

Abstract
We here address the question whether the unique capacity of mesenchymal stem cells to re‐establish tissue homeostasis depends on their potential to sense pathogen‐associated molecular pattern and, in consequence, mount an adaptive response in the interest of tissue repair. After injection of MSCs primed with the bacterial wall component LPS into murine wounds, an unexpected acceleration of healing occurs, clearly exceeding that of non‐primed MSCs. This correlates with a fundamental reprogramming of the transcriptome in LPS‐treated MSCs as deduced from RNAseq analysis and its validation. A network of genes mediating the adaptive response through the Toll‐like receptor 4 (TLR4) pathway responsible for neutrophil and macrophage recruitment and their activation profoundly contributes to enhanced wound healing. In fact, injection of LPS‐primed MSCs silenced for TLR4 fails to accelerate wound healing. These unprecedented findings hold substantial promise to refine current MSC‐based therapies for difficult‐to‐treat wounds.
Keywords: adaptive transcriptomic response, LPS sensing, mesenchymal stem cells, MSC‐based therapy, neutrophils, wound healing
Subject Categories: Immunology, Skin, Regenerative Medicine
Mesenchymal stem cells sense and shape their neighbourhood, accelerating tissue repair when injected into wounds. Exposure to LPS results in a profound transcriptomic shift in MSCs towards a multifaceted anti‐microbial defence and rapid wound closure.

Introduction
Mesenchymal stem cells (MSCs) are multipotent progenitor cells residing in a variety of tissues including bone and skin 1. Apart from self‐renewal and their differentiation capacity into several lineages, their paracrine interplay with distinct resident and non‐resident cells ensures effective tissue renewal and organ homeostasis 2. MSCs thus play a crucial role in maintaining the structural and functional integrity of the skin 1, 3. MSCs have also been implicated to improve tissue repair in preclinical animal models including several skin‐related pathologies like thermal burn 4, pressure ulcers 5, and full‐thickness skin wounds 6, 7 and in human studies 8, 9, 10, 11, 12. However, out of many hundred clinical trials with MSCs, only few were approved for clinical use 13, 14. We and others are therefore highly interested to advance the emerging concept that priming with danger molecules will enhance MSC potency and, in consequence, will improve MSC‐based therapies for accelerated tissue repair 15, 16.
Wound healing proceeds via sequential 17, partly overlapping stages including the inflammatory, proliferative, and remodeling phase 18, 19, 20. Tissue‐resident MSCs and immune cells migrate toward the wound site to efficiently repair damaged tissue in a well‐orchestrated manner 18. Among immune cells recruited to the wound site, activated neutrophils cleanse the wound site from tissue debris and effectively counteract invading pathogens. Neutrophils thereby play a crucial role during early phase of wound healing and—though currently unexplored—may even set the stage for subsequent phases of tissue repair 21. Activated neutrophils employ distinct strategies to immobilize and eventually kill invading microorganisms by oxidative burst released microbicidal reactive oxygen species (ROS), phagocytosis, proteolytic degradation, and the expulsion of their DNA decorated with proteolytic granules. This later process is referred to as neutrophil extracellular trap (NET) formation 21, 22, 23. Overactivation of neutrophils results in severe tissue damage. Therefore, temporal and spatial regulation of activated immune cells by MSCs and other resident cells surrounding tissue injury and, in consequence, the control of inflammation is essential for proper tissue repair. To regulate various immune cells, MSCs—via Toll‐like receptors (TLR)—are endowed with the capacity to respond to signals of their environment 15, 24. After TLR activation, MSCs adopt either a pro‐inflammatory or a anti‐inflammatory phenotype and thus control inflammation specifically modulating immune cells such as natural killer cells 25, 26, macrophages 16, 27, and T cells 28.
Though clinically highly important, it is currently unexplored how MSCs mount an adaptive response to shape the function and activation state of neutrophils during tissue injury and, importantly, how this may impact on overall wound healing. Previously, we have shown that MSCs suppress neutrophil activation by dampening their oxidative burst (the release of ROS), the release of proteolytic enzymes, and phagocytosis of activated neutrophils 29.
In the present study, we wished to investigate how MSCs adaptively regulate neutrophil functions under conditions reflecting wound infection, where MSC‐mediated suppression of neutrophil functions would rather be detrimental. To address this question, we evaluated the adaptive response of MSCs on activated neutrophil functions in the presence and absence of the bacterial lipopolysaccharide (LPS) which belongs to the family of pathogen‐associated molecular patterns (PAMP). Exposure of MSCs with LPS allowed us to mimic an infected wound environment. LPS binds to TLR4 present on MSCs and subsequently activates the downstream NF‐ΔκB and mitogen‐activated protein kinase (MAPK) pathways 30. Interestingly, LPS‐treated MSCs substantially augment neutrophil activation resulting in an increased NET formation and increased ROS production as opposed to MSC suppression of activated neutrophils under “non‐primed” conditions. Unexpectedly, injection of LPS‐primed MSCs into full‐thickness wounds significantly enhanced wound healing as opposed to remarkably less acceleration of wound repair following injection of non‐primed MSCs. We found that TLR4‐silenced MSCs upon LPS treatment failed to activate neutrophils with a profound deficiency to release neutrophil extracellular traps (NETs) and an impaired capacity for ROS generation. Most importantly, TLR4‐silenced MSCs were not able to accelerate wound healing. These data imply a causal role for TLR4‐dependent LPS sensing and the subsequent relay of this signal into MSCs, thus shaping an adaptive response to the LPS encounter. Employing unbiased and validated RNAseq analysis, we uncovered among other genes granulocyte chemotactic protein 2 (GCP2/CXCL6), epithelial‐derived neutrophil‐activating protein 78 (ENA78/CXCL5), interleukin‐1 beta (IL‐1β), and IL‐8, chemokines which are known to recruit and activate neutrophils. Collectively, our data uncovered MSCs—which due to their sensing capacity—act as “adaptive drug stores”. This is a clinically relevant finding, which is highly suited to be therapeutically exploited for the benefit of patients with difficult‐to‐treat and/or infected wounds.
Results
MSCs primed with LPS adaptively promote neutrophil activation
To investigate the adaptive response of LPS‐primed MSCs on neutrophil activation, NET formation was measured. MSCs were first challenged with bacterial LPS, an endotoxin and outer cell wall constituent of Gram‐negative bacteria that binds to the TLR4 receptor 31. MSCs were then co‐cultured with freshly isolated human neutrophils in the presence of phorbol 12‐myristate 13‐acetate (PMA). PMA is a constitutive protein kinase C activator, which is commonly used to activate neutrophils 32. NET formation was detected by immunostaining with an antibody directed against DNA‐histone which in case of NET formation stains expulsed DNA‐histone streaks. Consistent with earlier reports 29, 33, our results confirmed reduced NET formation in PMA‐activated neutrophils in the presence of MSCs (Fig 1A, lower row, left outer panel) as opposed to PMA‐treated neutrophils revealing enhanced NET formation (Fig 1A, upper row, third panel from left). By contrast to the complete suppression of NET formation in MSCs co‐cultured with PMA‐activated neutrophils (Fig 1A, lower row, left outer panel), treatment of MSCs with increasing LPS concentrations relieved the suppression of NET formation in a concentration‐dependent manner in MSCs co‐cultured with PMA‐activated neutrophils (Fig 1A, lower row, three outer right panels). Incubation of co‐cultures with an IgG antibody served as control for the specific histone staining (Fig 1A, upper row, outer left panel).
Figure 1. LPS‐primed MSCs activate neutrophils with enhanced NET .
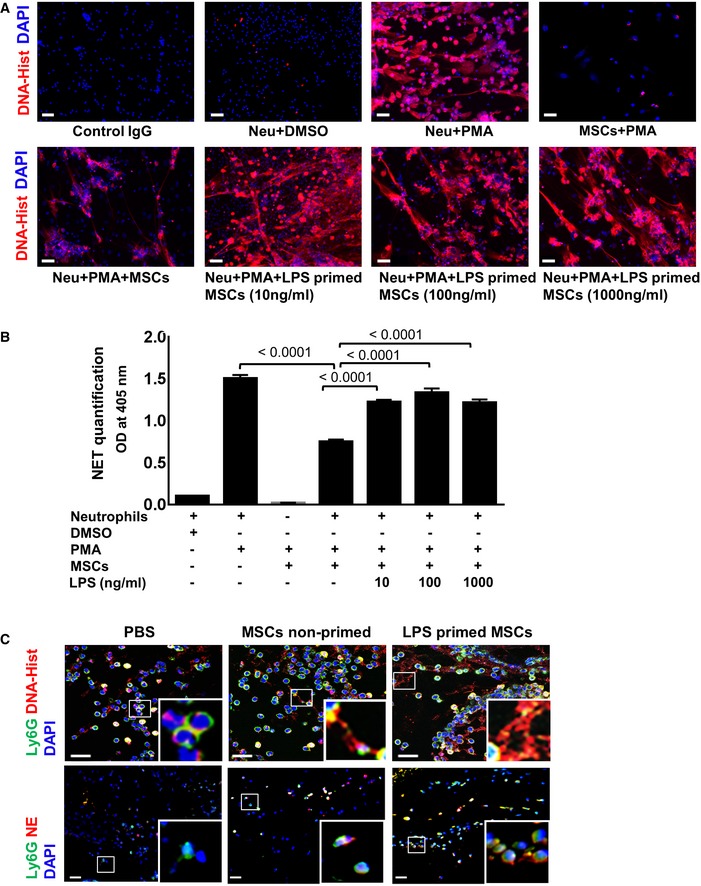
- MSCs were exposed to increasing LPS concentrations, thereafter washed and co‐cultured with PMA‐activated neutrophils. Incubation of neutrophils with PMA alone served as a positive control and DMSO‐treated neutrophils as a negative control. Scale bars: 50 μm.
- Quantification of NET‐bound elastase indicative of NET formation. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, n = 3 biological replicates.
- Representative microphotographs of murine wound sections immunostained for Ly6G (neutrophils, green) and DNA‐histone (displayed as expulsed streaks in red indicative of NETs). Wounds injected with LPS‐primed MSCs (upper row, outer right panel) depict enhanced NET formation (magnified inset) and increased expression of activation markers like neutrophil elastase (NE, lower panel, outer left panel) compared to wounds injected with non‐primed MSC (middle panels) or PBS‐injected wounds (outer left panels). Murine skin wounds treated with PBS injections served as control. Scale bar: 10 μm (upper row) and 50 μm (lower row). To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets.
Source data are available online for this figure.
