

Abstract
Pyrazoles are an important class of heterocycles found in a wide range of bioactive compounds and pharmaceuticals. Pyrazole synthesis often requires hydrazine or related reagents where an intact N–N bond is conservatively installed into a pyrazole precursor fragment. Herein, we report the multicomponent oxidative coupling of alkynes, nitriles, and Ti imido complexes for the synthesis of multisubstituted pyrazoles. This modular method avoids potentially hazardous reagents like hydrazine, instead forming the N–N bond in the final step via oxidation-induced coupling on Ti. The mechanism of this transformation has been studied in-depth through stoichiometric reactions of the key diazatitanacyclohexadiene intermediate, which can be accessed via multicomponent coupling of Ti imidos with nitriles and alkynes, ring opening of 2-imino-2H-azirines, or direct metalation of 4-azadiene-1-amine derivatives. The critical transformation in this reaction is the 2-electron oxidation-induced N–N coupling on Ti. This is a rare example of formal N–N coupling on a metal center, which likely occurs through an electrocyclic mechanism analogous to a Nazarov cyclization. Conveniently, these 2-electron-oxidized diazatitanacyclohexadiene intermediates can be accessed via disproportionation of the 1-electron-oxidized species, which allows utilization of weak oxidants such as TEMPO
Graphical Abstract
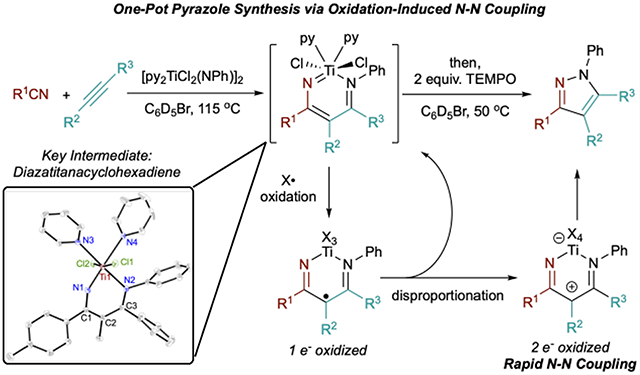
INTRODUCTION
Pyrazoles are an important class of N–N-bond-containing heterocycles found in flagship pharmaceuticals such as Eliquis, Viagra, Celebrex, and Acomplia.1–3 Multisubstituted pyrazoles are most commonly synthesized via the Knorr condensation of hydrazines onto 1,3-diketones4–6 or through a 1,3-dipolar cycloaddition of hydrazones with alkynes or electron-deficient alkenes after autoxidation.7–15 Direct N–N bond formation is an attractive alternative to these classical methods, as it would remove the need to use potentially toxic and/or explosive hydrazine reagents. However, N–N bond-forming methods are limited, particularly those proceeding through formal reductive elimination at a transition metal center. This is due to the unfavorable thermodynamics of N–N bond reductive elimination, which results in a weak N–N bond at the expense of two strong metal–N bonds. Despite this challenge, there are a few elegant examples of late transition metal facilitated N–N bond reductive eliminations: (1) Cu-catalyzed or -mediated synthesis of pyrazoles from nitriles and enamines (Figure 1a),16,17 as well as a few related transformations18–22 and (2) a stoichiometric example featuring a binuclear Ni complex which couples triazabicyclodecene ligands after oxidation with hypervalent iodine reagent PhICl2 (Figure 1b).23 In the pyrazole examples, the aromaticity gained provides driving force to overcome the otherwise weak N–N bond (approximately 24 kcal/mol for the parent pyrazole24). In the binuclear Ni example, generation of high-valent metal centers makes reductive elimination more favorable. Oxidatively induced reductive elimination is a common strategy employed in late-transition-metal organometallic chemistry to overcome otherwise challenging couplings.25–32 Moreover, increasing the oxidation state of the system as a whole has been shown to increase the electrophilicity of the ligands toward nucleophilic coupling.33 Other aerobically triggered reductive eliminations have been reported for the synthesis of hydrazines and azoarenes,34 and there is also an example of air oxidation of a d0 yttrium triamide complex that liberates aminyl radicals competent for N–N coupling.35
Figure 1.

(a) Cu-catalyzed oxidation-induced formal N–N reductive elimination for the synthesis of pyrazoles from electron-poor enamines and nitriles. (b) Ni-mediated N–N bond reductive elimination. (c) Formal [2+2+1] multicomponent synthesis of pyrazoles via oxidation-induced N–N coupling, and a proposed catalytic cycle for pyrazoles compared to the previously reported [2+2+1] synthesis of pyrroles.
Our group has produced several examples of early transition metal-catalyzed oxidative aminations of alkynes for the synthesis of pyrroles,36–40 which proceeds through an electrocyclic reductive elimination to form a C–N bond from an azatitanacyclohexadiene intermediate (Figure 1c, bottom right).41 We envisioned that reaction of an analogous diazatitanacyclohexadiene42‘43–formed from the reaction between a nitrile and the common azatitanacyclobutene intermediate44–54–could result in a formal N–N bond reductive elimination to produce multisubstituted pyrazoles, either through catalytic turnover with azobenzene as an oxidant or by further reaction with an exogenous oxidant (Figure 1c, bottom left). Herein, we report an example of oxidation-induced N–N bond reductive elimination, yielding pyrazoles from diazatitanacyclohexadienes (Figure 1c, top). These diazatitanacyclohexadienes can be generated from the coupling of an alkyne and a nitrile with a Ti imido complex, providing a practical and simple multicomponent [2+2+1] route to pyrazoles. Mechanistic studies indicate that N–N coupling is triggered by 2-electron oxidation of the diazatitana- cyclohexadiene, which generates an intermediate that can undergo electrocyclic ring closure in analogy to a Nazarov-type cyclization.
RESULTS AND DISCUSSION
Multicomponent Coupling for the Synthesis of Pyrazoles
Initial catalytic multicomponent coupling attempts of azobenzene, alkynes, and nitriles catalyzed by [py2TiCl2(NPh)]2, were met with limited success. For example, reaction of p-tolunitrile, 3-hexyne, and azobenzene for 20 h at 145 °C yields 16% of the desired 4,5-diethyl-N-phenyl-3-p-tolylpyrazole (l) in addition to 57% N-phenyl-3-hexanimine (2) and 9% 2,3,4,5-tetraethyl-N-phenylpyrrole (3) (Figure 2). Although the yield of 1 is lower than the catalyst loading, we were able to demonstrate catalytic turnover using a labeled azobenzene, ArNNAr (Ar = p-(CF3O)C6H4), which showed incorporation of both Ph (from the precatalyst) and Ar into the pyrazole product (Table S2).
