

Abstract
Calcium (Ca2+) release-activated Ca2+ (CRAC) channels mediated by STIM1/2 and ORAI (ORAI1–3) proteins form the dominant store-operated Ca2+ entry (SOCE) pathway in a variety of cells. Among these, the enamel-forming cells known as ameloblasts rely on CRAC channel function to enable Ca2+ influx, which is important for enamel mineralization. This key role of the CRAC channel is supported by human mutations and animal models lacking STIM1 and ORAI1, which results in enamel defects and hypomineralization. A number of recent reports have highlighted the role of the chanzyme TRPM7 (transient receptor potential melastanin 7), a transmembrane protein containing an ion channel permeable to divalent cations (Mg2+, Ca2+), as a modulator of SOCE. This raises the question as to whether TRPM7 should be considered an alternative route for Ca2+ influx, or if TRPM7 modifies CRAC channel activity in enamel cells. To address these questions, we monitored Ca2+ influx mediated by SOCE using the pharmacological TRPM7 activator naltriben and the inhibitor NS8593 in rat primary enamel cells and in the murine ameloblast cell line LS8 cells stimulated with thapsigargin. We also measured Ca2+ dynamics in ORAI1/2-deficient (shOrai1/2) LS8 cells and in cells with siRNA knock-down of Trpm7. We found that primary enamel cells stimulated with theTRPM7 activator potentiated Ca2+ influx via SOCE compared to control cells. However, blockade of TRPM7 with NS8593 did not decrease the SOCE peak. Furthermore, activation of TRPM7 in shOrai1/2 LS8 cells lacking SOCE failed to elicit Ca2+ influx, and Trpm7 knock-down had no effect on SOCE. Taken together, our data suggest that TRPM7 is a positive modulator of SOCE potentiating Ca2+ influx in enamel cells, but its function is fully dependent on the prior activation of the ORAI channels.
Keywords: Ca2+ signaling, CRAC channels, SOCE, TRPM7 channels, enamel cells
Graphical Abstract
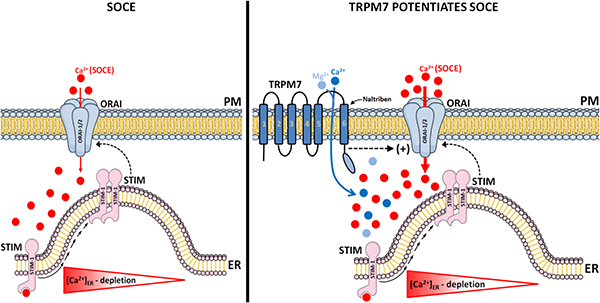
1. Introduction
It is well established that calcium (Ca2+) handling is one of the most widespread transducing systems, regulating a wide range of cell biology and pathophysiologic processes [1–5]. Enamel is the most calcified vertebrate tissue, requiring a steady supply of Ca2+ for its mineralization. Specialized epithelial cells known as ameloblasts first form, and then mineralize the enamel crystals, supported by adjacent cells of the enamel organ [6]. Enamel development is commonly defined in two stages: 1) a developmental (secretory); and 2) a mineralization (maturation) stage. The latter has been shown to feature a marked increase in ion transport, particularly Ca2+, as crystals grow in thickness [7–9]. Recent studies have shown that a key Ca2+ influx pathway in enamel cells is via store-operated Ca2+ entry (SOCE) mediated by the Ca2+ release-activated Ca2+ (CRAC) channels [10–13]. SOCE is widely expressed in many non-excitable cells [14–16]. Physiologically, a depletion in endoplasmic reticulum (ER) Ca2+ concentration ([Ca2+]ER) triggers the oligomerization and redistribution of the ER resident Ca2+ sensors stromal interaction molecules-STIM1 and STIM2 in close proximity to the plasma membrane. At these sites, STIM proteins couple to the highly Ca2+-selective conductance pore known as ORAI proteins (ORAI1–3), thereby opening the channel and enabling sustained Ca2+ influx [14,17,18].
Pharmacological and molecular data support the crucial role of SOCE in enamel cells. Selective ORAI inhibitors (synta-66, BTP2, GSK7975A) markedly reduce or nearly abolish SOCE in enamel cells, while mutations in STIM1 or ORAI1 genes in patients result in abnormal enamel with hypomineralization [7,11]. Mice lacking STIM1/2 or ORAI1 showed deficient SOCE and enamel defects [12,13]. Combined, these data highlight the critical role of SOCE as a key Ca2+ influx channel in enamel cells. However, recent reports have revealed that the transient receptor potential melastanin 7 (TRPM7) is a modulator of SOCE [19,20] and that Trpm7-inactive knock-in mutant mice exhibited hypomineralized enamel [21]. As the genetic disruption of TRPM7 (Trpm7−/−) is embryonic lethal in mice [21,22], analyses of the enamel phenotype performed in heterozygous mice showed deficient mineralization of their enamel and cranial bones [21]. These phenotypes were attributed to Mg2+ deficiency, which was required for alkaline phosphatase activity and mineralization [21]. In addition to reduced enamel volume, another study suggested that TRPM7 was involved in the early stages of amelogenesis via cAMP response element binding protein (CREB) [23]. Therefore, both TRPM7 and CRAC components are expressed in enamel cells, and it would be of interest to understand the potential functional links between these two distinct channels.
The ORAI family of proteins (ORAI1–3) bear little homology to other channels and are quite distinct from TRPM7 [16]. All ORAI proteins share a structure with both N- and C-termini located in the cytosol and display four transmembrane (TM) domains with two extra and two intracellular loops [14,16]. ORAI proteins likely form heteromeric units and it is generally considered that hexamers of the first TM of ORAI1 form the central pore [16], lined by a number of glutamate amino acids near the outer end that confer high Ca2+ selectivity [16,24]. TRPM7 is an ubiquitous transmembrane protein composed of an ion channel permeable to divalent cations (Mn2+, Ca2+, Mg2+, Co2+, Ni2+ and Zi2+) fused to a C-terminal α-kinase domain that phosphorylates downstream substrates [25,26]. TRPM7 contains six TM domains, with the channel pore found between TM5 and TM6 and the kinase domain in the C-terminus [27]. Ion channels exist in multiple highly dynamic conformational states associated with transitions between open, closed and inactivated steady-state [27–29]. A recent report showed lower cell proliferation in TRPM7 kinase-dead (KD) K1646R knock-in mice associated with inactivated (non-conducting state) or loss of pre-activated state of TRPM7 in K1646R knock-in mice [20].
Some of the main physiological functions of TRPM7 channels include maintaining the intracellular level of Mg2+ linked with cell proliferation and differentiation, regulating cell volume and playing a role in the immune system [22,30]. Recent evidence also indicates that TRPM7 not only contributes to Ca2+ homeostasis [19, 25, 31], but is also found in intracellular vesicles distinct from those of the endocytotic pathway [32]. Although several molecular targets have been postulated, the cellular and functional mechanisms through which TRPM7 activation occurs remain unclear [31]. TRPM7 have been linked to different downstream signaling pathways through the C-terminal α-kinase domain [25]. Previous evidence reported that the TRPM7 channel is activated by agonists of G protein-coupled receptors (GPCR) coupled to phospholipase C (PLC) [25,31]. From a pharmacologic viewpoint, a number of TRPM7 modulators, such as the selective agonist gating-modulator of the TRPM7 channel naltriben [33] and the reversible Mg2+-dependent TRPM7 channels antagonist NS8593, [34] allow us to tease apart the function of TRMP7 in enamel cells in vitro [33–35].
Given the important role of SOCE in enamel formation [10–12], and that TRPM7 is able to conduct Ca2+ as well [25], this raises the issue of whether TRPM7 can directly act as a Ca2+ influx channel or a modulator of SOCE in enamel cells. This will have implications for developing models of Ca2+ homeostasis in enamel cells and, potentially, for developing enamel regeneration models, as these depend on a full understanding of the physiological requirements of ameloblasts to induce mineralization. Using rat primary enamel cells from the secretory and maturation stages and the ameloblast cell line LS8 cells, we show that activation of the TRPM7 channel enhances Ca2+ influx, but only after SOCE stimulation. Moreover, TRPM7 activation cannot elicit Ca2+ influx in ORAI1/ORAI2-deficient ameloblasts and Trpm7 knock-down has no effect on Ca2+ influx via SOCE. Taken together, our findings suggest that TRPM7 is unable to stimulate Ca2+ influx in enamel cells in the absence of ORAI proteins. It can, however, positively modulate the effects of SOCE following CRAC channel activation.
2. Materials and methods
2.1. Animal use
All procedures employed in this study were conducted in accordance with guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of New York University College of Dentistry (protocol # IA16–00625).