In a complementary approach, NETs were quantified using a specific ELISA for NET‐bound elastase enzyme. Interestingly, neutrophil elastase activity indicative of NET formation was significantly increased in LPS‐primed MSCs co‐cultured with activated neutrophils as opposed to non‐primed MSCs co‐cultured with activated neutrophils (Fig 1B). The increase in NET formation was consistent with different LPS concentrations (10, 100, and 1,000 ng/ml) employed for MSC priming (Fig 1B), thus confirming data from NET immunostaining. Neutrophils treated with PMA alone served as a positive control for NET formation, while DMSO‐treated neutrophils served as a negative control (Fig 1A and B). More importantly and previously unreported, was the observation that LPS priming of MSCs can restore neutrophil activation and in consequence, their competence to remove tissue debris and pathogens after tissue trauma. These findings clearly suggest that MSCs are endowed with the unique capacity to sense pathogen‐associated molecular patterns, like LPS, and in response to such infection‐derived stimuli in their immediate microenvironment adaptively undergo a functional switch of MSCs, which—unlike their canonical immunosuppressive role—favors neutrophil activation in the interest of pathogen removal and tissue repair. Interestingly, the activation of neutrophils leads to a strong release of reactive oxygen species (ROS), mainly superoxide anion radicals. This microbicidal ROS release is suppressed by non‐primed MSCs as opposed to no suppression by LPS‐primed MSCs (Fig EV1). These are unprecedented findings as they imply that MSCs sense LPS, mimicking Gram‐negative bacterial infection, and, in consequence, MSCs adaptively refrain from microbicidal NET and ROS suppression. Of note, suppression of microbicidal NET formation was attenuated independent of whether LPS‐primed MSCs were co‐cultured with activated neutrophils or activated neutrophils were co‐cultured with MSCs in the presence of LPS (Appendix Fig S1A and S2B). The latter condition would reflect that neutrophils will be exposed to the same environment as MSCs. This ensures the efficient defense of a potential infection.
Figure EV1. LPS‐treated MSCs augment neutrophil activation and enhanced ROS production.
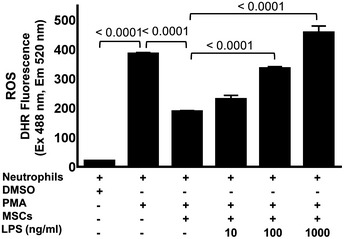
Graph shows quantification of ROS by measuring fluorescence intensity of DHR123 dye. The MSCs were primed with increasing concentrations of 10 ng/ml, 100 ng/ml, and 1,000 ng/ml LPS followed by co‐culture with neutrophils. The MSCs were then incubated with ROS‐specific dye DHR123, and the fluorescence was read after 60 min of incubation at 488/520 nm with spectrophotometer. Incubation of neutrophils with PMA alone served as a positive control and DMSO‐treated neutrophils as a negative control. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, three biological replicates.
To investigate whether our in vitro findings are at all relevant for wounds in vivo, LPS‐primed MSCs were intradermally injected into wound margins of mice and thereafter analyzed for the numbers of neutrophils and their activation state of infiltrating neutrophils using classical markers such as DNA‐histone indicative of NET formation. The number of neutrophils at the wound site increased significantly following injection of LPS‐primed MSCs with a maximum at 24 h (Fig EV2). We observed substantial NET formation in situ following injection of LPS‐primed MSCs into full thickness (Fig 1C, upper row, right outer panel) as opposed to distinctly suppressed NETs in non‐primed MSC‐injected wounds (Fig 1C, upper row, middle panel). Moreover, these findings were confirmed with the neutrophil‐specific activation marker neutrophil elastase (NE), a secreted enzyme degrading collagen and elastin which is also involved in macrophage recruitment. NE expression was enhanced in resident Ly6G‐positive neutrophils at the wound site indicative of neutrophil activation after injection of LPS‐primed MSCs (Fig 1C, lower row, outer right panel) as opposed to injection of non‐primed MSCs or PBS controls into the wound margin (Fig 1C, lower row, middle panel and lower row, outer left panel).
Figure EV2. Recruitment of neutrophils increases with time after induction of wounds in mice.
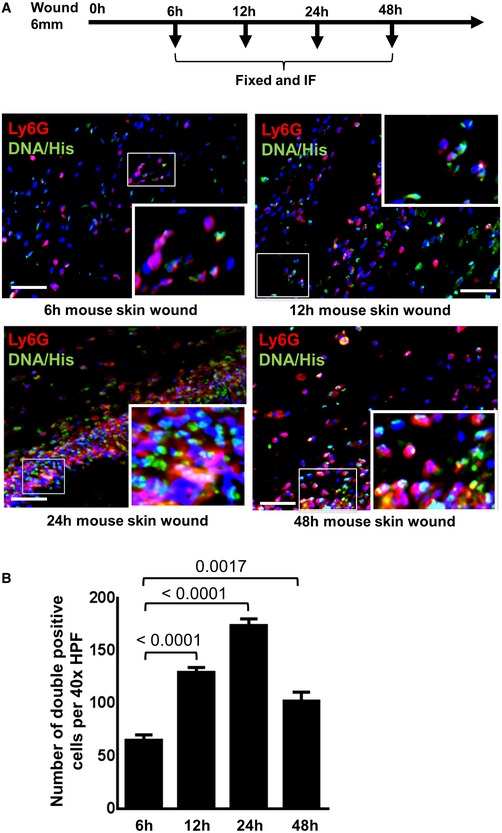
- Immunostaining of Ly6G (green) and DNA‐histone (DNA‐Hist, red) was performed at 6, 12, 24, and 48 h after induction of full‐thickness wounds. Nuclei were stained by DAPI (blue). Staining with Ly6G for neutrophils and DNA‐histone for NET formation allowed to study neutrophil recruitment and NET formation. To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Scale bars: 50 μm
- Quantitative analysis of Ly6G and DNA‐histone double‐positive cells at different time intervals. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
Together, these results indicate that MSCs sense bacterial infection in their microenvironment—adopt an unusual pro‐inflammatory phenotype—which for the sake of tissue protection from infection adaptively promotes neutrophil activation. Previous data of our laboratory 29 in conjunction with the herein reported findings imply that—depending on environmental cues of their direct neighborhood—MSCs are endowed with a remarkable, previously unreported plasticity. In terms of neutrophil activation, they adopt either an immunogenic or an immunosuppressive phenotype. Interestingly, the loss of immunosuppressive function of LPS‐primed MSCs is rather specific for neutrophils. By contrast, T‐cell activation and proliferation is similarly suppressed in a mixed lymphocyte reaction in the presence of either LPS‐primed or non‐primed MSCs (Appendix Fig S2).
Priming of MSCs with LPS fundamentally changes their transcriptome
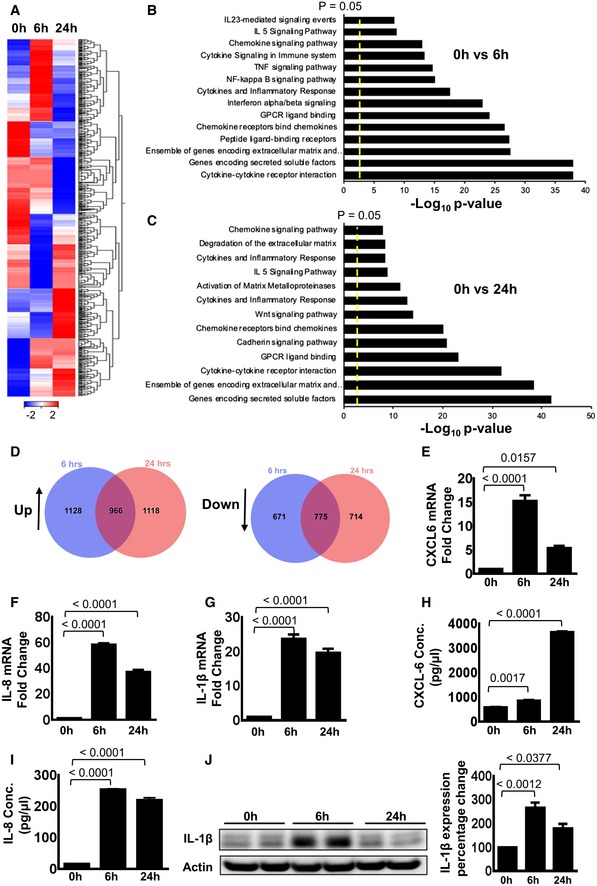
To further explore the underlying mechanisms driving MSCs to swiftly adopt a pro‐inflammatory, immunogenic state when exposed to pathogen‐associated molecular patterns like LPS, an unbiased transcriptome analysis strategy was employed. The MSC response to LPS exposure for 6 and 24 h was evaluated. Hierarchical clustering analysis of the pre‐ranked gene‐based Pearson correlation was performed for the RNAseq expression profile of LPS‐ or PBS‐exposed MSCs (Fig 2A).
Figure 2. LPS priming leads to profound transcriptomic reprogramming in MSCs.
-
AHierarchical clustering analysis of RNAseq expression profile from MSCs stimulated with LPS for 6 and 24 h. The heat‐map shows the gene expression profile of non‐primed MSCs and MSCs which were LPS primed for 6 and 24 h. Red color represents up‐regulation, while blue color depicts down‐regulation in gene expression; each data point represents FPKM in log2 value.
-
B, CPathway analysis depicts that cytokine–cytokine receptor interactions, genes encoding secreted soluble factors, an ensemble of genes encoding extracellular matrix, chemokine receptors which bind distinct chemokines, GPCR ligand binding, cytokine and inflammatory response, chemokine signaling pathway and IL‐5 signaling are the most dominant pathways as assessed by transcriptional changes when non‐primed and LPS‐primed MSCs were compared at 6 and 24 h after LPS priming.
-
DVenn diagram analysis displays up‐regulated and down‐regulated gene numbers indicated in blue and pink colors, respectively. Purple color indicates the number of shared genes of the up‐regulated and down‐regulated genes at 6 h and 24 h after LPS stimulation. This implies rapid changes of expression of the same genes.
-
E–GValidation of the selected genes uncovered from global RNAseq analyses by qRT–PCR analyses displays up‐regulation of CXCL‐6, IL‐8, and IL‐1β after LPS treatment. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, n = 3 biological replicates.
-
H, IELISA results depict an up‐regulated expression of CXCL‐6 after 24 h of LPS treatment, while a significantly increased IL‐8 expression after 6 and 24 h of LPS treatment. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, n = 3 biological replicates.
-
JWestern blot analysis showed up‐regulation of IL‐1β expression upon LPS priming of LPS‐primed MSCs. Actin served as control. Densitometry graph shows significant increase after 6 h of LPS stimulation compared to non‐primed MSCs. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, n = 3 biological replicates.
Source data are available online for this figure.
Interestingly, RNAseq analysis uncovered major changes in the expression of a variety of cytokines and chemokine receptors, implying a contribution in the regulation of cells of the immune system (Fig 2A and B, and Appendix Table S1). In particular, genes associated with chemotaxis and inflammatory response were significantly up‐regulated in MSCs after 6 h of LPS exposure (Fig 2A and B, Appendix Table S1). Both the extent of the fold‐change and the number of genes were significantly modulated in LPS‐primed MSCs compared to untreated MSC group (Appendix Tables S1). Gene expression of more than 3,000 genes significantly changed after LPS treatment. Transcriptome reprogramming involves cytokines or chemokines such as CCL1, CCL2, CCL3, CCL4, CCL5, CCL7, CCL8, CX3CL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, and CXCL10; interleukin and interleukin‐related genes such as IL‐1α, IL‐1β, IL‐6, IL‐15, IL‐15RA, IL‐17, IL‐32, IL‐33, IL‐34, IRAK2, and LIF; TNF and TNF‐related genes such as TNF, TNF‐AIP2, TNF‐AIP3, TNF‐AIP6, TNF‐AIP8, TNF‐RSF9, and TNF‐SF9; a prostaglandin‐related gene such as PTGS2; and interferon‐related genes such as IFI44L, IFIH1, IFIT1, IFIT2, IFIT3, IFIT5, IFITM1, IRG1, ISG15, ISG20, MX1, MX2, OAS1, OAS2, OAS3, and OASL. A major portion of these genes is involved in innate immune cell trafficking (IL‐8, CXCL6, IL‐6, IL‐1β, CXCL3, CXCL1, IL‐1α, C3a, and C5a, leukotrienes like LTB4 and LTB2) and localization of activated cells to distinct tissue compartments (CSF2, IL‐1β, and CCL5; Appendix Table S1).
