

Abstract
BACKGROUND: The INTERCEPT Blood System, a photochemical treatment (PCT) process, has been developed to inactivate pathogens in platelet concen‐trates. These studies evaluated the efficacy of PCT to inactivate pathogens in plasma and the effect of PCT on plasma function.
STUDY DESIGN AND METHODS: Jumbo (600 mL) plasma units were inoculated with high titers of test pathogens and treated with 150 µmol per L amotosalen and 3 J per cm2 long‐wavelength ultraviolet light. The viability of each pathogen before and after treatment was measured with biological assays. Plasma function was evaluated through measurement of coagulation factors and antithrombotic protein activities.
RESULTS: The levels of inactivation expressed as log‐reduction were as follows: cell‐free human immunodeficiency virus‐1 (HIV‐1), greater than 6.8; cell‐associated HIV‐1, greater than 6.4; human T‐lymphotropic virus‐I (HTLV‐I), 4.5; HTLV‐II, greater than 5.7; hepatitis B virus (HBV) and hepatitis C virus, greater than 4.5; duck HBV, 4.4 to 4.5; bovine viral diarrhea virus, 6.0; severe acute respiratory syndrome coronavirus, 5.5; West Nile virus, 6.8; bluetongue virus, 5.1; human adenovirus 5, 6.8; Klebsiella pneumoniae, greater than 7.4; Staphylococcus epidermidis and Yersinia enterocolitica, greater than 7.3; Treponema pallidum, greater than 5.9; Borrelia burgdorferi, greater than 10.6; Plasmodium falciparum, 6.9; Trypanosoma cruzi, greater than 5.0; and Babesia microti, greater than 5.3. Retention of coagulation factor activity after PCT was expressed as the proportion of pretreatment (baseline) activity. Retention was 72 to 73 percent of baseline fibrinogen and Factor (F)VIII activity and 78 to 98 percent for FII, FV, FVII, F IX, FX, FXI, FXIII, protein C, protein S, antithrombin, and α2‐antiplasmin.
CONCLUSION: PCT of plasma inactivated high levels of a wide range of pathogens while maintaining adequate coagulation function. PCT has the potential to reduce the risk of transfusion‐transmitted diseases in patients requiring plasma transfusion support.
ABBREVIATIONS:
- APTT
activated partial thromboplastin time
- BVDV
bovine viral diarrhea virus
- CAD
compound adsorption device
- DHBV
duck hepatitis B virus
- FACT
factor assay control plasma
- PC
protein C
- PCT
photochemical treatment
- PS
protein S
- PT
prothrombin time
- SARS‐CoV
severe acute respiratory syndrome coronavirus
- UVA
long‐wavelength ultraviolet (light)
- VWF:RCo
von Willebrand factor:ristocetin cofactor
- WNV
West Nile virus.
Approximately 3.3 million units of fresh‐frozen plasma (FFP) and 816,000 units of cryoprecipitate are transfused annually in the United States. 1 FFP is indicated for treatment of congenital and acquired coagulation factor deficiencies, coagulopathy resulting from liver disease, massive blood loss, and thrombotic thrombocytopenia purpura. In addition, FFP may be used to prepare cryoprecipitate for fibrinogen replacement and treatment of von Willebrand’s disease. Typical therapeutic use of FFP for correction of coagulopathy requires transfusion of approximately 10 to 20 mL per kg FFP per transfusion episode, necessitating exposure to multiple donors. 2 Plasma exchange therapy for thrombotic thrombocytopenia purpura patients may require repeated large volume FFP transfusions with even greater donor exposures.
Donor screening and postdonation testing have greatly reduced the risk of transfusion‐transmitted diseases in patients requiring transfusion of blood products. Residual risk persists, however, because only a limited number of pathogens are routinely screened, 3 new blood‐borne organisms continue to emerge, 4 and in many cases, the diagnostic tests available are insufficiently sensitive to detect low‐level contaminants and infections in the “window period.” 5 , 6 Even though quarantine plasma could potentially eliminate the window period by repeated donor testing within 4 to 6 months, the safety of this approach still depends on test sensitivity. Furthermore, testing remains a reactive approach to blood safety. The contaminating organisms must be identified before screening tests can be developed.
In contrast, pathogen inactivation is a proactive approach to blood safety. Since the 1990s, the transfusion community has become more receptive to this new approach provided that the method is efficacious, safe, easy to implement, and cost‐effective. For plasma, two inactivation technologies, methylene blue treatment 7 , 8 and solvent/detergent (S/D) treatment, 9 have received regulatory approval and are currently in clinical use in several countries in Europe. Neither of these methods, however, has a broad range of effectiveness against pathogens, because S/D treatment only inactivates lipid‐enveloped viruses, 10 and methylene blue is ineffective against intracellular viruses. 11 Furthermore, S/D treatment has been shown to compromise some of the in vitro coagulation function of plasma and is contraindicated by the FDA for patients undergoing liver transplant. 12
The INTERCEPT Blood System for platelets (PLTs) is the only pathogen inactivation technology for blood cellular components that is CE Marked and in clinical use in several countries in Europe. 13 The INTERCEPT Blood System is a photochemical treatment (PCT) process that utilizes amotosalen (also known as S‐59) and long‐wavelength ultraviolet(UVA; 320‐400 nm) light to permanently cross‐link helical regions of DNA and RNA. 14 PCT has been shown to inactivate a broad range of viruses, 15 , 16 , 17 bacteria, 18 and protozoa, 19 , 20 as well as white blood cells (WBCs) 21 in PLT concentrates. Because plasma has similar optical properties to PLT concentrate, it is expected that the PCT process developed for use with PLTs is applicable to plasma; thus supporting the synergy of use of the same PCT technology for two blood components in a blood center. These studies evaluated the efficacy of PCT of plasma for the preparation of pathogen inactivated fresh‐frozen plasma (FFP).
MATERIALS AND METHODS
Plasma collection
Apheresis plasma (approx. 600 ± 25 mL) was collected from volunteer donors by plasmapheresis on an automated plasmapheresis machine (Haemonetics PCS 2, Haemonetics Corp., Braintree, MA; or Autopheresis‐C, Baxter Healthcare Corp., Deerfield, IL) with sodium citrate anticoagulant. For the pathogen inactivation studies, apheresis FFP units were stored frozen (not greater than −18°C) before use. For the plasma function studies, apheresis plasma units were transported at room temperature and processed within 8 hours of collection.
