

Abstract
Abstract Air travel can rapidly transport infectious diseases globally. To facilitate the design of biosensors for infectious organisms in commercial aircraft, we characterized bacterial diversity in aircraft air. Samples from 61 aircraft high‐efficiency particulate air (HEPA) filters were analyzed with a custom microarray of 16S rRNA gene sequences (PhyloChip), representing bacterial lineages. A total of 606 subfamilies from 41 phyla were detected. The most abundant bacterial subfamilies included bacteria associated with humans, especially skin, gastrointestinal and respiratory tracts, and with water and soil habitats. Operational taxonomic units that contain important human pathogens as well as their close, more benign relatives were detected. When compared to 43 samples of urban outdoor air, aircraft samples differed in composition, with higher relative abundance of Firmicutes and Gammaproteobacteria lineages in aircraft samples, and higher relative abundance of Actinobacteria and Betaproteobacteria lineages in outdoor air samples. In addition, aircraft and outdoor air samples differed in the incidence of taxa containing human pathogens. Overall, these results demonstrate that HEPA filter samples can be used to deeply characterize bacterial diversity in aircraft air and suggest that the presence of close relatives of certain pathogens must be taken into account in probe design for aircraft biosensors.
Practical Implications
A biosensor that could be deployed in commercial aircraft would be required to function at an extremely low false alarm rate, making an understanding of microbial background important. This study reveals a diverse bacterial background present on aircraft, including bacteria closely related to pathogens of public health concern. Furthermore, this aircraft background is different from outdoor air, suggesting different probes may be needed to detect airborne contaminants to achieve minimal false alarm rates. This study also indicates that aircraft HEPA filters could be used with other molecular techniques to further characterize background bacteria and in investigations in the wake of a disease outbreak.
Keywords: Aircraft biosensor, Probe design, 16S, Microbial diversity, Infectious diseases, High‐efficiency particulate air filter
Introduction
In 2009, over 769 million airline passengers flew to and within the United States (US Department of Transportation 2010), and over 629 000 international flights landed in the United States (Hwang et al., 2011). The potential for these aircraft passengers to transport infectious diseases to non‐infected areas is a serious public health concern, as demonstrated by the severe acute respiratory syndrome (SARS) virus that originated in 2002 and rapidly spread into 25 countries on five continents, resulting in over 8000 cases and 774 fatalities (Peiris et al., 2003). The potential for intercontinental spread of infectious bacteria via passenger aircraft also exists, particularly for newly emerging strains such as multiple‐drug‐resistant tuberculosis (Udwadia et al., 2012; Velayati et al., 2009). While reports of bacterial transmissions during air travel are infrequent, rare instances of Mycobacterium tuberculosis and Neisseria meningitidis transmission within aircraft have been observed (Abubakar, 2010; Kenyon et al., 1996; Rachael et al., 2009; Wang, 2000). Deliberate infection of aircraft passengers with bacterial pathogens is also a threat (National Research Council, 2006). One measure that could mitigate infectious disease spread is rapid detection of pathogens in aircraft air prior to deplaning.
An aircraft is considered a high‐consequence environment in which false alarms lead to extraordinary costs for airlines, the traveling public, and governmental authorities. On the basis of 2009 flight statistics, a biosensor that could be deployed in commercial aircraft arriving into the United States would require a probability of false alarm of under 10−6, less than approximately one false alarm per year as calculated in the studies by Hwang et al. (2011). As such, a critical obstacle in deploying rapid onboard biosensors is to minimize false alarms that may be generated by diverse microbes in circulated air in aircraft, making the understanding of microbial background critical for biosensor design.
To date, several studies have assessed bacteria species present in air from aircraft (Dechow et al., 1997; La Duc et al., 2006, 2007; McKernan et al., 2008; Osman et al., 2008; Wick and Irvine, 1995). Collectively, these studies conclude that humans and their belongings are a major source of airborne microorganisms in aircraft cabins. However, these prior studies are likely to have identified only a small fraction of the bacterial diversity present in aircraft cabins for a few reasons. First, previous studies of aircraft air employed culture methods (Dechow et al., 1997; McKernan et al., 2008; Osman et al., 2008; Wick and Irvine, 1995) and/or cloning of 16S rRNA gene sequences (La Duc et al., 2007; Osman et al., 2008). Results from culture methods reveal only a small fraction of the total microbial reservoir (Osman et al., 2008), especially for aerosolized Gram‐negative bacteria (Heidelberg et al., 1997). The effectiveness of culture methods is likely to be further reduced in the aircraft environment because of drying caused by the low relative humidity (<20%) and the rapid air exchange rate (of ∼0.5 Kg per min per cabin occupant). In addition, prior studies used grab samples that provided limited sampling, with short sampling duration (usually 3 min or less) and discrete sampling strategies (Board on Environmental Studies and Toxicology (BEST) (2002). La Duc et al. (2007) and Osman et al. (2008) in their studies pooled samples, creating composite samples of up to 10 m3 of air.
One way to accomplish the more comprehensive, integrated sampling is by collecting air samples via HEPA ventilation filters used in the air‐handling systems onboard aircraft. HEPA filters are advantageous because they are expected to capture at least 99.9% of bacteria (Bull, 2008), are aboard aircraft for months, and may collect particles from on the order of 107 m3 of air during their typical 5000‐h use. Even though only a small extract of filter media may be used for analysis, the amount of air sampled is large. On the order of 70 m3 of air may flow through each square centimeter of media over the life of a typical HEPA recirculation filter. From ventilation filter material, DNA and RNA can be extracted to identify microbes present (Goyal et al., 2011; Korves et al., 2011; Noris et al., 2011). Molecular methods relying only on extracted DNA have been used extensively in many environments to obtain a comprehensive picture of the microbial community, including indoor air environments (Rintala et al., 2008; Tringe et al., 2008). One molecular method for sampling environments that is relatively fast and inexpensive is the use of DNA microarrays (Brodie et al., 2007; DeSantis et al., 2007). DNA microarrays make use of short oligonucleotides probe sequences adhered to glass surfaces to assay the gene sequences present in a sample. A microarray with probes for 16S rRNA gene sequences (used for bacterial classification), called the PhyloChip, has been used to analyze microbial communities in outdoor air (Brodie et al., 2007) and many other environments (Kelly et al., 2010; Sunagawa et al., 2009; Tsiamis et al., 2008; Weinert et al., 2011; Yergeau et al., 2008). The PhyloChip has been shown to expand the breadth of coverage of microbial diversity compared to both culture methods and cloning (DeSantis et al., 2007).
In this study, we used HEPA filter samples and the G2 PhyloChip to analyze microbial communities in aircraft air and compare them to bacterial communities in ambient outdoor air from North American cities. We focused especially on lineages related to pathogens that could pose challenges for biosensor probes. Our results reveal diverse bacteria in aircraft, differences in bacterial composition between aircraft and outdoor air samples, and the presence of bacterial lineages that could affect biosensor probes.
Materials and methods
Aircraft and outdoor air samples
Used aircraft HEPA filters removed during routine maintenance were obtained from undisclosed airlines. These 61 filters came from aircraft with intercontinental and North American routes and were collected during 2008 and 2009 (Table 1). Service hours for 30 filters were between 500 and 15 190 h. Service hours for the remaining 31 filters were not recorded, but filter removal times during standard maintenance are expected to vary from 6 to 18 months. Information about aircraft types or cities within the route types cannot be disclosed in compliance with non‐disclosure agreements. Typically, continuous removal of aircraft air occurs through cabin vents at the floor level on the side walls, which run along the entire length of the cabin (some airframes may exhaust air through the ceiling). Up to 55% of the exhausted cabin air is recirculated back into the cabin after it permeates the HEPA filter, while the rest of the exhausted cabin air is released out of the aircraft through outflow valve(s) (Hunt and Space, 1994).