Figure 2.

Multicomponent coupling of p-tolunitrile, 3-hexyne, and azobenzene catalyzed by py2TiCl2(NPh).
A potential mechanism for the multicomponent catalytic reaction was computed in analogy to our previous multicomponent synthesis of pyrroles (Figure 3). Computations indicate that the key intermediate diazatitanacyclohexadiene IM5-pyz is very stable (ΔG = −17.4 kcal/mol relative to py3TiCl2(NPh)) and the barrier to N–N reductive elimination (TS3-pyz 47.1 kcal/mol from IM5-pyz) is overwhelmingly high. In fact, 1H NMR monitoring of the reaction shows the formation of a new species consistent with the structure of IM5-pyz at 90% yield relative to catalyst within 10 min, suggesting the stability of IM5-pyz is inhibitive for productive catalysis. While the formation of 1a demonstrates that diazatitanacyclohexadiene IM5-pyz or another analogous species is capable of N–N coupling, we suspect that the high barrier to ring closure likely facilitates competitive acid/base chemistry to occur, ultimately leading to hydroamination. In fact, protonated diazatitanacycles crystallize from the reaction mixture (see Supporting Information). Thus, unlike previous examples of Ti-catalyzed nitrene transfer, PhNNPh may not be a strong enough oxidant to drive productive N–N bond-forming catalysis.
Figure 3.

Computed cycle for pyrazole formation via direct N–N bond coupling (M06/6–311G(d,p)/SMD, 145 °C, C6D5Br) compared to C–N bond formation in the analogous multicomponent pyrrole synthesis (M06/6–311G(d,p)/SMD, 115 °C, PhCF3) by Ti imido catalysts. Pyrrole computations and intermediate labels are adapted from ref 37. All free energies are referenced to py3TiCl2(NPh) = 0.0 kcal/mol.
Diazatitanacyclohexadiene Synthesis
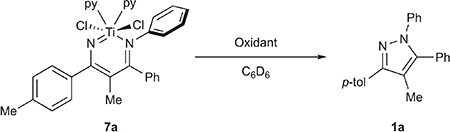
We next synthesized several diazatitanacyclohexadiene model complexes in order to study potential routes to productive N–N bond formation with other oxidants beyond PhNNPh (Figure 4). Protonolysis of TiCl2(NMe2)2 with a 4-azadiene-1-amine (H2ADA) (4a) gives (HNMe2)TiCl2(ADAPh) (5a) in 46% isolated yield. Reaction of H2ADA (4a, 4b) with TiCl4(THF)2 in THF followed by the addition of 2 equiv of LiHMDS (HMDS = hexamethyldisilazide) produces the dinuclear species [TiCl2(ADA)]2 (6a, 6b), which is bridged through the imido ligands. Treatment of 6a and 6b with pyridine generates py2TiCl2(ADA) (7a, 7b) in 30% crystallized yield. The X-ray crystal structures for 5a and 7a are shown in Figure 4. 5a, 7a, and 7b all have similar solid-state metrics and contain a titanium vinylimido with a pendent imine donor. For example, 7a has a Ti1–N1 distance of 1.768(1) Å, indicative of Ti–N imido multiple bonding, and a Ti1–N2 distance of 2.126(1) Å, indicative of a Ti–N imine dative bond. These bond lengths are consistent with the computed values for IM5-pyz (Figure 3, bottom), where Ti1–N1 and Ti–N2 were computed to be 1.71701 and 2.10562 Å, respectively. Further reactions were carried out with 7a and 7b because the HNMe2 ligand in 5a can potentially participate in deleterious acid/base reactions.
Figure 4.

Syntheses of diazatitanacyclohexadienes 5a, 7a, and 7b and ORTEP diagrams of 5a and 7a. Thermal ellipsoids are drawn at 50% probability and hydrogen atoms omitted for clarity. Full crystallographic data for 5a, 7a, and 7b is available in the Supporting Information.
Figure 5.

Reaction of 8a with a series of Ti complexes spanning TiII, TiIII, and TiIV oxidation states.
Figure 7.

Cyclic voltammogram of 9a (DCM solvent, 3.5 mM 9a, 0.1 M NBu4BPh4) at scan rates 1–200 mV/s (red-blue). Inset displays ipa/ipc as a function of scan rate.
Diazatitanacyclohexadiene Oxidation
Although 7a and 7b are both TiIV, we hypothesized that ligand-based oxidation could increase the electrophilicity of Nα and promote N–N coupling in analogy to late transition metal-based oxidations that induce reductive elimination. In fact, several oxidants promote facile, room temperature N–N coupling with 7a and 7b (Table 1). Reaction of 7a and 7b with 2 equiv of TEMPO resulted in 80% isolated yield of the N–N coupled pyrazole product 1a or 1b after 2 h (Table 1, entry 1), respectively. When the reaction is performed with 1 equiv of TEMPO, a 1:1 mixture of unreacted 7a and pyrazole 1a is observed, which is indicative of a net 2-electron stoichiometry requirement for the reaction (Table 1, entry 2). Likewise, hypervalent iodine reagents PhICl2, PhI(TFA)2, and PhI(OAc)2 all resulted in good yields of 1a or 1b (Table 1, entries 3–5). Interestingly, oxidation of 7a by Phl(OAc)2 (Table 1, entry 5) results in an initial mixture of 1a with a kinetic azirine product (8a), which slowly converts to 1a over time. Intermediacy of 8a could indicate potential free nitrene reactivity55–57 or transmetalation to I, but control experiments rule out these possibilities (Figures S90 and S96). The intermediacy of 8a was not evident in any other reaction. Other oxidants also engender productive N–N coupling, including FcPF6 (Fc = Cp2Fe+) and even O2, albeit in lower yields. Prolonged exposure of 7a to PhNNPh even at elevated temperatures (up to 145 °C) did not furnish tractable amounts of 1a, consistent with early catalytic attempts. Thermolysis of 7a in the absence of any oxidant also only led to trace amounts of 1a. N–N bond coupling was not observed when reacting with several common oxidants such as I2, NBS, NOBF4, and Me3NO, instead leading to intractable mixtures.