2.2. Primary cell culture and line
Male and female Sprague Dawley rats (100–120 g) were used to isolate secretory (SEC) and maturation (MAT) enamel organ (EO) cells from the lower incisors, as previously described [9,12,36]. To obtain single cell populations, isolated cell clumps from each stage were transferred to Eppendorf tubes containing 1mL of Hanks’ balanced salt solution (Thermo Fisher, USA; #:14065-056) with 1% Antibiotic-Antimycotic (Thermo Fisher, USA; #:15240-062). Subsequently, EO cells were digested with 0.25 mg/ml of Liberase TL (Roche, Germany; #414654) for 30 minutes at 37°C in a 5%-CO2 incubator, manually pipetted every 10 minutes to mechanically separate the cells. The enzymatic reaction was stopped by adding 2mL of X-Vivo™ 15 (Lonza Bioscince, USA; #: 04–418Q) cell media containing 10% FBS (Thermo Fisher, USA; #:12483-020) and 1% Penicillin-Streptomycin (Thermo Fisher, USA; #:15140-122). Cells were centrifuged at 500 X g for 5 minutes, washed twice and plated on glass cover slips coated with Corning™ Cell-Tak (Fisher Scientific, USA; #:CB-40240). To confirm the isolation of SEC and MAT cell populations, we measured the mRNA expression of enamelin (ENAM) and ameloblast-associated protein (ODAM) that are the major secreted products of secretory and maturation stage ameloblasts, respectively [7,9, Fig. S1, A and B]. Isolated SEC and MAT cells were used within 24 hours after dissection. To increase cell purity in primary EO cells, we labeled fibroblasts using a PE-conjugated monoclonal anti-rat CD90 antibody. The immortalized murine ameloblasts cell line LS8 cells [37] that have been used widely to study various aspects of enamel development [38] were also used. LS8 cells are a good model to study Ca2+ signaling in the context of enamel formation as they express functional SOCE and the molecular components of the CRAC channels [10,12].
2.3. Retroviral transduction and siRNA
shRNAs against Orai1, Orai2 or Renilla (as a negative control) were cloned into the shRNA scaffold of the pLMP-mirE11 retroviral backbone (expressing GFP and puromycin resistance), as recently described [12]. To generate siRNA against Trpm7, LS8 cells were seeded at a density of 1.106 cells/well in 6-well plates and grown in DMEM (Thermo Fisher, USA; #:11885-092) containing 10% FBS and antibiotics. One day after seeding, cells (60–80% confluent) were transfected with siRNA against mouse Trpm7 using ON-TARGETplus Mouse Trpm7 reagents (Dharmacon, USA; #:J-040716-06-0002), according to the manufacturer’s instructions. Briefly, the plasmid containing Trpm7 siRNA (6 μL) was diluted in DMEM (150 μL) and transfected with Lipofectamine™ RNAiMAX (Termo Fisher, USA; #:13778-075) at a 1:1 ratio. After mixing at room temperature, the Trpm7 siRNA-lipid complex (25 pM) was incubated drop-wise onto the cells and cultured for two days. Control cells were exposed to transfectant in the absence of siRNA using ON-TARGETplus Non-targeting Pool (Dharmacon, USA; #:D-001810-10-05). Transfection and knock-down efficiency were monitored at the mRNA levels by RT-PCR of Trpm7 expression.
2.4. Real-time PCR of Orai1, Orai2 and Trpm7
Total RNA was isolated using the RNeasy Micro Kit (Qiagen, USA; #:74004), as indicated by the manufacturer, followed by reverse transcription using the iScript™ cDNA Synthesis Kit (Bio-Rad, USA; #:1708890). For qRT-PCR, we used the SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad, USA; #:1725270) and performed the experiments in a CFX Connect thermocycler (Bio-Rad, USA). Gapdh and β-actin were used as housekeeping genes. All primers were used at 0.25 nM. Relative quantification of gene expression was determined by the 2−ΔΔCT method [11,12]. All primer sequences used are shown in Table S1.
2.5. [Ca2+]cyt measurements
Cytosolic Ca2+ concentration ([Ca2+]cyt) measurements of SEC and MAT and LS8 cells were performed as described [10–12]. Briefly, cells were incubated for one hour at room temperature with 1 μM of a ratiometric Ca2+ probe, Fura-2-AM (Thermo Fisher, USA; #:F-1221), in Ca2+-free Ringer’s solution (pH 7.4) composed as follows (mM): 155.0 NaCl; 4.5 KCl; 1.0 MgCl2; 10 D-glucose; and 10 HEPES (Sigma-Aldrich, USA). To increase cell purity in primary EO cells, we labeled fibroblasts using a PE-conjugated monoclonal anti-rat CD90 antibody (1:500 dilution) (BioLegend, USA; #:105201) and excluded the latter from further analysis, as previously described [11]. Fluorescence recordings were obtained by: 1) an inverted light microscope (Nikon 2000U Eclipse, Japan) equipped with an objective (Nikon S Fluor × 20; numerical aperture: 0.45) coupled to device camera (CoolSNAP HQ2 CCD Camera; Photometrics, USA), controlled by computer software (NIS Elements version 4.20.01, USA); or 2) an inverted light microscope (Nikon Ti2-E Eclipse, Japan) equipped with an objective (Nikon S Fluor × 20; numerical aperture: 0.75) and a digital SLR camera (DS-Qi2; Nikon, Japan) controlled by computer software (NIS Elements version 5.20.01, USA).
Cells were continuously perfused by a six- or eight-way perfusion system (VC-6/8 valve controller) at 5–6 ml per minute with a common outlet 0.28-mm tube driven by electrically controlled valves (Harvard Bioscience Inc., USA). A normal Ringer’s solution (the same Ca2+-free Ringer’s solution, however, containing 2mM of Ca2+) or a Ca2+-free solution were used to dissolve all drugs (at room temperature). Fura-2-AM was excited alternatively at 340 and 380 nm using a Lambda LS xenon-arc lamp (Sutter Instrument, USA) or pE-340 fura (Cool Led, USA). Emitted fluorescence was collected through a 510 nm emission filter. Fluorescence images were generated at 5-second intervals and the ratio values were calculated. A Fura-2 calcium imaging calibration kit (Thermo Fisher, USA; #:F6774) was used to estimate the [Ca2+]cyt, according to the manufacturer’s specifications. Standard control buffer (background fluorescence), zero free-Ca2+ buffer (free-Ca2+) and 39 μM free-Ca2+ buffer (saturating Ca2+) were used to convert the emission ratio at 340/380 nm excitation to estimate the free [Ca2+]cyt.
2.5.1. SOCE measurements
To measure SOCE, ER Ca2+ store depletion was stimulated by pre-incubation with 1 μM of thapsigargin (Sigma-Aldrich, USA; #:T9033) for 20 minutes prior to the experimental protocols in Ca2+-free Ringer’s solution. Subsequently, under fluorescence recordings, cells were continuously perfused for 120 seconds with Ca2+-free Ringer’s solution, followed by a re-addition of 2 mM of extracellular Ca2+ in normal Ringer’s solution to measure the delta (Δ) SOCE peak. To block SOCE, cells were also preincubated with pharmacological CRAC antagonists synta-66 (5 μM) (Sigma-Aldrich, USA; #:1949) or with 3,5-bis trifluoromethyl pyrazole-BTP-2 (20 μM) (Sigma-Aldrich, USA; #:203890) for two hours [10,14,39]. Synta-66 concentration was determined after the construction of the concentration-effect curve (Fig. S2, B). The TRPM7 agonist, naltriben (NAL, 100 μM) (Tocris Bioscience, USA; #:0892) [31,35], was perfused simultaneously with the Ca2+ re-addition for 720 seconds. In other sets of experiments, the TRPM7 antagonist NS8593 (30 μM) (Tocris Bioscience, USA; #:4597) was simultaneously perfused during the Ca2+ re-addition, in the absence or presence of naltriben (100 μM), as described [35].
2.6. Bioinformatic analyses
The Pfam protein families database release, 32.0 [40], was used to retrieve information about the various domains of TRPM7 (UniProt ID Q923J1). Release 32.0 improved refinement of domain boundaries and their functional annotation. In particular, we highlight two domains in the TRPM7 protein: 1) the ion transfer domain of 246 amino acids (855–1108 amino acid equivalent to alignment region 858–1104 aa in Q923J1) spanning over exons 19–24 (this domain has six transmembrane helices in which the last two helices flank a loop that determines ion selectivity); 2) the MHCK/EF2 kinase domain of 195 amino acids (1617–1814 amino acid equivalent to alignment region 1619–1814 aa in Q923J1) spanning over exons 34–38 of isoform Trpm7–202. The kinase domain is from the alpha-kinase family [41]. Ensembl genome browser 97 was used to visualize the gene and the localization of the domains identified by Pfam release 32.0 as well as other databases (Fig. S5).
2.7. Data analyses and statistics
All data, mathematical analyses and graphs were analyzed and/or generated using the GraphPad Prism software version 8.0 (Inc., California, USA). The Δ SOCE peak, SOCE slope and area under the curve (A.U.C.) were calculated by integrating the transients versus time during the stimulus duration for each experiment. The SOCE slope (increase or decrease) parameter was fitted by the GraphPad Prism software using the one-phase association equation [2]. Data represents the mean ± SEM of the minimum of three independent experiments. Differences between the means of the group data that fit a normal distribution were analyzed using a two-tailed unpaired Student’s t-test variance or, when indicated, a one-way ANOVA followed by a Bonferroni’s multiple comparison post-hoc test. The limit of significance was established at *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05 vs. SEC group; n.s., non-significant.