PCT disposable sets and UVA illumination device
The PCT disposable set for treatment of plasma (Baxter Healthcare Corp.) consisted of the following sequentially integrated components: 15 mL of 6 mmol per L amotosalen hydrochloride solution in saline packaged inside a PL 2411 plastic container and protected from UVA light; a 1.3‐L PL 2410 plastic container for illumination of plasma; a compound adsorption device (CAD) to reduce the concentration of amotosalen and its photoproducts, which consisted of an adsorbent disk composed of a copolymer of polystyrene and divinylbenzene particles fused with an ultrahigh‐molecular‐weight polyethylene plastic enclosed in an acrylic housing; and three 400‐mL PL 269 plastic containers for storage of the treated plasma.
Plasma was treated by passing through each component in a series of steps. In Step 1, the plasma unit was sterile connected to the amotosalen container and the plasma content passed through the amotosalen container into the illumination container. In Step 2, the plasma containing amotosalen was illuminated with UVA light. In Step 3, the illuminated plasma mixture was passed through the CAD by gravity into the storage containers (Step 4; Fig. 1). The residual amotosalen remaining in plasma after the CAD step is 1.5 µmol per L or less on average, which is consistently achieved.
Figure 1.
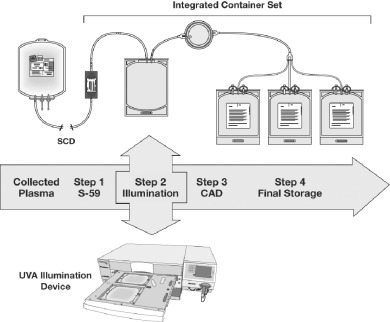
The PCT system for plasma. The PCT system consists of a UVA illumination device and an integral disposable set. The device can illuminate 2 units of plasma per processing cycle. The disposable set provides a single‐use, closed, integrated system for pathogen inactivation treatment of a plasma unit. The integrated disposable set is composed of the following sterile components: an amotosalen (S‐59) container, a plastic illumination container, a CAD, and three plastic storage containers. The processing steps are as described under Materials and Methods section. SCD = sterile connection device.
Illumination of plasma was performed in a UV illumination system (Baxter Model R4R4007, Nova Biomedical, Waltham, MA). The device was capable of illuminating 2 units of plasma per processing cycle. The illuminator delivered a 3 J per cm2 UVA treatment to each plasma unit in approximately 7 to 9 minutes. During illumination plasma units were reciprocally agitated at approximately 70 cycles per minute.
Inactivation procedures and viability assays for pathogens
Before addition of amotosalen, plasma units of approximately 585 mL (unless otherwise specified) were inoculated with the applicable pathogen to a final concentration of approximately 106 infectious organisms per mL whenever possible. In all cases, the inoculum volume consisted of not greater than 10 percent of the final plasma volume. Table 1 summarizes the strain and the supplier of each pathogen evaluated in these studies.
Table 1.
The origin of the test pathogens and established biological methods used to detect and quantify viability
| Organism | Strain | Origin (supplier) | Method of detection |
|---|---|---|---|
| HIV‐1, cell‐free | IIIB | Chronically infected H9 cells (gift from C.V. Hanson) | Microplaque in MT‐2 cells 42 |
| HIV‐1, cell‐associated | IIIB | Chronically infected H9 cells (gift from C.V. Hanson) | Micro‐plaque in MT‐2 cells 42 |
| HTLV‐I | 2060 | California Department of Health Services, Richmond, CA | β‐Galactosidase production by infected pA18GBHK‐21 cells 43 |
| HTLV‐II | C‐19 | California Department of Health Services, Richmond, CA | β‐Galactosidase production by infected pA18GBHK‐21 cells 43 |
| HBV | MS‐2 | NIH repository, Bethesda, MD | Infectivity in chimpanzee 44 |
| HCV | Hutchinson | NIH repository, Bethesda, MD | Infectivity in chimpanzee 45 |
| DHBV | P‐type | Congenitally infected ducks, Hepadnavirus Testing Laboratories, Menlo Park, CA | Infectivity in Legarth‐Pekin ducklings 46 |
| BVDV | NADL | ATCC, Rockville, MD | Plaque in bovine turbinate cells 16 |
| WNV | 3356 | Clone lineage I, pFL‐WNV (gift from K. Bernard) | Plaque in Vero cells 47 |
| SARS‐CoV | Urbani | CDC, Atlanta, GA | Plaque in Vero‐E6 cells 15 |
| Bluetongue virus | Station | ATCC, Rockville, MD | Plaque in embryonic bovine trachea cells 16 |
| Human adenovirus | 5 | Onyx Pharmaceuticals, Inc., Richmond, CA | Plaque on lung carcinoma cells (A549) 16 |
| S. epidermidis | Septicemia patient | California Department of Health Services, Richmond, CA | Colony formation on Luria Bertani agar 18 |
| K. pneumoniae | Septicemia patient | California Department of Health Services, Richmond, CA | Colony formation on Luria Bertani agar 18 |
| Y. enterocolitica | Septicemia patient | California Department of Health Services, Richmond, CA | Colony formation on Luria Bertani agar 18 |
| T. pallidum | Nichols | University of Washington, Seattle, WA | Infectivity in New Zealand white rabbits 18 |
| B. burgdorferi | CA4 | University of California, Berkeley, CA | Dark field microscopy of cultures in BSK‐H medium 18 |
| P. falciparum | FcB1 | Max‐Planck‐Institute (gift from H. Heidrich) | Infectivity in RBCs 48 |
| T. cruzi | Tulahuen | University of Washington, Seattle, WA | Infectivity in 3T3 cells 19 |
| B. microti | C3H/HcN adapted | Wild strain isolated from a field mouse at State University of NY, Stony Brook, NY | Infectivity in C3H/HcN mice 49 |
Inoculated plasma units were then treated with 150 µmol per L amotosalen and a 3 J per cm2 UVA treatment. Samples taken after addition of organism, but before addition of amotosalen, were used to determine the pre‐PCT input titer. These samples were serially diluted in assay medium or phosphate‐buffered saline before viability measurement. The post‐PCT samples were taken immediately after UVA illumination. The CAD step was not performed in the pathogen inactivation studies so that measurement of log reduction was a result of illumination of plasma treated with amotosalen only and not affected by potential pathogen affinity for the CAD. The post‐PCT samples were assayed for viable organisms undiluted when possible. In some cases, dilution up to 1:10 was required to prevent toxicity of plasma in the culture system. Table 1 also summarizes the biological assays used for quantifying the number of viable organisms. Detailed assay procedures are as described in the respective references. Pre‐ and post‐PCT titers were quantified with standard assays. The Reed‐Muench method was used where end‐dilutions were required for quantification. 22 Four replicate experiments (unless otherwise specified) for each organism were performed with four independent units (or pools) of plasma.