Table 1.
Oxidation of 7a with Various Oxidantsa
 | ||||
|---|---|---|---|---|
| entry | oxidant | time (h) | T (°C) | yield (%) |
| 1 | TEMPO (2 equiv) | 2 | 50 | 87(80b) |
| 2 | TEMPO (1 equiv) | 2 | 50 | 40 |
| 3c | PhICl2 | 3 | rt | 70 |
| 4c | Phl(O2CCF3)2 | 3 | rt | 76 |
| 5c | Phl(OAc)2 | 3 | rt | 44 |
| 6 | FcPF6 (2 equiv) | 16 | rt | 32 |
| 7 | FcPF6(2 equiv) | 70 | rt | 50 |
| 8 | O2(1 atm) | 3 | 60 | 17 |
Conditions: 7a (0.02 M), oxidant (1–2 equiv), 0.5 mL of benzene. Yields are determined in situ via 1H NMR against internal TMB standard.
Isolated yield.
Yields are determined via 1H NMR against internal TCE standard added after the reaction.
In Situ Multicomponent Pyrazole Synthesis
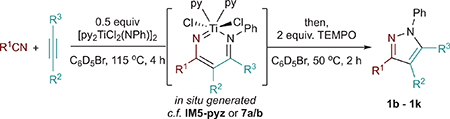
One-pot multicomponent synthesis via alkynes and nitriles is an attractive, unrealized, and simple route to pyrazoles. Given that diazatitanacycles have been observed as long-lived intermediates in our initial catalytic reaction attempts, it seemed plausible that they could be generated in situ by reaction of an alkyne and nitrile with a Ti imido and then oxidized to generate the desired pyrazole product in a one-pot procedure (Table 2). Indeed, diazatitanacycles analogous to 7a and 7b can be synthesized in 81% in situ yield (1H NMR) by reaction of [py2TiCl2(NPh)]2 with an excess of p-tolunitrile (6 equiv; 3 equiv with respect to [Ti]) and 3-hexyne (6 equiv) in C6D5Br after 1 h at 115 °C (Table 2, entry 1). C6D5Br solvent was chosen because it is an affordable-to-access polar NMR solvent, and was successful for our previous [2+2+1] pyrrole cyclizations.41 Addition of TEMPO after diazatitanacyclohexadiene formation, followed by heating at 50 °C for 2 h affords pyrazole 1 in 75% yield with respect to [py2TiCl2(NPh)]2 (98% yield with respect to IM5-pyz). Based off of this result, comparative reactions were next carried out at a 3:3:1 and 1:1:1 stoichiometry of benzonitrile, 3-hexyne, and Ti=NR (Table 2, entries 2 and 3). While the 1:1:1 stoichiometry results in a lower conversion to the metallacyclic intermediate (Figures S54 and S57), 1b is still generated in 56% overall yield with respect to alkyne (Table 2, entry 3). This reaction could be performed at 1 mmol scale, resulting in 65% yield of 1b (see Supporting Information). Inferring that the alkyne and nitrile fragments may often be the more precious components of this reaction, we next briefly explored the scope of this reaction at the 1:1:1 stoichiometry. In all cases, the desired pyrazole products were furnished in moderate to good yield. This method can tolerate both electron-rich and electron-poor nitriles (entries 3–5). p-(Trifluoromethyl)benzonitrile affects a slightly lower yield (49%) which is explained by detectable amounts of titanacycle decomposition (entry 5). Alkyl nitriles (entries 6–8) are also effective coupling partners in the reaction; however, the yield of diazatitanacycle oxidation is poor when coupling acetonitrile to 1-phenyl-1-propyne (entry 8). 1-Phenyl-1-propyne couples with both acetonitrile and benzonitrile, affording the 5-methyl-4-phenyl regioisomers selectively (entries 8 and 9); notably, 5-alkyl-3-arylpyrazoles are challenging to access via Knorr pyrazole synthesis due to the regioselectivity of the hydrazine condensation step. Tetraarylpyrazoles, which have shown the enhancement of aggregation-induced emission,58,59 are accessible in 37% in situ yield for pyrazole 1j (entry 11) or 42% isolated yield when diphenylacetylene (1k) is used (see Supporting Information). This scope demonstrates that multisubstituted pyrazoles can be accessed directly and generally in a single step through multicomponent coupling of Ti imidos, while avoiding prerequisite hydrazine or hydrazine-derived coupling partners. Conveniently, [py2TiCl2(NPh)]2 and derivatives thereof can be prepared in a single pot from TiCl4(THF)2, and demonstrated benchtop-compatible Ti imido generation.60 Thus, access to these multicomponent coupling reactions does not require specialized equipment or protocols.
Table 2.
Substrate Scope of One-Pot In Situ Pyrazole Synthesis from Alkynes and Nitrilesa
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | product | yield (%) |
| 1b | 4-Me-C6H4 | Et | Et | 1 | 75c |
| 2b | Ph | Et | Et | 1b | 79c |
| 3 | Ph | Et | Et | 1b | 56 |
| 4 | 4-MeO-C6H4 | Et | Et | 1c | 52 |
| 5 | 4-CF3-C6H4 | Et | Et | 1d | 49 |
| 6 | iPr | Et | Et | 1e | 54 |
| 7 | Me | Et | Et | 1f | 43 |
| 8 | Me | Ph | Me | 1g | 28 |
| 9 | Ph | Ph | Me | 1h | 54 |
| 10 | Ph | Me | Me | 1i | 52 |
| 11 | Ph | 4-t-BuC6H4 | 4-t-BuC6H4 | 1j | 37 |
Conditions: 0.025 mmol of [py2TiCl2(NPh)]2, 0.05 mmol (2 equiv) of alkyne, 0.05 mmol (2 equiv) of nitrile, 0.5 mL of PhBr, 115 °C, 4h; yields with respect to alkyne are determined in situ via 1H NMR against internal 1,3,5-trimethoxybenzene standard.