3. Results
3.1. TRPM7 activation enhances SOCE in enamel cells
To determine the maximum capacity of Ca2+ mobilization of the well-known pharmacological TRPM7 activator, naltriben [33,34], we first constructed concentration-effect curves in LS8 cells as a reference (Fig. 1, A–B). The ameloblasts LS8 cells have been widely used in enamel research and also show robust SOCE [10,12,38]. Naltriben peaks were shown at 100 μM, as also reported elsewhere [35,42]. We used this concentration in primary cells and analyzed the effects of naltriben (NAL,100 μM) in secretory and maturation -stage ameloblasts under resting levels (2 mM Ca2+) showing that naltriben elicited a very small elevation in Ca2+ influx (~16 nM) in cells of both stages (Fig. 1, C–D). In addition, we found a small increase (~12 nM) in cytosolic Ca2+ in naltriben-stimulated cells in Ca2+-free solution (Fig. 1, E–F), suggesting that TRPM7 channels might release Ca2+ from the ER.
Figure 1. Effects of the TRPM7 activator, naltriben (NAL), in ameloblast cell line LS8 and primary enamel cells.
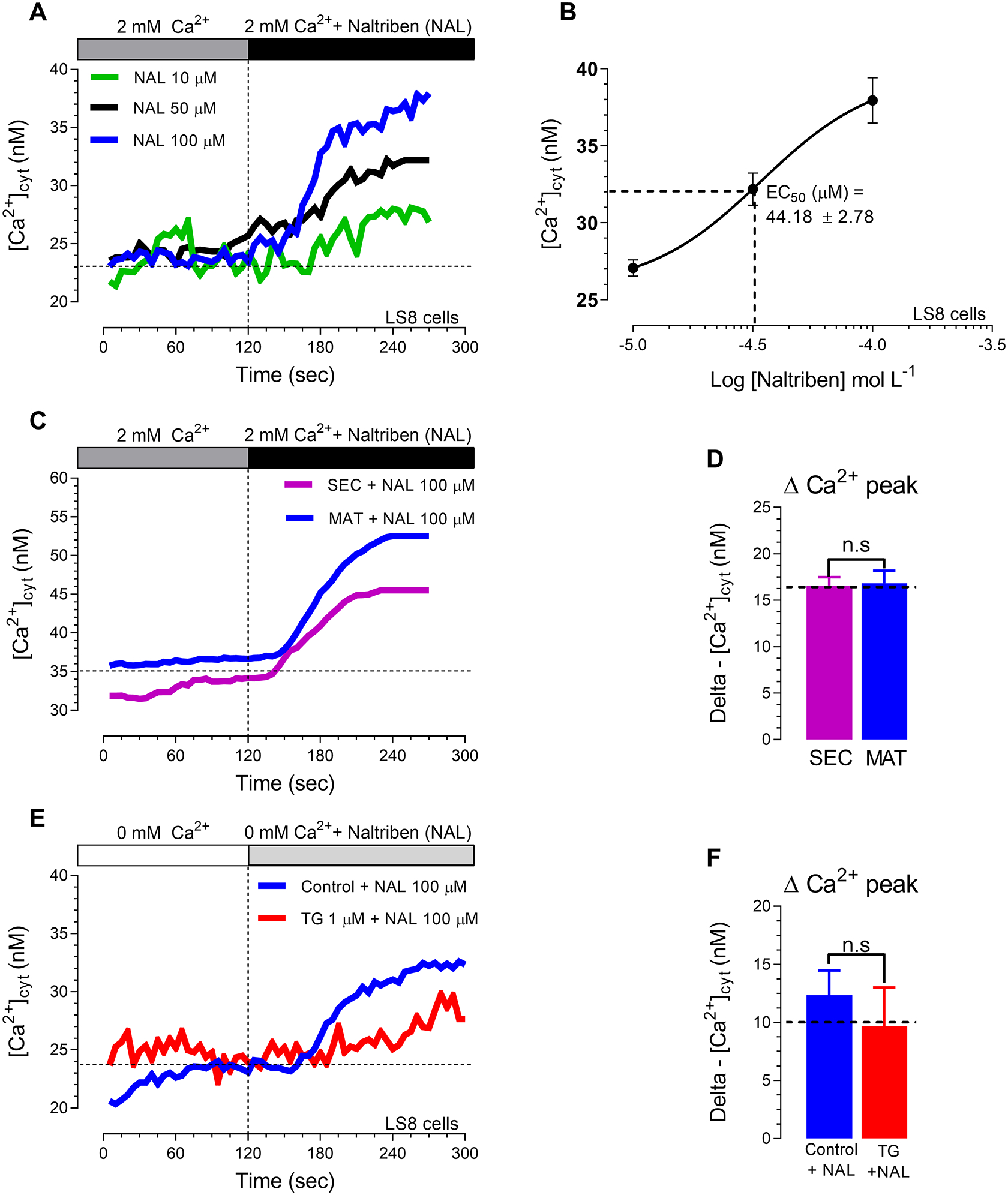
We constructed a concentration-effect curve in the enamel cell line (LS8 cells) using the well-known pharmacological TRPM7 activator, naltriben (A-B), to determine the maximum capacity of Ca2+ mobilization. Naltriben showed an EC50 around 45 μM (B) with Ca2+ peak in LS8 cells at 100 μM (A-B). We then used naltriben (100 μM) in primary ameloblasts from the secretory (SEC) and maturation (MAT) stages, (C-D). Changes in [Ca2+]cyt transients were monitored using naltriben (100 μM) after ER-Ca2+ depletion with thapsigargin (1 μM, 20 min, red trace) or without store depletion (blue trace) (E-F). Data represent the mean ± SEM of minimum three independent experiments. Data analyzed by two-tailed unpaired Student’s t-test at *P < 0.05; **P < 0.01; ***P < 0.001; n.s., non-significant.
To address whether TRPM7 is involved in Ca2+ influx mediated by SOCE in enamel cells, we first measured the expression level of Orai1 and Trpm7 by RT-PCR. Both Orai1 and Trpm7 expression levels were higher (~95% and ~65%, respectively) in maturation cells (Fig. 2, A–B). We also analyzed SOCE in primary enamel cells from the secretory and maturation stages without TRPM7 activation. All cells were pre-treated with thapsigargin (1 μ M, 20 minutes) to passively and maximally deplete the ER Ca2+ stores before measuring SOCE by re-addition of 2 mM Ca2+. Fura-2-AM-loaded cells showed significantly higher SOCE in maturation (Fig. 2, C–F). These data are also consistent with the increased expression and function of both STIM1 and ORAI1 in maturation stage by RT-PCR and protein level as we have reported elsewhere [10].
Figure 2. TRPM7 channel activation potentiates Ca2+ influx via SOCE in primary enamel cells.
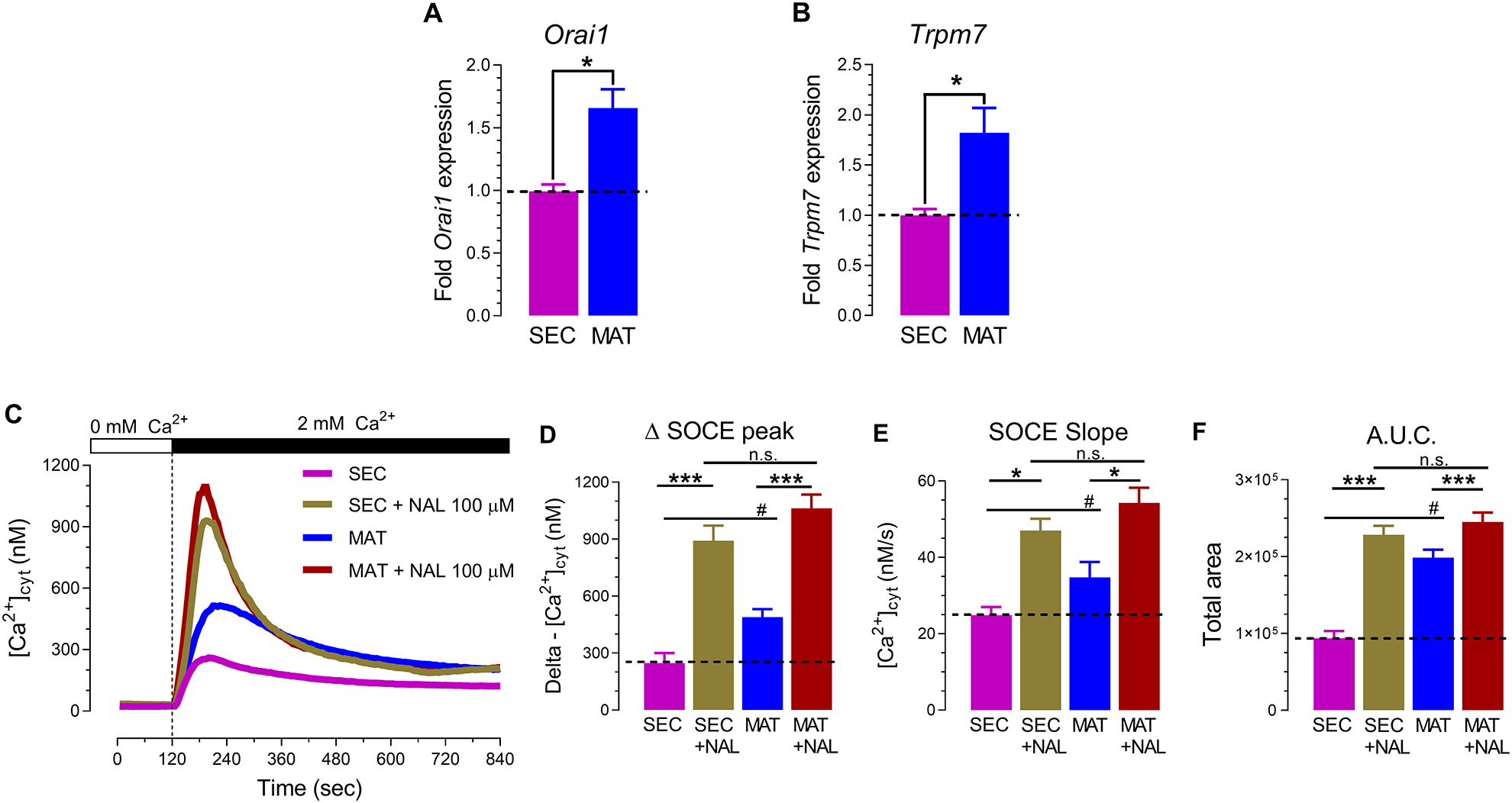
RT-PCR analyses of secretory (SEC) and maturation (MAT) stage enamel organ cells show increased expression of Orai1 (A) and Trpm7 (B) in maturation. (C) Representative original traces of [Ca2+]cyt transients in SEC and MAT ameloblasts recorded after pre-incubation with thapsigargin (20 minutes, 1 μM), followed by perfusion with a Ca2+-free Ringer’s solution (120 seconds) before simultaneous re-addition of 2 mM extracellular Ca2+ and naltriben (NAL,100 μM). (D) Quantification of delta SOCE peak, (E) SOCE slope, and (F) area under the curve (A.U.C.) data represent the mean ± SEM of minimum three independent experiments. Data analyzed by two-tailed unpaired Student’s t-test at *P < 0.05; **P < 0.01; ***P < 0.001 or one-way ANOVA followed by a Bonferroni’s multiple comparison post-hoc test at #P < 0.05 vs. SEC group; n.s., non-significant.
