

Abstract
Genetics is a significant factor contributing to congenital heart disease (CHD), but our understanding of the genetic players and networks involved in CHD pathogenesis is limited. Here, we searched for de novo copy number variations (CNVs) in a cohort of 167 CHD patients to identify DNA segments containing potential pathogenic genes. Our search focused on new candidate disease genes within 19 deleted de novo CNVs, which did not cover known CHD genes. For this study, we developed an integrated high-throughput phenotypical platform to probe for defects in cardiogenesis and cardiac output in human induced pluripotent stem cell (iPSC)-derived multipotent cardiac progenitor (MCPs) cells and, in parallel, in the Drosophila in vivo heart model. Notably, knockdown (KD) in MCPs of RPL13, a ribosomal gene and SON, an RNA splicing cofactor, reduced proliferation and differentiation of cardiomyocytes, while increasing fibroblasts. In the fly, heart-specific RpL13 KD, predominantly at embryonic stages, resulted in a striking ‘no heart’ phenotype. KD of Son and Pdss2, among others, caused structural and functional defects, including reduced or abolished contractility, respectively. In summary, using a combination of human genetics and cardiac model systems, we identified new genes as candidates for causing human CHD, with particular emphasis on ribosomal genes, such as RPL13. This powerful, novel approach of combining cardiac phenotyping in human MCPs and in the in vivo Drosophila heart at high throughput will allow for testing large numbers of CHD candidates, based on patient genomic data, and for building upon existing genetic networks involved in heart development and disease.
Introduction
Congenital heart defects are the most common human birth defects, accounting for almost one third of all infants with major congenital anomalies (1, 2). Despite advances in treatment and early detection that has helped improve outcome in congenital heart disease (CHD) patients, CHD remains an important cause of mortality and morbidity in children and adults (3–5). It is likely that most cases of CHD are due to a genetic etiology, but in the majority of patients, the specific cause is not known. Aneuploidies (9–18% incidence) (6) and copy number variations (CNVs) (10–15% incidence) (7) account for a significant proportion of genetic anomalies (8) linked to CHD (9).
Recently, array comparative genomic hybridization (array-CGH) has replaced karyotype analysis for the detection of chromosomal imbalances (10). It has been estimated that pathogenic CNVs are found in 15–20% of patients with CHDs and associated extra-cardiac anomalies, whereas the frequency is less in patients with isolated CHDs (4–14%) (11). Establishment of causality for an individual CNV can be challenging, particularly for those CNVs that are inherited from a clinically unaffected carrier parent and for rare CNVs that may be previously unreported polymorphisms. It is also generally accepted that de novo CNVs are more likely to be pathogenic compared with inherited CNVs (7, 12–16). Consequently, we hypothesize that a powerful approach to identify candidate disease-causing genes for CHD is through functional testing of the genes within de novo CNVs in CHD patients (see below).
Functional validation is essential to verify and elucidate the role of candidate genes in heart development and pathogenesis. The use of human multipotent cardiac progenitor (MCP) (17, 18) cells is a desirable high-throughput platform to screen a variety of phenotypic outputs because they are human in origin. Furthermore, treatment protocols, such as siRNA knockdown (KD) manipulations, can be performed in multi-gene combinations, which also address the likely oligogenic nature of CHD. However, they are an in vitro cell culture system. Thus, as a complementary in vivo approach, the Drosophila heart is a powerful genetic model suitable for efficient screening of gene candidates and providing a whole organism-based assessment of cardiac development, structure and function. The fly heart is ideal for elucidating conserved pathways driving cardiac development in the animal kingdom (19–21) because of low genetic redundancy and relative simplicity of genetic networks, thus allowing for efficient identification of novel genes that can provide insights into the genetics and pathogenesis of CHD. Furthermore, recent advances have established the Drosophila heart as a relatively high-throughput platform compared with other in vivo models, with extensive toolsets available to manipulate the fly genome (22–24).
In this study, we searched for de novo CNVs in 167 patients with CHDs. In order to identify novel, cardiac relevant and thus potentially CHD relevant genes within CNV segments, we prioritized a set of 19 deleted CNVs and tested the genes using a combination of in vitro (human MCP) and in vivo (Drosophila) systems. Combinatorial KD of 9 genes from 2 CNVs using the MCP assays identified the genes RPL13 (Ribosomal Protein L13) and SON (Son DNA binding protein) to be crucial for determination of cardiac differentiation and proliferation. Remarkably, in the fly, cardiac RpL13 KD resulted in a ‘no heart’ phenotype, even at larval stages, and Son KD caused reduced contractility. KD of another ribosomal gene, RPS15A, also reduced cell proliferation in MCPs, similar to RPL13 KD, and also caused heart loss in the fly, but of the anterior region only, suggesting specialized roles for ribosomal genes in cardiac disease. KD of several other CNV-derived genes in the fly heart, most notably Decaprenyl Diphosphate Synthase 2 (Pdss2), produced severely reduced contractility likely caused by altered cytoskeletal structures. These findings exemplify the power of this dual platform approach and are a novel method for efficient screening and validation of CHD candidate genes that can advance our understanding of the genetic mechanisms underlying human CHD.
Results
Identified de novo CNV segments contain known CHD genes
Meeting our criteria, a total of 167 patients were identified from the SG and DECIPHER databases with 14 common types of heart defects (Fig. 1, Supplementary Material, Tables 1 and 2). There was a slight female bias in the sex distribution. A total of 197 de novo CNV segments, 141 deletions and 56 duplications, were identified from the 167 patients, which were distributed throughout the genome (Supplementary Material, Table 3). Selection for smaller CNV regions (<1 Mb) or CNV segments with overlaps of any size or gene region between patients refined the list to 54 total CNV segments from 87 patients (Supplementary Material, Table 4). Following a comprehensive literature review on each of the genes within these regions, the identified genes were classified into four categories: (I) CNVs (segments) with previously reported genes known to be associated with human CHDs (Supplementary Material, Table 5), (II) CNVs (7 segments) with previously reported genes causing specific CHDs presenting with a different type of CHD not previously reported (Supplementary Material, Table 6), (III) CNVs (6 segments) with genes implicated in heart development not previously associated with human CHDs (Supplementary Material, Table 7) and (IV) CNVs (37 segments) with newly identified genes not previously reported to be involved in cardiac development or CHDs (Table 1 and Supplementary Material, Tables 8 and 9). Although some of these genes have been implicated to cause syndromes associated with CHD, the role of these genes in heart development is unknown. Among the CNV segments within category IV, 24 were deleted CNVs (Table 1 and Supplementary Material, Table 8), while 13 were duplicated (Supplementary Material, Table 9). In this study, we focused on the 24 de novo deleted CNVs in category IV, i.e. those not previously associated with heart development or human CHD. Potentially pathogenic single-nucleotide polymorphisms (SNP) in known CHD genes were not screened for in patients with category IV CNVs and therefore, patient phenotypes could be affected by additional SNP variants outside of CNV segments.
Figure 1.
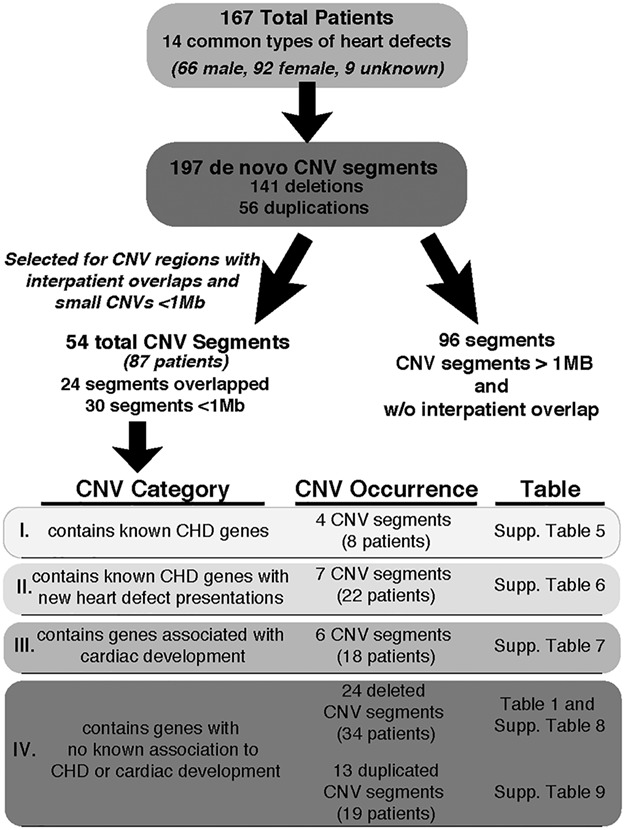
Flow chart highlighting the criteria and categorization used to identify de novo CNVs from CHD patients. List of 14 heart defects: Ventricular septal defect, atrial septal defect, AVCD, tetralogy of fallot, coarctation of aorta, aortic valve stenosis, pulmonic stenosis, truncus arteriosus, hypoplastic left heart, anomalous pulmonary venous return, Ebstein anomaly, transposition of the great arteries, bicuspid aortic valve and mitral valve prolapse.
Table 1.