Conditions: 0.025 mmol of [py2TiCl2(NPh)]2, 0.15 mmol (6 equiv) of alkyne, 0.15 mmol (6 equiv) of nitrile, 0.5 mL of PhBr, 115 °C, 1 h.
Yields reported with respect to [py2TiCl2(NPh)]2, which is the limiting reagent under condition B.
Mechanistic Studies of N–N Coupling
N–N coupling on Ti could plausibly occur through one (or more) of three possible oxidation states–either the “default” oxidation state, 1-electron-oxidized, or 2-electron-oxidized (Figure 5, top). In order to determine the system oxidation state that triggers N–N coupling on Ti, we sought to conduct N–N coupling reactions with the entire redox series. Previous work in our group has shown that Cp2Ti(BTMSA) (BTMSA = bis(trimethylsilyl)acetylene)61–65 rapidly ring-opens 2H-azirines at the C–N bond.66 Thus, we hypothesized 2-imino-2H-azirine 8a–an isomer of pyrazole 1a–could react with a TiII source to give a diazatitanacyclohexadiene in the “default” oxidation state. By extension of this logic, reacting 2-imino-2H-azirines with TiIII and TiIV precursors could allow for interrogation of 1- and 2-electron-oxidized diazatitana- cyclohexadiene species, respectively.
The reaction of TiII, TiIII, and TiIV precursors with the 2-imino-2H-azirine 8a is shown in Figure 5. Reaction of 8a and Cp2Ti(BTMSA) rapidly gives the expected diazatitanacyclohexadiene Cp2Ti(ADAPh) (9a) in 89% yield (Figure 5, reaction II). Interestingly, XRD analysis of 9a (Figure 6) shows a Til–N1 bond distance of 1.857(l) Å and a Til–N2 distance of 2.12l(l) Å, indicating an iminyl-enamide bonding description for this complex, contrasting the vinylimidoimine resonance form in complexes 5a, 7a, and 7b (Figure 4). Consistent with earlier results with 7a and 7b, no N–N coupling occurs from 9a in this oxidation state.
Figure 6.

Oxidation of 9a to 10a by PhICl2 followed by thermal elimination of Cp2TiCl2 to give 1a, and ORTEP diagrams of 9a and 10a. Thermal ellipsoids are drawn at 50% probability, and hydrogen atoms are omitted for clarity. One crystallographically independent molecule of 10a and a solvent Et2O have also been omitted. Full crystallographic data is available in the Supporting Information, including CIF files.
Having shown that TiII reacts cleanly with 8a to give diazatitanacyclohexadiene 9a, we moved to investigate the in situ formation of a diazatitanacycle radical via radical ring opening of 8a by TiCl3(THF)3 (Figure 5, reaction III) A benzene solution of 8a reacts instantaneously with a benzene slurry of TiCl3(THF)3 yielding a 1:1 mixture of pyrazole 1a and presumably the “default” oxidation state diazatitanacycle, (THF)xTiCl2(ADAPh) (7a-0e−). Addition of 2 equiv of pyridine generates 7a with the gradual precipitation of a yellow solid presumed to be (py)2TiCl4. The formation of equal parts pyrazole 1a, 7a, and TiCl4 strongly suggests the initial formation of diazatitanacycle radical TiCl3(ADAPh·) (7a-1e–), followed by disproportionation into the “default” oxidation state and the 2-electron-oxidized species 7a-2e–, which then couples to form pyrazole.
Given the disproportionation reactivity with TiIII, we then tested the reactivity of 2-imino-2H-azirines with TiIV (Figure 5, reaction IV). Treatment of azirine 8a with TiCl4(THF)2 generates an unidentified intermediate species, which upon heating to 50 °C results in conversion to 1a as the major product. Alper demonstrated that TiCl4 will ring open 2H-azirines, in which γ-chlorination occurs after C–N bond cleavage.67 By analogy, 2-imino-2H-azirines likely undergo similar reactivity to form 7a-Cl-2e−, which can then undergo N–N coupling upon chloride loss. Consistent with this mechanism, there are examples of late transition metal-catalyzed 2-acyl-2H-azirine or 2-imino-2H-azirine rearrangements to isoxazoles or pyrazoles, respectively, that may proceed through similar vinyl-nitrenoid intermediates.68–71
Reactivity of Cp2Ti(ADAPh) (9a) with PhICl2
The 2H-azirine ring-opening redox series in Figure 5 strongly suggests that N–N coupling to form pyrazoles occurs through the 2-electron-oxidized diazatitanacycle of the form 7a-2e–. We next aimed to synthesize analogues of the 7a-2e– intermediate and observe their direct N–N coupling reactivity. We hoped that the stability imbued by the titanocene moiety in 9a would allow for isolation of an intermediate in the PhICl2 oxidations of diazatitanacyclohexadienes. Mixing a dark claret C6D6 solution of 9a with PhICl2 at room temperature immediately furnishes a bright yellow solution, yielding 10a quantitatively (Figure 6), which exists as two isomers. The solid-state structure of 10a (Figure 6) reveals chlorination of Ti and the Cγ position of the diazatitanacyclohexadiene ring. 10a is a “2-electron-oxidized” organic moiety, similar to the proposed azirine ring-opened product from TiCl4(THF)2 7a-Cl-2e– (Figure 5, reaction IV) and Alper’s earlier observations of γ-chlorination. However, steric encumbrance imposed by the Cp rings in 10a likely prevents imine coordination to Ti. Interestingly, 9a does not react with TEMPO, which may indicate that reaction of TEMPO with 7a initially requires coordination to Ti.
Refluxing 10a in C6D6 for 22 h gives pyrazole 1a and Cp2TiCl2 as the major products (Figure 6), consistent with 2-imino-2H-azirine ring-opening by TiCl4(THF)2. The slower rate of decomposition of 10a into pyrazole compared to the reaction of 8a and TiCl4 is likely due to the hindered approach of the imine during N–N coupling. Further, cyclic voltammetry of 9a features a quasi-reversible 1-electon oxidation at –0.55 mV vs Fc0/+ which exhibits fully reversible behavior at scan rates ≥200 mV/s (Figure 7). A linear dependence of ipa on ν1/2 was observed, consistent with a mass-transfer-limited process. This suggests that the quasi-reversibility of 9a oxidation at low scan rates arises from a chemical event coupled to oxidation, which, based on the 2-imino-2H-azirine reactivity above, is likely related to disproportionation of the 1-electron-oxidized Cp2Ti(ADAPh•) en route to 1a formation. Notably, no other current deflection is observed in the anodic sweep before oxidation of the supporting electrolyte at ~700 mV.