When perfused simultaneously during Ca2+ re-addition, naltriben (100 μM) was able to enhance SOCE in both secretory and maturation stage cells (Fig. 2 C–F). The delta (Δ) peak of Ca2+ influx was ~3-fold and ~2-fold higher in secretory and maturation, respectively, when TRPM7 was activated. The speed of Ca2+ influx also increased (Fig. 2 E). Taken together, these functional data indicate that the pharmacological activation of TRPM7 channel activity potentiates SOCE in primary enamel cells of both stages, although the enhancing effects are higher in secretory cells despite elevated levels of Trpm7 expression in maturation.
This difference between gene expression levels and potentiation of Ca2+ influx is unclear, but could be linked to the buffering capacity of SOCE by mitochondria [43], as mitochondria are more abundant in maturation stage cells [44] and therefore could sequester a higher Ca2+ load. Alternatively, Broertjes et al., [45] showed that overexpression of TRPM7 increases Ca2+ influx when associated with upregulation of second messengers (PKC and PKA). We also analyzed the possible effects of naltriben (100 μM) in [Ca2+]cyt clearance (Fig. S3 A). Our data showed a decrease (~45%) in the slope of SOCE inactivation only in secretory stage cells (Fig. S3 C), suggesting faster inactivation kinetics of Ca2+ transients after naltriben stimulation.
3.2. Effects of ORAI and TRPM7 blockade in Ca2+ influx
We then compared the effects of blocking ORAI channels and TRPM7 using pharmacological inhibitors. First, we depleted the Ca2+ stores with thapsigargin to stimulate SOCE in the presence and absence of the ORAI blockers synta-66 or BTP-2. Both inhibitors decreased Ca2+ influx significantly, showing lower Ca2+ peaks in secretory and maturation stage cells (Fig. 3, A–B and E–F) as well as other parameters (Fig. 3, C–D and G–H).
Figure 3. Selective pharmacological inhibition of ORAI in primary enamel cells.
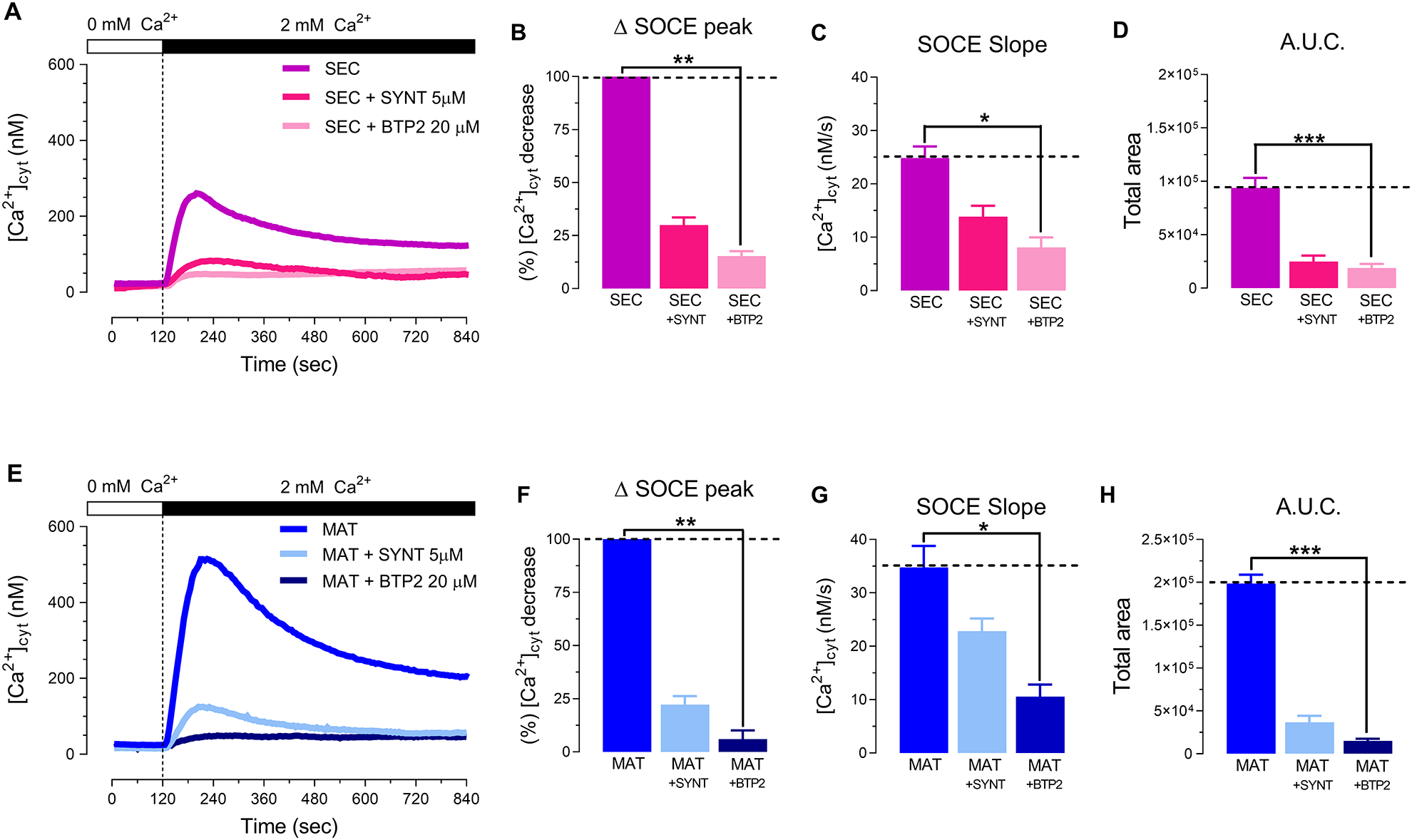
Representative original traces from secretory (SEC) (A) and maturation (MAT) (E) ameloblasts exposed to the CRAC channel antagonists (synta-66, 5 μM or BTP-2, 20 μM). Both ORAI blockers provoked a substantial reduction in delta SOCE peak (B, F), SOCE slope (C, G) and A.U.C. (D, H) Data represent the mean ± SEM of minimum three independent experiments. Data analyzed by one-way ANOVA followed by a Bonferroni’s multiple comparison post-hoc test at *P < 0.05; **P < 0.01; ***P < 0.001.
To assess whether TRPM7 contributes to SOCE or if TRPM7 is a positive modulator of SOCE, we measured Ca2+ influx in primary enamel cells after depleting the Ca2+ stores with thapsigargin to stimulate SOCE in the presence of the TRPM7 channel inhibitor NS8693. Delta SOCE peak, SOCE slope and A.U.C. parameters did not show a reduction in maturation stage cells when exposed to NS8693 (30 μM) (Fig. 4, A–D). However, NS8593-(30 μM) treated secretory cells showed an increase (~35%) in Ca2+ influx (Fig. 4, A–D). A plausible explanation for this could be related to the structural similarities between NS8593 with a benzimidazole homologue (NNC 55–0396), which may interact with a common ligand-binding site in TRPM7 with distinct functional consequences for channel function, as previously suggested [33–35]. Since TRPM7 blockade did not decrease SOCE, these data suggest that the TRPM7 channel is not a component of SOCE in enamel cells.
Figure 4. TRPM7 inhibition reverses Ca2+ influx potentiation, but does not decrease SOCE in primary enamel cells.