Table 1.
Aircraft filter samples
| Group | Collection Period | Continents Included in Routes | Number of Samples | Microarray Analysis Dates |
|---|---|---|---|---|
| Unknown 2008 | June–July 2008 | Unidentified | 5 | August 22 and October 30, 2008 |
| Intercontinental 2008 | August–September 2008 | North America, Europe, Asia, South Americaa | 26 | October 30, 2008 and July 17, 2009 |
| Intercontinental 2009 | July 2009 | North America, Europe, Asia | 11 | February 23, 2010 |
| North American 2009 | July–August 2009 | North America | 19 | February 23, 2010 |
aIn grouping, Hawaii was considered to be part of North America.
Used HEPA filters were de‐identified at MITRE (McLean, VA, USA) and then shipped to Kansas State University (Manhattan, KS, USA) for filter material recovery. Filters were disassembled and approximately 8 cm × 4 cm samples were cut using methods to limit contamination, as described by Korves et al. (2011). Filter samples were sent to Lawrence Berkeley National Laboratory and stored at room temperature for several weeks to months prior to DNA extraction. One sample was analyzed per filter.
Forty‐three outdoor air samples that were previously collected and processed for PhyloChip analysis [method described by Brodie et al. (2007)] were included here for comparative analysis. The sample collection occurred in the spring to fall seasons as follows: Austin (May–August 2003; 17 weeks), Detroit (first 3 weeks of August 2004), El Paso (last 3 weeks of June 2003), San Antonio (May–August 2003; 17 weeks), and San Jose (last 2 weeks of September/first week of October 2003). Detailed analysis of the microbial communities in relation to environmental data for the Austin and San Antonio samples has been published (Brodie et al., 2007), and digital *.CEL files created after PhyloChips were scanned for that study were used for comparison with aircraft samples as described below.
Filter and reagent blanks
To account for background DNA on filter material, two new, unused aircraft HEPA filters were obtained from the Pall Aeropower Corporation (New Port Richey, FL, USA). For the outdoor air sample set, an unused (unopened) filter was extracted in 2004 when the San Antonio samples were processed. The unused filters were extracted and analyzed in the same manner as the used filter samples and are referred to as filter blanks. To control for background potentially introduced during extraction and PCR, each set of filter samples processed included a ‘reagent blank’ (only reagents added to extraction tubes).
DNA extraction
After several methods were tried, DNA from aircraft filter samples was extracted using a modified Miller method (DeSantis et al., 2007; Miller et al., 1999), which provided template with undetectable PCR inhibition (see Supplemental Data S1). A 4‐cm2 piece (2 cm × 2 cm) of HEPA filter was cut into small pieces and placed in a Lysing Matrix E tube (MP Biomedicals, Solon, OH, USA). Three hundred fifty microlitre of Miller phosphate buffer and 350 μl of Miller SDS lysis buffer were added and mixed. Four hundred fifty microlitre of phenol–chloroform–isoamyl alcohol (25:24:1) was then added, and the tubes were bead‐beat at 5.5 m/s for 35 s in a bench‐top homogenizer (FastPrep‐24; MP Biomedicals). The tubes were spun at 10 000 × g for 5 min at 4°C. Five hundred fifty microlitre of supernatant was transferred to a 2‐ml tube, and an equal volume of chloroform was added. Tubes were mixed and then spun at 10 000 × g for 5 min. Four hundred eighty microlitre aqueous phase was transferred to another tube and two volumes of Solution S3 (MoBio, Carlsbad, CA, USA) was added and mixed by inversion. The rest of the clean‐up procedures followed the instructions in the MoBio Soil DNA extraction kit. Samples were recovered in 50‐μl Solution S5 and stored at −20°C. Extractions for the outdoor air samples were performed as described in (Brodie et al., 2007) except for Detroit samples, which differed only by starting from stored (room temperature) filters instead of filter washes (stored at −20°C). These extractions differed slightly from the methods used for the aircraft sample extractions described above (400 μl of Miller phosphate buffer, 400 μl of Miller SDS lysis buffer, bead‐beating at 6.5 m/s for 45 s, and first centrifugation at 16 000 × g) (Brodie et al., 2007).
PCR amplification of 16S rRNA genes
For aircraft samples, DNA extracts were amplified using 8‐temperature gradient PCR and universal 16S rRNA gene primers (27f 5′‐AGAGTTTGATCCTGGCTCAG‐3′, 1492r 5′‐GGTTACCTTGTTACGACTT‐3′). Three microlitre of template was used at each temperature. Twenty‐five microlitre reactions (final concentrations were 1× Ex Taq Buffer with 2 mmol/l MgCl2, 300 nmol/l each primer [27f and 1492r], 200 μmol/l each dNTP (TaKaRa), 25‐μg bovine serum albumin (Roche Applied Science, Indianapolis, IN, USA), and 0.625 U Ex Taq (TaKaRa Bio Inc., through Fisher Scientific, Pittsburg, PA, USA) were amplified using an iCycler (Bio‐Rad, Hercules, CA, USA) and the following thermocycling conditions: 95°C for 3 min for initial denaturation, 30 cycles of 95°C for 30 s, 48–58°C for 30 s, and 72°C for 2 min and then final extension for 10 min at 72°C. PCR products from each annealing temperature for a sample were combined and concentrated to 40 μl or less final volume using Microcon YM‐100 filters (Millipore, Billerica, MA, USA). Fourty microlitre water was added to the filter units and spun through prior to loading the PCR product for concentration. One microlitre of concentrated PCR product was quantified on a 2% agarose E‐gel using the Low Range Quantitative DNA Ladder (Invitrogen, Carlsbad, CA, USA). For the outdoor air samples, PCR conditions were as described by Brodie et al. (2007), including 35 cycles of amplification. For filter and reagent blanks, at least 150 μl PCR product was pooled and 1 μl quantified by gel.
PhyloChip (G2) analysis
Analyses were performed using the second‐generation PhyloChip (G2), designed from NCBI GenBank records for 16S rRNA gene sequences. A total of 9773 clusters of sequences were constructed as operational taxonomic units (OTU) within the domains Bacteria and Archaea. OTU were grouped into 3297 subfamilies; subfamilies can consist of more than one genus. Details of probe selection and chip design are presented elsewhere (Brodie et al., 2006, 2007; DeSantis et al., 2007). For aircraft samples, 500 ng of bacterial PCR product was applied to each PhyloChip. For outdoor air samples, up to 2 ug of amplified PCR product had been used; most had approximately 1.5 μg PCR product loaded. For filter and reagent blank samples, as much as 40 μl concentrated product were used for PhyloChip analysis for two reasons: (i) to optimize the chance of detecting amplicons at very low concentrations and (ii) to load volumes of blank samples similar to actual samples. Ordinarily, reagent blanks are run only on a PhyloChip if visible bands are observed when loading 5 μl PCR product on a gel from a test PCR (single annealing temperature). However, for the aircraft dataset, several reagent blanks that did not produce visible bands were amplified using gradient PCR and run on the PhyloChip to assess reagent background. Application of PCR product followed previously described procedures (Brodie et al., 2006, 2007; DeSantis et al., 2007), including the use of the same internal standard (spike) mix used to provide similarly scaled data for all samples being compared; see Supplemental Data S1 for a brief description.
