), 120.3 (s, 2C, m-tolyl C), 21.3 (s, 2C, tolyl Me), 12.3 (s, 10C, Cp* Me). Anal. Calcd for C34H44Zr: C, 75.08; H, 8.15. Found: C, 75.38; H, 8.21.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the National Institutes of Health (Grant 1R35GM119457) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow). Equipment for the Chemistry Department NMR facility was supported through a grant from the National Institutes of Health (Grant S10OD011952) with matching funds from the University of Minnesota.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b01082.
Spectroscopic (NMR) and quantitative GC-FID data (PDF)
REFERENCES
- (1).Kaim W; Schwederski B Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life: An Introduction and Guide; Wiley: Chichester, U.K., 1994. [Google Scholar]
- (2).Beaumier EP; Pearce AJ; See XY; Tonks IA Modern Applications of Low-Valent Early Transition Metals in Synthesis and Catalysis. Nat. Rev. Chem 2019, 3, 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Egorova KS; Ananikov VP Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar]
- (4).Davis-Gilbert ZW; Tonks IA Titanium Redox Catalysis: Insights and Applications of an Earth-Abundant Base Metal. Dalt. Trans 2017, 46, 11522–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pearce AJ; See XY; Tonks IA Oxidative Nitrene Transfer from Azides to Alkynes via Ti(II)/Ti(IV) Redox Catalysis: Formal [2 + 2+1] Synthesis of Pyrroles. Chem. Commun 2018, 54, 6891–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2 + 2 + 1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nat. Chem 2016, 8, 63–68. [DOI] [PubMed] [Google Scholar]
- (7).See XY; Beaumier EP; Davis-Gilbert ZW; Dunn PL; Larsen JA; Pearce AJ; Wheeler TA; Tonks IA Generation of Ti II Alkyne Trimerization Catalysts in the Absence of Strong Metal Reductants. Organometallics 2017, 36, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2 + 2 + 1] Pyrrole Synthesis. Angew. Chem., Int. Ed 2018, 57, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Desnoyer AN; See XY; Tonks IA Diverse Reactivity of Diazatitanacyclohexenes: Coupling Reactions of 2 H -Azirines Mediated by Titanium(II). Organometallics 2018, 37, 4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Davis-Gilbert ZW; Yao LJ; Tonks IA Ti-Catalyzed Multicomponent Oxidative Carboamination of Alkynes with Alkenes and Diazenes. J. Am. Chem. Soc 2016, 138, 14570–14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2 + 2 + 1] Pyrrole Synthesis from Alkynes and Azobenzene. J. Am. Chem. Soc 2018, 140, 7267–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Davis-Gilbert ZW; Kawakita K; Blechschmidt DR; Tsurugi H; Mashima K; Tonks IA In Situ Catalyst Generation and Benchtop-Compatible Entry Points for TiII/TiIV Redox Catalytic Reactions. Organometallics 2018, 37, 4439–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chiu H-C; See XY; Tonks IA Dative Directing Group Effects in Ti-Catalyzed [2 + 2 + 1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catal. 2019, 9, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yamamoto K; Nagae H; Tsurugi H; Mashima K Mechanistic Understanding of Alkyne Cyclotrimerization on Mononuclear and Dinuclear Scaffolds: [4 + 2] Cycloaddition of the Third Alkyne onto Metallacyclopentadienes and Dimetallacyclopentadienes. Dalt. Trans 2016, 45, 17072–17081. [DOI] [PubMed] [Google Scholar]