Discussion of the Mechanism of Oxidatively Induced N–N Coupling Mediated by Ti
We propose the mechanism for N–N coupling in Figure 8 based on the stoichiometric oxidations of 7a,b, the 2H-azirine 8a ring-opening reactivity with various Ti oxidation states, and the reactivity and electrochemistry of 9a. First, multicomponent coupling of a Ti imido with an alkyne and nitrile via the previously established [2+2]/insertion mechanism44,49 gives diazatitanacycle A, arbitrarily considered to be in the “default” oxidation state of this system. As demonstrated in the series of redox experiments above (Figure 5, reaction II), A can also be accessed via ring-opening of 2-imino-2H-azirines by a TiII precursor such as Cp2Ti(BTMSA). The barrier for N–N reductive elimination from A is too high, facilitating entry into competitive decomposition pathways. Single-electron oxidation of A leads to the 1-electron-oxidized diazatitanacycle B, which is unstable. From B, there are two possible reaction pathways: (1) disproportionation to A and 2-electron-oxidized C, which undergoes rapid N–N coupling to form 1 and a TiX4 coproduct, or (2) further oxidation to D, as seen in the oxidation of 9a to 10a by PhICl2. The reaction of 8a with TiCl3(THF)3, which results in 50% N–N coupled 1a and 50% of the “default” oxidation state 7a (Figure 5, reaction III), provides evidence for disproportionation. Further, weak oxidants such as TEMPO or FcPF6 result in product formation despite an inability to oxidize 7a by two electrons, indicating that a disproportionation mechanism is likely in play.
Figure 8.

Proposed mechanism for pyrazole formation from diazatitanacyclohexadienes.
Although D is also 2-electron-oxidized like C, C–X dissociation to C is likely necessary for rapid N–N coupling to occur. Evidence for this can be seen in several reactions. For example, reaction of TiCl4(THF)2 with 8a (Figure 5, reaction IV) results in full conversion to 1a, but the reaction is slower than that of TiCl3(THF)3 and requires heat to drive it to completion. Based on work from Alper, reaction IV likely generates an intermediate analogous to D. Thus, the major difference between the oxidized diazatitanacycles in reactions III and IV is the presence of an extra equivalent of Cl− in IV; indicating that dissociation of halide from D to make the carbocationic diazatitanacycle C is likely kinetically relevant. Further evidence for the role of X− dissociation from D may also be seen in the overall yields with hypervalent iodine oxidants presented in Table 1: reactions with anion equivalents that would be poor leaving groups (e.g., OAc−) generally result in lower yields than more weakly coordinating anion equivalents and/or better leaving groups (Cl−, O2CCF3−).
Based on the above evidence, the 2-electron-oxidized diazatitanacycle C is likely responsible for rapid N–N bond formation, which depending on oxidant strength can be accessed either through disproportionation of a 1-electron-oxidized species or directly through 2-electron oxidation. We have previously established in related [2+2+1] pyrrole syntheses that C–N bond reductive elimination from a 6-membered azatitanacycle proceeds through an electrocyclic mechanism. Here, oxidation of the diazatitanacycle should make the α-N significantly more electrophilic, prompting ringclosure in analogy to a Nazarov-like electrocyclization (Figure 9). The kinetic slowdown in the presence of coordinating anions is further evidence for an electrocyclic mechanism: intermediate C, with full π-conjugation, undergoes rapid reaction, whereas intermediate D lacks the conjugated π-system requisite for electrocyclization.
Figure 9.

Analogy between electrocyclic N–N coupling and the electrocyclic Nazarov cyclization of dienones.
CONCLUSION
In summary, we have shown that simple titanium imido complexes are capable of selective coupling with alkynes and nitriles to generate diazatitanacyclohexadienes, which can be oxidized under mild conditions to generate pyrazoles in good yield. By exploring an electrochemical series of 2-imino-2H-azirine ring openings with TiII, TiIII, and TiIV, we have demonstrated that Ti-mediated N–N coupling occurs from 2-electron-oxidized diazatitanacyclohexadienes via a potentially electrocyclic process. While oxidatively induced reductive elimination has been extensively explored, the concept of d0 transition metals mediating oxidatively induced couplings through ligand-centered oxidations may be a general strategy for accessing new reactivity and myriad new types of metal- mediated bond couplings, particularly via these types of electrocyclic pathways.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the results reported within this paper. A.J.P. would like to thank Victor Young Jr. for assistance with XRD analysis, as well as Evan P. Beaumier and Xin Yi See for enlightening discussions and generously sharing reagents.
Funding
Financial support was provided by the National Institutes of Health (1R35GM119457) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow). Instrumentation for the University of Minnesota Chemistry NMR facility was supported through a grant through the National Institutes of Health (S10OD011952). X-ray diffraction experiments were performed with a diffractometer purchased through a grant from NSF/MRI (1229400) and the University of Minnesota.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13173.