PhyCA processing of Affymetrix *.CEL files
CEL files were generated from scanned images of PhyloChips (Affymetrix GeneChip Scanner 3000 7G; Santa Clara, CA, USA). The CEL files provide probe intensity values and PhyCA processing compiles the individual probe intensities into OTU. Details of how probe intensities are used in PhyCA to generate information about each sample are included in the Supplementary Materials of the study by Hazen et al. (2010). Briefly, probe sets consisting of pairs of perfect and mismatch probes were used to distinguish real signals from background. PhyCA uses a two‐step process to first select a group of OTU with positive signals (Stage 1) and then to test those signals for cross‐hybridization potential (Stage 2). Probes with greater cross‐hybridization potential between neighboring OTU are more heavily penalized, reducing their contribution to the final output. The criteria used for Stage 1 and Stage 2 analysis for the G2 chips were Stage 1: rQ1 ≥ 0.379, rQ2 ≥ 0.565, rQ3 ≥ 0.82, pf ≥ 0.93, mean_without_hi_lo ≥ 500; Stage 2: r xQ3 ≥ 0.515 (pf is defined in the Supplemental Data S1). From Stage 2 processing results, any subfamily passing these criteria in at least one sample in the dataset is included in a subfamily report (binary calls for all samples). OTU that pass Stage 1 and are contained in subfamilies that pass Stage 2 are then included in OTU binary and intensity reports (trimmed mean intensity for perfect‐match probes in each OTU). The trimmed mean intensity values are referred to as mean fluorescence intensity (MFI).
Data analysis
All aircraft samples were treated as independent samples. It is possible that some of the filters in the intercontinental and unknown route groups were collected from the same aircraft, but limited collection of information from contributing airlines prevents disambiguation of sample replicates. Because filter collection depended on availability, samples do not represent a random sample of airline routes. Therefore, results of comparisons between the groups of aircraft samples apply to these specific samples and may not be more broadly construed as comparisons between categories.
Bacterial composition was assessed in three ways. First, the presence of taxa in a sample was evaluated using the Stage 1 and Stage 2 criteria stated above and an additional criterion to account for background, described below. Second, the prevalence of taxa was determined as the frequency at which taxa were determined to be present across samples. Third, relative abundance, a measure of the relative amounts of taxa present compared across samples, was assessed using MFI without regard to presence/absence calls (binary data) (Brodie et al., 2007).
To account for background, presence/absence was assessed with a criterion based on filter and reagent blank data. A signal‐to‐noise (SNR) ratio was computed for every sample‐OTU combination based on the raw MFI signal and a corresponding filter or reagent blank value (see Supplemental Data S1). An OTU was assessed to be absent in a sample if the SNR was <2. OTU that failed the SNR threshold for all samples were eliminated from the dataset; this eliminated approximately 100 OTU.
To account for background in relative abundance assessments, sample MFI values were adjusted using filter and reagent blank data. For samples in extraction sets in which the reagent blank yielded no visible band, the highest MFI from among the filter and reagent blank values was subtracted from the raw signal MFI sample value for each OTU. Owing to contamination observed in the reagent blank processed on 10 February 2010, all aircraft samples extracted on this date were corrected based on the maximum MFI of either the filter or the 10 February 2010 reagent blank samples. Non 10 February 2010 reagent blank samples were not used to correct the 10 February 2010 dataset and vice versa. The same procedure was implemented for the urban air dataset. Contamination was observed in the Detroit reagent blank, and thus, all Detroit samples were corrected based on the maximum of either the Detroit reagent blank or filter blank. Any negative values that resulted from the filter or reagent blank subtractions were set to zero.
To compare relative abundances of taxa across sample groups, OTU were ranked by adjusted MFI for each sample. Where two or more OTU shared the same MFI value, the average rank was used for both (e.g., if the third and fourth highest OTU tied for MFI, each was given rank 3.5). Intensity ranks were used instead of the original MFI values to facilitate comparisons between aircraft and outdoor air samples; the magnitude of fluorescence intensity values could not be compared directly because outdoor and aircraft air samples were collected with different methods and had different amounts of PCR product loaded onto chips. Furthermore, total richness estimates could not be compared directly because greater PCR loading could result in a greater pool from which to draw representatives, potentially affecting the breadth of taxa detected. To assess the relative abundance of subfamilies, the OTU that ranked the highest based on intensity was used to represent the subfamily. For characterizing the most abundant subfamilies, median ranks were calculated in Excel.
To examine differences in composition, OTU rank data were used to generate a Bray–Curtis similarity matrix, which was then used to generate non‐metric multidimensional scaling (NMDS) plots, or were tested using analysis of similarity (ANOSIM) in Primer (version 6 PRIMER‐E Ltd, Lutton, Ivybridge, UK) (Clarke and Gorley, 2006). ANOSIM is a permutation test analogous to analysis of variance (ANOVA) for nonparametric analysis (Clarke, 1993). To identify the OTU with the largest contributions to similarity and dissimilarity between groups, a similarity percentage (SIMPER) analysis was performed in Primer (version 6) (Clarke and Gorley, 2006). The ANOVA was performed in R 2.14.1 (R Development Core Team, 2011).
Results
Bacterial composition in aircraft air samples
Overall, 3763 operational taxonomic units (OTU) were detected in the set of aircraft filter samples. These OTU represented 606 subfamilies and 41 phyla‐level groups, as well as bacteria unclassified at the phylum level (Figure 1; Supplemental Table S1). Analyses based on fluorescence intensity rankings indicate that human‐associated bacteria as well as bacteria that were associated with outdoor environments were abundant in the aircraft samples (Table 2.) The highest ranked subfamilies included members associated with skin and oral, gastrointestinal and respiratory tracts, as well as subfamilies associated with oceans, lakes, hot springs, and soil (Table 2). The subfamilies with the highest ranked OTU were largely from the Firmicutes phylum (Table 2). Among the subfamilies in the top 10% by rank abundance, 43% were from Firmicutes, 20% were from Actinobacteria, and 14% were from Proteobacteria.
Figure 1.
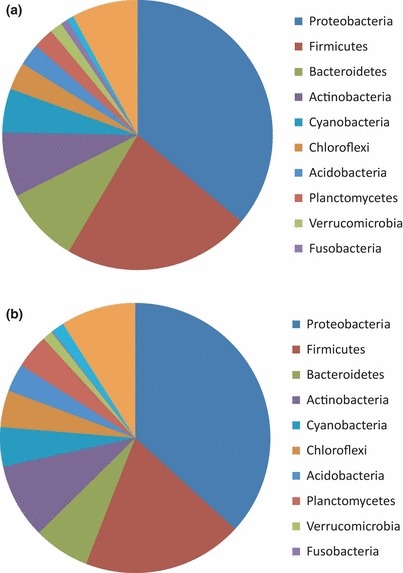
The relative number of subfamilies per phyla detected in (a) aircraft samples and (b) outdoor air samples
Table 2.