Deleted CNVs containing genes with no known association with CHD and unknown function in cardiac development (category IV). See complete list of genes in Supplementary Table 8. Patient ID numbers are included to allow for cross reference with Supplementary Tables 1 and 4 for additional phenotypic description
| CNV segment ID | Locus | Coordinates of region of overlap or single CNV | Syndrome | # of patients | Patient ID number | Heart defects | Genes |
|---|---|---|---|---|---|---|---|
| 1 | 10q25.3 | 117108537–117433657 | 4 | 4456, 248409, 254604, 256406 | APVRa, VSD, TOF, ASD | ATRNL1 | |
| 2 | 14q12 | 29904720–30316660 | 14q12 Microduplication | 1 | 252353 | VSD | MIR548AI, PRKD1 |
| 3 | 2p15 | 61567864–61568645 | 2p15-p16.1 Microdeletion | 1 | 2172 | AS | USP34 |
| 4 | 3p26.3 | 1145284–1237776 | 1 | 262200 | VSD | CNTN6 | |
| 5 | 21q11.2-21.1 | 16378214–16782361 | 1 | GC7008 | MVP | NRIP1 | |
| 6 | 1q44 | 244744915–244828276 | 1 | GC75902 | HLHS | C1orf101, DESI2 | |
| 7 | 3p25.3 | 11393089–11618468 | 1 | 274688 | ASD | ATG7, VGLL4 | |
| 8 | 6q21 | 107461798–107814267 | 1 | 254740 | VSD | PDSS2, SOBP | |
| 9 | 6p25.3 | 366332–771504 | 6p25.3 Microdeletion | 1 | 273668 | VSD | IRF4, EXOC2, HUS1B |
| 10 | 17p13.3 | 87009–490921 | Miller-Dieker syndrome (MDS) | 1 | 253979 | TOF | RPH3AL, LOC100506388, C17orf97, FAM101B, VPS53 |
| 11 | 16p13.3 | 2312956–2353159 | 1 | 2627 | ASD | RNPS1, MIR3677, MIR940, MIR4717, ABCA3 | |
| 12 | 16q24.3 | 89475450–89652148 | ANKRD11 haploinsufficiency/16q24.3 Microdeletion | 1 | 249412 | AVCD | ANKRD11, LOC101927817, SPG7, RPL13, SNORD68, CPNE7 |
| 13 | 21q22.11 | 34886796–35051669 | 21q deletion syndrome | 1 | GC52571 | ASD | GART, SON, MIR6501, DONSON, CRYZL1, ITSN1 |
| 14 | 1q44 | 246839121–247179291 | 1 | 271992 | VSD | SCCPDH, LINC01341, AHCTF1, ZNF695, ZNF670-ZNF695 | |
| 15 | 17q21.31 | 43868982–44378247 | 17q21.31 Microdeletion, 17q21.31 Microduplication | 1 | 252142 | VSD, ASD | CRHR1, MAPT-AS1, SPPL2C, MAPT, MAPT-IT1, STH, KANSL1, KANSL1-AS1, LOC644172, LRRC37A, ARL17A, ARL17B |
| 16 | 9q34.3 | 140403363–140596152 | 9q34 Microdeletion/Kleefstra | 2 | 250053 | ASD, COA, | PNPLA7, MRPL41, DPH7, ZMYND19, ARRDC1, ARRDC1-AS1, EHMT1 |
| 17 | 22q11.21-22 | 21721591–22423216 | 22q11.2 Distal microdeletion | 4 | 262483, 254238, 251833, 251228 | VSD (2), TOF, AVCD | HIC2, RIMBP3B, RIMBP3C, TMEM191C, PI4KAP2, UBE2L3, YDJC, CCDC116, SDF2L1, MIR130B, MIR301B, PPIL2, YPEL1, MAPK1, PPM1F, TOP3B, IGLC1 |
| 18 | 19p13.11 | 16517519–17477318 | 1 | 4101 | TOF | 26 | |
| 19 | 19p13.3 | 259395–1144343 | Peutz-Jeghers (PJS) | 1 | 258539 | ASD | 40 |
VSD = ventricular septal defect, AVCD = atrioventricular canal defect, ASD = atrial septal defect, TOF = tetralogy of fallot, HLHS = hypoplastic left heart syndrome, APVR, COA = coarctation of aorta, MVP = mitral valve prolapse, AS = aortic stenosis and APVR = anomalous pulmonary venous returna.
Functional screening in human Multipotent Cardiac Progenitors (MCPs) identifies novel candidate CHD genes
The bioinformatic analyses identified 11 genes within deleted CNVs that were previously associated with human CHD, demonstrating that by focusing on de novo CNVs in CHD patients, we were able to identify established pathogenic CHD genes (Supplementary Material, Tables 5 and 6) and may also enable us to identify novel candidate genes. Out of the 24 deleted de novo CNVs from category IV, five deleted CNV segments were not included because either no genes were present within the segment or reagents were not available, leaving 19 deleted CNV segments to be tested (Table 1, Supplementary Material, Table 8). These 19 segments were prioritized according to size, reasoning that smaller CNV segments containing fewer genes would increase the likelihood of identifying an individual candidate disease-causing gene within a specific locus. In order to identify novel candidate genes for CHD among the genes located within CNVs, we recently developed a high-throughput functional screening platform that enables rapid identification of gene candidates involved in human cardiac proliferation and differentiation using MCPs (17). We subjected the 19 deleted CNV segments (1–40 genes per CNV) to the MCP cardiac differentiation assay (17, 18) and assessed cell proliferation by measuring changes in total cell number and differentiation by calculating the proportion of cardiomyocytes, fibroblasts or vascular endothelial cells following siRNA mediated KD (Fig. 2A).
Figure 2.
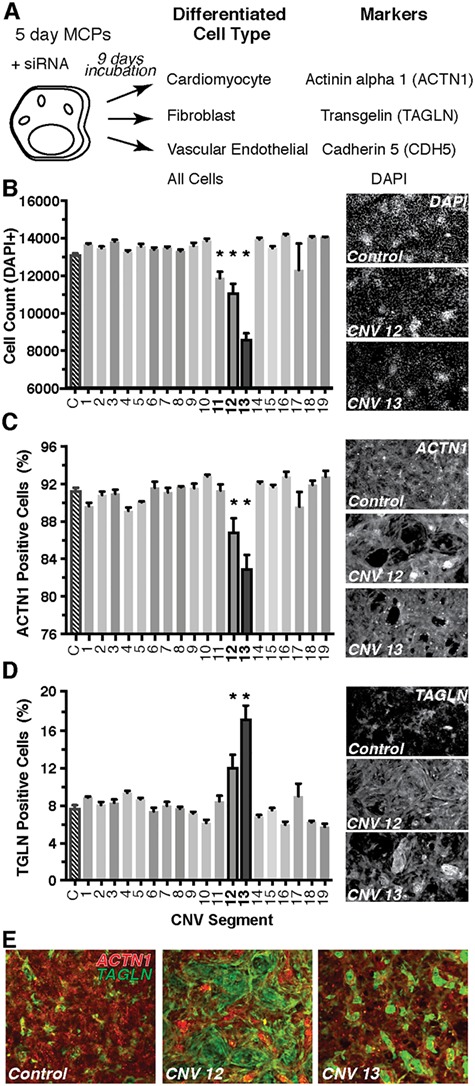
Screening of CNV segments for deviations in human MCP proliferation and differentiation. A differentiation assay of MCPs was used to screen through the deleted gene candidates within each CNV (A). Briefly, MCPs spontaneously differentiate into cells of mesodermal lineage including cardiomyocytes, fibroblasts or vascular endothelial. Any changes in the proportion of these various cell types following siRNA treatment would indicate a role in differentiation. All available siRNAs for the genes within each CNV segment were combined and tested. CNV segments 11–13 produced a decrease in total cell count as marked by nuclear DAPI staining (B). Only CNVs 12 and 13 produced a decrease in the proportion of cardiomyocytes (ACTN1, C) and an increase in fibroblasts (TAGLN, D). Representative images of each staining are to the right of each panel. Composite images depicting the shift in staining from cardiomyocytes (red) to fibroblast (green) in CNVs 12 and 13 cultures (E) *P < 0.05.
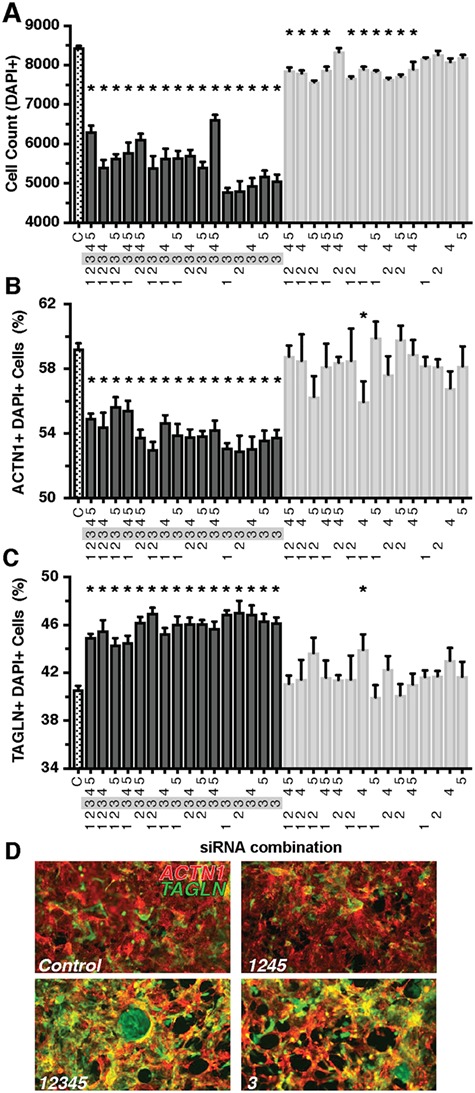
Initially, we combined the available siRNAs for all genes within each individual CNV segment, which included a range of 1–34 siRNAs (for list of targeted genes, see Supplementary Material, Table 8), and transfected day 5 MCPs. Overall, we tested 114 siRNAs directed against genes on our list, leaving 26 genes untested. MCPs were allowed to spontaneously differentiate for nine days. Total cell number (DAPI+), cardiomyocytes (DAPI+; Actinin-alpha1, ACTN1+), fibroblast (DAPI+; Transgelin, TAGLN+) and vascular endothelial (DAPI+; Cadherin 5, CDH5+) were quantified at day 14 of differentiation, 9 days post-siRNA transfection (Fig. 2A, see Materials and Methods). In this context, loss of function of segment 11 (RNPS1, ABCA3), 12 (ANKRD11, SPG7, RPL13, CPNE7) and 13 (GART, SON, DONSON, CRYZL1, ITSN1) significantly decreased total cell number (Fig. 2B) by day 14 of differentiation, whereas CNV-specific siRNA combinations of gene KD of the other CNV segments exhibited no change. Importantly, CNV segments 12 and 13 loss of function also caused a significant reduction in the proportion of cardiomyocytes (Fig. 2C) and a significant increase in the proportion of fibroblasts (Fig. 2D). Only CNV segment 13 also produced a decrease in the proportion of vascular endothelial cells that expressed CDH5 (Supplementary Material, Fig. 1). As a test of siRNA efficiency, we found that pooling the 5 siRNAs against genes within CNV 13 sufficiently knocked down the target mRNAs (Supplementary Material, Fig. 2). Consequently, we then focused on the genes found within CNV segments 12 and 13, due to their ability to shift the proportion of cardiomyocyte versus fibroblast differentiation (illustrated in Fig. 2E).