Remarkably, the identified factors play a significant role in neutrophil activation, differentiation, and survival. When specifically searching for genes impacting on neutrophil numbers, recruitment to the wound site, and their activation state, a number of different pro‐inflammatory cytokines and chemokines were detected which are known to be involved in granulopoiesis, the process which enhances the formation of neutrophils originating from hematopoietic stem cells (IL‐23 via IL‐17 and IL‐17 via GCSF induction), neutrophil recruitment IL‐8, IL‐1α, IL‐1β, CXCL5, CXCL6, and CCL5 along with the complement factor C3, and extravasation from the blood vessels to the wound site (Appendix Table S1). We also observed the release of macrophage‐recruiting factors released by the LPS‐primed MSCs (e.g., CCL5, C3, CCL4/MIP1β, IL‐1α, CXCL2/MIP2α, CXCL20, and CXCL1) 34, 35. Moreover, genes responsible for cell–cell contact during engulfment of neutrophils by macrophages (intercellular adhesion molecule 1, ICAM1) in the inflammation resolving phase of wound healing and genes responsible for activation of neutrophils at the wound site (IL‐8, IL‐1β, IL‐6, CXCL6) were found up‐regulated after LPS stimulation (Appendix Table S1). Of note, following LPS priming of MSCs, down‐regulation of IRAK3, involved in suppression of the TLR4 signaling, RGS7, a negative regulator of chemokine receptors of the G protein‐coupled receptors, and PDCD4, programmed cell death protein 4, was observed (Appendix Table S1).
This profound change of the transcriptomic signature of MSCs apparently promotes MSC survival and neutrophil activation at the wound site to fight putative infections. This notion is further supported by pathway enrichment analyses (PEA). In fact, PEA uncovered “cytokine–cytokine receptor interaction”, as most prominent signatures in LPS‐primed MSCs. Other important pathways “gene encoding secreted soluble factors”, “ensemble of genes encoding extracellular matrix and extracellular matrix‐associated protein”, “chemokine receptors bind chemokines”, “GPCR ligand binding”, “cytokine and inflammatory response”, “chemokine signaling pathway”, and “IL5 signaling pathways” were up at both 6‐ and 24‐h time point in MSCs after LPS exposure (Fig 2B and C). However, “TNF and NF‐κB signaling pathways” that are intimately interconnected are activated only at 6‐h time point (Fig 2B). TNF‐α is known to promote tissue inflammation by activating neutrophils and macrophages 20, 36. This process is mediated by the NF‐κB transcription factor. Following TNF‐α exposure, a latent cytoplasmic NF‐κB becomes activated, enters the nucleus, and induces expression of an inflammatory and anti‐apoptotic gene program. Some of the above‐mentioned genes are early response gene of this TNF‐α–NF‐κB axis 37, 38. In addition, “degradation of extracellular matrix pathway”, “Wnt signaling pathway”, “cadherin signaling pathway”, and “activation of matrix metalloproteinases pathway” were induced at 24‐h time point only (Fig 2C).
The Venn diagram depicts substantial overlap in genes expressed between 6 and 24 h after LPS priming of MSCs (Fig 2D). The results show that 1,128 genes were up‐regulated after 6 h of LPS exposure and 1,118 genes were up‐regulated after 24 h of LPS exposure, while 966 genes were up‐regulated at both time points. In addition, 671 genes were down‐regulated after 6 h of LPS treatment and 714 genes were down‐regulated after 24 h, while 775 genes were down‐regulated at both time points (Fig 2D). This implies global changes in the expression of genes following LPS challenge.
RNAseq data of selected genes were further validated at mRNA level by RT–PCR. The analyzed genes included CXCL6/GCP2, IL‐8, and IL‐1β (Fig 2E–G). IL‐1β has been shown to induce inflammation via a MyD88‐independent pathway that converges to NF‐κB‐induced transcription of inflammatory genes 39. LPS stimulation for 6 and 24 h resulted in a significant changes in CXCL6, IL‐8, and IL‐1β expression at the protein level as well (Fig 2H–J), whereas IL‐8 is a potent pro‐inflammatory cytokine and strong chemoattractant 40 for neutrophil recruitment and activation. LPS priming also enhanced the mRNA expression of MyD88 and IL‐6 in MSCs (Fig EV3A). These data were further supported by a time kinetic experiment following LPS challenge suggesting the induction of key TLR4 pathway proteins (Fig EV3B). Collectively, these findings indicate profound global changes in different pathways that most likely push MSCs to adopt a pro‐inflammatory state when sensing an infected microenvironment.
Figure EV3. LPS‐treated MSCs activate MyD88 and IL‐6 expression in the TLR4 signaling pathway.
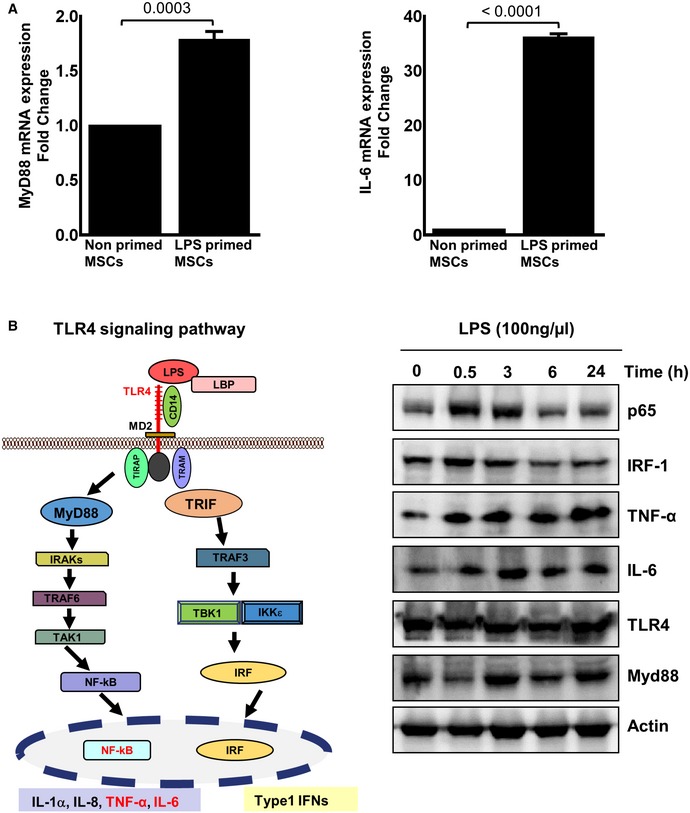
- Human MSCs were treated with LPS (100 ng/ml) for 24 h. The mRNA expression of MyD88 and IL‐6 was assessed by qRT–PCR analysis at 24 h. MyD88 (upper left panel) and IL‐6 gene expression (upper right panel) revealed an increase in MyD88 and IL‐6 in LPS‐primed MSCs. Statistical analysis was performed using unpaired t‐test, and values are represented as mean ± SEM, three biological replicates.
- The TLR4 signaling pathway is depicted. Upon LPS binding, conformational changes in the TLR4 receptor led to recruitment of intracellular TLR domains harboring adaptor proteins. A bifurcation in MyD88‐dependent signaling converges to NF‐κB and pro‐inflammatory cytokine release, among them IL‐6, and the TRIF signaling pathway relaying its signals to downstream effectors like the transcription factor IRF which transactivates type 1 interferons. The right panel depicts representative Western blot analyses with a time‐dependent regulation of TLR4 and components of the TLR4 signaling pathway after exposure of MSCs with LPS. p65, a component of the heterodimeric NF‐κB transcription factor, and the IRF‐1 transcription factor are swiftly induced at 0.5 h, but down‐regulated to basal levels at 6 h after LPS challenge of MSCs. The target genes of NF‐κB are induced at 0.5 h, and this induction is differentially maintained. The expression of indicated proteins was analyzed in the cell lysates from LPS (100 ng/ml)‐ or PBS‐treated MSCs, three biological replicates.
Source data are available online for this figure.
LPS‐induced distinct proteins belonging to the MSCs fingerprint in early wound healing in mice
Previously, we have shown that > 70% of MSCs were still present at the wound site at 24 h after MSC injection to the wound site 29.
To explore whether the LPS fingerprint observed in MSCs in vitro may also play a role in vivo, LPS‐primed MSC wounds were collected at 24 h after tissue injury and stained for selected proteins corresponding to those genes uncovered by RNAseq analysis. Human β2 microglobulin (h‐β2M) served as a marker to selectively stain injected human MSCs. MSC numbers did not significantly differ in wounds injected with non‐primed or with LPS‐primed MSCs (Appendix Fig S3). Sections were stained for CXCL6/GCP‐2, IL‐8, and IL‐1β, and double‐stained cells (hβ2M+/CXCL6+; hβ2M+/IL‐8+; and hβ2M+/IL‐1β+) were counted (Fig 3A). Intriguingly, we found that CXCL6/GCP‐2, IL‐8, and IL‐1β were significantly up‐regulated in LPS‐primed MSCs after injection into wounds compared to injection of non‐primed control MSCs (Fig 3A). CXCL6/GCP‐2 expression induces neutrophil recruitment to the wound site, while no expression of these proteins was found in PBS‐injected control wounds (Fig 3A). These data imply that the LPS fingerprint at the transcriptome level was at least in part also true for the corresponding protein expression under in vivo conditions of wounds injected with LPS‐primed MSCs.
Figure 3. LPS‐primed MSCs increase expression of neutrophil regulatory genes in vivo .
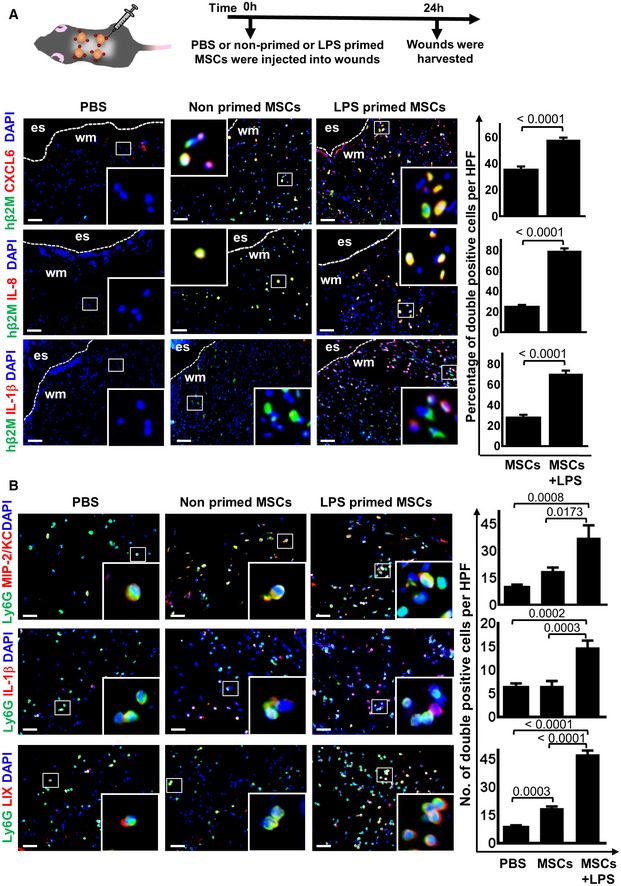
- The upper scheme depicts the experimental design. Representative microphotographs show that injection of LPS‐primed MSCs into wounds increased the expression of CXCL6, IL‐8, and IL‐1β. Graphs display numbers of double‐positive cells for h‐β2M + CXCL6, h‐β2M + IL‐8, and h‐β2M + IL‐1β, respectively. MSC‐injected wounds served as controls. Statistical analysis was performed using unpaired t‐test, and values are represented as mean ± SEM, six biological replicates. To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Es, eschar on wound margin; wm, wound margin; scale bars: 50 μm.