Subfamilies with the highest median fluorescence intensity ranks in aircraft air samples and corresponding subfamily ranks in outdoor air samplesa
| Subphylum, class, family, and subfamily | Associated habitats | Aircraft air median rank | Outdoor air median rank |
|---|---|---|---|
| Bacilli Halobacillus Halobacillus sfA | Marine | 1 | 24 |
| Bacilli Bacillus Bacillus sfA | Soil; water; human‐associated | 2 | 1 |
| Bacilli Unclassified Unclassified sfA | Soil; water; human‐associated | 3 | 5 |
| Bacilli Lactobacillales Streptococcaceae sfA | Respiratory, skin, oral; LAB | 4 | 53 |
| Bacilli Exiguobacterium Exiguobacterium sfA | Permafrost, sediment | 5 | 60 |
| Sulfobacillus Sulfobacillus Sulfobacillus sfA | Hydrothermal vents; metal; bioreactors | 6 | 39 |
| Clostridia Unclassified Unclassified sfA | Gastrointestinal tract | 7 | 2 |
| Actinobacteridae Intrasporangiaceae Intrasporangiaceae sfA | Oral; sludge | 8 | 3 |
| Bacilli Paenibacillaceae Paenibacillaceae sfA | Soil; water; human‐associated | 9 | 18 |
| Bacilli Marinococcus Marinococcus sfA | Soil | 10 | 145 |
| Bacilli Aneurinibacillus Aneurinibacillus sfA | Geothermal; soil; sludge | 11 | 67 |
| Bacilli Planococcaceae Planococcaceae sfA | Ice; soil; manure | 12 | 41 |
| Bacilli Geobacillus Geobacillus sfA | Soil; hot springs; petroleum | 13 | 92 |
| Bacilli Thermoactinomycetaceae Thermoactinomycetaceae sfA | Hydrothermal vents; air conditioners | 14 | 15 |
| Clostridia Clostridiales Acetivibrio sfA | Aquifers; cellulose‐degrading community | 15 | 19 |
| Bacilli Lactobacillales Lactobacillaceae sfA | Gastrointestinal tract; LAB | 16 | 29 |
| Clostridia Clostridiales Unclassified sfB | Gastrointestinal tract | 17 | 13 |
| Opitutae Opitutaceae Opitutaceae sfA | Soil; termite gut | 18 | 97 |
| Bacilli Marinococcus Marinococcus sfB | Saline soil | 19 | 12 |
| Bacilli Lactobacillales Unclassified sfA | Gastrointestinal tract, LAB | 20 | 113 |
| Alicyclobacillus Alicyclobacillus Alicyclobacillus sfA | Soil; hot springs | 21 | 83 |
| TM7‐3 EW055 Unclassified sfA | Soil; sludge | 22 | 30 |
| Bacilli Lactobacillales Carnobacteriaceae sfA | Food‐associated; equine manure clone; LAB | 23 | 178 |
| Actinobacteridae Gordoniaceae Gordoniaceae sfA | Skin; respiratory; sludge | 24 | 20 |
| Planctomycetacia Planctomycetales MPL7 Unclassified sfA | River biofilm | 25 | 331 |
aSubfamilies were ranked based on the operational taxonomic units within the subfamily with the highest median rank. 809 subfamilies were ranked.
LAB, Lactic Acid Bacteria.
Close relatives of human pathogens in aircraft samples
Operational taxonomic units that contain human pathogens and their close relatives were detected (Figure 2); because 16S rRNA gene sequences generally cannot differentiate pathogens from close non‐pathogenic relatives, these results do not establish the presence of pathogens. Detected OTU contain sequences from Bacillus anthracis, Brucella species, Burkholderia pseudomallei, Clostridium botulinum, Coxiella burnetii, Francisella tularensis, Salmonella enterica, and Escherichia coli, as well as sequences from closely related non‐ or weakly‐ pathogenic bacteria. Some of these OTU were detected at high frequencies. For example, the OTU that contains B. anthracis as well as the weaker pathogen B. cereus and insect pathogen B. thuringiensis and an OTU that contains Salmonella enterica strains were found in the majority of aircraft samples. OTU that contain other foodborne pathogens, such as Helicobacter pylori, Campylobacter jejuni, and Listeria monocytogenes, respiratory pathogens such as Klebsiella pneumoniae and Streptococcus pneumoniae, and pathogens that commonly cause hospital infections, such as Acinetobacter baumannii, Pseudomonas aeruginosa, and Staphylococcus aureus, also were detected. It is noteworthy that certain OTU containing important pathogens were not detected, including OTU for Burkholderia mallei and B. pseudomallei (OTU 3912), Chlamydophila psittaci (OTU 7198), Clostridium botulinum (OTU 5882), Coxiella burnetii (OTU 9373), Mycobacterium tuberculosis (OTU 250), Vibrio cholerae (OTU 1854), and Yersinia pestis (OTU 5861).
Figure 2.
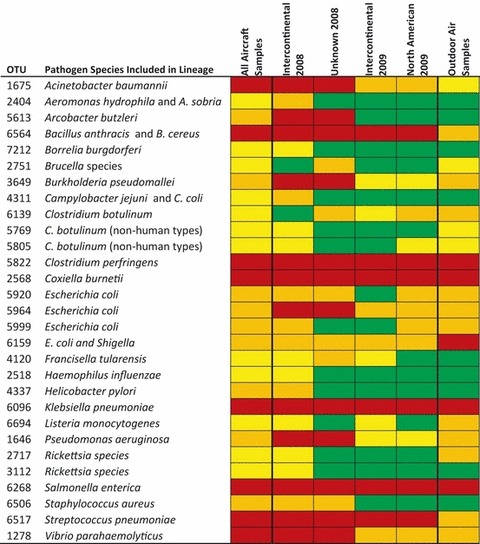
Percentages of samples in which pathogen‐containing operational taxonomic units (OTU) were detected with the PhyloChip. Red, >50%; Orange, >10–50%; Yellow, >0–10%; Green, not detected. Because OTU detections can be caused by either pathogens or non‐pathogenic bacteria in the same OTU, these results do not indicate the presence of the listed pathogens
Comparisons between groups of aircraft samples
Aircraft sample groups differed in the average number of subfamilies detected (ANOVA: F = 12.7, df = 3,57, P < 0.00001), with intercontinental samples collected in 2008 having on average a greater number of subfamilies than the North America‐only route samples and intercontinental samples collected in 2009 having the fewest (Supplemental Figure S1). Of the 606 subfamilies detected among aircraft samples, 30% were detected only in Intercontinental/Unknown route samples and not among North American samples. Most of these subfamilies were infrequently detected, and just seven of these subfamilies included OTU detected in at least 30% of the International 2008 samples.
Sample groups differed in overall composition, as shown by an ANOSIM of OTU fluorescence intensity rankings (Global R statistic = 0.413, P = 0.001; Figure 3). Pairwise R statistic values (Supplemental Table 2) indicate somewhat large differences between the Intercontinental 2008 and 2009 groups, and moderate differences between the Intercontinental 2008 and North American 2009 groups and between the Intercontinental 2009 group and the Unknown route 2008 group. Minor differences occurred between the North American and Intercontinental groups that were both collected in 2009. A SIMPER analysis revealed that the difference in composition between Intercontinental 2008 and 2009 groups was largely due to the greater relative abundance in the Intercontinental 2008 samples of Actinobacteriae class OTU (Actinobacteria phylum), Enterobacteriaceae and Pseudomonadaceae OTU (Gammaproteobacteria subphylum), and Betaproteobacteria subphylum OTU, and greater relative abundance in the Intercontinental 2009 samples of Firmicutes OTU (especially from Bacilli and Clostridia classes), Planctomycetes OTU, Solibacteres class OTU (Acidobacteria phylum), and Treponemaceae family OTU (Spirochaetes phylum).
Figure 3.