Deconvolution of genes within CNV segment 12 in MCPs
We next pursued a combinatorial approach testing every possible combination of genes within CNV segment 12 to determine which gene or genes are required to recapitulate the decrease in total cell number and the change in the proportion of cardiomyocytes and fibroblasts. Among the five genes within CNV segment 12, four genes had available siRNAs yielding a total of 15 combinations. Remarkably, all combinations that contained siRNAs against RPL13 (gene 3) caused a significant decrease in total cell number and the proportion of cardiomyocytes, while increasing the proportion of fibroblasts (Fig. 3A–C). There was also an increase or trend in the proportion of vascular endothelial cells in most combinations containing RPL13 (gene 3) (Supplementary Material, Fig. 3). Thus, these results indicate that the KD of ribosomal protein RPL13 alone is sufficient to recapitulate the decrease in cell number and change in the proportion of cell types among the genes tested within CNV segment 12.
Figure 3.
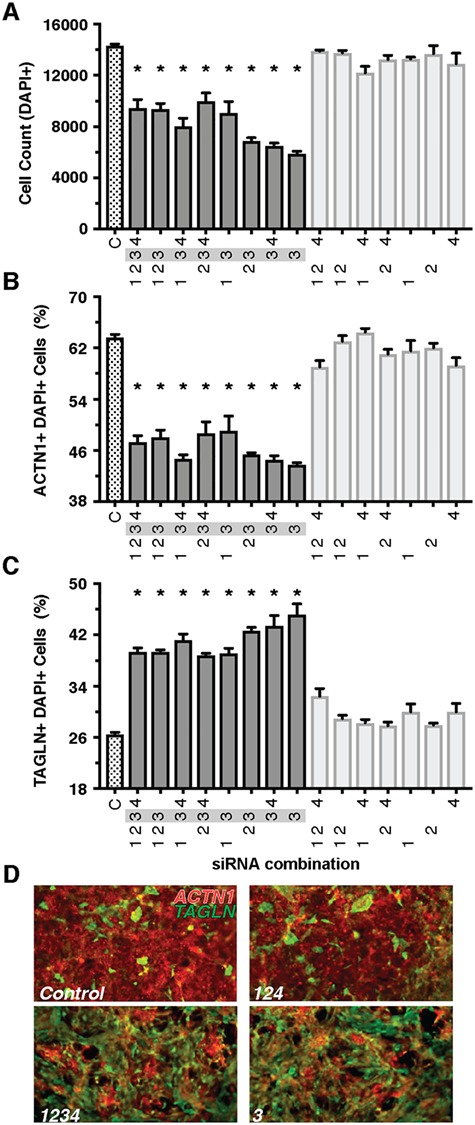
Combinatorial analysis of CNV Segment 12 implicates RpL13 as a candidate gene. Combinations that included the siRNA for RpL13 (gene 3) resulted in lower total cell count (A), decreased proportion of cardiomyocytes (B) and increased proportion of fibroblasts (C). Representative images of control (D, top) and treatment combinations that include RpL13 siRNA (D, bottom). A decrease in red ACTN1 staining and an increase in green TAGLN staining are observed in RpL13 treated conditions. Gene 1- Ankrd11; gene 2: Spg7; gene 3: RpL13; gene 4: Cpne7; Snord68 siRNA not available*P < 0.05.
Identification of novel candidate CHD genes from CNV segment 12 in the Drosophila heart
The use of the Drosophila heart has been a powerful tool in dissecting genetic players involved in cardiac development and function (23). Therefore, we used the fly heart as a complementary in vivo functional assay to assess the developmental, structural and functional effects of the KD of candidate genes. Any phenotypic consequences are considered indicative of a role for the gene candidate in the heart and warrants further mechanistic investigations. In the fly, genes were knocked down individually using available RNAi lines driven by a heart-specific driver (Hand4.2-GAL4) (25), which confers expression in post-mitotic heart progenitors at mid-embryonic stages and remains active in the heart throughout life. For the majority of genes, two RNAi lines were tested (for list of RNAi lines used, see Supplementary Material, Table 10). We assessed heart function and structure in 1-week-old adult hearts (Fig. 4A).
Figure 4.
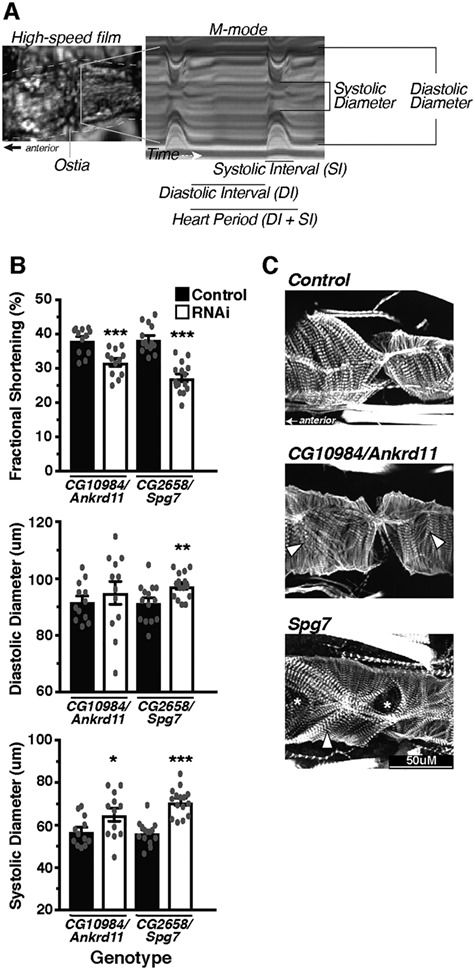
Within CNV segment 12, Ankrd11 and Spg7 produce moderate alterations in heart function and structure. The Drosophila heart was used as an in vivo model system to assess the effects of candidate gene KD specifically in the heart using Hand4.2-GAL4 on function and structure (A). Hearts were filmed with a high-speed camera and analyzed using SOHA analysis, following which hearts were fixed and stained for phalloidin to demarcate filamentous actin. Within CNV segment 12, a moderate decrease in FS (top) caused largely by increased SD (bottom) was detected in Ankrd11 and Spg7 KD hearts (B). Phalloidin staining of the hearts visualized the cytoskeletal structure, whereby KD of Spg7 produced actin filament disorganization, whereas Ankrd11 KD produced minimal changes (C). Arrow heads indicate myofibrillar disorganization, whereas asterisks indicate gaps/holes in the actin filament structure. *P < 0.05, **P < 0.01 and ***P < 0.001.
Among the five genes within CNV segment 12, three genes (ANKRD11, SPG7 and RPL13) had Drosophila orthologs and were knocked down in the heart individually (Table 2, Supplementary Material, Tables 10 and 11). Of these three genes, cardiac CG10984/ANKRD11 and Spg7 KD decreased contractility as measured by fractional shortening (FS), caused by an increased systolic diameter (SD) indicating a primarily systolic dysfunction upon loss-of-function (Fig. 4B). The hearts with CG10984/ANKRD11 and Spg7 KD also exhibited myofibrillar disorganization by staining filamentous actin with the fluorescent marker phalloidin (Fig. 4C), consistent with the observed reduction of contractility (Fig. 4B).
Table 2.
Effect of KD of candidate CHD genes on cardiac parameters in Drosophila. numbers indicate the percent change in cardiac temporal and structural parameters between RNAi KD and background control flies. Numbers highlighted in light gray have percent changes between 15 and 20. Numbers highlighted in dark gray have percent changes >20. *P < 0.05, **P < 0.01 and ***P < 0.001. Results from additional RNAi lines reported in Supplementary Table 11
| CNV segment | Human gene | Fly ortholog | Heart period | Diastolic interval | Systolic interval | Arrhythmic index | Diastolic diameter | Systolic diameter | Fractional shortening |
|---|---|---|---|---|---|---|---|---|---|
| 1 | ATRNL1 | dsd | +4 | +2 | +7 | +54 | –11* | –16** | +9 |
| 2 | PRKD1 | pkd | +3 | +1 | +4 | –20 | –13*** | –8 | –8* |
| MIR548AI | No ortholog | ||||||||
| 3 | USP34 | puf | –7 | –11 | + 5 | –62* | –2 | + 1 | –4 |
| 4 | CNTN6 | Cont | –5 | –10 | + 7 | –18 | –1 | –3 | +1 |
| 5 | NRIP | No ortholog | |||||||
| 6 | DESI2 | CG7222 | + 3 | –6 | +22** | +25 | +6 | +18* | –18** |
| CG12231 | 0 | –6 | +13* | –14 | +6* | –1 | +10** | ||
| C1orf101 | No ortholog | ||||||||
| 7 | ATG7 | Atg7 | +31* | +47* | +1 | +103 | +4 | +10 | – 9 |
| VGLL4 | CG1737 | –20 | –43* | +42** | –41 | –2 | +4 | +9 | |
| 8 | PDSS2 | CG10585 | +14 | +18 | +48* | 0 | +5 | +78*** | –88*** |
| SOBP | Sobp | –2 | –4 | +1 | –77* | +2 | +7 | –7 | |
| 9 | EXOC2 | Sec5 | +26 | +30 | +14 | +78 | –3 | 0 | –4 |
| HUS1B | Hus1-like | –2 | –7 | +7 | –51 | –3 | +7 | –15* | |
| IRF4 | No ortholog | ||||||||
| 10 | RPH3AL | Rph | +6 | +6 | +5 | –54 | –10* | –8 | –4 |
| VPS53 | Vps53 | +50 | +89*** | 0 | –77* | +2 | +15** | –15*** | |
| LOC100506388 | No ortholog | ||||||||
| C17orf97 | No ortholog | ||||||||
| FAM101B | No ortholog | ||||||||
| 11 | RNPS1 | RnpS1 | +0 | +1 | –2 | +5 | +14*** | +31*** | –18*** |
| ABCA3 | CG1718 | –7 | –7 | –6 | +26 | –1 | +2 | –6 | |
| CG6052 | +20 | +26 | +8 | +25 | +3 | +16*** | –15*** | ||
| MIR3677 | No ortholog | ||||||||
| MIR940 | No ortholog | ||||||||
| MIR4717 | No ortholog | ||||||||
| 12 | ANKRD11 | CG10984 | +10 | +9 | +11 * | –27 | +3 | +14* | –17*** |
| SPG7 | CG2658 | +22 | +37* | –5 | +53 | +6** | +26*** | –29*** | |
| RPL13 | RpL13 | No heart | |||||||
| SNORD69 | No ortholog | ||||||||
| CPNE7 | No ortholog | ||||||||
| LOC101927817 | No ortholog | ||||||||
| 13 | ITSN1 | Dap160 | –15 | –17 | –9 | +13 | –10** | –5 | –10* |
| GART | Gart | +69 | +97*** | +11 | +4 | –1 | +7 | –11** | |
| SON | Son | +12 | +10 | +17* | +10 | +1 | +11* | –13* | |
| DONSON | hd | +26* | +51* | –9* | +475* | –13*** | +1 | –28*** | |
| CRYZL1 | No ortholog | ||||||||
| MIR6501 | No ortholog | ||||||||
Large-ribosomal subunit gene RpL13 in CNV segment 12 is essential for heart formation
The third gene within CNV segment 12, RpL13, codes for a subunit of the large cytosolic ribosome complex. RpL13 KD in the heart throughout life using two different RNAi lines led to viable 3rd instar larvae that underwent pupation, but then succumbed to a major die off at the pupal stage, with some adult survivors. These adult survivors had ‘no heart’ tube structures (Fig. 5A and B) and consequently, phalloidin staining did not detect actin filamentous structures within the heart region (Fig. 5C and D, Supplementary Material, Fig. 4A and B). We tested whether embryonic heart development was already compromised upon cardiac RpL13 KD or if heart loss was occurring when the heart remodels during pupal stages. Cardiac examination of 3rd instar larvae also exhibited a ‘no heart’ phenotype (Fig. 5E and F), suggesting that heart development was already compromised at embryonic or early larval stages. Heart field-specific embryonic stage KD of RpL13 (TinD-GAL4; midline-GFP) (26–28) resulted in a mostly ‘no heart’ phenotype in adults. Some flies displayed a partial heart with significant constriction and aberrant cytoskeletal structures of the remaining segments (Fig. 5G and H). Cardiomyocytes were present in late stage 17 embryo and early L1-larvae, suggesting that cell-death is not an immediate effect of RpL13 KD (Supplementary Material, Fig. 4C and D). In support of this embryonic role, KD of RpL13 during embryonic and early L1 larval stages using a temperature-sensitive heart specific driver resulted in a ‘no-heart’ phenotype (Fig. 5I). However, KD of RpL13 starting at larval (mid L1) through pupal stages up until eclosion, developmental stages that involve cell growth and remodeling requiring considerable protein translation, did not cause any loss of heart structures (Fig. 5J), suggesting a temporally-targeted role for RpL13 in the embryonic fly heart. Mef2-GAL4 driving RpL13-RNAi in all muscle tissues led to lethality during L1–L2 larval stages suggesting RpL13 has developmental functions outside of the heart as well (data not shown). Based on these observations and the reduced MCP proliferation, we propose that RPL13 is essential for heart development and the likely candidate gene contributing to pathogenesis within CNV segment 12.