The level of inactivation was calculated as log‐reduction with the formula
| Log‐reduction = Log(pre‐PCT titer/post‐PCT titer). |
The pre‐PCT and post‐PCT titers were expressed in scientific notations. The mean level of inactivation and standard deviation (SD) were determined.
PCT of fresh plasma for plasma function studies
The total target volume in the illumination container was 600 ± 25 mL, composed of 585 ± 25 mL of plasma and 15 mL of amotosalen solution. The nominal amotosalen concentration was approximately 150 µmol per L. Each unit of plasma containing amotosalen was illuminated with a 3 J per cm2 UVA treatment on the R4R4007 illumination device. Plasma samples for evaluation of coagulation factor activity were taken before the addition of amotosalen (baseline, pre‐PCT) and after PCT including CAD treatment (post‐PCT). Samples were snap‐frozen and stored at or below −65°C before testing.
Measurement of in vitro coagulation function
Soluble fibrinogen (Factor [F]I) was measured with a modified Clauss assay in which the clotting time of a diluted plasma sample, after conversion by thrombin into insoluble fibrin, is compared to a standard curve prepared with reference plasma of known fibrinogen concentration. Coagulation factors were assayed with one‐stage prothrombin time (PT)‐based clotting assays (FII, FV, FVII, FX) or one‐stage activated partial thromboplastin time (APTT)‐based clotting assays (FVIII, F IX, FXI). The clotting time of a mixture of diluted test plasma sample and plasma deficient in the factor being quantified was compared with a reference curve constructed from the clotting times of five dilutions, ranging from 1:5 to 1:320, of plasma with known activity mixed with deficient plasma. These coagulation tests, as well as the PT and APTT, were performed on an automated coagulation analyzer (MLA Electra 1400C or 1600C, Instrumentation Laboratory Co., Lexington, MA). Reagents included brain thromboplastin (Hemoliance, Instrumentation Laboratory Co.), Platelin L (bioMérieux, Durham, NC), and congenital factor–deficient substrate. The endpoint of all tests was the formation of a clot detected photooptically and measured in seconds. The level of the factor being measured was inversely proportional to the time it takes for a clot to form. Factor assay control plasma (FACT; George King Biomedical, Inc., Overland Park, KS) was used as the reference standard for the coagulation factor assays.
FXIII was measured with the research‐use‐only FXIII kit (Berichrom, Dade Behring, Marburg, Germany). FXIII, activated by thrombin, releases an activation product that leads to a series of reactions resulting in a decrease in nicotinamide adenine dinucleotide (NADH), detected by monitoring absorbance at 340 nm. The assay was performed on a clot timer (BCT, Dade Behring), and standard human plasma (Dade Behring) was used as the reference standard.
The von Willebrand factor:ristocetin cofactor (VWF:RCo) activity was measured with the BC von Willebrand reagent (Dade Behring). In the assay, lyophilized PLTs are agglutinated by the VWF in the presence of ristocetin, resulting in a decrease in turbidity measured by the change in absorbance on the Behring clot timer. FACT was used as the reference standard.
Protein C (PC) and protein S (PS) were measured with kits (Staclot PC kit and the Staclot PS kit, respectively, both from Diagnostica Stago, Asnieres, France). PC and PS assays were based on the prolongation of the APTT resulting from inactivation of FV and FVIII by activated PC. The activator in the PC assay is an extract of Agkistrodon c. contortrix snake venom; the activator in the PS assay is activated PC. The tests were performed on the Behring clot timer. A unicalibrator (Diagnostica Stago) a multi‐parametric calibrator, was used as the reference standard.
Antithrombin was measured with a kit (Stachrom ATIII, Diagnostica Stago). Plasma containing antithrombin was incubated with a known excess of thrombin. A chromogenic substrate, imidolyzed by the remaining thrombin, was detected photooptically on the MLA Electra 1400C or 1600C coagulation analyzer. FACT was used as the reference standard.
α2‐Antiplasmin was quantified with reagents from Diagnostica Stago. In this chromogenic method, plasmin was added in excess to the test plasma, resulting in the formation of antiplasmin‐plasmin complexes. The concentration of residual plasmin is measured by its amidolytic activity on a chromogenic substrate measured at 405 nm. α2‐Antiplasmin concentration is inversely proportional to the residual plasmin concentration and is determined by color intensity. This analysis was performed by Esoterix Laboratories (Aurora, CO) with an analyzer (STA, Diagnostica Stago).
The mean and SD were determined for each coagulation parameter quantified. The activity of each coagulation parameter remaining after PCT was expressed as percent retention of the pretreatment (baseline) activity. Comparison of the PT and APTT was based on the prolongation of the clotting time after PCT relative to baseline.
RESULTS
PCT inactivation of viruses in plasma
PCT with 150 µmol per L amotosalen and a 3 J per cm2 UVA treatment inactivated a wide variety of viruses, including enveloped, nonenveloped, DNA, and RNA viruses in plasma (Table 2). Initial viral titers (pre‐PCT) of 104 to 106 infectious viruses per mL were achieved for all viruses except for hepatitis B virus (HBV) and hepatitis C virus (HCV). The highest available titers were used in all cases, whenever possible, to achieve the maximum dynamic range of infectivity.
Table 2.