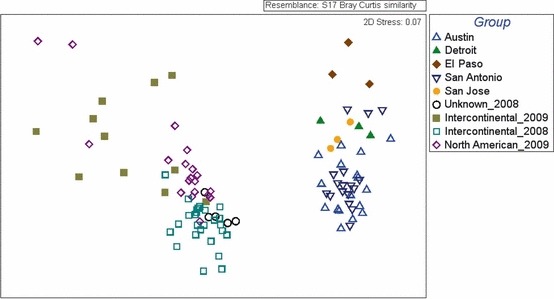
Non‐metric multidimensional scaling plot for aircraft and outdoor air samples based on relative abundance of operational taxonomic units (OTU). Plot was calculated using Bray–Curtis dissimilarity values of operational taxonomic units rank intensities
Sample groups also differed in whether or not certain pathogen‐containing OTU were detected (Table 2). In particular, thirteen pathogen‐containing OTU were detected among Intercontinental/Unknown samples but not in North American samples (Figure 2). Most of these OTU were detected infrequently (and therefore their absence in North American samples may be due to chance); only the OTU containing A. butzleri was detected in more than 30% of the samples within any Intercontinental/Unknown sample group. None of the pathogen‐containing OTU investigated were unique to North American samples.
Comparisons between aircraft and outdoor air samples
Comparisons between aircraft and outdoor samples indicate overlap in constituent subfamilies, and some differences. Of the 606 subfamilies detected in aircraft samples, 457 were also detected in outdoor air. Most of the 149 subfamilies detected only in aircraft samples were infrequently detected; just four of these subfamilies were detected in at least 30% of the aircraft samples, including a Firmicutes Acidaminococcaceae subfamily, a Peptostreptococcaceae subfamily, an Alphaproteobacteria Phyllobacteriaceae subfamily, and a chloroplasts subfamily. Of the 660 subfamilies detected in outdoor air, 203 subfamilies were unique to outdoor air samples. The outdoor air samples also included 10 phyla not detected in any aircraft air samples (Elusimicrobia_TG1, Coprothermobacteria, Lentisphaerae, OctSpA1‐106, a1b02,0 Natronoanaerobium, WS1, SC4, AC1, Thaumarchaeota). Just one phylum was detected in aircraft air samples but not in outdoor samples (Phylum OP1).
Aircraft and outdoor air samples differed slightly in the frequency of detection for several subfamilies (Supplemental Table 1). The mean number of subfamilies per sample differed modestly for families within the Firmicutes (higher frequency in aircraft samples) and Actinobacteria (higher frequency in outdoor samples) and a few other phyla (Bacteroidetes, Chloroflexi, Planctomycetes, Deltaproteobacteria). Families containing bacteria commonly associated with humans (Bacteroideaceae), anaerobic environments including vertebrate guts (Acidaminococcaceae), and food fermentation (e.g., dairy; Leuconostoc_FM) were more prevalent in aircraft samples, while families commonly associated with soil and/or water (e.g., Anaerolineae, Gemmata_FM, Kineosporiaceae, Myxococcus_FM, and Rubrobacteraceae) were more prevalent in the outdoor air samples.
The overall bacterial composition based on relative abundance of OTU differed substantially between aircraft and outdoor air samples (ANOSIM Global R statistic: 0.855, P < 0.001; Figure 3). SIMPER results were tabulated for the OTU that contributed to 20% of the cumulative total for either within‐group similarity or between‐group dissimilarity. SIMPER results showed that aircraft samples had 89.8% similarity and that the OTU that contributed the most to the overall similarity of aircraft samples largely belonged to the phylum Firmicutes (48% of 437 OTU), especially within the orders Bacillus, Halobacillus, Lactobacillales, Clostridiales, and Peptostreptococcaceae; the phylum Actinobacteria (26%), especially within the orders Arthrobacter, Micromonosporaceae, and Streptosporangineae; and the subphylum Gammaproteobacteria (13.5%), especially within the family Pseudomonadaceae. In outdoor air composition, Actinobacteria were particularly important (overall group similarity among outdoor air samples was 91%; 56% of the 434 OTU for group similarity belonged to Actinobacteria), and Firmicutes and Proteobacteria (especially Alphaproteobacteria and Betaproteobacteria) were important to a lesser degree (accounting for 24% and 16% of the OTU for group similarity, respectively).
The SIMPER results focusing on between‐group comparisons indicate that many OTU making large contributions to differences between aircraft and outdoor air samples were in the Pseudomonadaceae family of the Gammaproteobacteria subphylum (more abundant in aircraft samples; see Supplemental Table 3) and in the Comamonadaceae family of the Betaproteobacteria class (more abundant in outdoor air samples). In addition, OTU within several other Gammaproteobacteria families, Deltaproteobacteria families, and Firmicutes families had higher average intensity in aircraft samples, while OTU from several other Betaproteobacteria families and Actinobacteria families had higher average intensities in outdoor air samples. Of the 25 most abundant subfamilies in aircraft air, most were also abundant in urban outdoor air samples (Table 2).
While most pathogen‐containing OTU found in aircraft samples were also found in outdoor samples, nine pathogen‐containing OTU were unique to the aircraft samples (Figure 2). All of these OTU were also absent from all North American aircraft samples. In addition, a few pathogen‐containing OTU were detected more frequently in aircraft than in outdoor air samples, including OTU containing A. baumannii (62% aircraft vs. 5% outdoor samples), B. anthracis (97% aircraft vs. 33% outdoor samples), and S. pneumoniae (77% aircraft vs. 35% outdoor samples; for each of these three comparisons, Fisher’s exact test was P < 0.0001).
Discussion
Over 15 years ago, it was established that molecular methods provided more sensitive detection of bioaerosols compared with culture‐based methods (Alvarez et al., 1994), yet sample collection device designs remained focused on culture assay suitability for air sampling. Macnaughton et al. (1999) demonstrated that bioaerosols could be monitored using high‐volume air samplers and filters, from which the biomolecules of interest were recovered directly. Radosevich et al. (2002). demonstrated that DNA could be extracted directly from filters that had collected material for 24 h. In 2006, Farnsworth et al. (2006) showed that HVAC filters could be used to monitor bioaerosols, although they had not yet incorporated molecular detection methods. La Duc et al. (2007) and Osman et al. (2008) demonstrated the power of combining both high‐volume air sampling and molecular detection. The current study advances the science of indoor air sampling, specifically of aircraft air, by using both high‐volume, multimonth air sampling (via HEPA filters) and molecular assay methods (PhyloChip assay) that provide greater depth than traditional cloning and sequencing to identify the types of bacteria recoverable from commercial aircraft air and their relative abundances.
Nevertheless, the use of HEPA filters for microbial community profiling has limitations. First, ongoing sample collection over months does not provide for sample preservation over the long sampling period, and both degradation and growth of bacteria on the filters are possible. While desiccation would present problems for culture‐based assays, it is much less of a concern for DNA‐based assays. DNA can be extracted from very dry sample material (e.g., hyperarid soils, (Drees et al., 2006)) and the desiccation process may reduce enzymatic reactions, including DNAses, though there are likely some loses of DNA, too. Another challenge is DNA extraction optimization, including addressing PCR inhibition if observed.
This study identified substantially greater bacterial diversity in aircraft air than previous studies employing conventional molecular and culture techniques and shorter‐duration sampling. Prior 16S rRNA gene cloning studies identified approximately 100 bacteria lineages representing eight phyla (La Duc et al., 2007; Osman et al., 2008). In contrast, the current study detected 3763 OTU from 606 bacterial subfamilies representing 41 phyla. This greater diversity is probably due to the PhyloChip assay and the use of HEPA filters. The PhyloChip assay is capable of identifying up to 70‐fold more bacterial taxa than traditional cloning methods (La Duc et al., 2009) and can detect bacteria that represent as little as 0.01% of a community (Brodie et al., 2007). The most abundant lineages in our study overlapped with those found in previous aircraft studies. In particular, prior studies also found an abundance of Gram‐positive cocci (Dechow et al., 1997; McKernan et al., 2008), particularly Streptococcus (McManus and Kelley, 2005; Osman et al., 2008) and Bacillus (McKernan et al., 2008; Osman et al., 2008). As in prior studies, abundant bacteria types included bacteria associated with humans (La Duc et al., 2007; McKernan et al., 2008; McManus and Kelley, 2005; Osman et al., 2008), particularly the gastrointestinal, oral, and respiratory tracts.