- (15).Makar S; Saha T; Singh SK Naphthalene, a Versatile Platform in Medicinal Chemistry: Sky-High Perspective. Eur. J. Med. Chem 2019, 161, 252–276. [DOI] [PubMed] [Google Scholar]
- (16).Anthony JE Functionalized Acenes and Heteroacenes for Organic Electronics. Chem. Rev 2006, 106, 5028–5048. [DOI] [PubMed] [Google Scholar]
- (17).Al Kobaisi M; Bhosale SV; Latham K; Raynor AM; Bhosale SV Functional Naphthalene Diimides: Synthesis, Properties, and Applications. Chem. Rev 2016, 116, 11685–11796. [DOI] [PubMed] [Google Scholar]
- (18).Tadross PM; Stoltz BM A Comprehensive History of Arynes in Natural Product Total Synthesis. Chem. Rev 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]
- (19).Hoye TR; Baire B; Niu D; Willoughby PH; Woods BP The Hexadehydro-Diels−Alder Reaction. Nature 2012, 490, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sullivan JM Explosion during Preparation of Benzenediazonium-2-Carboxylate Hydrochloride. J. Chem. Educ 1971, 48, 419. [Google Scholar]
- (21).Huang W; Zhou X; Kanno K; Takahashi T Pd-Catalyzed Reactions of o -Diiodoarenes with Alkynes for Aromatic Ring Extension. Org. Lett 2004, 6, 2429–2431. [DOI] [PubMed] [Google Scholar]
- (22).Yoshikawa E; Radhakrishnan KV; Yamamoto Y Palladium-Catalyzed Controlled Carbopalladation of Benzyne. J. Am. Chem. Soc 2000, 122, 7280–7286. [Google Scholar]
- (23).Peña D; Pérez D; Guitián E; Castedo L Palladium- Catalyzed Cocyclization of Arynes with Alkynes: Selective Synthesis of Phenanthrenes and Naphthalenes. J. Am. Chem. Soc 1999, 121, 5827–5828. [Google Scholar]
- (24).Hsieh J-C; Cheng C -H. O-Dihaloarenes as Aryne Precursors for Nickel-Catalyzed [2 + 2 + 2] Cycloaddition with Alkynes and Nitriles. Chem. Commun 2008, No. 26, 2992. [DOI] [PubMed] [Google Scholar]
- (25).Fukutani T; Hirano K; Satoh T; Miura M Synthesis of Highly Substituted Naphthalene and Anthracene Derivatives by Rhodium-Catalyzed Oxidative Coupling of Arylboronic Acids with Alkynes. Org. Lett 2009, 11, 5198–5201. [DOI] [PubMed] [Google Scholar]
- (26).Kerr JA Bond Dissociation Energies by Kinetic Methods. Chem. Rev 1966, 66, 465–500. [Google Scholar]
- (27).Benson SW III - Bond Energies. J. Chem. Educ 1965, 42, 502. [Google Scholar]
- (28).Buchwald SL; Nielsen RB Group 4 Metal Complexes of Benzynes, Cycloalkynes, Acyclic Alkynes, and Alkenes. Chem. Rev 1988, 88, 1047–1058. [Google Scholar]
- (29).Majoral J-P; Meunier P; Igau A; Pirio N; Zablocka M; Skowronska A; Bredeau S Zirconocene [Cp2Zr] Synthon and Benzynezirconocene Complexes as Tools in Main Group Element Chemistry. Coord. Chem. Rev 1998, 178−180, 145–167. [Google Scholar]
- (30).Berkovich EG; Shur VB; Vol’pin ME; Lorenz B; Rummel S; Wahren M Die Reaktion von Stickstoff Mit Dehydrobenzol in Der Koordinationssphäre von Titan Chem. Ber. 1980, 113, 70–78. [Google Scholar]