Figure 5.
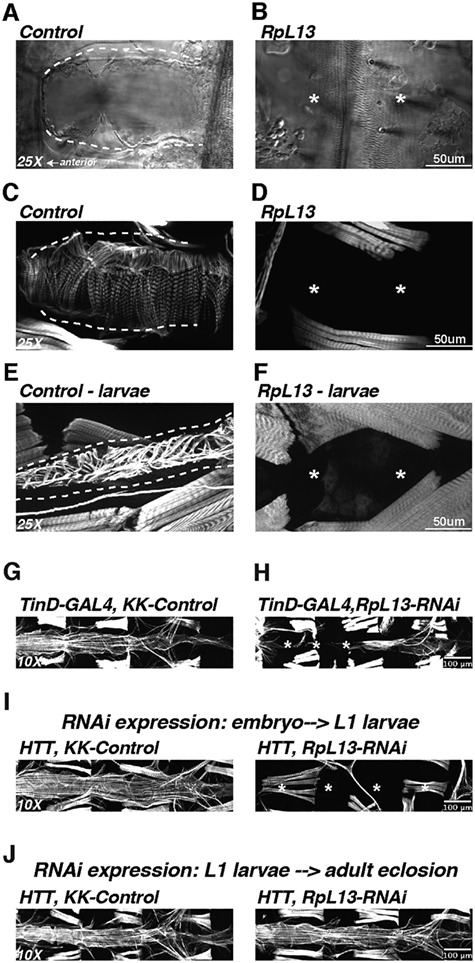
KD of RpL13 in Drosophila heart leads to absence of the heart. KD of the ribosomal subunit RpL13 in the fly heart leads to complete loss of the heart in adults as visualized by brightfield microscopy (A, B) as well as actin staining using fluorescently tagged phalloidin (C, D). Absence of the heart was also observed in 3rd instar larvae (E, F). Embryonic dorsal mesoderm expression of RpL13 RNAi using the TinD-GAL4; midline-GFP driver (26–28) led to a mostly absent heart with posterior remnants that were constricted (G,H). Using a temperature sensitive driver (HTT; Hand4.2-GAL4, Tubulin-GAL80ts, Tubulin-GAL80ts) (54) to regulate Hand4.2-GAL4 KD of RpL13 allowed us to temporally regulate RpL13 expression during development. RpL13-RNAi expression during embryonic and early L1-larval stages resulted in a no-heart phenotype (I), while RpL13-RNAi expression starting at L1 larval stages through adult eclosion did not lead to any loss of heart structure (J), suggesting a developmental role for RpL13 specifically during embryonic stages. Asterisk denotes absent heart structures.
Deconvolution of genes within CNV segment 13 in MCPs
Similar to segment 12, we tested in MCPs all 31 possible combinations of the 5 genes found within CNV segment 13. Remarkably again, in all combinations that included the siRNA against 1 of the 5 genes, SON (gene 3), a significant decrease in total cell count and a shift in the proportion of cardiomyocytes and fibroblasts were observed (Fig. 6A–D). Most combinations with SON siRNA resulted in a decrease of CDH5-positive cells (Supplementary Material, Fig. 5). We also detected a small but significant decrease in total cell number using siRNA combinations of the other 4 genes within CNV segment 13, suggesting a possible contribution of these genes to the overall phenotype (Fig. 6A). These data indicate that SON, a gene involved in pre-mRNA splicing (29, 30), is the main candidate pathogenic gene for CNV segment 13.
Figure 6.
Combinatorial Analysis of CNV Segment 13 implicates Son in impeding cardiomyocyte differentiation in MCP cells. Combinations that included the siRNA for Son (Gene 3) resulted in lower total cell count (A). Certain combinations of siRNAs against genes other than Son also produced small decreases in total cell count. Only Son produced a significant decrease in the proportion of cardiomyocytes (B) and increased proportion of fibroblasts (C). Representative images of control (D, top) and treatment combinations that include Son siRNA (bottom). A decrease in red ACTN1 staining and an increase in green TAGLN staining are observed in Son-treated conditions. Gene 1- Itsn1; Gene 2: Gart; Gene 3: Son; Gene 4: Donson; Gene 5: Cryzl1.*P < 0.05.
Identification of novel candidate CHD genes from CNV segment 13 in the Drosophila heart
Four of the five genes within CNV 13 had Drosophila orthologs (Table 2). All four genes produced a decrease in FS of varying magnitude, but most dramatically by hd/DONSON KD (Fig. 7A). Son KD reduced FS mainly by increasing SD, whereas dap160/ITSN1 and hd/DONSON KD resulted in a decrease in diastolic diameters (DD; Fig. 7A). Phalloidin staining revealed moderate myofibrillar disorganization and gaps with Son and hd/DONSON KD, but less so with dap160/ITSN1 and Gart (Fig. 7B). Within CNV segment 13, not any one gene produced a drastically abnormal phenotype in the fly heart, unlike in CNV segment 12, but all four appear to have mild to moderate effects on heart structure and function when knocked down, consistent with the idea that in an in vivo context more than one gene in CNV 13 may potentially be involved in CHD pathogenesis, and that KD of a combination of the genes may produce a more prominent phenotype in flies. This is consistent with the MCP data, where combinations of genes without Son were also able to decrease total cell number to some extent (Fig. 6A).
Figure 7.
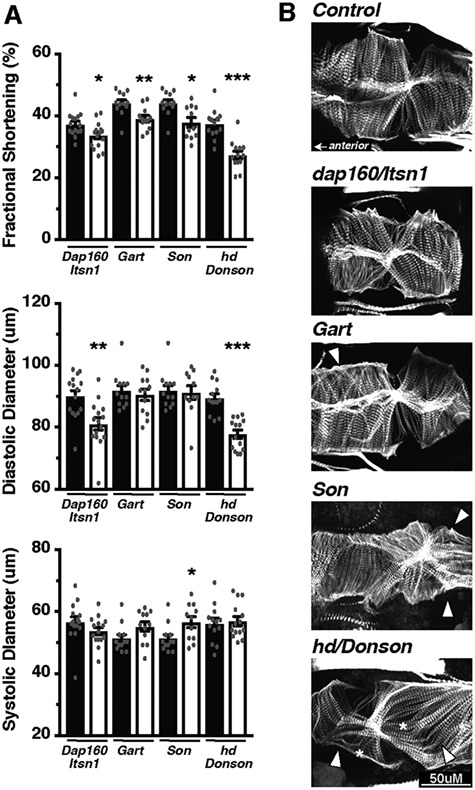
All genes within CNV segment 13 caused some degree of cardiac dysfunction in the fly. Within CNV segment 13 (A), the KD of genes individually in fly hearts produced a decrease in FS (top). This FS reduction was caused by a decrease in DD when Dap160/Itsn1 or hd/Donson was knocked down (middle), while Son caused an increase in SD (bottom). KD of Dap160/Itsn1 and Gart produced minimal changes to the actin cytoskeletal organization (B), while KD of Son and hd/Donson produced more disorganized actin fibers (B). Arrow heads indicate myofibrillar disorganization, whereas asterisks indicate gaps/holes in the actin filament structure. *P < 0.05, **P < 0.01 and ***P < 0.001.