- Results show that LPS‐primed MSCs injected into wounds provoke increased expression of MIP/KC in endogenous neutrophils. MIP/KC represents functional homologues of IL‐8 in mice and is known as neutrophil chemoattractant. Similar results were found for IL‐1β, which is a strong chemoattractant for neutrophil recruitment and bacterial clearance. In addition, LIX which shares 63% amino acid sequence identity with human GCP‐2/CXCL6, a chemoattractant for neutrophils. The graphs display numbers of double‐positive cells for Ly6G MIP/KC, IL‐1β, and LIX, respectively. PBS and non‐primed MSC‐injected wounds served as controls. To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates. Scale bars: 50 μm.
Next, we wished to explore whether pro‐inflammatory cytokines identified from RNAseq analysis of LPS‐primed MSCs in vitro were also up‐regulated in murine neutrophils during wound repair in vivo, and whether this may amplify pro‐inflammatory, microbicidal conditions at the wound site. Wounds were collected at 24 h after injection of LPS‐primed MSCs into wound margins and subjected to immunostaining for murine neutrophil chemokines including MIP2/KC, a homologue for human IL‐8 and macrophage‐inflammatory protein 2 (MIP2), IL‐1β, and the CXC chemokine LIX, the murine homologue of human CXCL6/GCP‐2. LPS‐primed MSCs—when injected into wounds—led to marked increase of MIP/KC, which is a strong neutrophil chemoattractant (Fig 3B, upper row, outer right panel). Similarly, protein expression of IL‐1β, involved in neutrophil recruitment and bacterial clearance, and LIX responsible for neutrophil recruitment were significantly up‐regulated in wounds injected with LPS‐primed MSCs (Fig 3B, middle and lower rows, outer right panel) as opposed to control wounds injected with non‐primed MSCs (Fig 3B, middle and lower rows, middle panel).
These findings are novel and may explain the observed extent of neutrophil activation and its beneficial effect for overall wound healing after a single injection of LPS‐primed MSCs. The following sequence of events is most likely. The adaptive MSC response following LPS priming may initiate the release of pro‐inflammatory chemokines and activating factors, which in a paracrine loop impact also on murine neutrophils at the wound site. In fact, the homology between the IL‐8, LIX/GCP‐2 receptors, and the CXCR in human and mice is high and in consequence can get activated by human molecules to activate neutrophils at the wound site 41. Also, some pro‐inflammatory factors as IL‐8 and IL‐1 can induce their own expression 42. In consequence, the secretion of chemokines from murine neutrophils in an autocrine loop may distinctly enhance the effective combat against the bacterial threat at the wound site.
Taken together, these findings confirmed our in vitro results suggesting that LPS‐primed MSCs—when injected into murine wounds—can enhance activation of resident neutrophils.
The TLR4‐dependent adaptive response of LPS‐primed MSCs is responsible for accelerated wound healing
To further study whether TLR4 sensing, in fact, results in neutrophil activation, TLR4‐silenced and non‐silenced MSCs (Fig EV4), which were either LPS‐primed or left non‐primed, were subjected to co‐cultures with neutrophils. Notably, TLR4‐silenced MSCs failed to activate neutrophils as indicated by significantly decreased NET formation (Fig 4A). This was in stark contrast to markedly enhanced NET formation in neutrophils co‐cultured with non‐silenced MSCs (Fig 4A). These observations were fully confirmed by the assessment of NET‐bound elastase enzyme (Fig 4B).
Figure EV4. TLR4‐silenced MSCs reveal suppressed expression of TLR4 at transcriptional and protein levels.
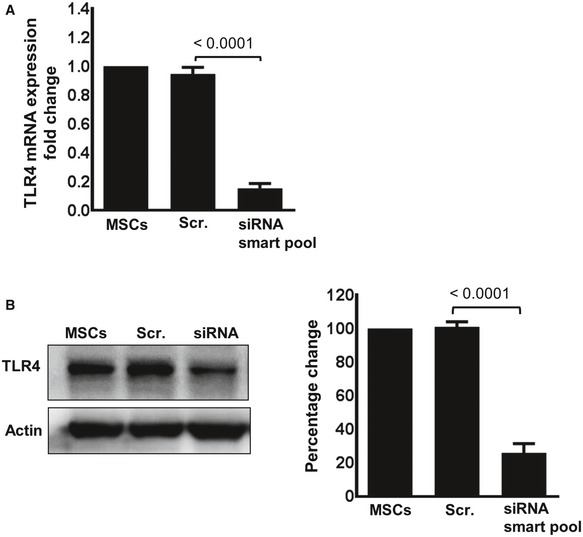
- MSCs were either treated with a scrambled control siRNA (Scr. siRNA) or with four different siRNAs targeting the TLR4 receptor (siRNA smart pool). Forty‐eight hours after transfection, TLR4‐silenced and non‐silenced MSCs were analyzed for RNA expression by qPCR. A representative experiment is depicted. Statistical analysis was performed using unpaired t‐test, and values are represented as mean ± SEM, three biological replicates.
- Seventy‐two hours after transfection, TLR4‐silenced and non‐silenced MSCs were treated with 100 ng/ml LPS for 16 h and subjected to Western blot analysis for protein expression. Densitometric assessment shows the percentage of the change in TLR4 expression in TLR4‐silenced and non‐silenced MSC. A representative experiment is depicted. Statistical analysis was performed using unpaired t‐test, and values are represented as mean ± SEM, three biological replicates.
Source data are available online for this figure.
Figure 4. TLR4‐silenced MSCs fail to activate neutrophils and to enhance NET formation.
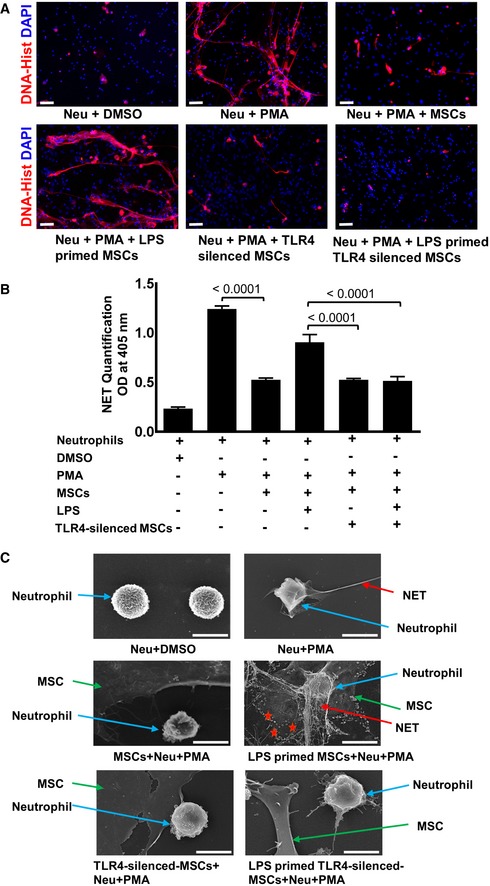
- Representative microphotographs of TLR4‐silenced and non‐silenced MSCs primed with LPS and co‐cultured with neutrophils. Immunostaining was performed with an antibody detecting NETs (DNA‐histone, red). Of note, TLR4‐silenced MSCs upon LPS priming cannot mount any adaptive response and fail to enhance NET formation (lower row, outer right panel) as compared to non‐silenced, LPS‐primed MSCs co‐cultured with PMA‐activated neutrophils (lower row, outer left panel). Neutrophils and DMSO served as a negative control (upper row, outer left panel), while neutrophils activated by the protein C kinase activator PMA served as a positive control depicting enhanced NET formation (upper row, middle panel). Co‐culture with MSCs suppressed enhanced NET formation (upper row, outer left panel) induced by PMA. Scale bars: 50 μm.
- Quantitative assessment of NET‐bound elastase employing a specific ELISA in identical experimental setting as described in Fig 1. Similarly, to immunostaining, a significant reduction of NET‐bound elastase was observed in TLR4‐silenced LPS‐primed MSCs when co‐cultured with activated neutrophils as opposed to high NET‐bound elastase in non‐silenced LPS‐primed MSCs co‐cultured with activated neutrophils. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, three biological replicates.
- High‐resolution scanning electron microscope analysis depicting enhanced NET formation (red arrows) expulsed from neutrophils (blue arrows) in the presence of LPS‐primed MSC co‐cultured with PMA‐activated neutrophils (middle row, outer left panel) as compared to reduced NETs in non‐primed MSCs co‐cultured with activated neutrophils (middle row, outer right panel). Of note, TLR4‐silenced LPS‐primed MSCs failed to activate neutrophils and NETs (lower row, outer left panel). TLR4‐silenced and non‐primed MSCs display reduced NETs in co‐cultures with activated neutrophils (lower row, outer right panel), suggesting that PMA activation shares TLR4 signaling components. PMA‐activated neutrophils alone served as a positive control with highly enhanced NETs (upper row, outer right panel) and DMSO‐treated neutrophils as negative controls (upper row, outer left panel). Red stars indicate neutrophil‐derived granules. Scale bars: 0.1 μm
Moreover, employing scanning electron microscopy, we studied different stages of NET formation (Fig 4C). Enhanced NET formation was obvious in LPS‐primed MSCs co‐cultured with activated neutrophils (Fig 4C). This effect was almost completely abolished in TLR4‐silenced, LPS‐primed MSCs co‐cultured with activated neutrophils (Fig 4C). These data further underscore the critical role of TLR4‐dependent LPS sensing in MSCs and its adaptive impact on NET formation which is key to pathogen elimination.
Together, these results suggest that TLR4 expressed on MSCs plays a crucial role in sensing an infected microenvironment and subsequently activate neutrophils.
To further assess whether MSCs in response to the PAMP molecule LPS change the course of wound healing, LPS‐primed, non‐primed MSCs, scrambled siRNA‐treated MSCs (scr siRNA) along with LPS‐primed TLR4‐silenced MSCs and LPS‐primed scr siRNA‐treated MSCs were injected into wound margins (Fig 5A and B). Wound closure was digitally measured and compared to PBS‐injected control wounds. Interestingly, wound closure was significantly accelerated in wounds injected with LPS‐primed MSCs as opposed to wounds injected with non‐primed MSCs at days 3 and 5 (Fig 5A and B). Injection of non‐primed MSCs still performed better in terms of wound closure when compared to PBS‐injected wounds (Fig 5A and B). Most interestingly, injection of TLR4‐silenced, LPS‐primed MSCs into wounds did not reveal any acceleration of wound closure at day 3 and day 5 when compared to wounds injected with LPS‐primed MSCs or with LPS‐primed, scr siRNA‐treated MSCs (Fig 5A and B). This was associated with an almost complete suppression of chemokines/cytokines involved in neutrophil recruitment and activation like CXCL6, IL‐1β, and IL‐8 in wounds injected with LPS‐primed TLR4‐silenced MSCs (Appendix Fig S4). These data confirm that TLR4 is mainly responsible for the release of CXCL6 and other cytokines.