The abundance of Firmicutes, and to a lesser extent Actinobacteria, in our aircraft samples also resembles results from other indoor air settings, including abundant human‐associated Gram‐positive bacteria in house dust (Taubel et al., 2009), in HVAC filter dust from occupied houses (Noris et al., 2011), in dust from offices (Rintala et al., 2008), and in air samples from office buildings (Stanley et al., 2008; Tsai and Macher, 2005). In particular, the abundance of subfamilies containing the genera Lactobacillus, Streptococcus, Corynebacterium, and Clostridia in our results corresponds to the abundance of these human‐associated genera in other indoor studies (Rintala et al., 2008; Taubel et al., 2009). Our results also included an abundance of bacteria associated with soil and aquatic habitats; in this respect, the results resembled a study of an indoor shopping center where bacteria associated with outdoor habitats were detected (Tringe et al., 2008), and a building study in which some abundant bacteria associated with soil were detected (Rintala et al., 2008).
The SIMPER results indicate that the OTU most responsible for differences between the aircraft and outdoor air samples included many members of the Pseudomonadaceae family (more prevalent in aircraft air) and the Comamonadaceae family (more prevalent in outdoor air). Pseudomonadaceae family members are associated with a wide environmental distribution, and Comamonadaceae family members are associated with water and soil. Other OTU that were more abundant in outdoor air were also soil‐associated, including several unclassified Betaproteobacteria OTU, a Rhizobiaceae OTU, and a Curtobacterium OTU. Soil suspended particles are a component of outdoor air (Andersen et al., 2009), and DeSantis et al. (2007) also demonstrated that many of the phyla detected in soil were also detected in outdoor air. In contrast, several OTU more abundant in aircraft were human or gastrointestinal associated, including OTU containing human floral bacterium Acinetobacter calcoaceticus, E. coli, and unclassified Clostridiales associated with animal intestines.
Among aircraft sample groups, there were no large differences in bacterial composition between groups of samples collected in the same year, suggesting that bacteria in commercial aircraft may be largely cosmopolitan (Figure 3; Supplemental Table S2). The differences between sample groups collected in 2008 and 2009 may reflect temporal differences in bacterial composition, route differences, and/or differences between aircraft types, but could also be due to batch effects because these samples were processed and analyzed at different times in the laboratory. The absence of large differences between the North American and intercontinental samples collected in the same year may result from US‐bound international flyers frequently flying on domestic flights to reach their final destinations. Nevertheless, the detection of multiple pathogen‐containing OTU in intercontinental or unknown route samples, but not in North American samples, suggests that some human‐associated microbes may be more prevalent in aircraft with particular intercontinental routes. Differences in the presence of pathogen‐containing OTU between geographical routes need to be taken into account in the selection of sensor probes and suggest that sampling different route types may be important despite the overall cosmopolitan pattern.
Our study identified many lineages containing known human pathogens (Figure 2). PhyloChip 16S rRNA gene sequence data cannot differentiate pathogens from their close, less pathogenic relatives. Even if the pathogens were present, they may not have been viable; DNA results do not distinguish infectious from non‐infectious particles. The results indicate that some lineages that contain pathogens, as well as close non‐ or weakly‐pathogenic relatives, were prevalent; the pathogens in these lineages include B. anthracis and B. cereus, gastrointestinal pathogen Salmonella enterica, pneumonia, ear infection, and meningitis‐causing Streptococcus pneumoniae, and the opportunistic nosocomial pathogen A. baumannii. The detection of most of these pathogen‐containing lineages is not surprising, given that they contain common human commensals (S. aureus, E. coli, and K. pneumoniae) or are a common cause of disease (S. pneumoniae). Some pathogen‐related lineages were also identified in previous studies of aircraft air, including Burkholderia cepacia, Salmonella enterica serovar Typhi, Neisseria meningitidis, and Mycobacterium species as well as opportunistic pathogens that affect immunocompromised persons (La Duc et al., 2007; Osman et al., 2008). The incidence of some pathogen‐containing OTU differed between the aircraft and outdoor air samples. In particular, the greater incidence of OTU containing B. anthracis and S. pneumoniae in aircraft parallels the association of human‐associated Firmicutes with aircraft and other indoor air.
As part of molecular biosensor development, it is important to document the occurrence and relative abundances of phylogenetic near‐neighbors to known human pathogens in the environment in which the biosensors are intended to be deployed; this study is a first step in that process. It is possible that in some cases, trace amounts of some pathogens could have been present and responsible for OTU detections. If trace amounts of some pathogens are present, then biosensor probes cannot merely detect presence or absence, but must take abundance, binding affinity, and/or viability into account. Other data, such as PCR tests for diagnostic (pathogenicity) genes, will be needed to evaluate the trace presence of pathogens. It is also important to note that even for bacterial taxa with similar 16S rRNA gene sequences in aircraft and outdoor air, there may be important differences in the bacterial genomes, including differences in the incidence of virulence factors and epitopes.
Our study demonstrates that molecular analyses of aircraft HEPA filters provide a way to deeply characterize bacteria in aircraft air. Molecular analyses of aircraft HEPA filters could also be used after an outbreak has occurred to investigate the source of the infectious organism; a similar use has recently been proposed for building ventilation filters (Ackelsberg et al., 2011). For biosensor development, characterizing background is critical for creating sensors with low false alarm rates. Further characterization of genetic background of aircraft and airport samples could be done using next‐generation sequencing techniques, and comparisons could be made to other indoor environments as more data becomes available (e.g., from the microBEnet project, http://www.microbe.net/). Identifying DNA sequences and other molecular targets specific to pathogens are among the many great challenges for biosensor research in the future.
Supporting information
Fig. S1 Boxplot for the number of subfamilies detected within the aircraft sample‐groups and in outdoor air samples.
Table S1 PhyloChip (G2) average subfamily richness (summarized at family) in cities and airplane cabins.
Table S2 Pair‐wise R statistics from ANOSIM analysis, which measures the amount of difference between groups of samples in OTU composition based on intensity rankings.
Table S3 Numbers of OTU in subfamilies that differentiate aircraft and outdoor air in rank abundance based on SIMPER results.
Data S1 Methods.
Supporting info item
Acknowledgements
We thank anonymous airlines for the contribution of used aircraft air filters, Michael Harkin for managing filter acquisition and transfer, Songeeta Palchaudhuri for initial HEPA filter extraction coordination, and Paul Magoha for recovering filter samples from the HEPA filter supports. We also thank Glenn Roberts and Richard Sciambi for discussions, which led to the initial set up this project, Marc Colosimo for 16S and PhyloChip advice, Jakk Wong for data analysis assistance, Joe Lundquist for useful discussions and Jean Watson for critical review of this manuscript. The authors also thank the anonymous reviewers for their comments and suggestions, which improved the quality of the data presented.
This research was funded, in part, by The MITRE Corporation. Work with the PhyloChip was supported, in part, by the U.S. Department of Energy under Contract No. DE‐AC02‐05CH11231 with the Lawrence Berkeley National Laboratory. Work performed at Kansas State University (KSU) was funded, in part, by the U.S. Federal Aviation Administration (FAA) Office of Aerospace Medicine through the National Air Transportation Center of Excellence for Research in the Intermodal Transport Environment (Cooperative Agreement 07‐C‐RITE‐KSU). The contents of this document reflect the views of the authors and The MITRE Corporation and do not necessarily reflect the views of the FAA or the U.S. Department of Transportation (DOT). Neither the United States Government, the FAA, nor the DOT makes any warranty or guarantee, expressed or implied, concerning the content or accuracy of these views.