PCT inactivates enveloped and nonenveloped viruses in plasma at high initial titers *
| Viruses | Pre‐PCT titer (n = 4) | Post‐PCT titer (n = 4) | Log‐reduction† |
|---|---|---|---|
| Enveloped | |||
| HIV‐1, cell‐free | 106.1±0.1 PFU/mL | <10−0.8±0.02 PFU/mL | >6.8 ± 0.1 |
| HIV‐1, cell‐associated | 105.9±0.2 PFU/mL | <10−0.5±0 PFU/mL | >6.4 ± 0.2 |
| HTLV‐I | 104.0±0.2 FFU/mL | ≤10−0.5±0.8 FFU/mL | ≥4.5 ± 0.7 |
| HTLV‐II | 104.7±0.1 FFU/mL | <10−1.0±0 FFU/mL | >5.7 ± 0.1 |
| HBV‡, § | 104.5 CID50/unit | None detected in 250 mL | >4.5 |
| HCV‡, § | 104.5 CID50/unit | None detected in 250 mL | >4.5 |
| DHBV | 105.6±0.2 ID50/mL | 101.2‐101.3 ID50/mL | 4.4‐4.5 |
| BVDV | 104.5±0.03 PFU/mL | <10−1.5±0 PFU/mL | ≥6.0 ± 0.03 |
| WNV | 106.7±0.3 PFU/mL | ≤10−0.1±0.2 PFU/mL | ≥6.8 ± 0.5 |
| SARS‐CoV | 104.0±0.1 PFU/mL | ≤10−1.5±0 PFU/mL | ≥5.5 ± 0.1 |
| Nonenveloped | |||
| Human adenovirus 5 | 105.5±0.5 PFU/mL | ≤10−1.3±0.3 PFU/mL | ≥6.8 ± 0.4 |
| Bluetongue virus | 104.0±0.2 PFU/mL | 10−1.0±0.3 PFU/mL | 5.1 ± 0.2 |
Results are reported as mean ± SD. n = number of replicates done for each virus; PFU/mL = plaque‐forming units per milliliter; FFU/mL = foci‐forming units per milliliter; ID50 = infectious dose necessary for infection of 50 percent of inoculated ducks; CID50 = infectious dose necessary for infection of 50 percent of inoculated chimpanzees.
Log‐reduction was calculated as the logarithm of the ratio of the pre‐PCT titer to the post‐PCT titer.
n = 3.
Inactivation was done in 250 mL instead of the 600 mL as specified under Materials and Methods.
PCT inactivated a mean of greater than 6.8 logs and greater than 6.4 logs of cell‐free and cell‐associated human immunodeficiency virus‐type 1 (HIV‐1), respectively. No biologically active HIV‐1 were detected in any of the test samples for four replicate experiments (Table 2). Similarly, human T‐cell lymphotropic virus‐I (HTLV‐I) and HTLV‐II were sensitive to PCT and mean log‐reductions of at least 4.5 and greater than 5.7, respectively, were obtained.
Inactivation of HBV and HCV by PCT was measured in nonimmune chimpanzees transfused with the entire unit of amotosalen‐ and UVA‐treated plasma that was previously inoculated with 104.5 CID50 of HBV or HCV (Table 2). Inactivation of each virus was assayed in two chimpanzees. None of the animals in either the HBV or the HCV study showed evidence of viral hepatitis during a 6‐month follow‐up with serologic, viral nucleic acid, biochemical, or histologic examination of liver biopsies, demonstrating an inactivation of greater than 4.5 logs for both viruses. Further evidence of PCT inactivation of hepadna virus and flaviviridae was demonstrated with, respectively, the model systems duck hepatitis B virus (DHBV) and bovine viral diarrhea virus (BVDV; Table 2). PCT inactivated 4.4 to 4.5 logs of DHBV and at least 6.0 logs of BVDV.
In plasma, PCT inactivated high levels of recently emerging viruses. Mean log‐reductions of at least 6.8 and at least 5.5 were achieved for West Nile virus (WNV) and severe acute respiratory syndrome coronavirus (SARS‐CoV), respectively (Table 2). PCT inactivated a mean of 5.1 logs of nonenveloped Bluetongue virus and at least 6.8 logs of nonenveloped human adenovirus 5 (Table 2).
PCT inactivation of bacteria in plasma
Gram‐positive Staphylococcus epidermidis and gram‐negative Klebsiella pneumoniae and Yersinia enterocoliticus were sensitive to PCT. High levels of inactivation were achieved in plasma (Table 3). Initial pre‐PCT bacterial levels of 106.6 to 106.7 colony‐forming units (CFU) per mL were achieved with all bacterial species. PCT with 150 µmol per L amotosalen and a 3 J per cm2 UVA treatment resulted in complete inactivation with no viable bacteria remaining in any of the test plasma samples in all four replicates. Mean log‐reductions achieved were greater than 7.3 for S. epidermidis and Y. enterocoliticus and greater than 7.4 for K. pneumoniae.
Table 3.
PCT inactivates gram‐positive and gram‐negative bacteria and spirochetes in plasma at high initial titers *
| Bacteria | Pre‐PCT titer (n = 4) | Post‐PCT titer (n = 4) | Log‐reduction† |
|---|---|---|---|
| Gram‐positive | |||
| S. epidermidis | 106.6±0.02 CFU/mL | <10−0.7±0 CFU/mL | >7.3 ± 0.02 |
| Gram‐negative | |||
| K. pneumoniae | 106.7±0.1 CFU/mL | <10−0.7±0 CFU/mL | >7.4 ± 0.1 |
| Y. enterocolitica | 106.6±0.01 CFU/mL | <10−0.7±0 CFU/mL | >7.3 ± 0.1 |
| Spirochetes | |||
| T. pallidum | 105.4±0.6 ID50/mL | <10−0.5±0 ID50/mL | >5.9 ± 0.6 |
| B. burgdorferi | ≥109.9±2.3 ID50/mL | <10−0.7±0 ID50/mL | >10.6 ± 2.3 |
Results are reported as mean ± SD. n = number of replicates done for each bacterium; ID50/mL = infectious dose necessary for infection of 50 percent of inoculated cells or animal hosts;
Log‐reduction was calculated as the logarithm of the ratio of the pre‐PCT titer to the post‐PCT titer.
Spirochetes Treponema pallidum, which causes syphilis, and Borrelia burgdorferi, which causes Lyme disease, were also sensitive to PCT. No viable organisms were detected after PCT in the test plasma samples in all four replicates, demonstrating mean log reductions of >5.9 and >10.6, respectively.
PCT inactivation of protozoa in plasma
PCT inactivation studies of Plasmodium falciparum, the protozoan that causes malaria, Trypanosoma cruzi, the pathogen that causes Chagas’ disease, and Babesia microti, an emerging protozoan causing babesiosis, exhibited mean log reductions of at least 6.9 for P. falciparum, greater than 5.0 for T. cruzi, and greater than 5.3 for B. microti (Table 4). In four replicate experiments, 150 µmol per L amotosalen and a 3 J per cm2 UVA treatment resulted in no detectable T. cruzi and B. microti in the test plasma samples.
Table 4.