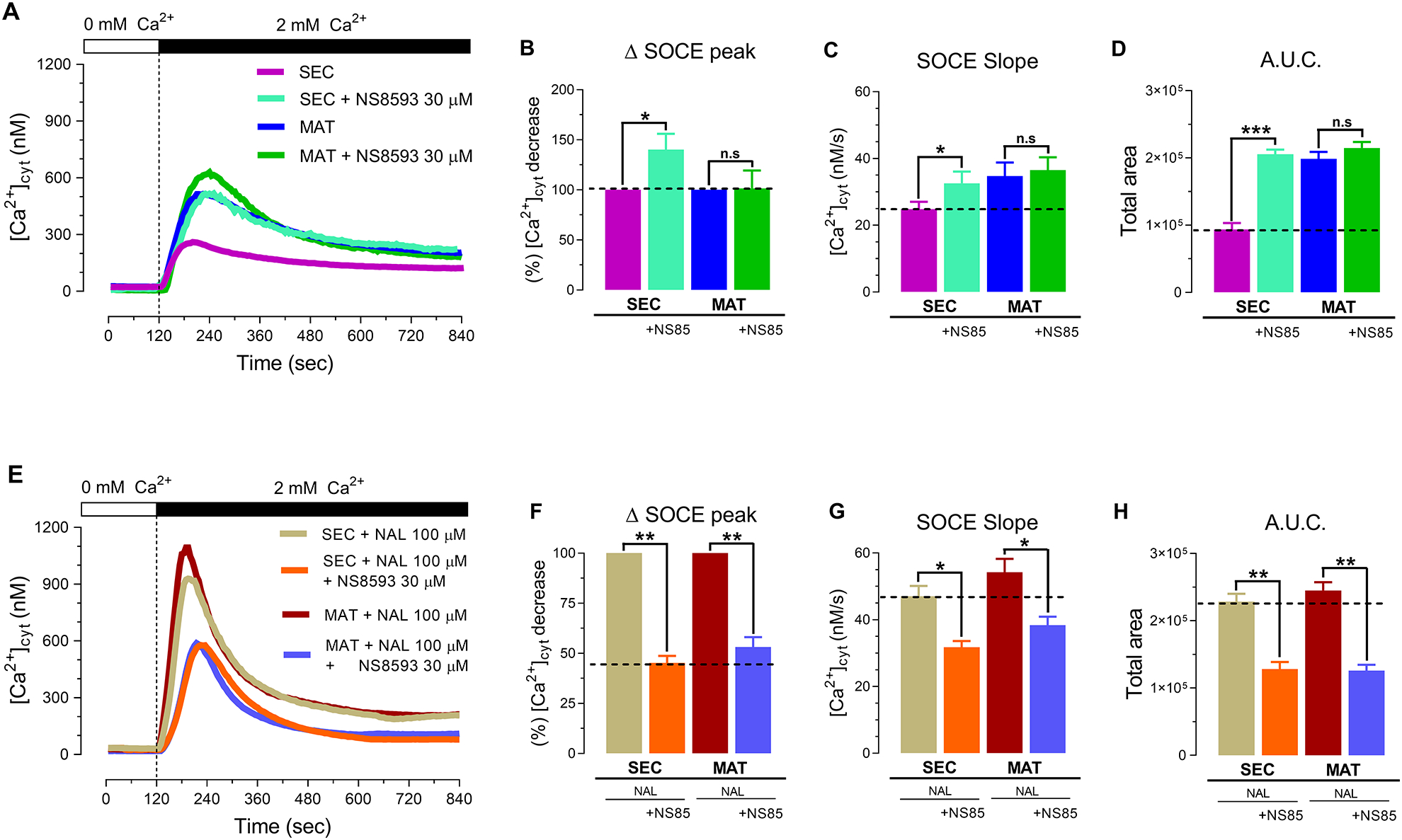
(A) Representative original traces of [Ca2+]cyt transients in control secretory (SEC) and maturation (MAT) stage ameloblasts and in cells exposed to the TRPM7 channel inhibitor NS8693 (30 μM). NS8693 did not elicit a reduction in delta SOCE peak (B), SOCE slope (C) or A.U.C. (D) in SEC or MAT cells. (E) Primary SEC and MAT cells stimulated with naltriben (NAL,100 μM) to active TRPM7 in the presence of NS8593 (30 μM) results in a significant decrease in delta SOCE peak (F), SOCE slope (G) and A.U.C. (H) Data represent the mean ± SEM of minimum three independent experiments. Data analyzed by two-tailed unpaired Student’s t-test at *P < 0.05; **P < 0.01; ***P < 0.001; n.s., non-significant.
We next addressed the question of whether potentiating TRPM7 in the presence of NS8593 (30 μM) modified the kinetics of Ca2+ influx. Primary enamel cells were simultaneously stimulated with naltriben (100 μM) and the antagonist NS8693 (30 μM), and were compared to cells stimulated only with naltriben. The combined treatment of naltriben and NS8693 showed a significant decrease in both secretory and maturation stage cells in the Δ peak (~53% and ~45%), slope (~37% and ~35%) and A.U.C. (~44% and ~49%) (Fig. 4, E–H). This suggests that NS8593 can inhibit a portion of Ca2+ influx in SOCE that is mediated by TRPM7. Combined, these data indicate that pharmacological blockade of TRPM7 does not decrease SOCE in enamel cells, but ORAI inhibition does. Furthermore, TRPM7 is not required for SOCE. This suggests that TRPM7 is not a component of SOCE in enamel cells, but can function as a positive modulator when activated, thereby enhancing SOCE. In addition, the SOCE enhancement mediated by TRPM7 activation can be reversed by blocking the channel with NS8593.
3.3. siRNA knock-down of TRPM7 has no effect on SOCE
We next generated a Trpm7 knock-down in the murine enamel cell line LS8 cells (siTrpm7) to test its effects on SOCE using a siRNA vector containing three strands (see Methods). Bioinformatics analyses revealed that two of these siRNA strands targeted the channel and the kinase domains of TRPM7 (Fig. S5). A knock-down of Trpm7 expression was confirmed by RT-PCR analysis, showing a dramatic decrease in mRNA of ~90% (Fig. 5, A). In siTrpm7 cells (Trpm7 KD), Orai1 and Orai2 expression was unchanged (Fig. 5, B–C). In addition, we tested the effects of naltriben (100 μM) during Ca2+ influx via SOCE in Trpm7 KD cells. As expected, control LS8 cells (non-targeting pool) stimulated with naltriben (100 μM) during Ca2+ re-addition revealed a potentiation in SOCE, represented by increased Δ Ca2+ influx peak (~70%), SOCE slope rate (~32%) and A.U.C (~40%) parameters (Fig. 5, D–G), confirming that TRPM7 positively modulates SOCE in this cell type. Despite significantly decreased TRPM7 expression in siTrpm7 cells, SOCE was unchanged compared to controls, even after stimulating these cells with naltriben (100 μM) (Fig. 5, D–G). These data support a non-essential role of TRPM7 in SOCE in enamel cells.
Figure 5. TRPM7 knock-down does not decrease SOCE in the ameloblast cell line LS8 cells.