©2012 The MITRE Corporation. All rights reserved. Approved for Public Release: 11‐4437, 12‐1951. Distribution Unlimited.
References
- Abubakar, I. (2010) Tuberculosis and air travel: a systematic review and analysis of policy, Lancet. Infect. Dis., 10, 176–183. [DOI] [PubMed] [Google Scholar]
- Ackelsberg, J. , Leykam, F.M. , Hazi, Y. , Madsen, L.C. , West, T.H. , Faltesek, A. , Henderson, G.D. , Henderson, C.L. and Leighton, T. (2011) The NYC native air sampling pilot project: using HVAC filter data for urban biological incident characterization, Biosecur. Bioterror., 9, 213–224. [DOI] [PubMed] [Google Scholar]
- Alvarez, A.J. , Buttner, M.P. , Toranzos, G.A. , Dvorsky, E.A. , Toro, A. , Heikes, T.B. , Mertikas‐Pifer, L.E. and Stetzenbach, L.D. (1994) Use of solid‐phase PCR for enhanced detection of airborne microorganisms, Appl. Environ. Microbiol., 60, 374–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, G.L. , Frisch, A.S. , Kellogg, C.A. , Levetin, E. , Lighthart, B. and Patemo, D. (2009) Aeromicrobiology/Air Quality. Encyclopedia of Microbiology, M. Schaechter, Oxford, Elsevier. [Google Scholar]
- Board on Environmental Studies and Toxicology (BEST) (2002) Biological Agents, The Airliner Cabin Environment and the Health of Passengers and Crew. Washington, DC, National Academy Press. [Google Scholar]
- Brodie, E.L. , DeSantis, T.Z. , Joyner, D.C. , Baek, S.M. , Larsen, J.T. , Andersen, G.L. , Hazen, T.C. , Richardson, P.M. , Herman, D.J. , Tokunaga, T.K. , Wan, J.M. and Firestone, M.K. (2006) Application of a high‐density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation, Appl. Environ. Microbiol., 72, 6288–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie, E.L. , DeSantis, T.Z. , Parker, J.P.M. , Zubietta, I.X. , Piceno, Y.M. and Andersen, G.L. (2007) Urban aerosols harbor diverse and dynamic bacterial populations, Proc. Natl Acad. Sci., 104, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull, K. (2008) Cabin air filtration: helping to protect occupants from infectious diseases. Travel Med Infect Dis, 6, 142–144. [DOI] [PubMed] [Google Scholar]
- Clarke, K. (1993) Non‐parametric multivariate analyses of changes in community structure, Aust. J. Ecol., 18, 117–143. [Google Scholar]
- Clarke, K.R. and Gorley, R.N. (2006) PRIMER v6: User Manual/Tutorial. Plymouth, PRIMER‐E. [Google Scholar]
- Dechow, M. , Sohn, H. and Steinhanses, J. (1997) Concentration of selected contaminants in cabin air of airbus aircrafts, Chemosphere, 35, 21–31. [DOI] [PubMed] [Google Scholar]
- DeSantis, T.Z. , Brodie, E.L. , Moberg, J.P. , Zubieta, I.X. , Piceno, Y.M. and Andersen, G.L. (2007) High‐density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment, Microb. Ecol., 53, 371–383. [DOI] [PubMed] [Google Scholar]
- Drees, K.P. , Neilson, J.W. , Betancourt, J.L. , Quade, J. , Henderson, D.A. , Pryor, B.M. and Maier, R.M. (2006) Bacterial community structure in the hyperarid core of the atacama desert, Chile, Appl Environ Microbiol, 72, 7902–7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth, J.E. , Goyal, S.M. , Kim, S.W. , Kuehn, T.H. , Raynor, P.C. , Ramakrishnan, M.A. , Anantharaman, S. and Tang, W. (2006) Development of a method for bacteria and virus recovery from heating, ventilation, and air conditioning (HVAC) filters, J. Environ. Monit., 8, 1006–1013. [DOI] [PubMed] [Google Scholar]
- Goyal, S.M. , Anantharaman, S. , Ramakrishnan, M.A. , Sajja, S. , Kim, S.W. , Stanley, N.J. , Farnsworth, J.E. , Kuehn, T.H. and Raynor, P.C. (2011) Detection of viruses in used ventilation filters from two large public buildings, Am. J. Infect. Control, 39, e30–e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen, T.C. , Dubinsky, E.A. , DeSantis, T.Z. , Andersen, G.L. , Piceno, Y.M. , Singh, N. , Jansson, J.K. , Probst, A. , Borglin, S.E. , Fortney, J.L. , Stringfellow, W.T. , Bill, M. , Conrad, M.E. , Tom, L.M. , Chavarria, K.L. , Alusi, T.R. , Lamendella, R. , Joyner, D.C. , Spier, C. , Baelum, J. , Auer, M. , Zemla, M.L. , Chakraborty, R. , Sonnenthal, E.L. , D’haeseleer, P. , Holman, H.‐Y.N. , Osman, S. , Lu, Z. , Van Nostrand, J.D. , Deng, Y. , Zhou, J. and Mason, O.U. (2010) Deep‐sea oil plume enriches indigenous oil‐degrading bacteria, Science, 330, 204–208. [DOI] [PubMed] [Google Scholar]
- Heidelberg, J.F. , Shahamat, M. , Levin, M. , Rahman, I. , Stelma, G. , Grim, C. and Colwell, R.R. (1997) Effect of aerosolization on culturability and viability of Gram‐ negative bacteria, Appl. Environ. Microbiol., 63, 3585–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, E.H. and Space, D.R. (1994) The airplane cabin environment: issues pertaining to flight attendant comfort, international in‐flight service management organization conference. Montreal, Canada. 1
- Hwang, G.M. , DiCarlo, A.A. and Lin, G.C. (2011) An analysis on the detection of biological contaminants aboard aircraft, PLoS One, 6, e14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, L. , Cockell, C. , Piceno, Y. , Andersen, G. , Thorsteinsson, T. and Marteinsson, V. (2010) Bacterial diversity of weathered terrestrial Icelandic volcanic glasses, Microb. Ecol., 60, 740–752. [DOI] [PubMed] [Google Scholar]
- Kenyon, T.A. , Valway, S.E. , Ihle, W.W. , Onorato, I.M. and Castro, K.G. (1996) Transmission of multidrug‐resistant Mycobacterium tuberculosis during a long airplane flight, N. Engl. J. Med., 334, 933–938. [DOI] [PubMed] [Google Scholar]
- Korves, T.M. , Johnson, D. , Jones, B.W. , Watson, J. , Wolk, D.M. and Hwang, G.M. (2011) Detection of respiratory viruses on air filters from aircraft, Lett. Appl. Microbiol., 3, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Duc, M. , Osman, S. , Dekas, A. , Stuecker, T. , Newcombe, D. , Piceno, Y. , Furhman, J. , Andersen, G. and Venkateswaran, K. (2006) A comprehensive assessment of biologicals contained within commercial airliner cabin air. Newyork, Pasedena, Jet Propulsion Laboratory, National Aeronautics and Space Administration. [Google Scholar]
- La Duc, M. , Stuecker, T. and Venkateswaran, K. (2007) Molecular bacterial diversity and bioburden of commercial airliner cabin air, Can. J. Microbiol., 53, 1259–1271. [DOI] [PubMed] [Google Scholar]