PCT inactivates protozoa in plasma at high initial titers *
| Protozoa | Pre‐PCT titer (n = 4) | Post‐PCT titer (n = 4) | Log‐reduction† |
|---|---|---|---|
| P. falciparum | 105.9±0 iRBCs/mL | ≤10−1.0±0.3 iRBCs/mL | ≥6.9 ± 0.3 |
| T. cruzi ‡ | 106.3±0.6 TCID50/mL | <101.3±0 TCID50/mL | >5.0 ± 0.6 |
| B. microti | 104.9±0.4 ID50/mL | <100.5±0.02 ID50/mL | >5.3 ± 0.4 |
Results are reported as mean ± SD. n = number of replicates done for each protozoa; iRBCs/mL = infected red blood cells per milliliter; TCID50/mL = infectious dose necessary for infection of 50 percent of inoculated cells; ID50/mL = infectious dose necessary for infection of 50 percent of inoculated mice.
Log‐reduction was calculated as the logarithm of the ratio of the pre‐PCT titer to the post‐PCT titer.
Inactivation was done in 250 mL of whole‐blood derived plasma instead of the 600 mL apheresis plasma as specified under Materials and Methods.
In vitro coagulation function of plasma after PCT
Evaluation of a comprehensive panel of coagulation parameters showed that PCT conserved coagulation function within levels suitable for transfusion support. 23 On average, the PT and APPT were prolonged after PCT by 1.0 and 4.3 seconds, respectively (Table 5). Fibrinogen, FVII, and FVIII were retained, on average, 72 to 78 percent of the initial pre‐PCT activity. All other coagulation parameters retained at least 82 percent of baseline activity, with FV, FXIII, and VWF:RCo retaining at least 92 percent. Antithrombotic proteins (PC, PS, antithrombin) demonstrated a high level of activity retention (Table 6), ranging between 95 and 98 percent of the initial pre‐PCT level. α2‐Antiplasmin was retained at 80 percent of baseline activity. Coagulation factor and antithrombotic protein activities fell within the reported reference ranges for conventional plasma. 24
Table 5.
Maintenance of clotting time and plasma coagulation factor activity after PCT *
| Coagulation parameter | Reference range† | Pre‐PCT | Post‐PCT | Post/pre (% retention) |
|---|---|---|---|---|
| PT (n = 14) | 11.1‐13.5 | 11.2 ± 0.3 sec | 11.6 ± 0.3 sec | 1.0 ± 0.1 sec‡ |
| APTT (n = 14) | 23.0‐35.0 | 26.8 ± 1.4 sec | 29.1 ± 1.7 sec | 4.3 ± 1.8 sec‡ |
| Fibrinogen (n = 91) | 167‐379 | 290 ± 40 mg/dL | 209 ± 36 mg/dL | 72 ± 5 |
| FII (n = 59) | 71‐127 | 96 ± 11 IU/dL | 85 ± 11 IU/dL | 88 ± 4 |
| FV (n = 91) | 77‐153 | 130 ± 23 IU/dL | 119 ± 19 IU/dL | 92 ± 7 |
| FVII (n = 91) | 58‐166 | 123 ± 32 IU/dL | 95 ± 20 IU/dL | 78 ± 6 |
| FVIII (n = 91) | 67‐235 | 157 ± 35 IU/dL | 115 ± 28 IU/dL | 73 ± 7 |
| F IX (n = 91) | 63‐143 | 108 ± 21 IU/dL | 88 ± 16 IU/dL | 82 ± 4 |
| FX (n = 59) | 66‐134 | 100 ± 13 IU/dL | 86 ± 11 IU/dL | 86 ± 3 |
| FXI (n = 91) | 62‐142 | 103 ± 22 IU/dL | 87 ± 18 IU/dL | 86 ± 5 |
| FXIII (n = 26) | NA | 110 ± 11 IU/dL | 102 ± 10 IU/dL | 93 ± 3 |
| VWF:RCo (n = 12) | NA | 114 ± 44 IU/dL | 111 ± 41 IU/dL | 97 ± 8 |
Results are reported as meant ± SD. n = number of replicates (treated plasma units) done for each parameter. (A higher number of replicates was performed for a basic testing panel, with additional parameters included in a subset of studies.) IU/dL = International Units/deciliter.
The reference range was calculated from the mean ± 2 SD of untreated, conventional plasma.
For PT and PTT, the effect of PCT was calculated by subtracting the pre‐PCT values from the post‐PCT values.
Table 6.
Maintenance of plasma antithrombotic protein activity after PCT *
| Protein | Pre‐PCT (IU/dL) | Post‐PCT (IU/dL) | Post/pre (% retention) |
|---|---|---|---|
| PC (n = 25) | 109 ± 15 | 102 ± 14 | 95 ± 9 |
| PS (n = 25) | 109 ± 12 | 107 ± 12 | 98 ± 5 |
| Antithrombin III (n = 26) | 94 ± 5 | 91 ± 6 | 96 ± 3 |
| α2‐Antiplasmin (n = 26) | 93 ± 5 | 75 ± 6 | 80 ± 4 |
Results are reported as mean ± SD. n = number of replicates done for each parameter; IU/dL = International Units/deciliter.
DISCUSSION
These studies showed that PCT with amotosalen and UVA light is effective against a broad range of pathogens in plasma. This finding is not surprising based on the pathogen inactivation efficacy previously demonstrated for PLTs with the same dose of amotosalen and same dose of UVA light. 15 , 16 , 17 , 18 , 19 , 20 The levels of inactivation for representative viruses, bacteria, and protozoa were measured with PCT conditions developed for the commercial application of FFP.
PCT inactivated high levels of enveloped viruses including HIV‐1, HBV, and HCV in plasma. Effectiveness against HBV was initially shown by inactivation of DHBV, an established infectivity model for human HBV. 25 A chimpanzee infectivity model was then used to confirm the level of inactivation of HBV in plasma. Effectiveness against HCV was initially shown by inactivation of BVDV, a flavivirus and model for human HCV. 26 Inactivation of HCV was then confirmed with the chimpanzee infectivity model as well. PCT is also effective against other flaviviruses such as WNV and, in preliminary studies, dengue virus (data not shown), examples of new pathogens that are of increasing concern to blood centers in North America.