CNV segment 8: KD of Pdss2 diminished the fly heart’s contractility
In the case of CNVs 12 and 13, critical genes located within these segments produced phenotypes in both MCPs and the fly heart upon KD, thus indicative of their cardiac requirement, and augmenting the likelihood for CHD relevance. Although the combined KD of the genes in each of the segments 1–11 did not exhibit any changes in MCPs proliferation or differentiation (Fig. 2), in the fly heart, several produced distinct cardiac functional defects (Table 2). Of them, KD of a gene in CNV segment 8 produced the most dramatic phenotype.
CNV segment 8 altered the copy number of Decaprenyl Diphosphate Synthase Subunit 2 (PDSS2), involved in the coenzyme Q10 biosynthetic pathway (31), and Sine Oculis Binding Protein (SOBP), which codes for a transcriptional cofactor (32). Cardiac KD of the fly orthologs (Table 2, Supplementary Material, Tables 10 and 11) indicate that Pdss2 is the likely pathogenic gene candidate of this relatively small CNV segment, as KD caused failure of most hearts to contract, and the remainder exhibited dramatically reduced contractility (FS), which was primarily due to systolic dysfunction (Supplementary Material, Fig. 6A). Phalloidin staining revealed disorganized myofibrillar alignment and significantly thinned actin filaments as evident in the cross-sectional view of the heart (Supplementary Material, Fig. 6B), which likely contributed to the reduced ability to contract. KD of Sobp produced minimal changes in FS or myofibrillar structure, implicating Pdss2 as the more likely candidate to contribute to disease progression.
Knockdown of RpL13 and Son in MCPs alters cell-cycle but not cell-death relevant gene expression
The combinatorial experiments using MCPs corroborated results from the initial screen and identified RPL13 and SON, as the specific genes most likely responsible for the decrease in total cell count and a shift in differentiation from cardiomyocytes to fibroblasts (illustrated in Figs 3D and 6D). These effects were caused by ~40 or 50% KD of RPL13 and SON mRNA levels, respectively (Supplementary Material, Fig. 7), when measured 2 days post-transfection. The mRNA levels returned to control levels by 5 days post-transfection indicating that the resulting effect on differentiation examined at day 9 (see Fig. 2A) is caused by a transient decrease in gene expression around day 2 post-transfection.
We examined further whether the decrease in cell number is due to reduced proliferation or an induction of apoptotic pathways by measuring the expression of cell-cycle and cell-death relevant genes. KD of either RPL13 or SON in MCPs caused a downregulation of a number of cell cycle genes (PCNA, CDK1, CCNB1 and CCNE1) at 2-days post-transfection (Fig. 8A), which recovered by day 9. Cell death related genes (CASP6 and CRADD), however, were not increased, but trended lower at day 2 following siRNA treatment before returning to control levels at day 9 (Fig. 8B). Three other cell-death related genes (CASP1, CIDEA, TNF) were measured, but did not reach detectable levels in any of the conditions.
Figure 8.
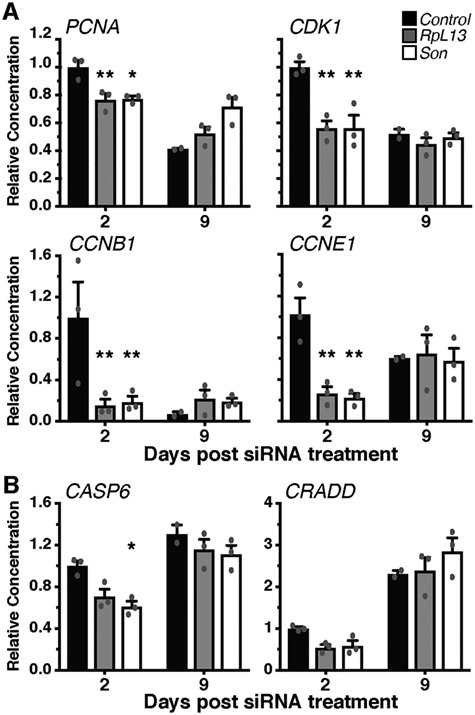
RpL13 and Son KD in MCPs transiently reduce mRNA expression of cell-cycle genes but not cell-death related genes. mRNA levels of cell cycle (A) and cell death (B) relevant genes following either RpL13 or Son KD. A decrease in all cell-cycle genes tested was detected two days post-transfection that returned to control levels at day 9. No changes in the expression of cell-death related genes were detected except for a decrease in Casp6 2 days after Son siRNA transfection. *P < 0.05 and **P < 0.01.
Knockdown of RpL13 and Son alters proliferation in differentiated cardiomyocytes and fibroblasts
We then tested the effect of RPL13 and SON KD on proliferation and cell death in cultures of differentiated cardiomyocytes derived from MCPs (day 25) (18) and foreskin primary fibroblasts by EdU incorporation and TUNEL assay, respectively. Three days post-siRNA transfection, the total number of both cardiomyocytes and fibroblasts were reduced (Fig. 9A). The number of EdU positive cardiomyocytes and fibroblasts was also reduced when RPL13 or SON were knocked down (Fig. 9B and D), indicating a reduction in the number of actively proliferating cells of either cell type. Quantification of DNA content/copy number in the nucleus using integrated density measurements of DAPI staining enabled cell-cycle staging of cells (G1, G2 and S-phase). Treatment with RPL13 or SON siRNA increased the proportion of cells in G1 stage (single DNA copy) and decreased the number of actively duplicating (S-phase) or duplicated cells (G2-phase), which suggests a block that inhibits cell-cycle entry rather than a broadly slowed proliferation rate (Supplementary Material, Fig. 8). There was no detectable increase in TUNEL positive cardiomyocytes or fibroblasts (Fig. 9C), suggesting that the reduced cell number was not due to cell death. Collectively, these results suggest that RPL13 and SON, play a role during heart formation by regulating cardiac progenitor and cardiomyocyte proliferation and differentiation, and not merely causing death.
Figure 9.
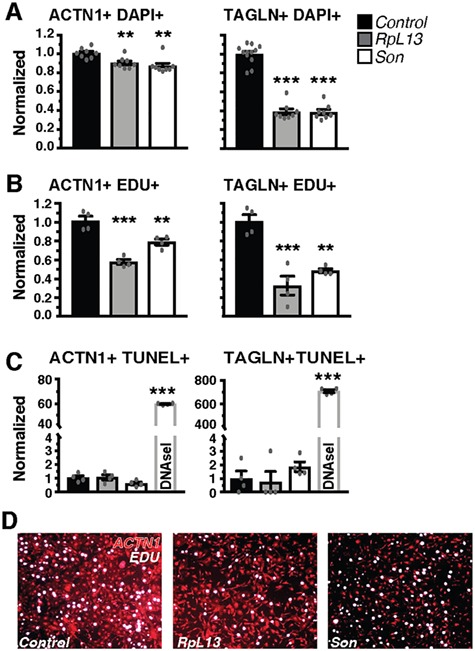
RpL13 and Son KD impedes proliferation but does not increase cell death. Effects on total cell count (A), cell proliferation (EdU incorporation, (B) and cell death (TUNEL, C) were assessed in more pure and mature cultures of cardiomyocytes (ACTN1+) and fibroblasts (TAGLN+). A decrease in total cell count (DAPI+, A) was caused primarily by a decrease in proliferation (EdU+, B) and not an induction of cell death (TUNEL+, C). As a positive control, DNase-I treatment of cells, caused a significant increase in TUNEL positive cells in both cardiomyocytes and fibroblasts. Representative images of EdU staining of cardiomyocyte cultures treated with RpL13 and Son siRNA (D). *P < 0.05, **P < 0.01 and ***P < 0.001.
Other Ribosomal protein subunits are associated with cardiac phenotypes and CHD
Previous studies have described cardiac dysfunction in the fly caused by haploinsufficiency of some ribosomal subunits including RpS15A (33). We tested whether cardiac KD of RpS15A alters heart formation and maintenance as severely as RpL13. Remarkably, cardiac-specific KD of RpS15A resulted in a significant loss of the anterior region of the heart in adults and 3rd instar larvae, whereas the posterior region of the heart was less affected (Supplementary Material, Fig. 9A–F). In MCP cultures, proliferation of mature cardiomyocytes (Differentiation Day 25) and fibroblasts, as measured by EdU incorporation, was also significantly reduced following RPS15A KD (Supplementary Material, Fig. 9G and H), similar to RPL13. Taken together, our results suggest that deficiency of ribosomal genes is likely candidates in the development of CHD. In support of this, other ribosomal genes were also hits in the Pediatric Cardiac Genomic Consortium (PCGC) CHD variant collections and had previously been associated with CHD as curated in the OMIM database (Supplementary Material, Table 12; 34).
Discussion
Pipeline development to identify novel CHD candidate genes
Several recent studies have been published describing CNVs in patients with CHD (11, 35, 36), and in most of these patients, establishment of causality has been challenging. Many CNVs are inherited from clinically unaffected parents, raising doubt about causality. In this study, we analyzed an unprecedented number of de novo CNVs identified in patients with CHD and focused our search on 19 deleted CNV segments that contained genes with no associations to CHD or cardiac development as a means to identify novel CHD causing genes. The genes located within the segments were functionally screened and tested using two complementary developmental assays utilizing human MCPs and the Drosophila heart, which resulted in the identification of novel CHD candidate genes. The platform utilizing MCPs enabled us to test combinations of siRNAs on proliferation and differentiation, which provided important insight on the additive effects of gene KDs addressing the multiple-hit hypothesis in cardiac pathogenesis. In the future, additional parameters, for example calcium and voltage transients (37), can be included to fully assess cardiomyocyte differentiation and quality, bolstering the power and utility of this platform. The use of the Drosophila heart captures and integrates the effects of gene KD on relevant biological processes involved in heart morphogenesis, in vivo, by driving gene KD in the heart throughout development, starting at post-mitotic embryonic stages and evaluating the consequences on heart function and structure at an adult stage. This wider developmental window may boost the sensitivity of this assay and enable the detection of genetic modifiers that alone produce subtle phenotypes in the heart, which may explain the wide range of alterations detected in various fly heart parameters and structural defects. Any structural or functional (contractility) phenotypes identified informs us of a role for the gene in the heart and allows us to prioritize gene candidates for further exploration of mechanism in CHD pathogenesis. In addition to corroborating results from the MCP assay, we identified additional CHD gene candidates with this in vivo model.