Figure 5. LPS‐primed MSCs accelerate wound healing in TLR4‐dependent manner.
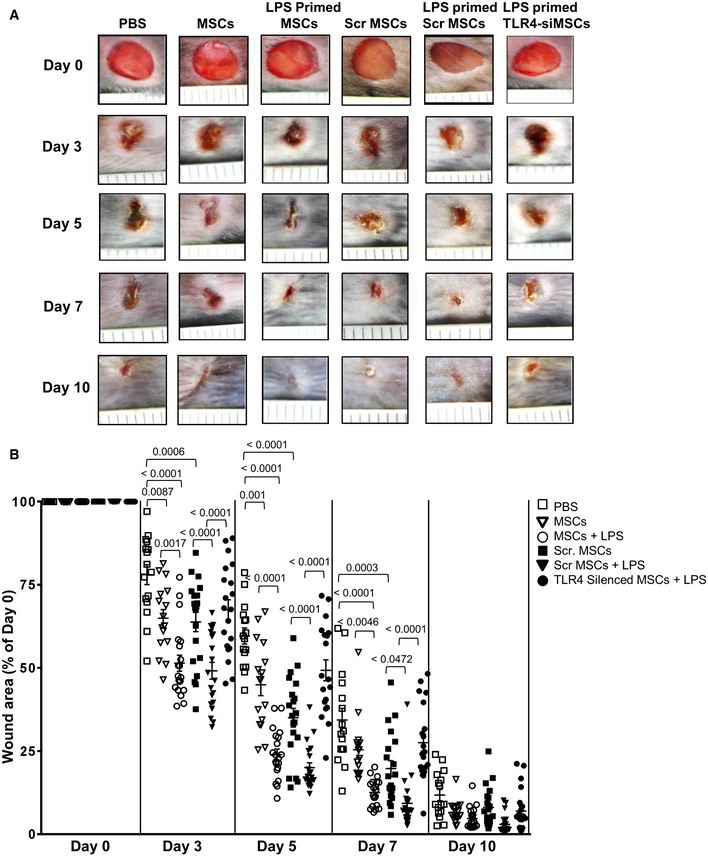
- Representative clinical pictures of murine wounds at 0, 3, 5, 7, and 10 days after wounding. Enhanced wound healing in the LPS‐primed MSCs group as opposed to all other groups.
- Statistical analysis of 20 wound areas per group at the indicated time points, expressed as percentage of the initial wound size (day 0), for PBS control, non‐primed MSCs, LPS‐primed MSCs, non‐primed scrambled siRNA‐treated (Scr) MSCs, LPS‐primed Scr MSCs, and LPS‐primed TLR4‐silenced MSCs. Results are mean ± SD of five biological replicates representing 1 of 3 independent experiments. Statistical analysis was performed using one‐way ANOVA.
Source data are available online for this figure.
Of note, unspecific activation of MSCs by scr siRNA does not appear to be predominantly responsible for acceleration of wound healing. In fact, the acceleration of wound healing injected with LPS‐primed scr MSCs is significantly better (Fig 5A, fifth row from left, and Fig 5B filled triangles) when compared to healing of wounds injected with non‐primed scr siRNA‐treated MSCs (Fig 5A, fourth row from left and Fig 5B, filled black squares). These data further confirm that TLR4 activation in MSCs plays a major role in enhancing wound healing.
These data are novel and imply that TLR4 sensing is not only causal for the adaptive MSC response, but also essentially required for enhancing wound healing. Our finding that TLR4‐silenced MSCs after priming did not even perform as good as non‐primed MSCs may suggest that TLR4—at least at an early wound phase—may mediate LPS‐independent responses to speed up wound healing. In fact, LPS is not the only TLR4 agonist, other danger signal ligands like S100A8/A9 released after tissue damage, also binds to TLR4 and may contribute to accelerate wound healing 15. Overall, these data suggest the following sequence of molecular and cellular events: Priming of MSCs with LPS results in a fundamental reprogramming of the transcriptome in MSCs with major impact on neutrophil activation, eventually after potential activation of other cell types enhancing wound closure in mice.
Acceleration of wound healing in LPS‐primed MSCs depends on enhanced TGFβ‐1 release from macrophages due to increased neutrophil engulfment
To explore whether neutrophil engulfment by macrophages was enhanced after injection of LPS‐primed MSCs at the wound site, double staining for neutrophils (Ly6G) and macrophages (F4/80) was performed using confocal microscopy (Fig 6A). This allowed us to assess numbers of macrophages with engulfed neutrophils. Interestingly, numbers of double‐positive cells were profoundly increased in day 1 wounds injected with LPS‐primed MSCs as opposed to significantly less double‐positive cells in wounds injected with non‐primed MSCs (Fig 6B). Moreover, single cell counts revealed highest numbers of neutrophils and macrophages in LPS‐primed MSCs injected day 1 wounds when compared to lower numbers in wounds injected with non‐primed MSCs (Fig 6C).
Figure 6. LPS priming of MSCs increased the expression of key proteins promoting granulation tissue formation in TLR4‐dependent manner.
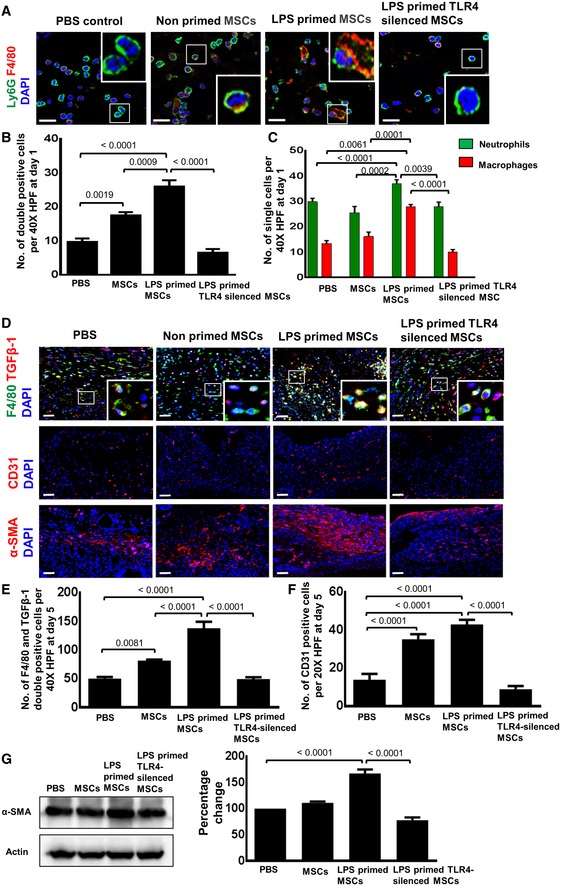
- Representative photomicrographs of confocal microscopy of sections from differently injected day 1 wounds stained for Ly6G+ neutrophils (green) and F4/80+ macrophages (red). Nuclei are stained with DAPI (blue). Double staining was performed for sections of day 1 wounds injected with PBS (control), non‐primed MSCs, LPS‐primed MSCs, and LPS‐primed TLR4‐silenced MSCs. Double‐stained cells indicate phagocytic engulfment of neutrophils by macrophages. To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Scale bar: 100 μm.
- Quantification of Ly6G and F4/80 double‐positive cells on sections of differently injected day 1 wounds. Wounds were injected as described in (A). Double‐positive cells were counted; statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
- Quantification of Ly6G+ neutrophils and F4/80+ macrophages on sections of differently injected day 1 wounds. Wounds were injected as described in (A). Single‐positive cells were counted; statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
- Representative photomicrographs of confocal microscopy of sections from differently injected day 3 wounds double‐stained for TGFβ‐1 (red) and for F4/80+ macrophages (green). Nuclei are stained with DAPI (blue). Double staining was performed for sections of day 5 wounds injected with PBS (control), non‐primed MSCs, LPS‐primed MSCs, and LPS‐primed TLR4‐silenced MSCs. Scale bar: 50 μm. Representative photomicrographs of sections of day 5 wounds immunostained for CD31 (indicative of endothelial cells and newly formed vessels) and for α‐SMA (indicative of myofibroblasts differentiation) after injection of LPS‐primed MSCs, non‐primed MSCs, LPS‐primed TLR4‐silenced MSCs or PBS (middle and lower panel). To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Scale bars: 50 μm.
- Double‐positive macrophages stained for TGFβ‐1 and F4/80 were counted; statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
- Quantitative analysis of CD31‐positive endothelial cells in sections of wounds injected with PBS, non‐primed MSCs, LPS‐primed MSCs, and LPS‐primed TLR4‐silenced LPS. Cell counting was performed on immunostained wound sections; statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
- Western blot analysis of lysates from day 5 wounds (left panel) and the corresponding densitometric analysis (right panel) depict enhanced α‐SMA protein expression in wounds injected with LPS‐primed MSCs as opposed to the respective control groups. Actin served as loading control. Statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, three biological replicates.
Source data are available online for this figure.
In order to understand why wound healing was profoundly accelerated following injection of LPS‐primed MSCs into wounds, and based on the finding of enhanced neutrophil activation, we hypothesized that recruitment and activation of neutrophils result in enhanced neutrophil apoptosis, which may lead to their enhanced phagocytosis by macrophages. Indeed, LPS‐primed MSCs markedly induced neutrophil apoptosis in vivo (Fig EV5). Engulfment of neutrophils by macrophages constitutes a strong signal for macrophages to release TGFβ‐1 43, 44. TGFβ‐1 is the strongest known inducer for differentiation of fibroblasts into myofibroblasts 45 which remodels their intermediate filaments toward the contractile α‐smooth muscle actin (α‐SMA). Myofibroblasts occurring during physiologic wound healing between day 3 and day 7 after wound injury were responsible for contracting wound margins to help wound closure 44.
Figure EV5. A significant increase in the caspase 3 activity of neutrophils at an early stage of wound healing in wounds injected with LPS‐primed MSCs.
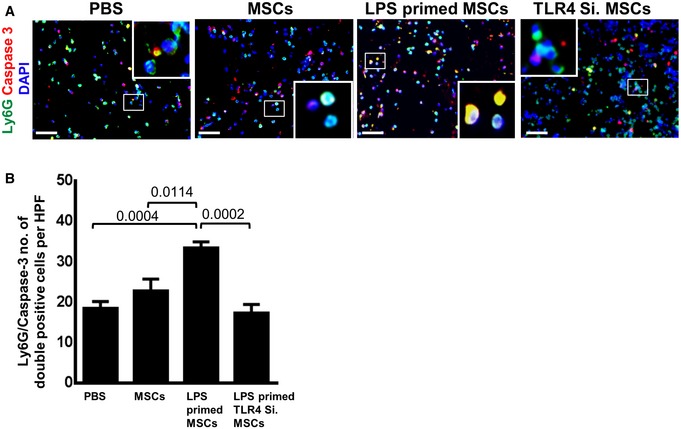
- Representative photomicrographs of sections from differently injected day 3 wounds stained for Ly6G+ neutrophils (green) and caspase 3 (red). Nuclei are stained with DAPI (blue). Double staining was performed for sections of day 3 wounds injected with PBS, non‐primed MSCs, LPS‐primed MSCs, and LPS‐primed TLR4‐silenced MSCs. Double‐stained cells indicate apoptotic neutrophils. To facilitate comparison, areas inside the rectangles are shown at 5× magnification in the insets. Scale bars: 50 μm.