PCT with amotosalen is very effective against cell‐associated HIV‐1 and against cell‐associated HTLV‐I and HTLV‐II viruses in plasma, demonstrating that amotosalen molecules readily permeate the cell membrane. The ability of the photochemical reagent to permeate the cell membrane as well as nuclear membrane is crucial because some blood‐borne viruses exist partially or largely associated with cells. Methylene blue treatment of plasma, for example, is not effective against cell‐associated or intracellular viruses. 11 The efficacy of the methylene blue system against intracellular viruses depends on the efficacy of filtration or freeze‐thaw to quantitatively disrupt WBCs. 8 , 27 Recent studies, however, demonstrate that conventional WBC filters fail to eliminate cell‐associated cytomegalovirus and HTLV‐I from PLT products. 28 , 29 Thus the methylene blue system is ineffective against these viruses.
The cumulative viral inactivation data in PLTs and plasma would predict that PCT is effective against the majority of enveloped viruses, DNA, or RNA, extracellular or intracellular. Thus PCT has the potential to inactivate new and emerging enveloped viruses and prevent them from entering the blood supply. Indeed, it was found that SARS‐CoV was inactivated by PCT in both PLTs and plasma. 15
Nonenveloped viruses are less common as labile blood component transfusion‐transmitted pathogens. Human parvovirus B19, and in very rare cases hepatitis A virus (HAV), however, have been transmitted by component blood transfusion. 30 , 31 A ubiquitous nonenveloped virus, TT virus, has been reported to be transmitted through the blood supply in Japan, but has yet to be directly linked with a specific disease state. 32
The efficacy of PCT against nonenveloped viruses in plasma continues to be evaluated and is expected to be similar to the results obtained for PLTs. 16 In these studies, inactivation experiments were only performed with two nonenveloped viruses, bluetongue virus and human adenovirus 5, as examples. Inactivation results comparable to the PLT experiments were obtained. Owing to the low permeability of the capsid, some nonenveloped viruses show little or no inactivation by PCT. The picornaviruses are known to be the most difficult to inactivate by chemical or physical means. 33 The tight capsid structure of picornaviruses such as HAV, polio virus, and encephalomyocarditis virus are thought to exclude even low‐molecular‐weight compounds such as psoralens from the interior of the virus and PCT is ineffective against these viruses. Human parvovirus B19 and human adenovirus 5, however, were found sensitive to PCT in PLTs. 16 , 34 The level of inactivation for these nonenveloped viruses can be increased by incubating the spiked blood products with amotosalen before illumination with UVA light. 34
The methylene blue system also showed variable efficacy against nonenveloped viruses, ranging from ineffective to complete inactivation. 8 Another system for treatment of plasma, the S/D system, is ineffective against nonenveloped viruses. S/D inactivates viruses by disrupting the viral membrane. 10
PCT is effective for inactivation of gram‐positive S. epidermidis and gram‐negative K. pneumoniae and Yersinia enterocolitica. Y. enterocolitica are cryophilic bacteria that can grow in cold temperatures. High levels of inactivation were achieved for all of these bacteria in plasma. Although transfusion‐transmitted bacteremia is not considered a serious problem for transfusion of FFP, it is reassuring to have a pathogen inactivation process that inactivates bacteria as well as viruses. In these studies, inactivation of two spirochetes, T. pallidum and B. burgdorferi, in plasma was also demonstrated. T. pallidum, which causes syphilis, is the only bacteria routinely screened for in blood banks. Under experimental conditions, B. burgdorferi has been reported to remain viable in frozen plasma for more than 1 month. 35 PCT has been shown to be effective against these agents and high levels of inactivation were obtained in plasma.
On an international level, other organisms such as T. cruzi (Chagas’ disease), P. falciparum (malaria), and B. microti (babesiosis) may be more of a concern than bacteria for frozen plasma because, like viruses, they do not require growth during storage to be a transfusion risk. P. falciparum and B. microti are intracellular pathogens, as discussed above, and are resistant to methylene blue treatment. 11
Experiments with plasma are ongoing to measure the level of WBC inactivation by PCT. Based on previous studies with PLTs, it is anticipated that high levels of WBC inactivation could be demonstrated in plasma. 21 Overall, the results presented here show that PCT offers the benefit of a broad spectrum of pathogen inactivation. Neither the methylene blue system nor the S/D system for plasma has shown this broad pathogen inactivation profile.
These studies also confirmed maintenance of adequate plasma coagulation and antithrombotic protein function after PCT. Because FFP is transfused primarily as a replacement for the liver‐derived coagulation factors FII, FV, FVII, F IX, FX, and FXI, in vitro studies to evaluate plasma function after processing with PCT were conducted for a panel of coagulation factors, antithrombotic proteins, and clotting time. The proteins most affected by PCT were fibrinogen and FVIII. The changes in coagulation proteins observed in plasma treated with PCT were also associated with slight prolongation in PT and APTT. The slight changes in PT and APTT after PCT, however, were not associated with any adverse clinical observations. 36 , 37 , 38 , 39
Levels of the anticoagulant PC and PS and antithrombin were relatively unaffected by PCT and α2‐antiplasmin was conserved by 80 percent. Previous studies on cryoprecipitate prepared from PCT plasma yielded approximately 95 and 88 percent activity retention for fibrinogen and FVIII, respectively, compared to cryoprecipitate prepared from untreated plasma. 40 Furthermore, cryosupernatant prepared from PCT plasma retained adequate levels of critical plasma proteins for plasma exchange therapy in acute thrombocytopenic purpura. The data indicate good preservation of hemostasis control proteins such as PS, α2‐antiplasmin, and VWF‐cleaving protease activity. 41
From a blood center perspective, the adoption of PCT would enhance safety of blood products and potentially decrease the number of donors rejected. PCT is easily integrated into the plasma preparation system by its ability to treat individual units of fresh apheresis plasma and small pools (three) of whole blood–derived plasma. For further ease of use, PCT is compatible with the existing plasma collection procedures, FFP processing, and distribution methods in use in blood centers. This PCT system allows rapid illumination and processing of plasma units and utilizes the same illumination device developed for pathogen inactivation of PLT concentrates. Thus, both plasma and PLTs can be treated with a similar processing system and the same illumination device.
In summary, photochemically treated plasma is functionally similar to untreated conventional plasma with the added benefit of pathogen inactivation.