Our phenotypical platforms are a powerful means of efficiently and broadly screening the function of genes in two complementary cardiac developmental contexts to score the potential pathogenicity of genes and generate a list of high-priority candidate genes for further testing in lower throughput mammalian models. Further study of candidate genes may uncover new pathways and mechanisms involved in cardiac development, ultimately providing important new fundamental insights into the pathogenesis of human CHDs. This pipeline, which takes advantage of the strengths of two unique cardiac model systems, could eventually be adapted as a module for initial pre-natal diagnosis to provide useful feedback to patients and their health providers.
Identification of novel candidate CHD genes
In our initial screen using MCPs, we combined available siRNAs against genes within each CNV segment. We identified two CNV segments that produced significant alterations in both proliferation and differentiation (CNVs 12 and 13), whereas KD of genes within CNV segment 11 caused a decrease in total cell number only. Although we focused on exploring genes within CNV segments 12 and 13, the genes in CNV segment 11 would be interesting to pursue to identify novel CHD genes that appear to involve different biological mechanisms as reflected by the distinct phenotypic outcome. Combinatorial and individual testing of the genes within CNVs 12 and 13 identified two novel candidate CHD genes, RPL13 (CNV 12) and SON (CNV 13) that alone were able to reproduce reduced cardiomyocyte proliferation and altered differentiation phenotype observed in the initial screen. These two genes are highly intolerant of loss-of-function variation and are likely haploinsufficient as scored by the pLI measure (Supplementary Material, Table 13).
In the Drosophila heart, KD of RpL13 produced third instar larvae and adult flies with no heart, suggesting that among the genes within CNV 12, RpL13 was the gene likely contributing to the disease phenotype, corroborating the results with human MCP. KD of RpL13 specifically in the embryo appears to be sufficient to produce the ‘no heart’ phenotype in adults and does not cause immediate cell death as an overall intact heart is still present at late embryonic stages. Survival of cardiomyocytes through embryonic stages could be due to inefficient KD of RpL13 attributable to maternal contribution and high zygotic expression of RpL13 in the embryo (38), or maternal contribution of RPL13 protein. Nevertheless, even if efficient KD of RpL13 occurs only towards end-stage embryos and early L1-larvae, when cardiomyocytes are already well-established, the short window of RpL13 KD thereafter remains sufficient to induce heart loss in adults suggesting interference with the cardiogenic differentiation program going forward. Furthermore, KD of RpL13 after the embryonic developmental stage, starting at 1st instar (L1) larval stage, through pupal stages up until adult eclosion, which involves considerable levels of protein translation due to growth and remodeling, did not cause cell death nor any gross morphological or functional differences in adult heart. These data suggest that KD of RpL13 does not cause immediate cell-death in cardiac progenitors and is not absolutely required for cell maintenance at that early stage, but possibly has a specific role in early development in programming cardiac cells for maintenance/homeostasis during later developmental stages.
Among the genes within CNV segment 13, the MCP assay identified SON as the primary candidate gene influencing proliferation and differentiation. In the fly, Son KD reduced cardiac contractility pointing to the possibility that this gene could pose cardiac risks. The MCP results also suggest that the other four genes may contribute to the disease phenotype as their combination decreased total cell number, which is consistent with the fly heart phenotypes, whereby the KD of the other three genes within the CNV also produced some degree of cardiac dysfunction. It is also interesting to note that the positional order of SON, GART and DONSON in the genome is conserved in several higher-order model organisms, and therefore could have concordant functions and interactions (39). This positional order is not conserved in flies, but their functional interactions could be.
Within CNV segment 8, the KD of PDSS2 caused a dramatic reduction in FS likely due to thinned circumferential myofibrils, but did not alter differentiation or proliferation in the MCP assay. This substantiates the use of the fly heart as a complementary assay to detect candidate gene functions potentially relevant to heart development and thus CHD (see Table 2). PDSS2 encodes a subunit of the decaprenyl diphosphate synthase, which is the first enzyme of the CoQ10 biosynthetic pathway, and mutations in PDSS2 lead to CoQ10 deficiency, defects of the mitochondrial respiratory chain and in a subset of tissues an increase in ROS production (31). Although enzymes are rarely haploinsufficient, stress conditions could elicit haploinsufficiency (40); in the heart, the high adenosine triphosphate (ATP) demand required for cardiomyocyte contraction and maturation could impose a stress and elicit developmental consequences on myofibrillar structure when PDSS2 is knocked down. Interestingly, seven patients have been identified with a PDSS2 mutation and all individuals presented with nephrotic syndrome as well as cardiomyopathy (41). In CHD patients, reductions in PDSS2 functionality may lead to altered cytoskeletal structures and energy metabolism, which could interact with the effects of other gene variants leading to a more severe morphological defect.
Potential roles for RpL13 and Son in CHD pathogenesis
The knock down of either RPL13 or SON caused a significant decrease in total cell number. This reduction was due to impeded replication/cell cycle rather than an induction of cell death, as measured by reduced levels of cell-cycle mRNA, reduced entry into G1 and S phases of the cell cycle, and a lack of cell-death gene induction. Furthermore, KD of RPL13 and SON reduced the number of EdU positive cells but did not increase TUNEL staining in cultures. In addition to the effect on replication, the KD of these genes caused a shift in the proportion of cell types: promoting fibroblast and hampering cardiomyocyte differentiation.
SON is a splicing cofactor necessary for proper splicing of a subset of genes involved in a number of cellular pathways including cell-cycle and apoptosis regulation, TGF-β signaling, Wnt signaling and integrin-mediated cell adhesion (29, 42), processes that when disrupted could mediate developmental mishaps. Furthermore, depletion in SON leads to loss of embryonic stem cell pluripotency and disruption of mitotic spindle formation (43, 44), which could explain the decreased proliferation and total cell number of MCP following SON KD. Loss of function mutations in SON has previously been associated with intellectual disability and developmental delay (45). A quarter of these cases also present CHDs that include mostly atrial and ventricular septal defects, which is consistent with the atrial septal defect found in the patient carrying the CNV 13 segment that harbors the SON gene. Thus, determining cardiac specific pre-mRNA targets of SON may uncover mechanisms leading to CHD.
RPL13 is a component of the large ribosomal subunit involved in protein synthesis. In adult flies, haploinsufficiency of other RpL and RpS genes often leads to cardiac dysfunction (33). However, ~25% of the genes tested did not produce a heart phenotype suggesting a degree of specificity in the influence of ribosomal subunit KD on heart function. There is emerging evidence that heterogeneity of ribosomal subunit composition can differentially regulate gene expression in different cell types potentially affecting development. A study examining the developmental consequences of Rpl38 mutations in mice demonstrated that decreased Rpl38 levels led to reduced translation of a specific subset of Hox mRNAs that are critical in patterning and development of the axial skeleton, without changing global protein translation (46, 47). Mice with loss-of-function mutations in the targeted Hox genes phenocopied Rpl38 mutant mice patterning defects, indicating that reduced translation of these Hox genes are the likely mediators of the developmental defect in Rpl38 mutants. Additionally, ribosomal paralogs, previously thought of as redundant, are emerging as specialized regulators of downstream targets, further supporting the idea that ribosomal heterogeneity acts as a gatekeeping mechanism for differential protein translation in cells (48). In our hands, we found that RpL13 and RpS15a KD both caused significant heart defects in Drosophila larvae and adults, with a complete loss of the heart following RpL13 KD, and an anterior focus of heart loss following RpS15a KD, suggesting spatial specificity in targeting of ribosomal subunit activity. Furthermore, we demonstrate temporal specificity in RpL13’s role in development, whereby its activity appears crucial during early heart development in the embryo, but it is not required for later developmental stages in the larvae and pupae.
The patient carrying CNV 12 that harbors the RPL13 gene has a complete atrioventricular canal defect (AVCD), a complex CHD that occurs in 7.4% of all infants born with CHD (49). Although there is strong evidence implicating a genetic etiology, the underlying genetic and pathogenic mechanisms are poorly understood. AVCDs are characterized by a defect in the development of the atrial and ventricular septa, as well as in the mitral and tricuspid valves. The development of this atrioventricular canal likely requires multiple cellular lineages through a precisely coordinated gene regulatory network. A number of rare inherited and de novo variants within other ribosomal protein genes have been identified in CHD probands from the PCGC by exome sequencing (34). Probands were diagnosed with a wide range of cardiac anomalies and conditions, including atrial septal defect, tetralogy of fallot and hypoplastic left heart syndrome. A subset of ribosomal protein subunits that have been curated and implicated in human CHDs are associated with the condition Diamond-Blackfan Anemia (DBA; MIM number 105650) (34). Mechanisms implicated in DBA may also be relevant in CHD, including p53 activation that could lead to cell-cycle arrest, senescence and apoptosis (50). These data implicate RPL13 as well as other ribosomal proteins as potential modulators of CHD that may involve a novel post-translational mechanism specifying the proteome driving the patterning and function of progenitor heart cells required for the normal development of the heart.
Materials and Methods
Bioinformatic analysis
We retrospectively analyzed patient data from two databases: (1) Database of chromosomal imbalance and phenotype in humans using Ensembl resources (DECIPHER) (51), (2) The Signature Genomics (SG) proprietary database of patients undergoing clinical array-CGH. The SG database was comprised of patients tested between mid-2011 and mid-2013.
As data in DECIPHER are reported from many different clinical genetic laboratories, a wide variety of CGH and SNP-based arrays were used to identify CNVs in the patients. Inheritance pattern status is available for each patient when parental samples have been analyzed. Patients analyzed by SG were tested using oligonucleotide-based, 135 K, whole-genome microarrays (SignatureChipOS versions 2–3), custom-designed by SG and manufactured by Roche NimbleGen (Madison, WI). De novo status of the CNVs was determined using either fluorescence in situ hybridization (FISH) or array-CGH on samples from parents, depending on the ability of FISH to visualize the abnormality found in the proband.
Using the two databases, we analyzed 167 patients with 14 common types of heart defects, carrying single or multiple de novo CNVs (Fig. 1, Supplementary Material, Tables 1–3). For all patients, the specific CHD was that described by the ordering health care professional. In order to compare CNVs from different patients, the coordinates were converted to that of the Genome Reference Consortium Human Build 37 (GRCh37) database. To limit the number of candidate genes, only small de novo CNVs (<1 Mb) and de novo CNVs with interpatient overlapping sections were studied further. Overlapping sections were of any size and region. We used IBM SPSS Statistics to determine overlapping CNVs and small CNVs. Using the OMIM and UCSC databases of genes, we identified genes contained within each section (either individual or overlapping). Using PubMed, we performed a comprehensive literature review on each of the genes in these regions. Using the database of genomic variants, we searched the segments containing genes not previously reported to be involved in CHDs for common CNVs (http://dgv.tcag.ca).