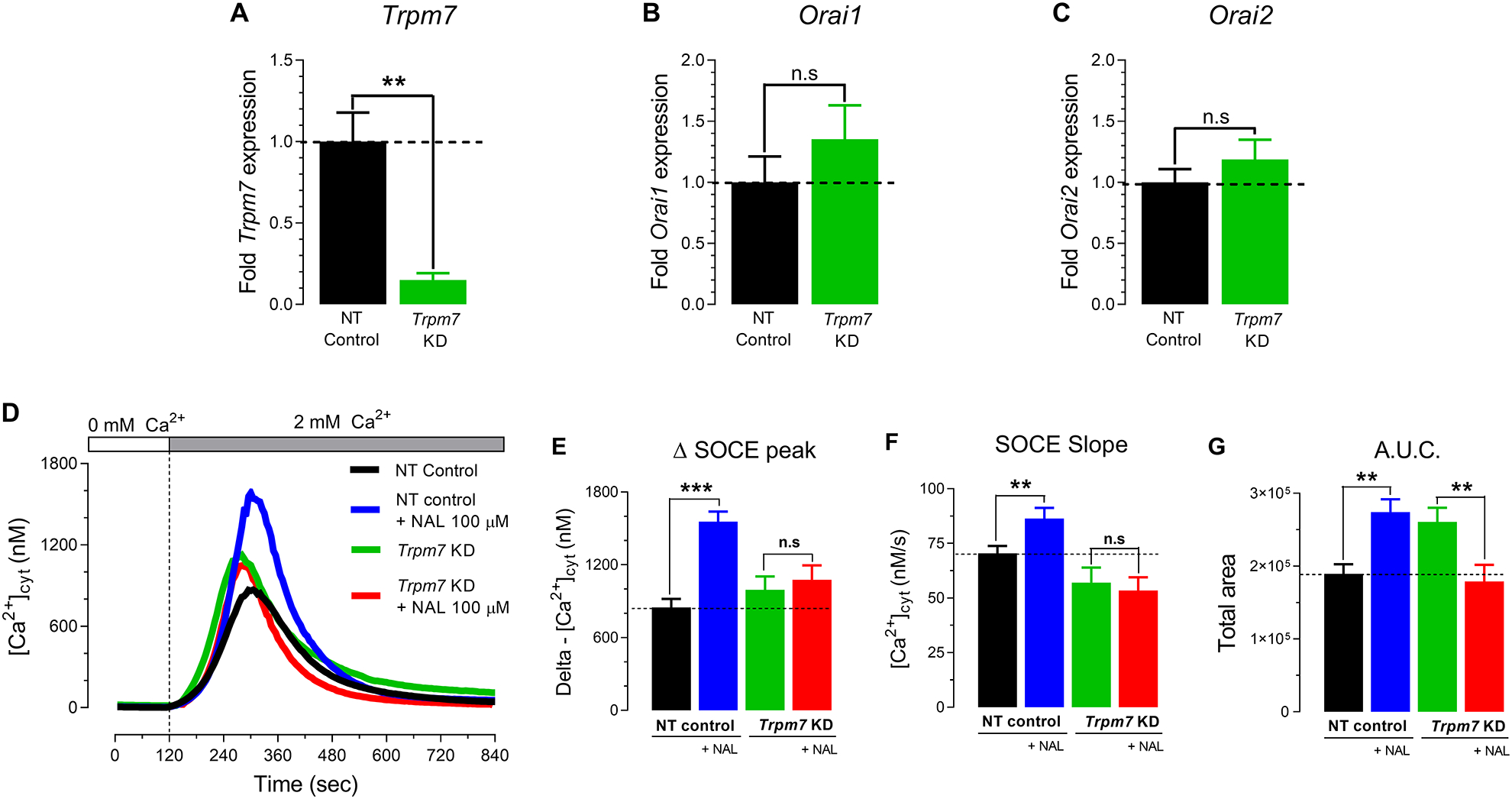
(A-C) mRNA expression of Trpm7, Orai1 and Orai2 and in Trpm7 knock-down (siTrpm7) LS8 cells by RT-PCR. (D) Representative original tracings of [Ca2+]cyt transients in LS8 cells measured after pre-incubation with thapsigargin (20 minutes, 1 μM), followed by perfusion with a Ca2+-free Ringer’s solution (120 seconds) before the simultaneous re-addition of 2 mM extracellular Ca2+ to stimulate SOCE in the presence or absence of naltriben (NAL,100 μM). TRPM7 activation elicited by NAL (100 μM) does not potentiate the delta SOCE peak (E), SOCE slope (F) or A.U.C. (G) in siTrpm7 LS8 cells. Data represent the mean ± SEM of minimum three independent experiments indicated in the histogram bars. Data analyzed by two-tailed unpaired Student’s t-test at *P < 0.05; **P < 0.01; ***P < 0.001; n.s., non-significant.
3.4. TRPM7 potentiation is dependent on the prior activation of ORAI
To more directly address possible links between ORAI proteins and TRPM7 in mediating Ca2+ influx in enamel cells, we used our previously developed ORAI1/ORAI2-deficient (shOrai1/2) LS8 cells [12] to measure Ca2+ influx following SOCE stimulation in the presence of naltriben. As predicted, RT-PCR analysis revealed decreased expression of Orai1 (~66%) and Orai2 (~75%), and in LS8 cells of shOrai1/2 (Fig. 6, A–B). Interestingly, Trpm7 expression was also significantly decreased (~42%) in shOrai1/2 cells (Fig. 6, C). shOrai1/2 LS8 cells were shown to have minimally residual SOCE (Fig. 6, E–G), possibly mediated by ORAI3, as discussed elsewhere [12]. Importantly, we show that shOrai1/2 LS8 cells stimulated with naltriben (100 μM) during the Ca2+ re-addition failed to elicit a raise in [Ca2+]cyt mediated by SOCE (Fig. 6, D). The Δ Ca2+ influx peak SOCE slope rate and A.U.C. remained unchanged (Fig. 6, E–G).These results suggest an essential role of ORAI proteins in Ca2+ influx. They also indicate the potentiation of SOCE by TRPM7 activation is dependent on ORAI proteins.
Figure 6. Ca2+ influx via the TRPM7 channel depends on ORAI in the ameloblast cell line LS8 cells.
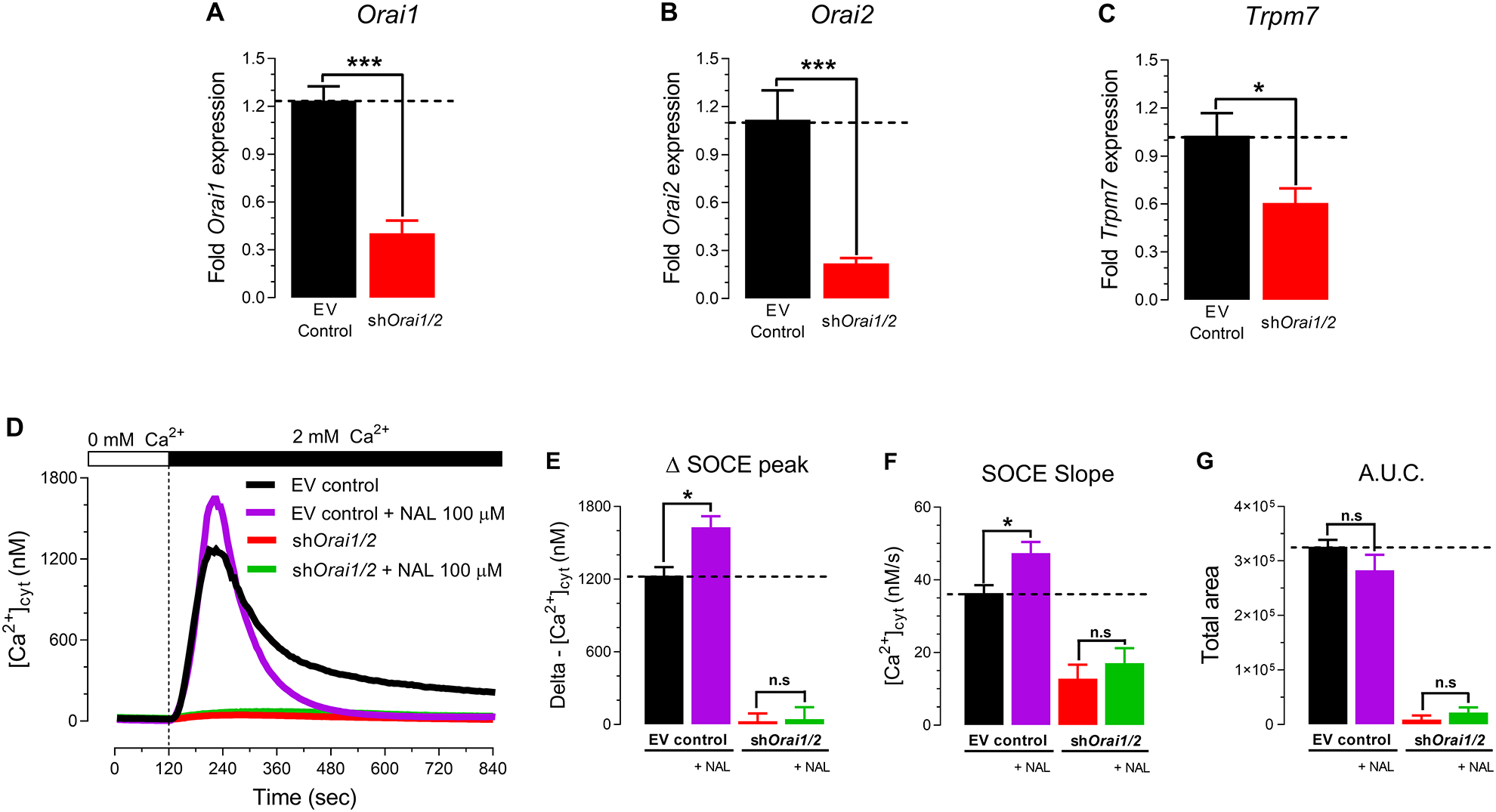
(A-C) Expression of Orai1, Orai2 and Trpm7 in shOrai1/2 LS8 cells, a murine ameloblast cell line, by RT-PCR. (D) Representative original traces of [Ca2+]cyt transients in shOrai1/2 cells and controls (transfected with empty vector) measured after pre-incubation with thapsigargin (20 minutes, 1 μM), followed by perfusion with a Ca2+-free Ringer’s solution (120 seconds) before simultaneous re-addition of 2 mM extracellular Ca2+ or with 2 mM Ca2+ and naltriben (NAL,100 μM). TRPM7 activation triggered by naltriben (NAL, 100 μM) of shOrai1/2 LS8 cells failed to elicit a rise in delta SOCE peak (E), SOCE slope (F) or A.U.C. (G). Data represent the mean ± SEM of minimum three independent experiments. Data analyzed by two-tailed unpaired Student’s t-test at *P < 0.05; **P < 0.01; ***P < 0.001; n.s., non-significant.