ACKNOWLEDGMENTS
The authors thank the following investigators for conducting the pathogen inactivation studies: Bernard Guillemain, MD, HTLV; Krishna Murthy, VD, and Harvey Alter, MD, HBV and HCV; Patricia Marion, PhD, DHBV; Kristen Bernard, PhD, WNV; Sheila Lukehart, PhD, T. pallidum; Robert Lane, MD, B. burgdorferi; Phillippe Grellier, MD, P. falciparum; and Wesley Van Voorhis, PhD, T. cruzi; Jorge Benach, PhD, B. microti. We also appreciate the following individuals for their contributions in the development of the INTERCEPT plasma product: Jocelyn Smyers, Betsy Donnelly, Lainie Corten, Meisa Propst, Reydan Mababangloob, Adam Sampson‐Johannes, John Kinsey, Debbie Hanson, Treva Peterson, Melchor Dolor, Margaret Rheinschmidt, MT, and George Cimino, PhD.
These studies were supported in part by Cerus Corp., Baxter Healthcare Corp., and Grants DAMD17‐01‐20007, DAMD17‐02‐20042, and DAMD17‐03‐20039.
The U.S. Army Medical Research Acquisition Activity (Fort Detrick, MD) is the awarding and administering acquisition office for Grants DAMD17‐01‐20007, DAMD17‐02‐20042, and DAMD17‐03‐20039. The contents of this report do not necessarily reflect the position or policy of the government and no official endorsement is implied by this award.
REFERENCES
- 1. Sullivan MT, McCullough J, Schreiber GB, Wallace EL. Blood collection and transfusion in the United States in 1997. Transfusion 2002;42:1253‐60. [DOI] [PubMed] [Google Scholar]
- 2. Practice Guidelines for blood component therapy: a report by the American Society of Anesthesiologists Task Force on Blood Component Therapy. Anesthesiology 1996;84:732‐47. [PubMed] [Google Scholar]
- 3. Dodd RY, Notari EP 4th, Stramer SL. Current prevalence and incidence of infectious disease markers and estimated window‐period risk in the American Red Cross blood donor population. Transfusion 2002;42:975‐9. [DOI] [PubMed] [Google Scholar]
- 4. Barbara J. Why “safer than ever” may not be safe enough. Transfus Med Hemother 2004;31(Suppl 1):2‐10. [Google Scholar]
- 5. Phelps R, Robbins K, Liberti T, et al. Window‐period human immunodeficiency virus transmission to two recipients by an adolescent blood donor. Transfusion 2004;44:929‐33. [DOI] [PubMed] [Google Scholar]
- 6. Fiebig EW, Heldebrant CM, Smith RI, et al. Intermittent low‐level viremia in very early primary HIV‐1 infection. J Acquir Immune Defic Syndr 2005;39:133‐7. [PubMed] [Google Scholar]
- 7. Mohr H, Lambrecht B, Knuever‐Hopf J. Virus inactivated single‐donor fresh plasma preparations. Infusionsther Transfusionsmed 1992;19:79‐83. [DOI] [PubMed] [Google Scholar]
- 8. Williamson LM, Cardigan R, Prowse CV. Methylene blue‐treated fresh‐frozen plasma: what is its contribution to blood safety? Transfusion 2003;43:1322‐9. [DOI] [PubMed] [Google Scholar]
- 9. Horowitz B, Bonomo R, Prince AM, et al. Solvent/detergent‐treated plasma: a virus‐inactivated substitute for fresh frozen plasma. Blood 1992;79:826‐31. [PubMed] [Google Scholar]
- 10. Horowitz B. Inactivation of viruses found with plasma proteins. Biotechnology 1991;19:417‐30. [DOI] [PubMed] [Google Scholar]
- 11. Wagner SJ, Robinette D, Storry J, et al. Differential sensitivities of viruses in red cell suspensions to methylene blue photosensitization. Transfusion 1994;34:521‐6. [DOI] [PubMed] [Google Scholar]
- 12. Alford BL. Important new drug warning [monograph on the Internet]. Watertown (MA): V.I. Technologies, Inc. Vitex); 2002. Available from: http://www.fda.gov/medwatch/safety/2002/plassd_deardoc.pdf [Google Scholar]
- 13. Osselaer JC, Doyen C, Sonet A, et al. Routine use of platelet components prepared with photochemical treatment (INTERCEPT platelets): impact on clinical outcomes and costs. Blood 2004;104:987a. [Google Scholar]
- 14. Wollowitz S. Fundamentals of the psoralen‐based Helinx technology for inactivation of infectious pathogens and leukocytes in platelets and plasma. Semin Hematol 2001;38(4 Suppl 11):4‐11. [DOI] [PubMed] [Google Scholar]
- 15. Pinna D, Sampson‐Johannes A, Clementi M, et al. Amotosalen photochemical inactivation of severe acute respiratory syndrome coronavirus in human platelet concentrates. Transfus Med 2005;15:269‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin L, Hanson CV, Alter HJ, et al. Inactivation of viruses in platelet concentrates by photochemical treatment with amotosalen and long‐wavelength ultraviolet light. Transfusion 2005;45:580‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jauvin V, Alfonso RD, Guillemain B, et al. In vitro photochemical inactivation of cell‐associated human T‐cell leukemia virus Type I and II in human platelet concentrates and plasma by use of amotosalen. Transfusion 2005;45:1151‐9. [DOI] [PubMed] [Google Scholar]
- 18. Lin L, Dikeman R, Molini B, et al. Photochemical treatment of platelet concentrates with amotosalen and long‐wavelength ultraviolet light inactivates a broad spectrum of pathogenic bacteria. Transfusion 2004;44:1496‐504. [DOI] [PubMed] [Google Scholar]
- 19. Van Voorhis WC, Barrett LK, Eastman RT, et al. Trypanosoma cruzi inactivation in human platelet concentrates and plasma by a psoralen (amotosalen HCl) and long‐wavelength UV. Antimicrob Agents Chemother 2003;47:475‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eastman RT, Barrett LK, Dupuis K, et al. Leishmania inactivation in human pheresis platelets by a psoralen (amotosalen HCl) and long‐wavelength ultraviolet irradiation. Transfusion 2005;45:1459‐63. [DOI] [PubMed] [Google Scholar]
- 21. Grass JA, Hei DJ, Metchette K, et al. Inactivation of leukocytes in platelet concentrates by photochemical treatment with psoralen plus UVA. Blood 1998;91:2180‐8. [PubMed] [Google Scholar]