Full experimental and computational details, including Figures S1-S109 and Tables S1 and S2 (PDF) CIF files for 5a, 6a, 7a, 7b, 9a, 10a, and S1 (ZIP)
Contributor Information
Adam J. Pearce, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States
Robin P. Harkins, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States
Benjamin R. Reiner, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States
Alexander C. Wotal, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States
Rachel J. Dunscomb, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States
Ian A. Tonks, Department of Chemistry, University of Minnesota–Twin Cities, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Ansari A; Ali A; Asif M; Shamsuzzaman S Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar]
- (2).Karrouchi K; Radi S; Ramli Y; Taoufik J; Mabkhot Y; Al- Aizari F; Ansar M Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fustero S; Sánchez-Roselló M; Barrio P; Simón-Fuentes A From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [DOI] [PubMed] [Google Scholar]
- (4).Knorr L Einwirkung von Acetessigester Auf Phenylhydrazin. Ber. Dtsch. Chem. Ges. 1883, 16, 2597–2599. [Google Scholar]
- (5).Silva VLM; Silva AMS; Pinto DCGA; Cavaleiro JAS; Elguero J 3(5)-(2-Hydroxyphenyl)-5(3)-Styrylpyrazoles: Synthesis and Diels-Alder Transformations. Eur. J. Org. Chem. 2004, 2004, 4348–4356. [Google Scholar]
- (6).Heller ST; Natarajan SR 1,3-Diketones from Acid Chlorides and Ketones: A Rapid and General One-Pot Synthesis of Pyrazoles. Org. Lett. 2006, 8, 2675–2678. [DOI] [PubMed] [Google Scholar]
- (7).Snider BB; Conn RSE; Sealfon S Reactions of Phenylhydrazones with Electron-Deficient Alkenes. J. Org. Chem. 1979, 44, 218–221. [Google Scholar]
- (8).Gómez Guillén M; Conde Jiménez JL Reaction of D-Galactose Phenylhydrazone with Nitroalkenes: Synthesis of Pentahydroxypentylpyrazoles. Carbohydr. Res. 1988, 180, 1–17. [Google Scholar]
- (9).Gómez-Guillén M; Hans-Hans F; Simon JML; Martín-Zamora ME New Pentahydroxypentylpyrazoles from the Reactions of D-Mannose and d-Galactose Methylhydrazones with Nitroalkenes. Carbohydr. Res. 1989, 189, 349–358. [Google Scholar]
- (10).Arrieta A; Ramón Carrillo J; Cossío FP; Díaz-Ortiz A; JoséGómez-Escalonilla M; de la Hoz A; Langa F; Moreno A Efficient Tautomerization Hydrazone-Azomethine Imine under Microwave Irradiation. Synthesis of [4,3’] and [5,3’]Bipyrazoles. Tetrahedron 1998, 54, 13167–13180. [Google Scholar]
- (11).Deng X; Mani NS Reaction of N-Monosubstituted Hydrazones with Nitroolefins: A Novel Regioselective Pyrazole Synthesis. Org. Lett. 2006, 8, 3505–3508. [DOI] [PubMed] [Google Scholar]
- (12).Tang M; Wang Y; Wang H; Kong Y Aluminum Chloride Mediated Reactions of N-Alkylated Tosyl-hydrazones and Terminal Alkynes: A Regioselective Approach to 1,3,5-Trisubstituted Pyrazoles. Synthesis 2016, 48, 3065–3076. [Google Scholar]
- (13).Kong Y; Tang M; Wang Y Regioselective Synthesis of 1,3,5-Trisubstituted Pyrazoles from N -Alkylated Tosylhydrazones and Terminal Alkynes. Org. Lett. 2014, 16, 576–579. [DOI] [PubMed] [Google Scholar]
- (14).Ma C; Li Y; Wen P; Yan R; Ren Z; Huang G Copper(I)-Catalyzed Synthesis of Pyrazoles from Phenylhydrazones and Dialkyl Ethylenedicarboxylates in the Presence of Bases. Synlett 2011, 2011, 1321–1323. [Google Scholar]
- (15).Deng X; Mani NS Regioselective Synthesis of 1,3,5-Tri- and 1,3,4,5-Tetrasubstituted Pyrazoles from N -Arylhydrazones and Nitroolefins. J. Org. Chem. 2008, 73, 2412–2415. [DOI] [PubMed] [Google Scholar]
- (16).Neumann JJ; Suri M; Glorius F Efficient Synthesis of Pyrazoles: Oxidative C-C/N-N Bond-Formation Cascade. Angew. Chem., Int. Ed. 2010, 49, 7790–7794. [DOI] [PubMed] [Google Scholar]
- (17).Suri M; Jousseaume T; Neumann JJ; Glorius F An Efficient Copper-Catalyzed Formation of Highly Substituted Pyrazoles Using Molecular Oxygen as the Oxidant. Green Chem. 2012, 14, 2193. [Google Scholar]
- (18).Ueda S; Nagasawa H Facile Synthesis of 1,2,4-Triazoles via a Copper-Catalyzed Tandem Addition-Oxidative Cyclization. J. Am. Chem. Soc. 2009, 131, 15080–15081. [DOI] [PubMed] [Google Scholar]
- (19).Tang X; Huang L; Yang J; Xu Y; Wu W; Jiang H Practical Synthesis of Pyrazoles via a Copper-Catalyzed Relay Oxidation Strategy. Chem. Commun. 2014, 50, 14793–14796. [DOI] [PubMed] [Google Scholar]
- (20).Chen B; Zhu C; Tang Y; Ma S Copper-Mediated Pyrazole Synthesis from 2,3-Allenoates or 2-Alkynoates, Amines and Nitriles. Chem. Commun. 2014, 50, 7677. [DOI] [PubMed] [Google Scholar]
- (21).Dai G; Yang L; Zhou W Copper-Catalyzed Oxidative Dehydrogenative N-N Bond Formation for the Synthesis of N, N’-Diarylindazol-3-Ones. Org. Chem. Front 2017, 4, 229–231. [Google Scholar]
- (22).Ryan MC; Kim YJ; Gerken JB; Wang F; Aristov MM; Martinelli JR; Stahl SS Mechanistic Insights into Copper-Catalyzed Aerobic Oxidative Coupling of N–N Bonds. Chem. Sci. 2020, 11, 1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Diccianni JB; Hu C; Diao T N-N Bond Forming Reductive Elimination via a Mixed-Valent Nickel(n)-Nickel(IH) Intermediate. Angew. Chem., Int Ed. 2016, 55, 7534–7538. [DOI] [PubMed] [Google Scholar]