- Quantification of Ly6G and caspase double‐positive cells on sections of differently injected day 3 wounds. A significant increase in double‐positive cells (Ly6G and caspase 3) in sections of wounds injected with LPS‐primed MSCs as opposed to wounds injected with LPS‐primed TLR4‐silenced MSCs and PBS control. Double‐positive cells were counted; statistical analysis was performed using one‐way ANOVA, and values are represented as mean ± SEM, six biological replicates.
To test this hypothesis, double immunostaining of macrophages was performed with F4/80, a macrophage marker, and with TGFβ‐1 which—according to previous data—predicts to be increased in macrophages after enhanced neutrophil phagocytosis. In fact, an increase in double‐positive F4/80+/TGFβ‐1+ macrophages in wounds injected with LPS‐primed MSCs as opposed to less double‐positive macrophages in wounds injected with non‐primed MSCs was observed (Fig 6D and E). Only few double‐stained macrophages were observed in wounds injected with PBS at day 5 after wounding (Fig 6D and E). Most interestingly, the number of double‐positive macrophages was severely reduced in wounds injected with LPS‐primed MSCs which were silenced for TLR4 (Fig 6D and E). This was indeed a strong evidence that TGFβ‐1 produced by macrophages causally depends on TLR4‐dependent MSC sensing which were mandatory for subsequent cellular events and eventually accelerates wound healing. In fact, we observed enhanced expression of CD31, indicative of new vessel formation, and α‐SMA, indicative of myofibroblasts in wounds injected with LPS‐primed MSCs when compared to wounds injected with non‐primed MSCs or PBS (Fig 6F and G).
Taken together, our results strongly indicate that MSCs sense infections in their microenvironment through TLR4 which relays the signal downstream and initiates a fundamental change in their transcriptome to enforce activation of neutrophils. Neutrophils thus are fully equipped to fight tissue infection. Apoptotic neutrophils at a later stage were phagocytosed by macrophages with enhanced release of TGFβ‐1. This, in consequence, stimulates myofibroblast differentiation and wound contraction, most likely angiogenesis, overall accelerating tissue repair (Fig 7).
Figure 7. LPS sensing of MSCs shapes the function of neutrophils at the wound site.
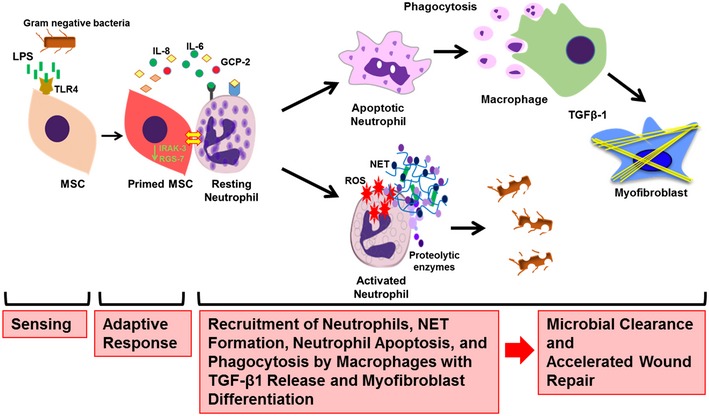
Graphical summary, depicting the molecules involved in sensing, signaling, and raising an adaptive response in MSCs. MSCs sense bacterial intruders as modeled by the key molecule LPS, a widely distributed PAMP and wall component of Gram‐negative bacteria. LPS is sensed via the TLR4 receptor, and the signal is relayed via MyD88 to downstream effectors which, in consequence, shape the adaptive MSC response. This response is enforced by fundamental transcriptomic reprogramming which is responsible for the release of factors critical for neutrophil and macrophage recruitment to the wound site. The adaptive response of LPS‐primed MSCs depicts a bifurcation with the activation of neutrophils with enhanced microbicidal NET formation and ROS release to directly counteract invading bacteria. In addition, in a second line of tissue protection and repair, apoptotic neutrophils are phagocytosed by macrophages. Neutrophil engulfment constitutes a strong signal for macrophages to release TGFβ‐1 which subsequently enhances differentiation of myofibroblasts and accelerates wound contraction and thus wound closure. Acceleration of wound closure together with enhanced NET formation and ROS release effectively counteracts microbial invasion. These data may stimulate new avenues to refine MSC‐based therapies for difficult‐to‐heal wounds and/or infected wounds.
Discussion
The major finding of this report is that a distinctly shaped adaptive response of MSCs—when confronted with an infected microenvironment—refines the innate immune system in particular neutrophils to accelerate tissue repair. These unprecedented data show how MSCs sense their neighborhood and, in consequence, undergo a fundamental transcriptomic reprogramming which adaptively regulates the recruitment and the activation state of neutrophils and macrophages at the wound site leading to a significant acceleration of wound healing. Employing complementary experimental approaches, we uncovered that priming of MSCs with bacterial wall LPS not only triggers neutrophil activation, but also initiates a cascade of cellular events which contributes to accelerated repair of skin wound injury (Fig 7).
Our data thus advance the emerging concept that MSCs sense exogenous and endogenous microenvironmental cues like LPS and, in consequence, mount a TLR4‐dependent adaptive response which augments neutrophil function and their activation, and following uptake by macrophages and enhanced TGFβ‐1 profoundly accelerates wound healing. Previously, MSC‐enforced modulation of inflammation was reported 46, 47, 48. In addition, a TLR‐dependent response of MSCs has been shown to modulate the function of natural killer cells, macrophages, and T cells 47, 48.
LPS‐primed MSCs when injected into wounds resulted in a significant acceleration of wound closure. Enhanced wound closure, in addition to the microbicidal activation of neutrophils, in fact, constitutes a major protection against further bacterial or other pathogen invasion. Thus, LPS‐primed MSCs raise a previously unknown, multifaceted effective defense and repair response. These findings together with earlier reports 49 show that TLR4 expressed on MSCs serves as a critical sensor that triggers LPS‐induced cell signaling via MyD88 to downstream pro‐inflammatory genes finally shaping the adaptive MSC response.
The fingerprint of this LPS‐induced adaptive MSC response not only enhances neutrophil recruitment to the wound site, but also promotes their activation in terms of enhanced NET formation, proteolytic enzymes, and ROS release. These events lead to a full microbicidal neutrophil activation response including strong oxidative burst, which earlier has been shown to initiate NET formation and the apoptosis of neutrophils 50, 51. Macrophage‐recruiting factors released by the LPS‐primed MSCs and by resident neutrophils themselves 52, 53, 54 result in the recruitment of macrophages to the wound site, which engulf apoptotic neutrophils, and this initiates the release of TGFβ‐1, which earlier was reported to induce myofibroblast formation and wound contraction 43, 44.
These data imply a beneficial contribution of LPS‐primed MSCs in accelerating mammalian tissue repair in vivo and, thus, are unprecedented. Of major interest and in its consistency previously unreported is that LPS‐primed MSCs silenced for TLR4 completely failed to accelerate tissue repair and down‐regulate the complete sequence of cellular events required for enhanced wound healing. Accordingly, LPS‐primed MSCs silenced for TLR4 lost their capacity to enhance neutrophil and macrophage recruitment/activation and, in consequence, to stimulate TGFβ‐1 expression, angiogenesis, and myofibroblast‐dependent wound contraction.
Additional mechanisms that may play a role in accelerating wound healing in wounds injected with LPS‐primed MSCs can currently not be excluded. In a recent report, injection of cell‐free exosomes from LPS‐treated bone marrow‐derived MSCs led to an attenuation of impaired wound healing in diabetic mice due to let‐7b present in the cargo of the applied exosomes. Let‐7b apparently switches pro‐inflammatory M1 macrophages which are predominant in murine diabetic wounds into anti‐inflammatory regenerative M2 macrophages 16.
So far, activation of neutrophils has widely been associated with chronic, difficult‐to‐treat conditions, including immune complex‐induced vasculitis 29, 55, lupus erythematodes 56, and other neutrophil‐dominated skin disorders 57, all characterized by chronic wounds and tissue breakdown due to persisting and unrestrained neutrophil activation. Our data are novel and interesting in that they prove that a transient, well‐controlled activation of neutrophils—by contrast to persisting conditions of chronically activated neutrophils—rather exerts beneficial effects on tissue repair upon injection of LPS‐primed MSCs into wounds. Our findings thus distinctly support the recently emerging concept of regenerative inflammation where neutrophils are suggested to beneficially impact on tissue repair 53.
Consistent with herein reported findings, a recent report proposed the existence of an inflammatory memory in inflammation‐experienced keratinocytes and demonstrates that inflammation trains the skin to heal faster 58. The authors further showed that the first bout of inflammation sensitizes cells to maintain chromosomal accessibility at key stress response genes. Upon a second inflammatory stimulus, genes governed by these domains are transcribed rapidly and hasten tissue regeneration 58. This may also apply for MSCs, and it is well possible that LPS priming also enhances the accessibility of neutrophil‐activating and other described genes which upon wound induced inflammation are faster transcribed, thus promoting enhanced wound healing.
In aggregate, our findings identify a novel concept of MSC priming which holds promise to refine MSC‐based therapies in clinical routine for difficult‐to‐treat wounds.
Materials and Methods
Mice and human samples
C57BL/6 male mice 8–10 weeks of age were used. Mice were held under specific pathogen‐free conditions and kept with free access to food and water at the animal care facility at Ulm University in compliance with the German Law for Welfare of Laboratory Animals. All animal experiments were approved by the responsible authorities at Ulm University. Human adipose‐derived mesenchymal stem cells were purchased from the American Type Culture Collection (ATCC) (LGC Standards GmbH, Wesel Germany). MSCs were seeded at a density of 3 × 103 cells/cm2 in mesenchymal stem cell basal medium with mesenchymal stem cell growth kit, low serum (ATCC), and cultured at 37°C under 5% CO2. MSCs were harvested with Accutase (PAA Laboratories, Pasching, Austria) at 70–80% confluence. MSCs at passages 3–5 were used for subsequent experiments.
Human peripheral neutrophils were isolated from buffy coats (German Red Cross, Ulm, Germany) using the Human Neutrophil Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's instructions.
Flow cytometry
Mesenchymal stem cells were harvested, washed with PBS, and incubated with antigen‐specific antibodies for 1 h at 4°C in the dark. The non‐specific staining was controlled by isotype‐matched antibodies for the corresponding fluorochrome channels (Appendix Fig S5A). Flow cytometry was performed on a Cytomics FC500 Flow Cytometer (Beckman Coulter, Brea, California). Data were analyzed with FlowJo (FlowJo, LLC, USA).