4. Discussion
Ca2+ is an essential component of mineralized enamel. Transepithelial Ca2+ transport by ameloblasts requires Ca2+ uptake prior to its delivery to the extracellular space. In enamel cells, the bulk of Ca2+ influx is mediated by SOCE [6,10,46]. The chanzyme TRPM7 has been recently described as being a SOCE modulator [19], while other studies show abnormal enamel in Trpm7-deficient mice [20], raising the possibility that TRPM7 may act as a modulator of Ca2+ influx in enamel cells. Here, we show that pharmacological activation of TRPM7 in enamel cells positively enhances SOCE (Fig. 1, C–F), thereby participating in Ca2+ influx. These effects, however, depend on the prior activation of SOCE. In TRPM7-deficient enamel cells, SOCE was unaffected (Fig. 5, D–G), suggesting that TRPM7 is a positive modulator but non-essential for SOCE.
Besetty et al., [20] showed that inactivation of the kinase domain of TRPM7 results in reduced SOCE in T cells of TRPM7 kinase-dead mice. Specifically, Faouzi and colleagues suggested that the downregulation of SOCE in Trpm7-deficient chicken B-lymphocytes was mediated by the phosphorylation of STIM2 by the kinase domain of TRPM7 [19]. In our study of enamel cells, we have addressed two main questions using Ca2+ imaging data: 1) is the TRPM7 channel a component of SOCE; and 2) can TRPM7 modulate SOCE?
Using rat enamel organs to isolate ameloblasts from secretory and maturation stage [46], we show that both cells have SOCE, with significantly higher Ca2+ influx in maturation (Fig. 2, C–F), in keeping with our previous findings [10,11]. To ascertain if TRPM7 affected Ca2+ influx following SOCE activation, secretory and maturation cells were perfused with the TRPM7 agonist naltriben, which elicited an increase of the SOCE signal (Fig 2, C–F). Naltriben is a selective gating modulator of the TRPM7 channel insensitive to intracellular Mg2+ levels [35]. It is also known as a delta (Δ) opioid receptor antagonist. A previous mRNA screen did not identify the expression of this opioid receptor in enamel cells [47]. These findings implicate TRPM7 as a novel physiological channel functionally involved in Ca2+ handling in enamel cells.
To confirm that the enhancement of SOCE mediated by naltriben was associated with TRPM7, we used the TRPM7 channel blocker NS8593. This compound is a potent TRPM7 channel inhibitor [48] and Faouzi et al., showed that NS8593 significantly inhibited SOCE in rat basophilic leukemia cells when used at the same concentration as in our study (30 μM) [19]. It is also known to block Ca2+ responses in mouse eggs during fertilization [49]. In enamel cells, however, NS8593 did not inhibit SOCE. Rather, it inhibited only the TRPM7-induced enhancement of SOCE mediated by the agonist naltriben (Fig. 4, E–H). These data confirm that naltriben activate the TRPM7 channel to participate in Ca2+ influx. By contrast, primary enamel cells treated with the CRAC channel blockers BTP-2 and synta-66 significantly reduced Ca2+ influx by SOCE (Fig. 3, A–H). These pharmacological approaches indicate that inhibition of TRPM7 does not affect SOCE in enamel cells, but blockade of ORAI channels significantly diminishes Ca2+ influx, confirming that SOCE is the main mediator of Ca2+ uptake. These results highlight that the TRPM7 channel is a positive modulator of Ca2+ influx via SOCE in enamel cells, but not a component of SOCE. This conclusion is in agreement with the Faouzi et al., results in DT40 chicken cells [19].
The efficiency of the TRPM7 channel in Ca2+ influx in enamel cells was further investigated using a molecular approach. Using our previously [12] developed enamel cell line LS8 cells lacking Orai1 and Orai2 (shOrai1/2), we first tested the expression of Trpm7 in these cells by RT-PCR. The expression of Trpm7 was downregulated in shOrai1/2 cells (Fig. 6, C), suggesting a molecular link between these two proteins (see also below). However, naltriben stimulation of TRPM7 in shOrai1/2 cells, which show nearly abolished Ca2+ influx, failed to elicit a raise in Ca2+ uptake. This was not the case in control LS8 cells where, similar to primary cells, naltriben enhanced SOCE (Fig. 6, D–G). The inability of the TRPM7 channel agonist to elevate cytosolic Ca2+ in shOrai1/2 cells indicates that the channel function of TRPM7 is dependent on previous activation of the ORAI pore. Given that Trpm7 expression was significantly decreased in shOrai1/2 cells, however, the possibility that TRPM7 could not be effectively activated in shOrai1/2 cells remained unclear.
To address this, we generated a knock-down of TRPM7 by siRNA in LS8 cells (siTrpm7). The double-stranded RNA molecules in the vector used here contained a mixture of four siRNA as a single reagent, including strands that targeted the channel and kinase regions of TRPM7 (see methods and Fig. S5). The expression of Trpm7 was dramatically decreased in siTrpm7 LS8 cells, but Orai1 or Orai2 expression was unchanged (Fig 5, A–C). As noted above, disrupting Orai1/Orai2 results in a decrease of Trpm7 expression, but the opposite does not appear to have a bearing on Orai1/Orai2 expression. This might indicate a dependence of TRPM7 expression on ORAI, but not the opposite. This scenario requires further analysis that is beyond the scope of the present study. Despite the significant reduction of Trpm7 expression in siTrpm7 LS8 cells, however, our data clearly show that SOCE was unaffected (Fig 5, A–G). This contrasts with data on splenocytes of TRPM7 kinase-dead K1646R knock-in mice, in which SOCE was significantly reduced [20]. However, the TRPM7 agonist, naltriben (100 μM), showed a decrease in A.U.C. in TRPM7 KD cells (Fig. 5 G), indicating a reduction in the total Ca2+ mobilization due a faster inactivation kinetics after stimulation. Similarly, Inoue et al., [50] showed that naltriben (50 μM) increases a TRPM7-like current and [Ca2+]cyt in adipocytes, that they associated with changes in Ca2+ clearance. This suggests that naltriben (50–100 μM) may affect clearance mechanisms (PMCAs or Na+/Ca2+ exchangers).
The possibility that TRPM7 is found in intracellular vesicles of enamel cells and the potential of these vesicles to contribute to changes in cytosolic Ca2+ were not directly addressed here. However, the small of changes in cytosolic Ca2+ in naltriben-stimulated cells in Ca2+-free solution (Fig. 1, E–F) suggests that, if these TRPM7-expressing vesicles are to be found in enamel cells, their contribution to Ca2+ signaling is minor. The possibility that naltriben is inefficient in activating intracellular TRPM7 channels cannot be discounted.
In summary, this study used pharmacological and molecular approaches to investigate the role of the TRPM7 channel on SOCE in enamel cells. We showed that the TRPM7 Ca2+-permeable channel itself is not a SOCE component. We also show that the enhancement of SOCE potentiated by TRPM7 activation depends on the previous activation of ORAI. These data indicate an indirect, but positive potentiating role of the TRPM7 channel on SOCE in enamel cells.
Supplementary Material
Highlights.
SOCE mediated by CRAC channels is enhanced upon activation of TRPM7 in enamel cells
TRPM7 blockade or TRPM7 knock-down does not affect SOCE in enamel cells
Stimulation of TRPM7 in ORAI1/2-deficient enamel cells failed to elevate [Ca2+]i
TRPM7 is not essential for SOCE but can enhance it
Acknowledgements
This work was supported by the National Institute of Dental and Craniofacial Research (NIDCR) (grants DE027679 and DE025639) to RSL. YI is funded by New York University Abu Dhabi research grant AD105. Computational analysis is supported by CGSB Core Bioinformatics resources. We are thankful to Dr. Bimal Desai for discussions and two anonymous reviewers for their comments that helped improve the manuscript.