- (24).Cyrański MK Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [DOI] [PubMed] [Google Scholar]
- (25).Bour JR; Camasso NM; Sanford MS Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl-CF3 Coupling. J. Am. Chem. Soc. 2015, 137, 8034. [DOI] [PubMed] [Google Scholar]
- (26).Lau W; Huffman JC; Kochi JK Electrochemical Oxidation-Reduction of Organometallic Complexes. Effect of the Oxidation State on the Pathways for Reductive Elimination of Dialkyliron Complexes. Organometallics 1982, 1, 155–169. [Google Scholar]
- (27).Kim J; Shin K; Jin S; Kim D; Chang S Oxidatively Induced Reductive Elimination: Exploring the Scope and Catalyst Systems with Ir, Rh, and Ru Complexes. J. Am. Chem. Soc. 2019, 141, 4137–4146. [DOI] [PubMed] [Google Scholar]
- (28).Lanci MP; Remy MS; Kaminsky W; Mayer JM; Sanford MS Oxidatively Induced Reductive Elimination from (tBu2Bpy)Pd(Me)2: Palladium(IV) Intermediates in a One-Electron Oxidation Reaction. J. Am. Chem. Soc. 2009, 131, 15618–15620. [DOI] [PubMed] [Google Scholar]
- (29).Kraatz H-B; van der Boom ME; Ben-David Y; Milstein D Reaction of Aryl Iodides with (PCP)Pd(II)-Alkyl and Aryl Complexes: Mechanistic Aspects of Carbon-Carbon Bond Formation. Isr. J. Chem. 2001, 41, 163–172. [Google Scholar]
- (30).Watson MB; Rath NP; Mirica LM Oxidative C–C Bond Formation Reactivity of Organometallic Ni(II), Ni(III), and Ni(IV) Complexes. J. Am. Chem. Soc. 2017, 139, 35–38. [DOI] [PubMed] [Google Scholar]
- (31).Tsou TT; Kochi JK Mechanism of Biaryl Synthesis with Nickel Complexes. J. Am. Chem. Soc. 1979, 101, 7547–7560. [Google Scholar]
- (32).Tsou TT; Kochi JK Reductive Coupling of Organometals Induced by Oxidation. Detection of Metastable Paramagnetic Intermediates. J. Am. Chem. Soc. 1978, 100, 1634–1635. [Google Scholar]
- (33).Camasso NM; Sanford MS Design, Synthesis, and CarbonHeteroatom Coupling Reactions of Organometallic Nickel(IV) Complexes. Science 2015, 347, 1218–1220. [DOI] [PubMed] [Google Scholar]
- (34).Kinoshita K On the Mechanism of Oxidation by Cuprous Chloride, Pyridine and Air. I. The Properties of the Reaction. Bull. Chem. Soc. Jpn. 1959, 32, 777–780. [Google Scholar]
- (35).Zhang L; Xia J; Li Q; Li X; Wang S Fast Synthesis of Hydrazine and Azo Derivatives by Oxidation of Rare-Earth-Metal-Nitrogen Bonds. Organometallics 2011, 30, 375–378. [Google Scholar]
- (36).Pearce AJ; See XY; Tonks IA Oxidative Nitrene Transfer from Azides to Alkynes via Ti(II)/Ti(IV) Redox Catalysis: Formal [2+2+1] Synthesis of Pyrroles. Chem. Commun. 2018, 54, 6891–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nat. Chem 2016, 8, 63–68. [DOI] [PubMed] [Google Scholar]
- (38).Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem., Int. Ed. 2018, 57, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chiu H-C; See XY; Tonks IA Dative Directing Group Effects in Ti-Catalyzed [2+2+1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catal 2019, 9, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kawakita K; Beaumier EP; Kakiuchi Y; Tsurugi H; Tonks IA; Mashima K Bis(Imido)Vanadium(V)-Catalyzed [2+2+1] Coupling of Alkynes and Azobenzenes Giving Multisubstituted Pyrroles. J. Am. Chem. Soc. 2019, 141, 4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2+2+1] Pyrrole Synthesis from Alkynes and Azobenzene. J. Am. Chem. Soc. 2018, 140, 7267–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Doxsee KM; Farahi JB; Hope H Synthesis and Formal [4 + 2] Cycloaddition Reactions of Vinylimido Complexes of Titanocene. J. Am. Chem. Soc. 1991, 113, 8889–8898. [Google Scholar]
- (43).Doxsee KM; Farahi JB Reductive Coupling of Nitriles via Formal [2+2] Cycloadditions to the Titanium-Carbon Double Bond. J. Am. Chem. Soc. 1988, 110, 7239–7240. [Google Scholar]
- (44).Schofield AD; Nova A; Selby JD; Schwarz AD; Clot E; Mountford P Reaction Site Diversity in the Reactions of Titanium Hydrazides with Organic Nitriles, Isonitriles and Isocyanates: Ti = Nα Cycloaddition, Ti = Nα Insertion and Nα-Nβ Bond Cleavage. Chem. - Eur. J 2011, 17, 265–285. [DOI] [PubMed] [Google Scholar]
- (45).Manßen M; De Graaff S; Meyer MF; Schmidtmann M; Beckhaus R Direct Access to Titanocene Imides via Bis(η5:η1-Penta-Fulvene)Titanium Complexes and Primary Amines. Organometallics 2018, 37, 4506–4514. [Google Scholar]
- (46).Groom LR; Schwarz AD; Nova A; Clot E; Mountford P Synthesis and Reactions of a Cyclopentadienyl-Amidinate Titanium Tert-Butoxyimido Compound. Organometallics 2013, 32, 7520–7539. [Google Scholar]
- (47).Pugh SM; Trösch DJM; Wilson DJ; Bashall A; Cloke FGN; Gade LH; Hitchcock PB; McPartlin M; Nixon JF; Mountford P Cycloaddition Reactions of the Titanium Imide [Ti(NBut)(MeC(2-C5H4N)(CH2NSiMe3)2)(Py)] with ButCP and MeCN. Organometallics 2000, 19, 3205–3210. [Google Scholar]
- (48).Tiong PJ; Groom LR; Clot E; Mountford P Synthesis, Bonding and Reactivity of a Terminal Titanium Alkylidene Hydrazido Compound. Chem. - Eur. J 2013, 19, 4198–4216. [DOI] [PubMed] [Google Scholar]