Adipogenic, osteogenic, and chondrogenic differentiation of MSCs
To assess the potential for lineage differentiation, adipogenic, osteogenic, and chondrogenic differentiation as a major part for the definition MSCs (Appendix Fig S5B–D), differentiation assays were performed as described previously 59. Briefly, MSCs at a density of 1 × 102/ml were seeded per well on 6‐well plate in respective differentiation media for 21 days. MSCs were subjected to Oil‐Red‐O staining indicative of adipogenic differentiation and Alizarin Red staining indicative of osteogenic differentiation after fixation 4% formaldehyde for 1 h. For chondrogenic differentiation, 2.5 × 105 MSCs were suspended in chondrogenic differentiation medium supplemented with TGFβ‐3 (Lonza, Basel, Switzerland) in 15‐ml tubes, and centrifuged at 150 g for 5 min, and the pellets were kept at 37°C, 5% CO2 incubator for 21 days with loosen caps. Media were replenished every 4 days. The pellets kept in growth medium were used as control. Pellets were embedded in OCT (Tissue‐TEK, CA, USA), and cryosections (5 μm) were prepared.
Histology and immunofluorescence staining
For immunostaining, 6‐μm‐thick cryosections were fixed in cold acetone for 10 min and blocked with 5% BSA in PBS for 1 h. Sections were incubated overnight with goat anti‐human aggrecan polyclonal antibody (R&D) at 5 μg/ml. Goat IgG (Life Technologies, NY, USA) was used as control. AF555‐conjugated rabbit anti‐goat was used as secondary antibody. Paraffin‐embedded sections were rehydrated and subjected to hematoxylin (Sigma‐Aldrich) and eosin (Sigma‐Aldrich) staining. For immunostaining, sections were subjected to antigen retrieval with target retrieval solution (DAKO) in a steam cooker for 5–7 min, followed by permeabilization with 0.25% Triton X‐100 (Sigma‐Aldrich) for 10 min and blocking with 5% BSA and 5% goat serum in PBS for 1 h. Thereafter, sections were incubated with anti‐mouse Ly6G (Abcam), anti‐DNA‐histone‐1 (Merck Millipore), anti‐MPO (R&D), and anti‐NE (Abcam) antibodies at 4°C overnight. In addition, we employed primary antibodies against α‐SMA (Progen), CD206 (Biorbyt), CXCL5 (Gene Tex), CXCL6 (Biorbyt), IL‐1β (Abcam), IL‐6 (R&D), IL‐8 (R&D), β2M (Signaling), F4/80 (eBioscience), TGFβ‐1 and CD31 (Cell Signaling), MIP2/KC and GCSF (R&D), and LIX (Biorbyt). Respective isotypes served as negative controls, and AF488‐ and AF555‐conjugated donkey IgG (Life Technologies) served as secondary antibodies. Nuclei were counterstained with DAPI. Coverslips were mounted with fluorescence mounting medium (DAKO). Photomicrographs were taken by using a Zeiss Axio Imager M1 microscope with an AxioCam digital color camera and AxioVision software v4.8 (Carl Zeiss) and confocal laser scanning microscope (Leica TCS SP8).
NET formation assay
1 × 105 MSCs were seeded into 6‐well plates. MSCs were treated with different LPS concentrations (10 ng/ml, 100 ng/ml, and 1,000 ng/ml) (Sigma‐Aldrich, St. Louis). MSCs were washed with phosphate‐buffered saline (PBS) and incubated at 37°C with 5% CO2. Neutrophils at a ratio of 1:10 were co‐cultured with MSCs for 2 h. Thereafter, 10 ng/ml phorbol 12‐myristate 13‐acetate (PMA) (Sigma‐Aldrich, St. Louis) was added and cells were incubated for 2.5 h at 37°C. Neutrophils treated with DMSO served as non‐activated controls. Cells were fixed with 4% PFA overnight. NET formation was assessed by the NET Elastase Activity Assay Kit (Cayman Chemical, USA) or by NET staining.
NET staining
For immunostaining of chromatin, human neutrophils and MSCs were co‐cultured in glass chamber slides (Millicell EZ slide, Merck Millipore, Germany) under conditions as described above. Cells were fixed overnight at 4°C in 1% paraformaldehyde (PFA) solution. Samples were blocked with 3% BSA for 1 h. Thereafter, samples were subjected to incubation with the primary antibody, anti‐DNA‐histone 1 antibody (1:500 dilution, Merck Millipore) for 2 h followed by AF555‐conjugated goat anti‐mouse IgG (1:200 dilution, Life Technologies) for 1 h. Nuclei were counterstained with DAPI. Photomicrographs were taken using a Zeiss Axio Imager M1 microscope as described previously.
Western blot
Western blot analyses were performed as previously described 60.
RNA isolation and quantitative RT–PCR
Cell pellets were incubated with 100 μl of RNA later (Ambion, Cat # AM7020). mRNA was isolated from MSCs either treated or non‐treated with LPS at a concentration of 100 ng/ml for 6 and 24 h, respectively, using Qiagen reagent (Life Technologies, Grand Island, NY, USA), and samples were stored at −80°C until further analysis. Quantitative RT–PCR was then performed employing specific primer sets (Appendix Table S2). For RNAseq analysis, RNA quality was verified with Bioanalyzer and RNA samples were considered for further analysis when showing RIN numbers between 9 and 10. Non‐primed MSCs served as controls.
Lymphocyte isolation and mixed lymphocyte reaction (MLR)
PBMCs were obtained from peripheral blood of healthy donors. Isolation was performed by density centrifugation on Biocoll separating solution (1.077 g/ml, Biochrom). MLRs were carried out in RPMI 1640, 5% fetal calf serum (Sigma‐Aldrich), 2 mM l‐glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin–streptomycin, and 0.05 mM 2‐mercaptoethanol at 37°C and 7.5% CO2. 2 × 106 PBMCs/ml were labeled with 5 μM CFSE (Thermo Fisher Scientific) for 10 min at 37°C followed by several washing steps with ice‐cold PBS‐5% FCS. 2.5 × 105 CFSE‐labeled PBMCs were stimulated with 2.5 × 105 irradiated allogeneic PBMCs (30 Gy) in the presence or absence of 2.5 × 105 MSCs. After 7 days, cells were stained for CD2‐APC (cl. RPA‐2.10, eBioscience) and 7‐AAD (Sigma) and proliferation was determined on CD2+ 7‐AAD− cells.
Wound healing model
Mice were anesthetized by ketamine (10 g/l) and xylazine (8 g/l) intraperitoneal injections (10 μl/g body weight). After shaving and cleaning the dorsal skin, four full‐thickness excisional wounds were generated on the back of each mice using 6‐mm round punchers (STIEFEL, Offenbach, Germany). Thereafter, 1 × 106 of either LPS‐primed, non‐primed or LPS‐primed TLR4‐silenced MSCs were injected into wound margins. PBS injections served as control. Each wound area was digitally photographed at 0, 3, 7, and 10 days. Wound areas were calculated using Photoshop software (version 7.0; Adobe Systems, San Jose, CA). Wound tissue was harvested and stored either in −80°C or fixed in 4% paraformaldehyde.
Scanning electron microscopy
Samples were fixed in 2.5% glutaraldehyde, 0.1% phosphate–cacodylate buffer, and 1% saccharose solution overnight. Samples were processed in the core facility of electron microscopy at Ulm University and analyzed under a field emission scanning electron microscope (ZEISS DSM 962) at 10 kV.
Small interfering RNA (siRNA) transfection
Mesenchymal stem cells were seeded at 1 × 105 cells per well in six‐well plate in antibiotic‐free medium overnight at 37°C. Human TLR4 siRNA smart pool and negative control were purchased from Accell, Germany. Samples were diluted in 1 × siRNA buffer according to the manufacturer's instructions. MSCs were transfected with human TLR4 siRNAs or control siRNA at 1 μM concentration in siRNA delivery media (Accell, Germany) for 72 h at 37°C with 5% CO2.
RNAseq library preparation and sequencing
Total RNA was isolated from the MSCs, and rRNA was removed using Ribo‐Zero Gold (Illumina). The rRNA‐depleted RNA was used for library preparation using TruSeq Directional RNAseq Kit (Illumina). Libraries were then sequenced in Illumina NextSeq 500. For data processing demultiplexed FASTQ reads were aligned to reference genome using Tophat2/Bowtie2 index. The aligned reads were then used for Cufflinks‐based transcript assembly and Cuffdiff‐based quantification of gene expression as well as differential gene expression (PRJNA469379; GEO: GSE114189; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114189).
Pathway impact analysis
The pathway plot was analyzed via Ingenuity Pathway Analysis (IPA), QIAGEN, using impact analysis that looks at over‐representation of differentially expressed genes in a given pathway and the total gene accumulation in a given pathway. The underlying pathway topologies, comprised of genes and their directional interactions, are obtained from the IPA database.
Hierarchical clustering
The hierarchical clustering and Venn diagram were carried out using various packages in RStudio environment (https://rstudio.com/) employing differentially expressed genes.
Statistical calculations
Data were analyzed with GraphPad Prism software (GraphPad Software, Inc., San Diego, CA). Quantification data are presented as the mean ± SEM from three independent experiments. Significance was defined with P values.
Author contributions
SM was involved in the design of this study and the experiments, performed experiments, and drafted the manuscript. KS designed many experiments and was involved in data analysis. MW, GS, and HG furthered the conceptual view and were involved in designing specific experiments. PM, DJ, and AB technically supported this work and helped with conceptual discussion of experiments. PH and LK supported this study technically. KS‐K conceived and supervised the study and wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
KS‐K is supported by the Graduate Training Group GRK 1789 “Cellular and Molecular Mechanisms in Aging (CEMMA)”, the Förderlinie Perspektivförderung “Zelluläre Entscheidungs‐ und Signalwege bei der Alterung” of the Ministerium für Wissenschaft, Forschung und Kunst Baden‐Württemberg, Germany, and the Collaborative Research Center 1149 for trauma research at Ulm. SM is supported by Deutscher Akademischer Austauschdienst (DAAD). We thank Dr. Angelika Rück (confocal and multiphoton microscopy) and Prof. Paul Walther (electron microscopy) from core facility of Ulm University for their support.
EMBO Reports (2020) 21: e48777
Contributor Information
Karmveer Singh, Email: karmveer.singh@uni-ulm.de.
Karin Scharffetter‐Kochanek, Email: karin.scharffetter-kochanek@uniklinik-ulm.de.
Data availability
The RNASeq data used in this present study were deposited in the Gene Expression Omnibus (GEO) with accession number GSE114189 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114189).