Footnotes
Declaration of interest: None
References
- [1].Lacruz RS, Feske S, Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci 1356, 45–79 (2015). DOI: 10.1111/nyas.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Souza Bomfim GH, Mendez-Lopez I, Arranz-Tagarro JA, et al. , Functional Upregulation of STIM-1/Orai-1-Mediated Store-Operated Ca2+ Contributing to the Hypertension Development Elicited by Chronic EtOH Consumption. Curr Vasc Pharmacol. 2017;15(3):265–281. DOI: 10.2174/1570161115666170201122750. [DOI] [PubMed] [Google Scholar]
- [3].Berridge MJ, Lipp P, Bootman MD, The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000. October;1(1):11–21. DOI: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- [4].Carafoli E, Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A. 2002. February 5;99(3):1115–22. DOI: 10.1073/pnas.032427999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Clapham DE, Calcium signaling. Cell. 2007. December 14;131(6):1047–58. DOI: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- [6].Lacruz RS, Enamel: Molecular identity of its transepithelial ion transport system. Cell Calcium 65, 1–7 (2017). DOI: 10.1016/j.ceca.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Eckstein M, Aulestia FJ, Nurbaeva MK, Lacruz RS, Altered Ca2+ signaling in enamelopathies. Biochim. Biophys. Acta 1865, 1778–1785 (2018). DOI: 10.1016/j.bbamcr.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smith CE, Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med. 1998;9(2):128–61. DOI: 10.1177/10454411980090020101. [DOI] [PubMed] [Google Scholar]
- [9].Lacruz RS, Smith CE, Bringas PJ, et al. , Identification of novel candidate genes involved in mineralization of dental enamel by genome-wide transcript profiling. J. Cell. Physiol 227, 2264–2275. DOI: 10.1002/jcp.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nurbaeva MK, Eckstein M, Concepcion AR, et al. , Dental enamel cells express functional SOCE channels. Sci. Rep 5, 15803 (2015). DOI: 10.1038/srep15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nurbaeva MK, Eckstein M, Devotta A, et al. , Evidence That Calcium Entry Into Calcium-Transporting Dental Enamel Cells Is Regulated by Cholecystokinin, Acetylcholine and ATP. Front Physiol. 2018. Jul 2;9:801 DOI: 10.3389/fphys.2018.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Eckstein M, Vaeth M, Aulestia FJ, et al. , Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization. Sci Signal. 2019. April 23;12(578). pii: eaav4663. DOI: 10.1126/scisignal.aav4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eckstein M, Vaeth M, Fornai C, et al. , Store-operated Ca2+ entry controls ameloblast cell function and enamel development. JCI Insight. 2017. March 23;2(6):e91166 DOI: 10.1172/jci.insight.91166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Putney JW, Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv Biol Regul. 2018. May;68:88–96. DOI: 10.1016/j.jbior.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Parekh AB, Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010. May;9(5):399–410. DOI: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- [16].Prakriya M, Lewis RS, Store-Operated Calcium Channels. Physiol Rev. 2015. October;95(4):1383–436. DOI: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang SL, Yu Y, Roos J, et al. , STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005. October 6;437(7060):902–5. DOI: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW, Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010. October;14(10):2337–49. DOI: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Faouzi M, Kilch T, Horgen FD, Fleig A, Penner R, The TRPM7 channel kinase regulates store-operated calcium entry. J Physiol. 2017. May 15; 595(10): 3165–3180. DOI: 10.1113/JP274006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Beesetty P, Wieczerzak KB, Gibson JN, et al. , Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci Rep. 2018; 8: 3023 DOI: 10.1038/s41598-018-21004-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nakano Y, Le MH, Abduweli D, et al. , A Critical Role of TRPM7 As an Ion Channel Protein in Mediating the Mineralization of the Craniofacial Hard Tissues. Front Physiol. 2016. July 6;7:258 DOI: 10.3389/fphys.2016.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ryazanova LV, Rondon LJ, Zierler S, et al. , TRPM7 is essential for Mg2+ homeostasis in mammals. Nat Commun. 2010. November 2; 1: 109 DOI: 10.1038/ncomms1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ogata K, Tsumuraya T, Oka K, et al. , The crucial role of the TRPM7 kinase domain in the early stage of amelogenesis. Sci Rep. 2017; 7: 18099 DOI: 10.1038/s41598-017-18291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fahrner M, Schindl R, Romanin C, Studies of Structure-Function and Subunit Composition of Orai/STIM Channel Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 2. [PubMed] [Google Scholar]
- [25].Zou Z, Rios FJ, Montezano AC, Touyz RM, TRPM7, Magnesium, and Signaling. Int J Mol Sci. 2019. April; 20(8): 1877 DOI: 10.3390/ijms20081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cohen RM, Moiseenkova-Bell VY, Structure of Thermally Activated TRP Channels. Curr Top Membr. 2014; 74: 181–211. DOI: 10.1016/B978-0-12-800181-3.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nadler MJ, Hermosura MC, Inabe K, et al. , LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001. May 31;411(6837):590–5. DOI: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- [28].Lau C, Hunter MJ, Stewart A, Perozo E, Vandenberg JI, Never at rest: insights into the conformational dynamics of ion channels from cryo-electron microscopy. J Physiol. 2018. April 1; 596(7): 1107–1119. DOI: 10.1113/JP274888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Minor DL, Let It Go and Open Up, an Ensemble of Ion Channel Active States. Cell. 2016. February 11;164(4):597–8. DOI: 10.1016/j.cell.2016.01.037. [DOI] [PubMed] [Google Scholar]
- [30].Sahni J, Tamura R, Sweet IR, Scharenberg AM, TRPM7 regulates quiescent/proliferative metabolic transitions in lymphocytes. Cell Cycle. 2010. September 1; 9(17): 3565–3574. DOI: 10.4161/cc.9.17.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chubanov V, Mittermeier L, Gudermann T, Role of kinase-coupled TRP channels in mineral homeostasis. Pharmacol Ther. 2018. April;184:159–176. DOI: 10.1016/j.pharmthera.2017.11.003. [DOI] [PubMed] [Google Scholar]
- [32].Abiria SA, Krapivinsky G, Sah R, et al. , TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc Natl Acad Sci U S A. 2017. July 25; 114(30): E6079–E6088. DOI: 10.1073/pnas.1707380114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chubanov V, Ferioli S, Gudermann T, Assessment of TRPM7 functions by drug-like small molecules. Cell Calcium. 2017. November;67:166–173. DOI: 10.1016/j.ceca.2017.03.004. [DOI] [PubMed] [Google Scholar]
- [34].Chubanov V, Schäfer S, Ferioli S, Gudermann T, Natural and Synthetic Modulators of the TRPM7 Channel. Cells. 2014. December; 3(4): 1089–1101. DOI: 10.3390/cells3041089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schäfer S, Ferioli S, Hofmann T, Zierler S, Gudermann T, Chubanov V, Mibefradil represents a new class of benzimidazole TRPM7 channel agonists. Pflugers Arch. 2016. April;468(4):623–34. DOI: 10.1007/s00424-015-1772-7. [DOI] [PubMed] [Google Scholar]
- [36].Smith CE, Nanci A, A method for sampling the stages of amelogenesis on mandibular rat incisors using the molars as a reference for dissection. Anat Rec. 1989. November;225(3):257–66. DOI: 10.1002/ar.1092250312. [DOI] [PubMed] [Google Scholar]
- [37].Chen LS, Couwenhoven RI, Hsu D, Luo W, Snead ML, Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch. Oral Biol 37, 771–778 (1992). DOI: 10.1016/0003-9969(92)90110-T. [DOI] [PubMed] [Google Scholar]
- [38].Sarkar J, Simanian EJ, Tuggy SY, et al. , Comparison of two mouse ameloblast-like cell lines for enamel-specific gene expression. Front. Physiol 5, 277 (2014). DOI: 10.3389/fphys.2014.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zitt C, Strauss B, Schwarz EC, et al. , Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004. March 26;279(13):12427–37. DOI: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- [40].El-Gebali S, Mistry J, Bateman A, et al. , The Pfam protein families database in 2019. Nucleic Acids Res. 2019. January 8;47(D1):D427–D432. DOI: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ryazanov AG, Pavur KS, Dorovkov MV MV, Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol. 1999. January 28;9(2):R43–5. DOI: 10.1016/s0960-9822(99)80006-2. [DOI] [PubMed] [Google Scholar]
- [42].Hofmann T, Schäfer S, Linseisen M, Sytik L, Gudermann T, Chubanov V, Activation of TRPM7 channels by small molecules under physiological conditions. Pflugers Arch. 2014. December;466(12):2177–89. DOI: 10.1007/s00424-014-1488-0. [DOI] [PubMed] [Google Scholar]
- [43].Malli R, Graier WF, The Role of Mitochondria in the Activation/Maintenance of SOCE: The Contribution of Mitochondrial Ca2+ Uptake, Mitochondrial Motility, and Location to Store-Operated Ca2+ Entry. Adv Exp Med Biol. 2017;993:297–319. DOI: 10.1007/978-3-319-57732-6_16. [DOI] [PubMed] [Google Scholar]
- [44].Ohshima H, Maeda T, Takano Y, Cytochrome oxidase activity in the enamel organ during amelogenesis in rat incisors. Anat Rec. 1998. December;252(4):519–31. DOI: 10.1002/(SICI)1097-0185(199812)252:4<519::AID-AR3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [45].Broertjes J, Klarenbeek J, Habani Y, Langeslag M, Jalink K, TRPM7 residue S1269 mediates cAMP dependence of Ca2+ influx. PLoS One. 2019; 14(1): e0209563 DOI: 10.1371/journal.pone.0209563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lacruz RS, Habelitz S, Wright JT, Paine ML ML, Dental Enamel Formation and Implications for Oral Health and Disease. Physiol Rev. 2017. July 1;97(3):939–993. DOI: 10.1152/physrev.00030.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Won J, Vang H, Kim JH, Lee PR, Kang Y, Oh SB, TRPM7 Mediates Mechanosensitivity in Adult Rat Odontoblasts. J Dent Res. 2018. August;97(9):1039–1046. DOI: 10.1177/0022034518759947. [DOI] [PubMed] [Google Scholar]
- [48].Chubanov V, y Schnitzler MM, Meißner M, Schäfer S, Abstiens K, Hofmann T, Gudermann T, Natural and synthetic modulators of SK (Kca2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br J Pharmacol. 2012. June; 166(4): 1357–1376. DOI: 10.1111/j.1476-5381.2012.01855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bernhardt ML, Padilla-Banks E, Stein P, Zhang Y, Williams CJ, Store-operated Ca2+ entry is not required for fertilization-induced Ca2+ signaling in mouse eggs. Cell Calcium. 2017. July;65:63–72. DOI: 10.1016/j.ceca.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Inoue H, Inazu M, Konishi M, Yokoyama U, Functional expression of TRPM7 as a Ca2+ influx pathway in adipocytes. Physiol Rep. 2019. October; 7(20): e14272 DOI: 10.14814/phy2.14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.