MCP cell culture and siRNA transfection
siRNAs were taken from the human siGENOME library from Dharmacon, Inc., which contain a pool of 4 unique siRNA sequences targeting different regions of the gene. Frozen 5-day-old human MCPs (17, 18) were thawed and transfected with siRNA (5 nM final concentration) using Lipofectamine RNAiMAX transfection reagent (Invitrogen). Approximately 20 000 cells were plated in each well of a 384-well plate (Greiner Bio-One) coated with Matrigel Basement Membrane Matrix (Corning). Cells were incubated at 37°C and media refreshed every second day. Cell-type composition was assessed by immunostaining (see below) 9 days following siRNA transfection. Each experiment contained quadruplicate technical replicates per condition. Each experiment was performed on different batches of MCP clones which were programmed independently.
EdU and TUNEL assay
Mature cardiomyocytes (day 25 of differentiation from MCPs) were seeded in matrigel-coated plates at a density of 5000 cells per well, whereas foreskin primary fibroblasts (American Type Culture Collection, CCD-1079SK) were seeded in gelatin-coated plates with a density of 500 cells per well. Cells were transfected with siRNA (5 nM) using lipofectamine RNAiMAX transfection reagent (Invitrogen). Cell proliferation was assessed using the Click-it EdU Plus Imaging Kit (Invitrogen), whereby cardiomyocytes were incubated with EdU 24 h prior to fixation, whereas fibroblasts were incubated with EdU for 2 h before final fixation. Cell death was assessed using a fluorescein-based in situ cell death detection kit (Roche). Positive control samples were incubated with DNase I for 20 min to induce DNA breaks. Cardiomyocytes and fibroblasts were fixed 3 days after siRNA transfection following which, cardiomyocyte cultures were stained with ACTN1, whereas fibroblast cultures were stained with TAGLN antibodies to identify cell type. DAPI was used to identify cell nuclei and quantify total cell number (see staining protocol below).
Cell culture immunostaining and imaging
Cells were fixed with warmed 4% paraformaldehyde solution without agitation for 30 min., followed by an additional 30 min Incubation with agitation. Following 3 washes with phosphate buffered saline (PBS), blocking solution was added to cells for 30 min (10% horse serum, 2.5% triton x-100, 10% gelatin). Cultures were incubated with the following primary antibodies for 1 h at room temperature (RT): ACTN1 (Sigma, A7811), TAGLN (Abcam, Ab14106), CDH5 (R&D Systems, AF938). Cells were washed with PBS 3 times and then incubated with Alexa-conjugated secondary antibodies (Life Technologies) including DAPI for 1 h at RT. All antibodies were diluted in blocking solution. The cells were washed three times with PBS and then imaged using a high-throughput microscope (ImageXpress, Molecular Devices). Fluorescence was quantified using custom MetaXpress software (Molecular Devices), whereby total cell number (DAPI) and percentage of ACTN1, TAGLN and CDH5 positive cells in each well were quantified (17, 18). For cell-cycle staging and analysis, DNA content was quantified by measuring total integrated intensity of DAPI staining (n = 4). Cardiomyocytes in the different cell cycle phases were identified by applying selected cutoffs based on the DNA content profiles using imaging analysis software MetaXpress (Molecular Devices).
RNA isolation and qRT-PCR
Human MCPs from 4 wells were pooled at day 2, 5 or 9 following siRNA transfection. A total of 3 replicates for each condition were collected. Total RNA was extracted using TRIzol (Invitrogen) and RNA (500 ng) was reverse transcribed to cDNA using QuantiTect Reverse Transcription Kit (Qiagen) and subject to qRT-PCR using the FastStart Essential DNA Green Master reagents (Roche) and LightCycler 96 Instrument (LC96, Roche). The data were analyzed using the ΔΔCt method using Hypoxanthine guanine phosphoribosyl transferase (Hprt-1) as a normalization control. Primer sequences are listed in Supplementary Material, Table 14.
Drosophila strains
A heart-specific GAL4 driver (Hand4.2-GAL4) was used to drive RNAi constructs in the Drosophila heart, including all cardioblasts/cardiomyocytes, pericardial cells and wing hearts throughout development starting at post-mitotic, mid-embryonic stages through adulthood (25, 52, 53). To temporally regulate Hand4.2-GAL4 activation of UAS-constructs, we combined two copies of a temperature-sensitive Tubulin-GAL80ts construct (HTT; Hand4.2-GAL4, Tubulin-GAL80ts, Tubulin-GAL80ts) (54). At 18°C, the presence of GAL-80 prevents induction of RNAi expression by GAL4. A shift to 29°C destabilizes GAL-80 allowing GAL-4 to induce RNAi expression. TinD-GAL4; midline-GFP driver was used to drive constructs in the embryonic heart (dorsal mesoderm-containing cardiac progenitor cell; 26, 27). Mef2-GAL4 was used to express RNAi in all muscle tissue (55). Drosophila GD and KK RNAi collection lines were obtained through the Vienna Drosophila Resource Center (56), TRiP RNAi lines generated by the Transgenic RNAi Project were obtained from the Bloomington Drosophila Stock Center and NIG-FLY stocks were obtained from the National Institute of Genetics. For specific transgenic line numbers, see Supplementary Material, Table 10.
Semiautomatic Optical Heartbeat Analysis to assess heart function
Assessment of Drosophila heart function and structure is as previously described (22). Briefly, semi-intact fly heart preparations of 1-week-old adult flies were prepared by exposing the beating heart within the abdomen. Hearts were kept oxygenated using artificial hemolymph and placed on an Olympus BX61WI microscope while being filmed through a 10x water immersion lens with a high-speed digital camera (Hamamatsu Photonics C9300 digital camera) using HCI image capture software (Hamamatsu). High-speed movies were analyzed using the semi-automated optical heartbeat analysis (SOHA) software (22). Parameters measured include heart period, diastolic interval, systolic interval (SI), arrhythmicity index (AI), DD, SD and FS. FS, a measure of contractility, is calculated using the following equation FS = (DD – SD)/DD. AI is calculated as the HP standard deviation normalized to the median HP (22, 57).
Immunostaining of adult Drosophila hearts
One-week adult fly hearts were dissected, treated with 10 mM EGTA in PBS + Triton (PBT; 0.03% Triton X-100) and then fixed with 4% PFA for 20 min. Following PBT washes, hearts were stained with Alexa Fluor 594 phalloidin (1300, Life Technologies) overnight in 4°C. Hearts were washed with PBT 3X followed by PBS 1X and mounted using ProLong Gold Mountant with DAPI (Life Technologies). Immunostained preparations were visualized with an Imager Z1 equipped with an Apotome (Carl Zeiss), Hamamatsu Orca Flash4.0 camera and Zen imaging software (Carl Zeiss).
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, La Jolla, USA). Data are presented as the mean ± SEM. Human MCP, cardiomyocyte and fibroblast data were analyzed using a one-way ANOVA analysis followed by a Dunnett’s multiple comparison post-hoc test. A two-way ANOVA analysis was used to analyze gene-expression data followed by Tukey multiple comparison post-hoc test. A student’s unpaired t-test was used to analyze Drosophila heart function data.
Conflict of Interest Statement. None declared.
Funding
This work was supported by National Institutes of Health (NRSA F32 HL131425 to A.M.S. and R01 HL054732 to R.B.). This work was also supported by a grant from the Wanek Foundation at Mayo Clinic in Rochester, M.N., to J.L.T., T.J.N., T.M.O., R.B. and A.R.C.