- La Duc, M.T. , Osman, S. , Vaishampayan, P. , Piceno, Y. , Andersen, G. , Spry, J.A. and Venkateswaran, K. (2009) Comprehensive census of bacteria in clean rooms by using DNA microarray and cloning methods, Appl. Environ. Microbiol., 75, 6559–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnaughton, S.J. , Cormier, M.R. , Jenkins, T.L. , Davis, G.A. and White, D.C. (1999) Quantitative sampling of indoor air biomass by signature lipid biomarker analysis, J. Ind. Microbiol. Biotechnol., 22, 80–87. [Google Scholar]
- McKernan, L.T. , Wallingford, K.M. , Hein, M.J. , Burge, H. , Rogers, C.A. and Herrick, R. (2008) Monitoring microbial populations on wide‐body commercial passenger aircraft, Ann. Occup. Hyg., 52, 139–149. [DOI] [PubMed] [Google Scholar]
- McManus, C.J. and Kelley, S.T. (2005) Molecular survey of aeroplane bacterial contamination, J. Appl. Microbiol., 99, 502–508. [DOI] [PubMed] [Google Scholar]
- Miller, D.N. , Bryant, J.E. , Madsen, E.L. and Ghiorse, W.C. (1999) Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples, Appl. Environ. Microbiol., 65, 4715–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (2006) Defending the U.S. air Transportation System Against Chemical and Biological Threats, Washington, DC, National Academic Press. [Google Scholar]
- Noris, F. , Siegel, J.A. and Kinney, K.A. (2011) Evaluation of HVAC filters as a sampling mechanism for indoor microbial communities, Atmos. Environ., 45, 338–346. [Google Scholar]
- Osman, S. , La Duc, M.T. , Dekas, A. , Newcombe, D. and Venkateswaran, K. (2008) Microbial burden and diversity of commercial airline cabin air during short and long durations of travel, ISME J., 2, 482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris, J.S.M. , Yuen, K.Y. , Osterhaus, A.D.M.E. and StÃhr, K. (2003) The severe acute respiratory syndrome, N. Engl. J. Med., 349, 2431–2441. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2011) R A Language and Environment for Statistical Computing. Vienna, Austria, R Foundation for Statistical Computing. [Google Scholar]
- Rachael, T. , Schubert, K. , Hellenbrand, W. , Krause, G. and Stuart, J.M. (2009) Risk of transmitting meningococcal infection by transient contact on aircraft and other transport, Epidemiol. Infect., 137, 1057–1061. [DOI] [PubMed] [Google Scholar]
- Radosevich, J.L. , Wilson, W.J. , Shinn, J.H. , DeSantis, T.Z. and Andersen, G.L. (2002) Development of a high‐volume aerosol collection system for the identification of air‐borne micro‐organisms, Lett. Appl. Microbiol., 34, 162–167. [DOI] [PubMed] [Google Scholar]
- Rintala, H. , Pitkaranta, M. , Toivola, M. , Paulin, L. and Nevalainen, A. (2008) Diversity and seasonal dynamics of bacterial community in indoor environment, BMC Microbiol., 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, N.J. , Kuehn, T.H. , Kim, S.W. , Raynor, P.C. , Anantharaman, S. , Ramakrishnan, M.A. and Goyal, S.M. (2008) Background culturable bacteria aerosol in two large public buildings using HVAC filters as long term, passive, high‐volume air samplers, J. Environ. Monit., 10, 474–481. [DOI] [PubMed] [Google Scholar]
- Sunagawa, S. , DeSantis, T.Z. , Piceno, Y.M. , Brodie, E.L. , DeSalvo, M.K. , Voolstra, C.R. , Weil, E. , Andersen, G.L. and Medina, M. (2009) Bacterial diversity and White Plague Disease‐associated community changes in the Caribbean coral Montastraea faveolata , ISME J., 3, 512–521. [DOI] [PubMed] [Google Scholar]
- Taubel, M. , Rintala, H. , Pitkaranta, M. , Paulin, L. , Laitinen, S. , Pekkanen, J. , Hyvarinen, A. and Nevalainen, A. (2009) The occupant as a source of house dust bacteria, J. Allergy Clin. Immunol., 124, 834–840. e47. [DOI] [PubMed] [Google Scholar]
- Tringe, S.G. , Zhang, T. , Liu, X. , Yu, Y. , Lee, W.H. , Yap, J. , Yao, F. , Suan, S.T. , Ing, S.K. , Haynes, M. , Rohwer, F. , Wei, C.L. , Tan, P. , Bristow, J. , Rubin, E.M. and Ruan, Y. (2008) The airborne metagenome in an indoor urban environment, PLoS One, 3, e1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, F.C. and Macher, J.M. (2005) Concentrations of airborne culturable bacteria in 100 large US office buildings from the BASE study, Indoor Air, 15(Suppl. 9), 71–81. [DOI] [PubMed] [Google Scholar]
- Tsiamis, G. , Katsaveli, K. , Ntougias, S. , Kyrpides, N. , Andersen, G. , Piceno, Y. and Bourtzis, K. (2008) Prokaryotic community profiles at different operational stages of a Greek solar saltern, Res. Microbiol., 159, 609–627. [DOI] [PubMed] [Google Scholar]
- Udwadia, Z.F. , Amale, R.A. , Ajbani, K.K. and Rodrigues, C. (2012) Totally drug‐resistant tuberculosis in India, Clin. Infect. Dis., 54, 579–581. [DOI] [PubMed] [Google Scholar]
- US Department of Transportation (2010) Summary 2009 traffic data for U.S and foreign airlines: Total passengers down 5.3 percent from 2008.
- Velayati, A.A. , Masjedi, M.R. , Farnia, P. , Tabarsi, P. , Ghanavi, J. , ZiaZarifi, A.H. and Hoffner, S.E. (2009) Emergence of new forms of totally drug‐resistant Tuberculosis bacilli, Chest, 136, 420–425. [DOI] [PubMed] [Google Scholar]
- Wang, P.D. (2000) Two‐step tuberculin testing of passengers and crew on a commercial airplane, Am. J. Infect. Control, 28, 233–238. [DOI] [PubMed] [Google Scholar]
- Weinert, N. , Piceno, Y. , Ding, G.‐C. , Meincke, R. , Heuer, H. , Berg, G. , Schloter, M. , Andersen, G. and Smalla, K. (2011) PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: many common and few cultivar‐dependent taxa, FEMS Microbiol. Ecol., 75, 497–506. [DOI] [PubMed] [Google Scholar]
- Wick, R.L. and Irvine, L.A. (1995) The microbiological composition of airliner cabin air, Aviat. Space Envir. Med., 66, 220–224. [PubMed] [Google Scholar]
- Yergeau, E. , Schoondermark‐Stolk, S.A. , Brodie, E.L. , Dejean, S. , DeSantis, T.Z. , Goncalves, O. , Piceno, Y.M. , Andersen, G.L. and Kowalchuk, G.A. (2008) Environmental microarray analyses of Antarctic soil microbial communities, ISME J., 3, 340–351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Boxplot for the number of subfamilies detected within the aircraft sample‐groups and in outdoor air samples.
Table S1 PhyloChip (G2) average subfamily richness (summarized at family) in cities and airplane cabins.
Table S2 Pair‐wise R statistics from ANOSIM analysis, which measures the amount of difference between groups of samples in OTU composition based on intensity rankings.
Table S3 Numbers of OTU in subfamilies that differentiate aircraft and outdoor air in rank abundance based on SIMPER results.
Data S1 Methods.
Supporting info item