- (31).Vaughan GA; Hillhouse GL; Rheingold AL Syntheses, Structures, and Reactivities of Unusual Four-Membered Metallacycles Formed in Insertion Reactions of N:N:O, N:N:NR, and N:N:CR2 with (.Eta.5-C5Me5)2Zr(C2Ph2). J. Am. Chem. Soc 1990, 112, 7994–8001. [Google Scholar]
- (32).Schock LE; Brock CP; Marks TJ Intramolecular Thermolytic Carbon-Hydrogen Activation Processes. Solid-State Structural Characterization of a Mononuclear. Eta.6-Me4C5CH2 Zirconium Complex and a Mechanistic Study of Its Formation from (Me5C5)2Zr(C6H5)2. Organometallics 1987, 6, 232–241. [Google Scholar]
- (33).Campora J; Buchwald SL Synthesis and Reactivity of a Titanocene-Benzyne Complex. Organometallics 1993, 12, 4182–4187. [Google Scholar]
- (34).Legrand C; Meunier P; Petersen JL; Tavares P; Bodiguel J; Gautheron B; Dousse G Unexpected Synthesis of Ortho-Substituted Diselenophenylenezirconocenes from the Para-Substituted Diphenylzirconocenes. Chemical and Structural Evidence of the Participation of a Cyclometalated Intermediate That Behaves as a Benzyne Zirconocene Equivalent. Organometallics 1995, 14, 162–169. [Google Scholar]
- (35).Kern RJ Tetrahydrofuran Complexes of Transition Metal Chlorides. J. Inorg. Nucl. Chem 1962, 24, 1105–1109. [Google Scholar]
- (36).Tung H; Brubaker CH Photochemical Decomposition of (Diphenyl)Bis(H5-Cyclopentadienyl) Titanium, (Diphenyl)Bis(H5-Pentamethylcyclopentadienyl) Titanium and the Zirconium Analogs. Inorg. Chim. Acta 1981, 52, 197–204. [Google Scholar]
- (37).Simoes JAM; Beauchamp JL Transition Metal-Hydrogen and Metal-Carbon Bond Strengths: The Keys to Catalysis. Chem. Rev 1990, 90, 629–688. [Google Scholar]
- (38).DiFranco SA; Maciulis NA; Staples RJ; Batrice RJ; Odom AL Evaluation of Donor and Steric Properties of Anionic Ligands on High Valent Transition Metals. Inorg. Chem 2012, 51, 1187–1200. [DOI] [PubMed] [Google Scholar]
- (39).Okamoto S; Yamada T; Tanabe Y; Sakai M Alkyne [2 + 2 + 2] Cyclotrimerization Catalyzed by a Low-Valent Titanium Reagent Derived from CpTiX 3 (X = Cl, O- i -Pr), Me 3 SiCl, and Mg or Zn. Organometallics 2018, 37, 4431–4438. [Google Scholar]
- (40).Taft RW Linear Free Energy Relationships from Rates of Esterification and Hydrolysis of Aliphatic and Ortho-Substituted Benzoate Esters. J. Am. Chem. Soc 1952, 74, 2729–2732. [Google Scholar]
- (41).Taft RW Polar and Steric Substituent Constants for Aliphatic and O-Benzoate Groups from Rates of Esterification and Hydrolysis of Esters 1. J. Am. Chem. Soc 1952, 74, 3120–3128. [Google Scholar]
- (42).Taft RW Linear Steric Energy Relationships. J. Am. Chem. Soc 1953, 75, 4538–4539. [Google Scholar]
- (43).Charton M Steric Effects. I. Esterification and Acid-Catalyzed Hydrolysis of Esters. J. Am. Chem. Soc 1975, 97, 1552–1556. [Google Scholar]
- (44).Charton M The Nature of the Electrical Effect of Alkyl Groups. 1. The Validity of the. Sigma.* Constants. J. Am. Chem. Soc 1977, 99, 5687–5688. [Google Scholar]
- (45).Clavier H; Nolan SP Percent Buried Volume for Phosphine and N-Heterocyclic Carbene Ligands: Steric Properties in Organometallic Chemistry. Chem. Commun 2010, 46, 841. [DOI] [PubMed] [Google Scholar]