- (49).McGrane PL; Jensen M; Livinghouse T Intramolecular [2+2] Cycloadditions of Group IV Metal-Imido Complexes. Applications to the Synthesis of Dihydropyrrole and Tetrahydropyridine Derivatives. J. Am. Chem. Soc. 1992, 114, 5459–5460. [Google Scholar]
- (50).Nguyen TT; Kortman GD; Hull KL Synthesis, Cycloaddition, and Cycloreversion Reactions of Mononuclear Titanocene-Oxo Complexes. Organometallics 2016, 35, 1713–1725. [Google Scholar]
- (51).Barnea E; Majumder S; Staples RJ; Odom AL One-Step Route to 2,3-Diaminopyrroles Using a Titanium-Catalyzed Four-Component Coupling. Organometallics 2009, 28, 3876–3881. [Google Scholar]
- (52).Cao C; Shi Y; Odom AL A Titanium-Catalyzed Three- Component Coupling To Generate α,β-Unsaturated β-Iminoamines. J. Am. Chem. Soc. 2003, 125, 2880–2881. [DOI] [PubMed] [Google Scholar]
- (53).Odom AL; McDaniel TJ Titanium-Catalyzed Multicomponent Couplings: Efficient One-Pot Syntheses of Nitrogen Heterocycles. Acc. Chem. Res. 2015, 48, 2822–2833. [DOI] [PubMed] [Google Scholar]
- (54).Basuli F; Aneetha H; Huffman JC; Mindiola DJ A Fluorobenzene Adduct ofTi(IV), and Catalytic Carboamination to Prepare α,β-Unsaturated Imines and Triaryl-Substituted Quinolines. J. Am. Chem. Soc. 2005, 127, 17992–17993. [DOI] [PubMed] [Google Scholar]
- (55).Padwa A; Smolanoff J; Tremper A Photochemical Transformations of Small Ring Heterocyclic Systems. LXV. Intramolecular Cycloaddition Reactions of Vinyl-Substituted 2H-Azirines. J. Am. Chem. Soc. 1975, 97, 4682–4691. [Google Scholar]
- (56).Nishiwaki T; Fujiyama F Studies on Heterocyclic Chemistry. Part XIII. Cleavage of 5-Benzyl-Amino-Oxazoles, Photoproducts of N-Benzyl-2H-Azirine-2-Carboxamides, by Dialkyl Phosphite. J. Chem. Soc., Perkin Trans. 1 1972, 1456. [Google Scholar]
- (57).Nishiwaki T; Nakano A; Matsuoka H Studies on Heterocyclic Chemistry. Part VII. Thermally Induced Dimerization of 5-Aminoisoxazoles and 2H-Azirines and Photochemistry of 5-Aminoisoxazoles. J. Chem. Soc. C 1970, 1825. [Google Scholar]
- (58).Mukherjee S; Salini PS; Srinivasan A; Peruncheralathan S AIEE Phenomenon: Tetraaryl vs. Triaryl Pyrazoles. Chem. Commun. 2015, 51, 17148–17151. [DOI] [PubMed] [Google Scholar]
- (59).Mukundam V; Kumar A; Dhanunjayarao K; Ravi A; Peruncheralathan S; Venkatasubbaiah K Tetraaryl Pyrazole Polymers: Versatile Synthesis, Aggregation Induced Emission Enhancement and Detection of Explosives. Polym. Chem. 2015, 6, 7764–7770. [Google Scholar]
- (60).Davis-Gilbert ZW; Kawakita K; Blechschmidt DR; Tsurugi H; Mashima K; Tonks IA In Situ Catalyst Generation and Benchtop-Compatible Entry Points for TiII/TiIV Redox Catalytic Reactions. Organometallics 2018, 37, 4439–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Rosenthal U; Pellny PM; Kirchbauer FG; Burlakov VV What Do Titano- and Zirconocenes Do with Diynes and Polyynes? Ace. Chem. Res 2000, 33, 119–129. [DOI] [PubMed] [Google Scholar]
- (62).Rosenthal U; Burlakov VV; Arndt P; Baumann W; Spannenberg A The Titanocene Complex of Bis(Trimethylsilyl)-Acetylene: Synthesis, Structure, and Chemistry. OrganometaUics 2003, 22, 884–900. [Google Scholar]
- (63).Burlakov VV; Polyakov AV; Yanovsky AI; Struchkov YT; Shur VB; Vol’pin ME; Rosenthal U; Gorls H Novel Acetylene Complexes ofTitanocene and Permethyltitanocene without Additional Ligands. Synthesis Spectral Characteristics and X-Ray Diffraction Study. J. Organomet. Chem. 1994, 476, 197–206. [Google Scholar]
- (64).Peulecke N; Lefeber C; Ohff A; Baumann W; Tillack A; Kempe R; Burlakov VV; Rosenthal U Ansa-Titanocene and -Zirconocene η2-Alkyne Complexes -Synthesis, Spectral Characteristics, and X-Ray Crystal Structure. Chem. Ber. 1996, 129, 959–962. [Google Scholar]
- (65).Ohff A; Pulst S; Lefeber C; Peulecke N; Arndt P; Burkalov VV; Rosenthal U Unusual Reactions of Titanocene- and Zirconocene-Generating Complexes. Synlett 1996, 1996, 111–118. [Google Scholar]
- (66).Desnoyer AN; See XY; Tonks IA Diverse Reactivity of Diazatitanacyclohexenes: Coupling Reactions of 2H-Azirines Mediated by Titanium(ll). OrgsanometaUics 2018, 37, 4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Alper H; Prickett JE; Wollowitz S Intermolecular and Intramolecular Cycloaddition Reactions of Azirines by Group 6 Metal Carbonyls and by Titanium Tetrachloride. J. Am. Chem. Soc. 1977, 99, 4330–4333. [Google Scholar]
- (68).Li X; Du Y; Liang Z; Li X; Pan Y; Zhao K Simple Conversion of Enamines to 2H-Azirines and Their Rearrangements under Thermal Conditions. Org. Lett. 2009, 11, 2643–2646. [DOI] [PubMed] [Google Scholar]
- (69).Pinho e Melo TMVD; Lopes CSJ; Rocha Gonsalves AM; Storr RC Reactivity of 2-Halo-2H-Azirines. Part II. Thermal Ring Expansion Reactions: Synthesis of 4-Haloisoxazoles. Synthesis 2002, 2002, 605–608. [Google Scholar]
- (70).Padwa A; Stengel T Transition Metal Catalyzed Ring Opening Reactions of 2-Phenyl-3-Vinyl Substituted 2H-Azirines. Tetrahedron Lett. 2004, 45, 5991–5993. [Google Scholar]
- (71).Alper H; Sakakibara T An Interesting Azirine Induced Reaction of the Cyclopentadienyliron Dicarbonyl Dimer. Can. J. Chem. 1979, 57, 1541–1543. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.