- 22. Reed LJ, Muench H. A simple method of estimating fifty‐per cent endpoints. Am J Hyg 1938;27:493‐7. [Google Scholar]
- 23. Heiden M, Seitz R. Quality of therapeutic plasma‐requirements for marketing authorization. Thromb Res 2002;107 (Suppl. 1):S47‐51. [DOI] [PubMed] [Google Scholar]
- 24. Beeck H, Hellstern P. In vitro characterization of solvent/detergent‐treated human plasma and of quarantine fresh frozen plasma. Vox Sang 1998;74(Suppl 1):219‐23. [DOI] [PubMed] [Google Scholar]
- 25. Ganem D, Varmus HE. The molecular biology of the hepatitis B viruses. Annu Rev Biochem 1987;56:651‐93. [DOI] [PubMed] [Google Scholar]
- 26. Choo QL, Kuo G, Weiner AJ, et al. Isolation of a cDNA clone derived from a blood‐borne non‐A, non‐B viral hepatitis genome. Science 1989;244:359‐62. [DOI] [PubMed] [Google Scholar]
- 27. Abe H, Yamada‐Ohnishi Y, Hirayama J, et al. Elimination of both cell‐free and cell‐associated HIV infectivity in plasma by a filtration/methylene blue photoinactivation system. Transfusion 2000;40:1081‐7. [DOI] [PubMed] [Google Scholar]
- 28. Visconti MR, Pennington J, Garner SF, et al. Assessment of removal of human cytomegalovirus from blood components by leukocyte depletion filters using real‐time quantitative PCR. Blood 2004;103:1137‐9. [DOI] [PubMed] [Google Scholar]
- 29. Pennington J, Taylor GP, Sutherland J, et al. Persistence of HTLV‐I in blood components after leukocyte depletion. Blood 2002;100:677‐81. [DOI] [PubMed] [Google Scholar]
- 30. Siegl G, Cassinotti P. Presence and significance of parvovirus B19 in blood and blood products. Biologicals 1998;26:89‐94. [DOI] [PubMed] [Google Scholar]
- 31. Vrielink H, Reesink HW. Transfusion‐transmissible infections. Curr Opin Hematol 1998;5:396‐405. [DOI] [PubMed] [Google Scholar]
- 32. Yokozaki S, Toyoda H, Nakano I, et al. Infection with TT virus, a novel transfusion‐transmissible DNA virus, in haemophiliacs and in blood products. Br J Haematol 1999;105:1114‐9. [DOI] [PubMed] [Google Scholar]
- 33. Hanson CV. Photochemical inactivation of viruses with psoralens: an overview. Blood Cells 1992;18:7‐25. [PubMed] [Google Scholar]
- 34. Sawyer L, Hanson D, Dupuis K. Inactivation of parvovirus B19 in platelets by Helinx technology. Transfusion 2004;44:102A. [Google Scholar]
- 35. McQuiston JH, Childs JE, Chamberland ME, Tabor E. Transmission of tick‐borne agents of disease by blood transfusion: a review of known and potential risks in the United States. Transfusion 2000;40:274‐84. [DOI] [PubMed] [Google Scholar]
- 36. De Alarcon P, Benjamin R, Dugdale M, et al. Fresh frozen plasma prepared with amotosalen HCl (S‐59) photochemical pathogen inactivation: transfusion of patients with congenital coagulation factor deficiencies. Transfusion 2005;45:1362‐72. [DOI] [PubMed] [Google Scholar]
- 37. Hambleton J, Wages D, Radu‐Radulescu L, et al. Pharmacokinetic study of FFP photochemically treated with amotosalen (S‐59) and UV light compared to FFP in healthy volunteers anticoagulated with warfarin. Transfusion 2002;42:1302‐7. [DOI] [PubMed] [Google Scholar]
- 38. Mintz P, Neff A, MacKenzie M, et al. Therapeutic plasma exchange (TPE) for thrombotic thrombocytopenic purpura (TTP) using plasma prepared with photochemical treatment (INTERCEPT plasma). Blood 2004;104:239a. [Google Scholar]
- 39. Mintz P, Steadman R, Blackall D, et al. Pathogen inactivated fresh frozen plasma prepared using HelinxTM technology is efficacious and well tolerated in treatment of end‐stage liver disease patients—the STEP AC trial. Blood 2001;98(Suppl 1):709a. [Google Scholar]
- 40. Mintz PD, Avery NL, Moulaison SL, et al. Preparation of cryoprecipitate from photochemically treated fresh frozen plasma. Transfusion 2000;40(Suppl 1):63S. [Google Scholar]
- 41. Yarranton H, Lawrie AS, Mackie IJ, et al. Coagulation factor levels in cryosupernatant prepared from plasma treated with amotosalen hydrochloride (S‐59) and ultraviolet A light. Transfusion 2005;45:1453‐8. [DOI] [PubMed] [Google Scholar]
- 42. Hanson CV, Crawford‐Miksza L, Sheppard HW. Application of a rapid microplaque assay for determination of human immunodeficiency virus neutralizing antibody titers. J Clin Microbiol 1990;28:2030‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Astier‐Gin T, Portail JP, Lafond F, Guillemain B. Identification of HTLV‐I‐ or HTLV‐II‐producing cells by cocultivation with BHK‐21 cells stably transfected with a LTR‐lacZ gene construct. J Virol Meth 1995;51:19‐29. [DOI] [PubMed] [Google Scholar]
- 44. Alter HJ, Creagan RP, Morel PA, et al. Photochemical decontamination of blood components containing hepatitis B and non‐A, non‐B virus. Lancet 1988;2:1446‐50. [DOI] [PubMed] [Google Scholar]
- 45. Feinstone SM, Alter HJ, Dienes HP, et al. Non‐A, non‐B hepatitis in chimpanzees and marmosets. J Infect Dis 1981;144:588‐98. [DOI] [PubMed] [Google Scholar]
- 46. Marion PL, Cullen JM, Azcarraga RR, et al. Experimental transmission of duck hepatitis B virus to Pekin ducks and to domestic geese. Hepatology 1987;7:724‐31. [DOI] [PubMed] [Google Scholar]
- 47. Shi PY, Tilgner M, Lo MK, et al. Infectious cDNA clone of the epidemic west nile virus from New York City. J Virol 2002;76:5847‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grellier P, Santus R, Mouray E, et al. Photosensitized inactivation of Plasmodium falciparum‐ and Babesia divergens‐infected erythrocytes in whole blood by lipophilic pheophorbide derivatives. Vox Sang 1997;72:211‐20. [DOI] [PubMed] [Google Scholar]
- 49. Etkind P, Piesman J, Ruebush TK 2nd, et al. Methods for detecting Babesia microti infection in wild rodents. J Parasitol 1980;66:107‐10. [PubMed] [Google Scholar]