Supplementary Material
References
- 1. Dolk H., Loane M., Garne E. and European Surveillance of Congenital Anomalies Working, G (2011) Congenital heart defects in Europe: prevalence and perinatal mortality, 2000 to 2005. Circulation, 123, 841–849. [DOI] [PubMed] [Google Scholar]
- 2. Acuna-Hidalgo R., Veltman J.A. and Hoischen A. (2016) New insights into the generation and role of de novo mutations in health and disease. Genome Biol., 17, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khoshnood B., de Vigan C., Vodovar V., Goujard J., Lhomme A., Bonnet D. and Goffinet F. (2006) Trends in antenatal diagnosis, pregnancy termination and perinatal mortality in infants with congenital heart disease: evaluation in the general population of Paris 1983-2000. J. Gynecol. Obstet. Biol. Reprod. (Paris), 35, 455–464. [DOI] [PubMed] [Google Scholar]
- 4. Gilboa S.M., Salemi J.L., Nembhard W.N., Fixler D.E. and Correa A. (2010) Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Circulation, 122, 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khairy P., Ionescu-Ittu R., Mackie A.S., Abrahamowicz M., Pilote L. and Marelli A.J. (2010) Changing mortality in congenital heart disease. J. Am. Coll. Cardiol., 56, 1149–1157. [DOI] [PubMed] [Google Scholar]
- 6. Hartman R.J., Rasmussen S.A., Botto L.D., Riehle-Colarusso T., Martin C.L., Cragan J.D., Shin M. and Correa A. (2011) The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr. Cardiol., 32, 1147–1157. [DOI] [PubMed] [Google Scholar]
- 7. Soemedi R., Wilson I.J., Bentham J., Darlay R., Topf A., Zelenika D., Cosgrove C., Setchfield K., Thornborough C., Granados-Riveron J. et al. (2012) Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet., 91, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaidi S. and Brueckner M. (2017) Genetics and genomics of congenital heart disease. Circ. Res., 120, 923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monroy-Munoz I.E., Perez-Hernandez N., Vargas-Alarcon G., Ortiz-San Juan G., Buendia-Hernandez A., Calderon-Colmenero J., Ramirez-Marroquin S., Cervantes-Salazar J.L., Curi-Curi P., Martinez-Rodriguez N. et al. (2013) Changing the paradigm of congenital heart disease: from the anatomy to the molecular etiology. Gac. Med. Mex., 149, 212–219. [PubMed] [Google Scholar]
- 10. Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J. et al. (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet., 86, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andersen T.A., Troelsen Kde L. and Larsen L.A. (2014) Of mice and men: molecular genetics of congenital heart disease. Cell. Mol. Life Sci., 71, 1327–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez-Revenga L., Mila M., Rosenberg C., Lamb A. and Lee C. (2007) Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet. Med., 9, 600–606. [DOI] [PubMed] [Google Scholar]
- 13. Glessner J.T., Bick A.G., Ito K., Homsy J., Rodriguez-Murillo L., Fromer M., Mazaika E., Vardarajan B., Italia M., Leipzig J. et al. (2014) Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res., 115, 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warburton D., Ronemus M., Kline J., Jobanputra V., Williams I., Anyane-Yeboa K., Chung W., Yu L., Wong N., Awad D. et al. (2014) The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet., 133, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomita-Mitchell A., Mahnke D.K., Struble C.A., Tuffnell M.E., Stamm K.D., Hidestrand M., Harris S.E., Goetsch M.A., Simpson P.M., Bick D.P. et al. (2012) Human gene copy number spectra analysis in congenital heart malformations. Physiol. Genomics, 44, 518–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lalani S.R., Shaw C., Wang X., Patel A., Patterson L.W., Kolodziejska K., Szafranski P., Ou Z., Tian Q., Kang S.H. et al. (2013) Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur. J. Hum. Genet., 21, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunningham T.J., Yu M.S., McKeithan W.L., Spiering S., Carrette F., Huang C.T., Bushway P.J., Tierney M., Albini S., Giacca M. et al. (2017) Id genes are essential for early heart formation. Genes Dev., 31, 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu M.S., Spiering S. and Colas A.R. (2018) Generation of first heart field-like cardiac progenitors and ventricular-like Cardiomyocytes from human pluripotent stem cells. J. Vis. Exp., 136. doi: 10.3791/57688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cripps R.M. and Olson E.N. (2002) Control of cardiac development by an evolutionarily conserved transcriptional network. Dev. Biol., 246, 14–28. [DOI] [PubMed] [Google Scholar]
- 20. Frasch M. (2016) Genome-wide approaches to Drosophila heart development. J. Cardiovasc. Dev. Dis., 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bodmer R. and Frasch M. (2010), Development and Aging of the Drosophila Heart. In Rosenthal N. and Harvey R.P (ed.)Heart Development and Regulation. Elsevier Inc. Vol. 1 pp. 47–86. [Google Scholar]
- 22. Fink M., Callol-Massot C., Chu A., Ruiz-Lozano P., Izpisua Belmonte J.C., Giles W., Bodmer R. and Ocorr K. (2009) A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques, 46, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ocorr K., Vogler G. and Bodmer R. (2014) Methods to assess Drosophila heart development, function and aging. Methods, 68, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wolf M.J. and Rockman H.A. (2008) Drosophila melanogaster as a model system for genetics of postnatal cardiac function. Drug Discov. Today Dis. Models, 5, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han Z., Yi P., Li X. and Olson E.N. (2006) Hand, an evolutionarily conserved bHLH transcription factor required for Drosophila cardiogenesis and hematopoiesis. Development, 133, 1175–1182. [DOI] [PubMed] [Google Scholar]
- 26. Reim I. and Frasch M. (2005) The Dorsocross T-box genes are key components of the regulatory network controlling early cardiogenesis in Drosophila. Development, 132, 4911–4925. [DOI] [PubMed] [Google Scholar]
- 27. Yin Z., Xu X.L. and Frasch M. (1997) Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development, 124, 4971–4982. [DOI] [PubMed] [Google Scholar]
- 28. Guida M.C., Birse R.T., Dall'Agnese A., Toto P.C., Diop S.B., Mai A., Adams P.D., Puri P.L. and Bodmer R. (2019) Intergenerational inheritance of high fat diet-induced cardiac lipotoxicity in Drosophila. Nat. Commun., 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn E.Y., DeKelver R.C., Lo M.C., Nguyen T.A., Matsuura S., Boyapati A., Pandit S., Fu X.D. and Zhang D.E. (2011) SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol. Cell, 42, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu X., Ng H.H. and Bubulya P.A. (2014) The role of SON in splicing, development, and disease. Wiley Interdiscip. Rev. RNA, 5, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinzii C.M., Garone C., Emmanuele V., Tadesse S., Krishna S., Dorado B. and Hirano M. (2013) Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J., 27, 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z., Montcouquiol M., Calderon R., Jenkins N.A., Copeland N.G., Kelley M.W. and Noben-Trauth K. (2008) Jxc1/Sobp, encoding a nuclear zinc finger protein, is critical for cochlear growth, cell fate, and patterning of the organ of corti. J. Neurosci., 28, 6633–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Casad M.E., Abraham D., Kim I.M., Frangakis S., Dong B., Lin N., Wolf M.J. and Rockman H.A. (2011) Cardiomyopathy is associated with ribosomal protein gene haplo-insufficiency in Drosophila melanogaster. Genetics, 189, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin S.C., Homsy J., Zaidi S., Lu Q., Morton S., DePalma S.R., Zeng X., Qi H., Chang W., Sierant M.C. et al. (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet., 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Derwinska K., Bartnik M., Wisniowiecka-Kowalnik B., Jagla M., Rudzinski A., Pietrzyk J.J., Kawalec W., Ziolkowska L., Kutkowska-Kazmierczak A., Gambin T. et al. (2012) Assessment of the role of copy-number variants in 150 patients with congenital heart defects. Med. Wieku. Rozwoj., 16, 175–182. [PubMed] [Google Scholar]
- 36. Hitz M.P., Lemieux-Perreault L.P., Marshall C., Feroz-Zada Y., Davies R., Yang S.W., Lionel A.C., D'Amours G., Lemyre E., Cullum R. et al. (2012) Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet., 8, e1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKeithan W.L., Savchenko A., Yu M.S., Cerignoli F., Bruyneel A.A.N., Price J.H., Colas A.R., Miller E.W., Cashman J.R. and Mercola M. (2017) An automated platform for assessment of congenital and drug-induced arrhythmia with hiPSC-derived Cardiomyocytes. Front. Physiol., 8, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Celniker S.E., Dillon L.A., Gerstein M.B., Gunsalus K.C., Henikoff S., Karpen G.H., Kellis M., Lai E.C., Lieb J.D., MacAlpine D.M. et al. (2009) Unlocking the secrets of the genome. Nature, 459, 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wynn S.L., Fisher R.A., Pagel C., Price M., Liu Q.Y., Khan I.M., Zammit P., Dadrah K., Mazrani W., Kessling A. et al. (2000) Organization and conservation of the GART/SON/DONSON locus in mouse and human genomes. Genomics, 68, 57–62. [DOI] [PubMed] [Google Scholar]
- 40. Deutschbauer A.M., Jaramillo D.F., Proctor M., Kumm J., Hillenmeyer M.E., Davis R.W., Nislow C. and Giaever G. (2005) Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics, 169, 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ivanyi B., Racz G.Z., Gal P., Brinyiczki K., Bodi I., Kalmar T., Maroti Z. and Bereczki C. (2018) Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr. Nephrol., 33, 439–446. [DOI] [PubMed] [Google Scholar]
- 42. Sharma A., Markey M., Torres-Munoz K., Varia S., Kadakia M., Bubulya A. and Bubulya P.A. (2011) Son maintains accurate splicing for a subset of human pre-mRNAs. J. Cell Sci., 124, 4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chia N.Y., Chan Y.S., Feng B., Lu X., Orlov Y.L., Moreau D., Kumar P., Yang L., Jiang J., Lau M.S. et al. (2010) A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature, 468, 316–320. [DOI] [PubMed] [Google Scholar]
- 44. Lu X., Goke J., Sachs F., Jacques P.E., Liang H., Feng B., Bourque G., Bubulya P.A. and Ng H.H. (2013) SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat. Cell Biol., 15, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim J.H., Shinde D.N., Reijnders M.R.F., Hauser N.S., Belmonte R.L., Wilson G.R., Bosch D.G.M., Bubulya P.A., Shashi V., Petrovski S. et al. (2016) De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet., 99, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kondrashov N., Pusic A., Stumpf C.R., Shimizu K., Hsieh A.C., Ishijima J., Shiroishi T. and Barna M. (2011) Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell, 145, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi Z., Fujii K., Kovary K.M., Genuth N.R., Rost H.L., Teruel M.N. and Barna M. (2017) Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell, 67, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Komili S., Farny N.G., Roth F.P. and Silver P.A. (2007) Functional specificity among ribosomal proteins regulates gene expression. Cell, 131, 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Digilio M.C., Pugnaloni F., De Luca A., Calcagni G., Baban A., Dentici M.L., Versacci P., Dallapiccola B., Tartaglia M. and Marino B. (2019) Atrioventricular canal defect and genetic syndromes: the unifying role of sonic hedgehog. Clin. Genet., 95, 268–276. [DOI] [PubMed] [Google Scholar]
- 50. Boultwood J., Pellagatti A. and Wainscoat J.S. (2012) Haploinsufficiency of ribosomal proteins and p53 activation in anemia: diamond-Blackfan anemia and the 5q-syndrome. Adv. Biol. Regul., 52, 196–203. [DOI] [PubMed] [Google Scholar]
- 51. Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M. and Carter N.P. (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am. J. Hum. Genet., 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hallier B., Hoffmann J., Roeder T., Togel M., Meyer H. and Paululat A. (2015) The bHLH transcription factor hand regulates the expression of genes critical to heart and muscle function in Drosophila melanogaster. PLoS One, 10, e0134204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Togel M., Meyer H., Lehmacher C., Heinisch J.J., Pass G. and Paululat A. (2013) The bHLH transcription factor hand is required for proper wing heart formation in Drosophila. Dev. Biol., 381, 446–459. [DOI] [PubMed] [Google Scholar]
- 54. McGuire S.E., Le P.T., Osborn A.J., Matsumoto K. and Davis R.L. (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science, 302, 1765–1768. [DOI] [PubMed] [Google Scholar]
- 55. Ranganayakulu G., Schulz R.A. and Olson E.N. (1996) Wingless signaling induces nautilus expression in the ventral mesoderm of the Drosophila embryo. Dev. Biol., 176, 143–148. [DOI] [PubMed] [Google Scholar]
- 56. Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature, 448, 151–156. [DOI] [PubMed] [Google Scholar]
- 57. Ocorr K., Fink M., Cammarato A., Bernstein S. and Bodmer R. (2009) Semi-automated optical heartbeat analysis of small hearts. J. Vis. Exp., 31. doi: 10.3791/1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.