References
- 1. Hocking AM, Gibran NS (2010) Mesenchymal stem cells: paracrine signaling and differentiation during cutaneous wound repair. Exp Cell Res 316: 2213–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bernardo ME, Fibbe WE (2013) Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 13: 392–402 [DOI] [PubMed] [Google Scholar]
- 3. Kfoury Y, Scadden DT (2015) Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 16: 239–253 [DOI] [PubMed] [Google Scholar]
- 4. Foubert P, Barillas S, Gonzalez AD, Alfonso Z, Zhao S, Hakim I, Meschter C, Tenenhaus M, Fraser JK (2015) Uncultured adipose‐derived regenerative cells (ADRCs) seeded in collagen scaffold improves dermal regeneration, enhancing early vascularization and structural organization following thermal burns. Burns 41: 1504–1516 [DOI] [PubMed] [Google Scholar]
- 5. Strong AL, Bowles AC, MacCrimmon CP, Frazier TP, Lee SJ, Wu X, Katz AJ, Gawronska‐Kozak B, Bunnell BA, Gimble JM (2015) Adipose stromal cells repair pressure ulcers in both young and elderly mice: potential role of adipogenesis in skin repair. Stem Cells Transl Med 4: 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang D, Qi Y, Walker NG, Sindrilaru A, Hainzl A, Wlaschek M, MacNeil S, Scharffetter‐Kochanek K (2013) The effect of adipose tissue derived MSCs delivered by a chemically defined carrier on full‐thickness cutaneous wound healing. Biomaterials 34: 2501–2515 [DOI] [PubMed] [Google Scholar]
- 7. Lau K, Paus R, Tiede S, Day P, Bayat A (2009) Exploring the role of stem cells in cutaneous wound healing. Exp Dermatol 18: 921–933 [DOI] [PubMed] [Google Scholar]
- 8. Badiavas EV, Falanga V (2003) Treatment of chronic wounds with bone marrow‐derived cells. Arch Dermatol 139: 510–516 [DOI] [PubMed] [Google Scholar]
- 9. Dash NR, Dash SN, Routray P, Mohapatra S, Mohapatra PC (2009) Targeting nonhealing ulcers of lower extremity in human through autologous bone marrow‐derived mesenchymal stem cells. Rejuvenation Res 12: 359–366 [DOI] [PubMed] [Google Scholar]
- 10. Falanga V, Iwamoto S, Chartier M, Yufit T, Butmarc J, Kouttab N, Shrayer D, Carson P (2007) Autologous bone marrow‐derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng 13: 1299–1312 [DOI] [PubMed] [Google Scholar]
- 11. Lu D, Chen B, Liang Z, Deng W, Jiang Y, Li S, Xu J, Wu Q, Zhang Z, Xie B et al (2011) Comparison of bone marrow mesenchymal stem cells with bone marrow‐derived mononuclear cells for treatment of diabetic critical limb ischemia and foot ulcer: a double‐blind, randomized, controlled trial. Diabetes Res Clin Pract 92: 26–36 [DOI] [PubMed] [Google Scholar]
- 12. Yoshikawa T, Mitsuno H, Nonaka I, Sen Y, Kawanishi K, Inada Y, Takakura Y, Okuchi K, Nonomura A (2008) Wound therapy by marrow mesenchymal cell transplantation. Plast Reconstr Surg 121: 860–877 [DOI] [PubMed] [Google Scholar]
- 13. Martin I, Galipeau J, Kessler C, Le Blanc K, Dazzi F (2019) Challenges for mesenchymal stromal cell therapies. Sci Transl Med 11: eaat2189 [DOI] [PubMed] [Google Scholar]
- 14. Galipeau J, Sensebe L (2018) Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell 22: 824–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Basu A, Munir S, Mulaw MA, Singh K, Crisan D, Sindrilaru A, Treiber N, Wlaschek M, Huber‐Lang M, Gebhard F et al (2018) A Novel S100A8/A9 induced fingerprint of mesenchymal stem cells associated with enhanced wound healing. Sci Rep 8: 6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ti D, Hao H, Tong C, Liu J, Dong L, Zheng J, Zhao Y, Liu H, Fu X, Han W (2015) LPS‐preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome‐shuttled let‐7b. J Transl Med 13: 308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anderson GR, Posokhova E, Martemyanov KA (2009) The R7 RGS protein family: multi‐subunit regulators of neuronal G protein signaling. Cell Biochem Biophys 54: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008) Wound repair and regeneration. Nature 453: 314–321 [DOI] [PubMed] [Google Scholar]
- 19. Eming SA, Wynn TA, Martin P (2017) Inflammation and metabolism in tissue repair and regeneration. Science 356: 1026–1030 [DOI] [PubMed] [Google Scholar]
- 20. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A et al (2011) An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 121: 985–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175 [DOI] [PubMed] [Google Scholar]
- 22. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13: 463–469 [DOI] [PubMed] [Google Scholar]
- 23. Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 22: 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abdi J, Rashedi I, Keating A (2018) Concise Review: TLR pathway‐miRNA interplay in mesenchymal stromal cells: regulatory roles and therapeutic directions. Stem cells 36: 1655–1662 [DOI] [PubMed] [Google Scholar]
- 25. Petri RM, Hackel A, Hahnel K, Dumitru CA, Bruderek K, Flohe SB, Paschen A, Lang S, Brandau S (2017) Activated tissue‐resident mesenchymal stromal cells regulate natural killer cell immune and tissue‐regenerative function. Stem Cell Reports 9: 985–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas H, Jager M, Mauel K, Brandau S, Lask S, Flohe SB (2014) Interaction with mesenchymal stem cells provokes natural killer cells for enhanced IL‐12/IL‐18‐induced interferon‐gamma secretion. Mediators Inflamm 2014: 143463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiossone L, Conte R, Spaggiari GM, Serra M, Romei C, Bellora F, Becchetti F, Andaloro A, Moretta L, Bottino C (2016) Mesenchymal stromal cells induce peculiar alternatively activated macrophages capable of dampening both innate and adaptive immune responses. Stem Cells 34: 1909–1921 [DOI] [PubMed] [Google Scholar]
- 28. Rashedi I, Gomez‐Aristizabal A, Wang XH, Viswanathan S, Keating A (2017) TLR3 or TLR4 activation enhances mesenchymal stromal cell‐mediated treg induction via notch signaling. Stem Cells 35: 265–275 [DOI] [PubMed] [Google Scholar]
- 29. Jiang D, Muschhammer J, Qi Y, Kugler A, de Vries JC, Saffarzadeh M, Sindrilaru A, Beken SV, Wlaschek M, Kluth MA et al (2016) Suppression of neutrophil‐mediated tissue damage‐A novel skill of mesenchymal stem cells. Stem Cells 34: 2393–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaisho T, Akira S (2006) Toll‐like receptor function and signaling. J Allergy Clin Immunol 117: 979–987; quiz 988 [DOI] [PubMed] [Google Scholar]
- 31. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F (1999) Toll‐like receptor‐4 mediates lipopolysaccharide‐induced signal transduction. J Biol Chem 274: 10689–10692 [DOI] [PubMed] [Google Scholar]
- 32. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM (2014) Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell 13: 690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y (2008) Mesenchymal stem cell‐mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2: 141–150 [DOI] [PubMed] [Google Scholar]
- 34. Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D (2010) CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol 30: 39–45 [DOI] [PubMed] [Google Scholar]
- 35. Tan X, Fujiu K, Manabe I, Nishida J, Yamagishi R, Nagai R, Yanagi Y (2015) Choroidal neovascularization is inhibited via an intraocular decrease of inflammatory cells in mice lacking complement component C3. Sci Rep 5: 15702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N, Stratis A, Renkl AC, Sunderkotter C, Wlaschek M et al (2006) Activated macrophages are essential in a murine model for T cell‐mediated chronic psoriasiform skin inflammation. J Clin Invest 116: 2105–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schutze S, Wiegmann K, Machleidt T, Kronke M (1995) TNF‐induced activation of NF‐kappa B. Immunobiology 193: 193–203 [DOI] [PubMed] [Google Scholar]
- 38. Liu T, Zhang L, Joo D, Sun SC (2017) NF‐κB signaling in inflammation. Signal Transduct Target Ther 2: 17023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN (2011) IL‐1alpha and IL‐1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187: 4835–4843 [DOI] [PubMed] [Google Scholar]
- 40. Akiyama K, You YO, Yamaza T, Chen C, Tang L, Jin Y, Chen XD, Gronthos S, Shi S (2012) Characterization of bone marrow derived mesenchymal stem cells in suspension. Stem Cell Res Ther 3: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neves CS, Gomes SS, dos Santos TR, de Almeida MM, de Souza YO, Garcia RM, Otoni WC, Chedier LM, Viccini LF, de Campos JM (2016) The phytoecdysteroid beta‐ecdysone is genotoxic in Rodent Bone Marrow Micronuclei and Allium cepa L. Assays. J Ethnopharmacol 177: 81–84 [DOI] [PubMed] [Google Scholar]
- 42. Tecchio C, Micheletti A, Cassatella MA (2014) Neutrophil‐derived cytokines: facts beyond expression. Front Immunol 5: 508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest 101: 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peters T, Sindrilaru A, Hinz B, Hinrichs R, Menke A, Al‐Azzeh EA, Holzwarth K, Oreshkova T, Wang H, Kess D et al (2005) Wound‐healing defect of CD18(‐/‐) mice due to a decrease in TGF‐beta1 and myofibroblast differentiation. EMBO J 24: 3400–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G (1993) Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122: 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vander Beken S, de Vries JC, Meier‐Schiesser B, Meyer P, Jiang D, Sindrilaru A, Ferreira FF, Hainzl A, Schatz S, Muschhammer J et al (2019) Newly defined ATP‐binding cassette subfamily B member 5 positive dermal mesenchymal stem cells promote healing of chronic iron‐overload wounds via secretion of interleukin‐1 receptor antagonist. Stem Cells 37: 1057–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Le Blanc K, Mougiakakos D (2012) Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol 12: 383–396 [DOI] [PubMed] [Google Scholar]
- 48. Prockop DJ, Oh JY (2012) Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther 20: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM (2010) A new mesenchymal stem cell (MSC) paradigm: polarization into a pro‐inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One 5: e10088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kirchner T, Moller S, Klinger M, Solbach W, Laskay T, Behnen M (2012) The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm 2012: 849136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lundqvist‐Gustafsson H, Bengtsson T (1999) Activation of the granule pool of the NADPH oxidase accelerates apoptosis in human neutrophils. J Leukoc Biol 65: 196–204 [DOI] [PubMed] [Google Scholar]
- 52. Soehnlein O (2018) Decision shaping neutrophil‐platelet interplay in inflammation: from physiology to intervention. Eur J Clin Invest 48: e12871 [DOI] [PubMed] [Google Scholar]
- 53. Soehnlein O, Steffens S, Hidalgo A, Weber C (2017) Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol 17: 248–261 [DOI] [PubMed] [Google Scholar]
- 54. Wantha S, Alard JE, Megens RT, van der Does AM, Doring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL et al (2013) Neutrophil‐derived cathelicidin promotes adhesion of classical monocytes. Circ Res 112: 792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, Luscinskas FW, Mayadas TN (2001) Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune‐mediated inflammation. Immunity 14: 693–704 [DOI] [PubMed] [Google Scholar]
- 56. Kahlenberg JM, Carmona‐Rivera C, Smith CK, Kaplan MJ (2013) Neutrophil extracellular trap‐associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J immunol 190: 1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cohen PR (2009) Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol 10: 301–312 [DOI] [PubMed] [Google Scholar]
- 58. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, Polak L, Kulukian A, Chai S, Fuchs E (2017) Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550: 475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Singh K, Krug L, Basu A, Meyer P, Treiber N, Vander Beken S, Wlaschek M, Kochanek S, Bloch W, Geiger H et al (2017) Alpha‐ketoglutarate curbs differentiation and induces cell death in mesenchymal stromal precursors with mitochondrial dysfunction. Stem Cells 35: 1704–1718 [DOI] [PubMed] [Google Scholar]
- 60. Singh K, Maity P, Krug L, Meyer P, Treiber N, Lucas T, Basu A, Kochanek S, Wlaschek M, Geiger H et al (2015) Superoxide anion radicals induce IGF‐1 resistance through concomitant activation of PTP1B and PTEN. EMBO Mol Med 7: 59–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The RNASeq data used in this present study were deposited in the Gene Expression Omnibus (GEO) with accession number GSE114189 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114189).