- (46).Hartwig JF; Andersen RA; Bergman RG Synthesis of a Highly Reactive (Benzyne)Ruthenium Complex. Carbon-Carbon, Carbon-Hydrogen, Nitrogen-Hydrogen and Oxygen-Hydrogen Activation Reactions. J. Am. Chem. Soc 1989, 111, 2717–2719. [Google Scholar]
- (47).Erker G The Reaction of Intermediate Zirconocene-Aryne Complexes with C-H Bonds in the Thermolysis of Diarylzirconocenes. J. Organomet. Chem. 1977, 134, 189–202. [Google Scholar]
- (48).Herwig PT; Enkelmann V; Schmelz O; Müllen K Synthesis and Structural Characterization of Hexa-Tert-Butyl- Hexa-Peri-Hexabenzocoronene, Its Radical Cation Salt and Its Tricarbonylchromium Complex. Chem. - Eur. J 2000, 6, 1834–1839. [DOI] [PubMed] [Google Scholar]
- (49).Shintani R; Takagi C; Ito T; Naito M; Nozaki K Rhodium-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic Dibenzosiloles by Enantioselective [2 + 2+2] Cycloaddition. Angew. Chem., Int. Ed 2015, 54, 1616–1620. [DOI] [PubMed] [Google Scholar]
- (50).Wang C; Morimoto T; Kanashiro H; Tanimoto H; Nishiyama Y; Kakiuchi K; Artok L Rhodium(I)-Catalyzed Carbonylative Arylation of Alkynes with Arylboronic Acids Using Formaldehyde as a Carbonyl Source. Synlett 2014, 25, 1155–1159. [Google Scholar]
- (51).Bodach A; Hebestreit R; Bolte M; Fink L Syntheses and Crystal Structures of Phenyl-Lithium Derivatives. Inorg. Chem 2018, 57, 9079–9085. [DOI] [PubMed] [Google Scholar]
- (52).Honjo Y; Shibata Y; Kudo E; Namba T; Masutomi K; Tanaka K Room Temperature Decarboxylative and Oxidative [2 + 2+2] Annulation of Benzoic Acids with Alkynes Catalyzed by an Electron-Deficient Rhodium(III) Complex. Chem. - Eur. J 2018, 24, 317–321. [DOI] [PubMed] [Google Scholar]
- (53).Kabalka GW; Ju Y; Wu Z A New Titanium Tetrachloride Mediated Annulation of α-Aryl-Substituted Carbonyl Compounds with Alkynes: A Simple and Highly Efficient Method for the Regioselective Synthesis of Polysubstituted Naphthalene Derivatives. J. Org. Chem 2003, 68, 7915–7917. [DOI] [PubMed] [Google Scholar]
- (54).Hu F; Lei X A Nickel Precatalyst for Efficient Cross-Coupling Reactions of Aryl Tosylates with Arylboronic Acids: Vital Role of Dppf. Tetrahedron 2014, 70, 3854–3858. [Google Scholar]
- (55).Kong W-J; Finger LH; Oliveira JCA; Ackermann L Rhodaelectrocatalysis for Annulative C-H Activation: Polycyclic Aromatic Hydrocarbons through Versatile Double Electrocatalysis. Angew. Chem., Int. Ed 2019, 58, 6342–6346. [DOI] [PubMed] [Google Scholar]
- (56).Geng K; Fan Z; Zhang A Rhodium-Catalyzed Oxidative Coupling of N-Acyl Anilines with Alkynes Using an Acylamino Moiety as the Traceless Directing Group. Org. Chem. Front 2016, 3, 349–353. [Google Scholar]
- (57).Kitamura T; Yamane M; Inoue K; Todaka M; Fukatsu N; Meng Z; Fujiwara Y A New and Efficient Hypervalent Iodine-Benzyne Precursor, (Phenyl)[ o -(Trimethylsilyl)Phenyl]Iodonium Triflate: Generation, Trapping Reaction, and Nature of Benzyne. J. Am. Chem. Soc 1999, 121, 11674–11679. [Google Scholar]
- (58).Fukutani T; Hirano K; Satoh T; Miura M Synthesis of Highly Substituted Acenes through Rhodium-Catalyzed Oxidative Coupling of Arylboron Reagents with Alkynes. J. Org. Chem 2011, 76, 2867–2874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.